

Abstract
Ferroptosis is an iron-dependent form of regulated nonapoptotic cell death, which contributes to damage in models of acute kidney injury (AKI). Heme oxygenase-1 (HO-1) is a cytoprotective enzyme induced in response to cellular stress, and is protective against AKI because of its antiapoptotic and anti-inflammatory properties. However, the role of HO-1 in regulating ferroptosis is unclear. The purpose of this study was to elucidate the role of HO-1 in regulating ferroptotic cell death in renal proximal tubule cells (PTCs). Immortalized PTCs obtained from HO-1+/+ and HO-1−/− mice were treated with erastin or RSL3, ferroptosis inducers, in the presence or absence of antioxidants, an iron source, or an iron chelator. Cells were assessed for changes in morphology and metabolic activity as an indicator of cell viability. Treatment of HO-1+/+ PTCs with erastin resulted in a time- and dose-dependent increase in HO-1 gene expression and protein levels compared with vehicle-treated controls. HO-1−/− cells showed increased dose-dependent erastin- or RSL3-induced cell death in comparison to HO-1+/+ PTCs. Iron supplementation with ferric ammonium citrate in erastin-treated cells decreased cell viability further in HO-1−/− PTCs compared with HO-1+/+ cells. Cotreatment with ferrostatin-1 (ferroptosis inhibitor), deferoxamine (iron chelator), or N-acetyl-l-cysteine (glutathione replenisher) significantly increased cell viability and attenuated erastin-induced ferroptosis in both HO-1+/+ and HO-1−/− PTCs. These results demonstrate an important antiferroptotic role of HO-1 in renal epithelial cells.
Keywords: iron, erastin, ferrostatin-1, acute kidney injury, proximal tubule
acute kidney injury (AKI) is a major complication in critically ill patients with incidence ranging from 3 to 22% (37, 46). Unresolved kidney injury can progress to chronic kidney disease and/or end-stage renal disease. The proximal tubule of the nephron, particularly the S3 segment, is highly sensitive to injury and consequent cell death following both ischemic and nephrotoxic insults (45). Therefore, there is considerable interest in the mechanisms underlying cell death in proximal tubule cells (PTCs) to identify specific molecular targets for amelioration or prevention of AKI.
Ferroptosis is one of many recently identified mechanisms of regulated cell death. It is nonapoptotic and associated with accumulation of lipid reactive oxygen species (ROS) due to increased lipid peroxidation (42). Ferroptosis was discovered in cancer cell models in which treatment with erastin in vitro results in increased cell death characterized by shrunken mitochondria and increased membrane density (8). The features of ferroptosis are morphologically, biochemically, and genetically distinct from known mechanisms of cell death such as apoptosis and necrosis, as well as newly identified mechanisms such as pyroptosis and necroptosis (5, 8, 52).
Ferroptosis is resistant to inhibition by small molecules that target conventional cell death pathways (8, 49, 53). It is triggered under conditions of glutathione depletion or inactivity of the glutathione peroxidase 4 (GPX4) enzyme (42, 43), indicating an important relationship with cellular redox status. In contrast, treatment with either an iron chelator or inhibitor of lipid peroxidation attenuates ferroptosis (8, 10). These findings indicate that ferroptosis is a process dependent on both iron and ROS. After oxidative stress, cells respond through inherent adaptive defense mechanisms to restore healthy cellular redox homeostasis. One such mechanism involves the heme oxygenase (HO) enzyme system, particularly its inducible isoform, HO-1, a cytoprotective, anti-inflammatory, and antioxidant enzyme that is robustly induced in renal proximal tubules after AKI. The enzymatic reaction of HO-1 is the rate-limiting step in the breakdown of heme into equimolar quantities of iron, biliverdin, and carbon monoxide (24). Therefore, it is a potential source of intracellular iron on which ferroptosis is dependent. Although expression of HO-1 is upregulated during ferroptosis in cancer cells (22), it is not clear whether HO-1 in this context potentiates ferroptosis or is induced as a protective response. Furthermore, the role of HO-1 in ferroptosis in the kidney has not yet been addressed.
The purpose of this study was to determine the potential role of HO-1 in the regulation of ferroptotic cell death in renal PTCs by using immortalized mouse PTCs derived from wild-type and HO-1-deficient mice. We show that the absence of HO-1 enhances ferroptosis, suggesting that the free iron generated by HO-1 does not facilitate ferroptosis per se and that HO-1 has an anti-ferroptotic effect in renal epithelial cells.
METHODS
Cell culture.
Primary proximal tubular cells were isolated from HO-1+/+ or HO-1−/− mice (8 wk old) characterized as described previously (4, 20, 58), and subsequently immortalized by transfection with an SV40 plasmid. The immortalized mouse PTCs were cultured in renal epithelial cell growth basal medium 2 (PromoCell, Heidelberg, Germany) supplemented with recombinant human epidermal growth factor (10 ng/ml), recombinant human insulin (5 μg/ml), epinephrine (0.5 μg/ml), hydrocortisone (36 ng/ml), human holo-transferrin (5 μg/ml), triiodo-l-thyronine (4 pg/ml), and 0.5% fetal calf serum. In addition, 1% antibiotic-antimycotic (GIBCO Life Technologies, Grand Island, NY) was added to prevent bacterial and fungal contamination.
For time course and dose response studies, HO-1+/+ cells were cultured for 1 day and then treated with either 0.1 or 1 μM erastin (Sigma-Aldrich, St. Louis, MO), or with different doses of RSL3 (Selleckchem, Houston, TX), both potent ferroptosis inducers. Treated cells were harvested at different time points over a 24-h time frame. Subsequent studies in both HO-1+/+ and HO-1−/− PTCs entailed cell treatment for 16 h with erastin in the presence or absence of N-acetyl-l-cysteine (NAC, glutathione source; Sigma-Aldrich), ferric ammonium citrate (FAC, iron source; Sigma-Aldrich), deferroxamine mesylate (DFO, iron chelator; Sigma-Aldrich), or ferrostatin-1 (Fer-1, ferroptosis inhibitor; Sigma-Aldrich).
Phase contrast microscopy.
Images of PTCs were captured using a Leica DM I600B microscope with the Leica Application Suite V4.2 (Leica Microsystems, Buffalo Grove, IL) and analyzed using ImagePro Plus 5.1 software (Media Cybernetics, Rockville, MD).
Real-time PCR analysis.
RNA was isolated from cells by using TRIzol Reagent (Ambion, Life Technologies) according to the manufacturer's protocol. One microgram of RNA was converted to cDNA by using the Quantitect cDNA Synthesis kit (Qiagen, Germantown, MD) according to the manufacturer’s protocol. Quantitative real-time PCR was performed with PowerUp SYBR Green Master Mix (ThermoFisher Scientific, Grand Island, NY) and primers for mouse HO-1 and GAPDH. Reactions were performed in triplicate, and specificity was monitored using melting curve analysis after cycling. Primers (Invitrogen, Grand Island, NY) used were as follows (5′-3′): mouse HO-1 forward 5′-GGTGATGGCTTCCTTGTACC-3′ and reverse 5′-AGTGAGGCCCATACCAGAAG-3′; and GAPDH forward 5′-ATCATCCCTGCATCCACT-3′ and reverse 5′-ATCCACGACGGACACATT-3′. Relative mRNA expression was quantified using the ΔΔ Ct method (1, 28), and GAPDH was used as an internal control. Results were expressed as fold change over vehicle-treated cells.
Western blot analysis.
Immunoblot analyses were performed as described previously (3). Briefly, cell cultures were lysed in radioimmunoprecipitation assay buffer (50 mM Tris/HCl, 1% vol/vol Nonidet P-40, 0.25% wt/vol deoxycholic acid, 150 mM NaCl, 1 mM EGTA, 1 mM sodium orthovanadate, and 1 mM sodium fluoride) with protease inhibitors (no. P2714, Sigma-Aldrich) and phosphatase inhibitors (no. B15001-A, Biotool.com, Houston, TX). Protein concentrations were quantified using the bicinchoninic acid protein assay (Thermo Fisher Scientific, Rockford, IL). Cell lysates were subjected to SDS-PAGE on a 4–20% acrylamide gradient. The resulting protein bands were transferred onto methanol-activated PVDF transfer membranes (Immobilon P). Membranes were blocked for 1 h in 5% wt/vol dehydrated milk and 0.1% vol/vol Tween-20 in PBS, then incubated with polyclonal rabbit anti-HO-1 antibody (1:5,000 dilution; SPA-894, Enzo LifeSciences) or anti-cleaved caspase-3 antibody (1:2,000 dilution; no. 9664L, Cell Signaling), followed by a peroxidase-conjugated goat anti-rabbit IgG antibody (1:5,000; Jackson ImmunoResearch Laboratories). Horseradish peroxidase activity was detected using enhanced chemiluminescence. The membrane was stripped and probed with mouse anti-GAPDH antibody (1:10,000 dilution; MAB374, EMD Millipore, Darmstadt, Germany). Densitometric analysis was performed using the Image-J software package (40) and normalized using GAPDH as a loading control.
HO enzyme activity measurement.
HO enzyme activity was measured by bilirubin generation as described previously (4). HO-1+/+ PTCs were treated with either vehicle or 1 µM erastin for 16 h after which cell lysates were obtained. Lysates were centrifuged at 18,800 g at 4°C for 10 min. The supernatant was then incubated with rat liver cytosol (2 mg protein—a source of biliverdin reductase), hemin (20 μM), glucose-6-phosphate (2 mM), glucose-6-phosphate dehydrogenase (0.2 μM), and NADPH (0.8 mM) for 1 h at 37°C in the dark. Bilirubin was extracted with chloroform, and the optical density of the resulting solution was measured at 464 and 530 nm. Enzyme activity was expressed as picomoles of bilirubin formed per hour per mg of protein.
Cell viability analysis.
Cell viability was evaluated with an AlamarBlue Cell Viability Assay Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. In brief, PTCs were plated onto collagen-coated 96-well plates, and after exposure to treatment conditions, 10 μl of AlamarBlue reagent was added to each well and incubated at 37°C in 5% CO2 for 4 h. Absorbance was measured at 570 and 600 nm using an EL800 universal microplate reader (Biotek Instruments). Viability was assessed by calculating the decrease in chemical reduction of the AlamarBlue reagent between treatment and vehicle control groups. Reduction of AlamarBlue reflects mitochondrial reductive capacity and therefore cellular metabolic health.
Statistical analysis.
At least three independent experiments were conducted for each experimental condition. Data are expressed as means ± SE. To compare means of two groups, the unpaired Student’s t-test was used. Analysis of variance was used for comparison of means among more than two groups with the appropriate post hoc analyses.
RESULTS
Erastin increases HO-1 gene expression and protein levels in PTCs.
To investigate the effects of erastin-induced ferroptosis on HO-1 gene expression and protein levels, immortalized HO-1+/+ PTCs were treated with either 0.1 or 1 µM erastin for 4, 8, 16, or 24 h. We observed a dose- and time-dependent increase in HO-1 mRNA levels at both concentrations of erastin, with the greatest HO-1 induction at the 16-h treatment time point (Fig. 1A). Increase in HO-1 protein levels correlated with mRNA expression, as the greatest induction in HO-1 was observed 16 h posttreatment (1 µM erastin) (Fig. 1, B and C). Furthermore, we observed a twofold increase in HO activity when measured 16 h after treatment with erastin (control vs. 1 µM erastin; 162.3 ± 29.5 vs. 339.5 ± 80.5 pg bilirubin/mg protein/h; means ± SE, n = 6–7/treatment).
Fig. 1.

Erastin increases heme oxygenase-1 (HO-1) expression in renal proximal tubular epithelial cells (PTCs). A: HO-1 mRNA expression in cultured immortalized HO-1+/+ PTCs after 0.1 or 1 μM erastin treatment at the indicated time points. Representative Western blot (B) and densitometry showing protein levels (C) of HO-1 in HO-1+/+ PTCs treated with erastin over time. Representative Western blot (D) and densitometry showing protein levels (E) of HO-1 in HO-1+/+ PTCs treated with RSL3 over time. Data shown represent the means ± SE of three independent experiments performed each time in triplicate; *P < 0.05 and **P < 0.01 compared with lower concentration at each time point.
RSL3 treatment increases HO-1 protein levels in a dose-dependent manner.
To assess HO-1 response to ferroptosis induction via GPX4 inhibition, we also treated immortalized HO-1+/+ PTCs with either 0.1 and 1 µM RSL3 and harvested cells at 4, 8, 16, and 24 h after treatment. We observed significant induction of HO-1 protein levels after 8 h of RSL3 treatment, and these levels began to revert to baseline by 24 h (Fig. 1, D and E).
Erastin treatment reduces cell viability in HO-1+/+ PTCs in the absence of apoptosis.
We validated the induction of ferroptosis in HO-1-proficient PTCs treated with erastin over time and observed that cell death was greater with increased duration of erastin treatment. Treatment of HO-1+/+ PTCs with 0.1 µM erastin did not induce detectable cellular toxicity following 8 h of treatment but resulted in a reduction in cell viability at 16 h (Fig. 2, A and B). However, cell viability was reduced at both 8 and 16 h after treatment with 1 µM erastin. Notably, induction of HO-1 gene expression was observed as early as 4 h after erastin treatment (Fig. 1A), a time point at which no discernible cytotoxicity was observed by phase contrast microscopy. To examine whether the reduction in viability observed with erastin treatment in HO-1+/+ PTCs is due to apoptosis, we assessed cleaved caspase 3 levels after 16 h of erastin (0.1, 1, or 10 µM) treatment. Cleaved caspase 3 expression in erastin-treated HO-1+/+ PTCs was undetectable by Western blotting (Fig. 2C), indicating that apoptosis was not a predominant mechanism for the decreased viability observed. HO-1-deficient PTCs treated with cisplatin (50 μM) for 16 h were used as a positive control in these experiments. These results also indicate that erastin-induced HO-1 expression is unique and not a generalized response to cells dying from an inducer of apoptosis per se.
Fig. 2.
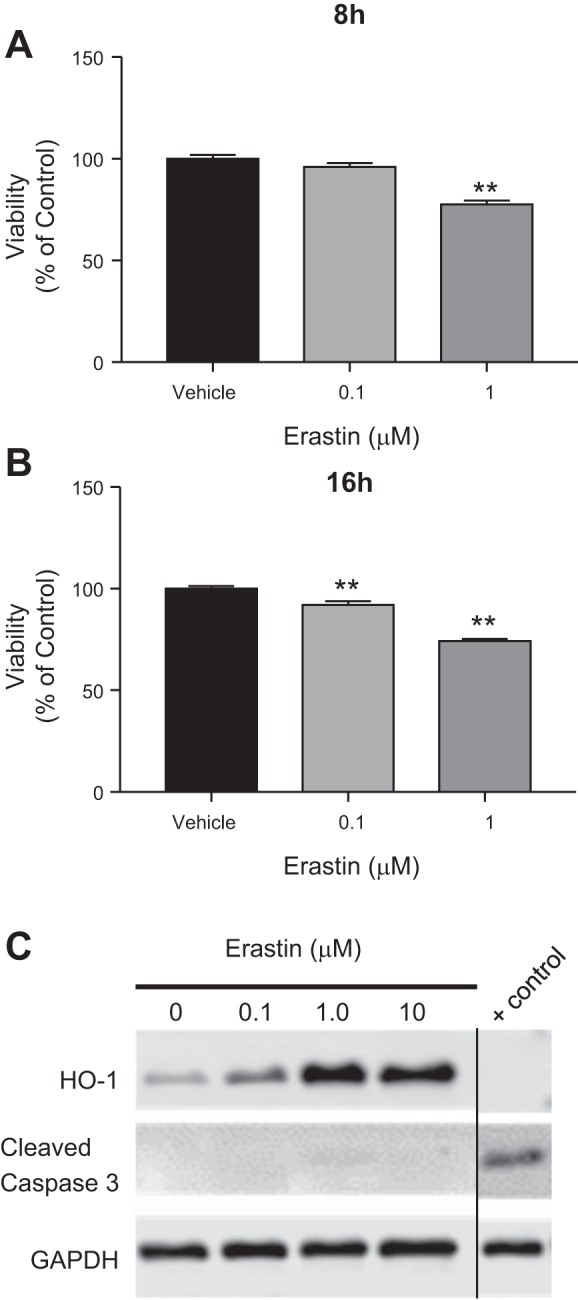
Erastin treatment reduces cell viability in a time- and dose-dependent manner. Cell viability in HO-1+/+ PTCs after treatment with either 0.1 or 1 μM erastin for 8 h (A) or 16 h (B). C: representative Western blot showing absence of cleaved caspase 3 in HO-1+/+ PTCs treated with the indicated doses of erastin for 16 h. HO-1−/− PTCs treated with 50 μM cisplatin (a known inducer of apoptosis) for 16 h were used as a positive control. All lanes are from the same blot. “Control” label indicates where the blot was cropped to include relevant lanes. Data are represented as means ± SE from three independent experiments with seven or eight replicates in each experiment; **P < 0.01 compared with vehicle-treated controls.
Similarly, to ascertain the effect of HO-1 on RSL3-induced ferroptosis, we treated both HO-1+/+ and HO-1−/− PTCs with either 0.1 or 1 µM RSL3 and assessed cell viability after 16 h. There was minimal ferroptotic cell death observable after treatment with 0.1 µM RSL3 (results not shown), but significant reduction in cell viability was observed with 1 µM RSL3 in HO-1−/− PTCs compared with wild-type cells. At higher concentrations (using 10 µM RSL3), we observed further increased cell death in HO-1−/− compared with HO-1+/+ PTCs (Fig. 3C). These findings indicate that HO-1-deficient PTCs are more sensitive also to RSL3-induced ferroptosis compared with PTCs proficient in HO-1.
Fig. 3.

Lack of HO-1 increases PTC sensitivity to erastin-induced ferroptosis. A: phase contrast microscopy images of HO-1+/+and HO-1−/− PTCs treated with either 0.1 or 1 μM erastin for 16 h. B: cell viability after treatment of PTCs with 0.1 or 1 μM erastin or 1; *P < 0.05 compared with 0.1 μM erastin-treated HO-1+/+ PTCs; **P < 0.01 compared with 0.1 μM erastin-treated HO-1−/− PTCs; #P < 0.01 compared with erastin-treated HO-1+/+ PTCs. C: cell viability after treatment of PTCs with 10 μM RSL3 for 16 h; *P < 0.01 compared with 1 μM RSL3 treatment; #P < 0.05 compared with RSL3-treated HO-1+/+ PTCs. Data shown represent means ± SE of three independent experiments with four to six replicates in each experiment.
HO-1 deficiency promotes erastin-induced ferroptosis.
To ascertain the role of HO-1 in erastin-induced cell death, we treated HO-1+/+ and HO-1−/− PTCs with either 0.1 or 1 µM erastin for 16 h and examined morphological changes and cell viability. We observed reduction in cell viability in both HO-1+/+ and HO-1−/− PTCs (Fig. 3, A and B). However, the viability observed in HO-1−/− PTCs after 0.1 or 1 µM erastin treatment was significantly reduced compared with HO-1+/+ PTCs. These results suggest the presence of HO-1 may attenuate erastin-induced cell death.
Iron chelation attenuates erastin-induced ferroptosis and reduces HO-1 levels.
To determine if erastin-induced cell death in PTCs is iron dependent, we examined the effect of iron chelation on cell viability of immortalized HO-1+/+ and HO-1−/− PTCs. To ensure adequate iron chelation, cells were pretreated for 4 h with deferroxamine mesylate (DFO), followed by erastin and DFO cotreatment for 16 h. As observed previously, erastin treatment resulted in a dose-dependent increase in cell death in both HO-1+/+ and HO-1−/− PTCs as indicated by phase contrast microscopy (Fig. 4, A and B) and quantitative assessment of cell viability (Fig. 5, A and B). Interestingly, iron chelation led to a significant reduction in cell death following erastin treatment. Furthermore, under these conditions, we examined the expression of HO-1 protein in response to iron chelation in erastin-treated HO-1+/+ PTCs to assess whether iron chelation in erastin-treated cells mitigates HO-1 induction. DFO resulted in reduced HO-1 protein expression in erastin-treated cells (Fig. 5C). These findings indicate that erastin-induced cell death is iron dependent in both HO-1+/+ and HO-1−/− PTCs and that chelating iron while subjecting PTCs to ferroptosis may decrease cellular stress and the need for HO-1 induction.
Fig. 4.

Iron chelation attenuates erastin-induced ferroptosis. Representative phase contrast microscopy images of HO-1+/+ (A) and HO-1−/− (B) PTCs treated with either 0.1 or 1 μM erastin with or without 0.1 mM deferroxamine mesylate (DFO, iron chelator) for 16 h.
Fig. 5.
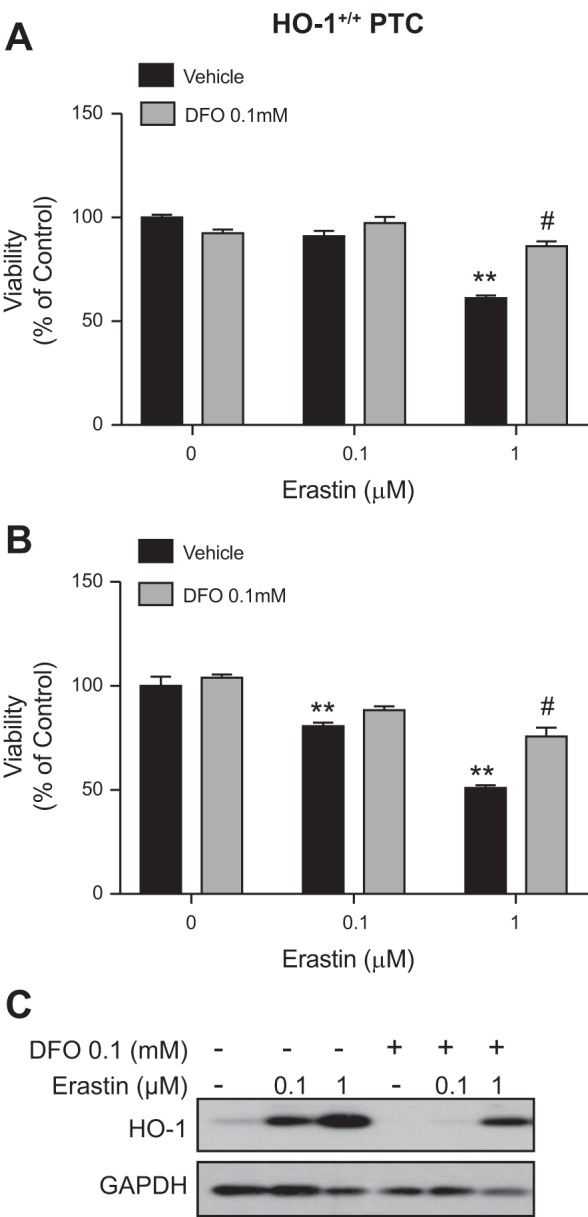
Iron chelation increases cell viability in erastin-treated cells. Cell viability in HO-1+/+ (A) and HO-1−/− (B) PTCs treated with either 0.1 or 1 μM erastin with or without 0.1 mM DFO for 16 h. C: representative Western blot showing HO-1 protein levels in HO-1+/+ PTCs treated with erastin in the presence or absence of DFO for 16 h. Data represented as means ± SE of three independent experiments with six to eight replicates in each experiment; **P < 0.01 compared with vehicle-treated cells; #P < 0.01 compared with 1 μM erastin-treated cells (HO-1+/+ or HO-1−/−).
Iron supplementation and HO-1 deficiency exacerbate erastin-induced ferroptosis.
As ferroptosis is an iron-dependent form of cell death, we examined whether iron supplementation augments erastin-induced ferroptosis in HO-1+/+ and HO-1−/− PTCs. Treatment of cells with 10 µg/ml of FAC (iron source) increased cell death in 1 µM of erastin-treated HO-1+/+ PTCs (Fig. 6A). However, in HO-1−/− PTCs, the addition of FAC alone, even in the absence of erastin treatment, was sufficient to induce cell death (Fig. 6B). In addition, cotreatment of HO-1−/− PTCs with 0.1 or 1 µM erastin and FAC (10 µg/ml) resulted in further reduction in cell viability as observed via phase contrast microscopy (Fig. 6B). We observed significant reduction in viability in HO-1+/+ and HO-1−/− PTCs cotreated with erastin and FAC (10 µg/ml). Importantly, this reduction in viability was greater in FAC and erastin cotreated HO-1−/− PTCs (Fig. 7B), supporting the hypothesis that in HO-1-deficient PTCs, iron supplementation augments ferroptosis and does so at lower concentrations of erastin when compared with HO-1+/+ PTCs. This indicates that HO-1 expression may partially preserve PTC viability during ferroptosis and that HO-1 deficiency increases susceptibility to ferroptosis in PTCs.
Fig. 6.

Iron supplementation exacerbates erastin-induced ferroptosis. Representative phase contrast microscopy images of HO-1+/+ (A) and HO-1−/− (B) PTCs treated with either 0.1 or 1 μM erastin with or without 10 μg/ml ferric ammonium citrate (FAC, iron source) for 16 h.
Fig. 7.
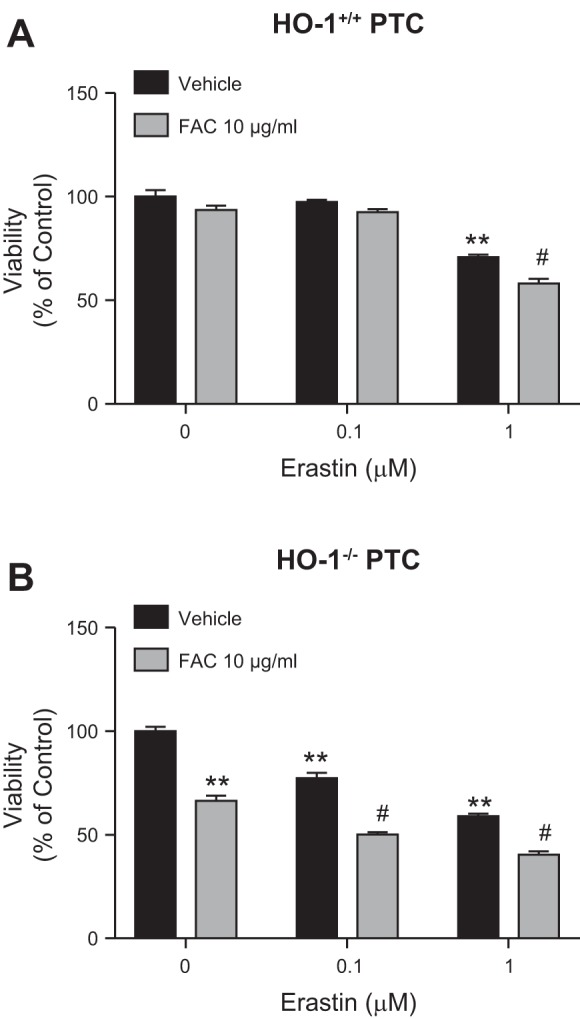
Iron supplementation and/or HO-1 deficiency potentiate erastin-induced ferroptosis. Cell viability in HO-1+/+ (A) and HO-1−/− (B) PTCs treated with either 0.1 or 1 μM erastin with or without 10 μg/ml FAC (ferric ammonium citrate) for 16 h compared with vehicle-treated cells. Data represented as means ± SE of four to six independent experiments with three to eight replicates in each experiment; **P < 0.01 compared with vehicle-treated PTCs; #P < 0.01 compared with 0.1 or 1 μM erastin-treated cells.
Glutathione attenuates erastin-induced ferroptosis and reduces HO-1 levels.
Since erastin treatment induces oxidative stress and subsequent ferroptosis by blocking the system xc- antiporter thus resulting in glutathione depletion (9), we examined whether glutathione replenishment would attenuate erastin-induced ferroptosis in the presence or absence of HO-1. We treated HO-1+/+ and HO-1−/− PTCs with erastin (0.1 or 1 µM) in the presence or absence of either 0.5 or 1 mM NAC (glutathione source). Cotreatment with NAC (0.5 or 1 mM) significantly increased cell viability in both the HO-1+/+ and HO-1−/− PTCs (Figs. 8, A and B, and 9, A and B), thus demonstrating that glutathione depletion and subsequent cell death induced by erastin can be completely rescued by cotreatment with NAC. In addition, we examined the levels of HO-1 in HO-1+/+ PTCs in response to cotreatment with erastin and NAC, and we observed significant reduction in HO-1 protein levels in cells cotreated with both erastin and NAC (Fig. 9C).
Fig. 8.

N-acetyl-l-cysteine (NAC) cotreatment rescues cells from erastin-induced ferroptosis. Representative phase contrast microscopy images of HO-1+/+ (A) and HO-1−/− (B) PTCs treated with either 0.1 or 1 μM erastin with or without 0.1 mM NAC (glutathione reducing agent) for 16 h.
Fig. 9.
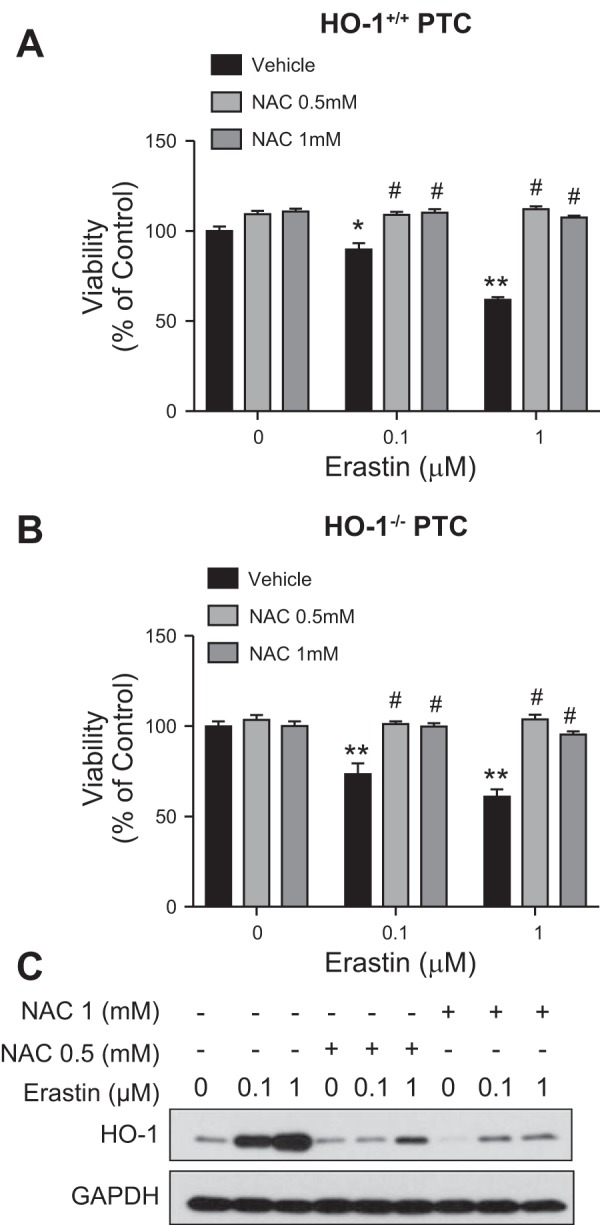
Glutathione supplementation rescues renal epithelial cells from erastin-induced ferroptosis. Cell viability of HO-1+/+ (A) and HO-1−/− (B) PTCs treated with either 0.1 or 1 μM erastin with or without 0.5 or 1 mM NAC for 16 h (magnification ×10). C: representative Western blot showing protein levels of HO-1+/+ PTCs treated with either 0.1 or 1 μM erastin with or without NAC cotreatment. Data represented as means ± SE of three or four independent experiments with four to eight replicates in each experiment; *P < 0.05 and **P < 0.01 compared with vehicle-treated cells; #P < 0.01 compared with erastin-only treatments.
Ferrostatin-1, a ferroptosis inhibitor, attenuates erastin-induced ferroptosis and reduces HO-1 levels.
Ferrostatin-1 was the first specific ferroptosis inhibitor to be developed, and it has been shown to inhibit ferroptosis by reducing lipid peroxidation in a variety of cell types as well as in vivo (29, 35, 41, 56). We treated HO-1+/+ and HO-1−/− PTCs with erastin in the presence or absence of 0.1 or 1 µM ferrostatin-1. We observed that ferrostatin-1 cotreatment resulted in mitigation of cell death in both HO-1+/+ and HO-1−/− PTCs. (Fig. 10, A and B); although, this response was more modest in HO-1−/− PTCs compared with HO-1+/+ (Fig. 11, A and B). Furthermore, from Western blots of cell lysates there appeared to be a reduction in HO-1 protein expression in cells cotreated with erastin and ferrostatin-1 compared with erastin alone (Fig. 11C).
Fig. 10.

Ferrostatin-1, a ferroptosis inhibitor, attenuates erastin-induced ferroptosis. Representative phase contrast microscopy images of HO-1+/+ (A) and HO-1−/− (B) PTCs treated with either 0.1 or 1 μM erastin with or without 0.1 or 1 μM ferrostatin for 16 h.
Fig. 11.
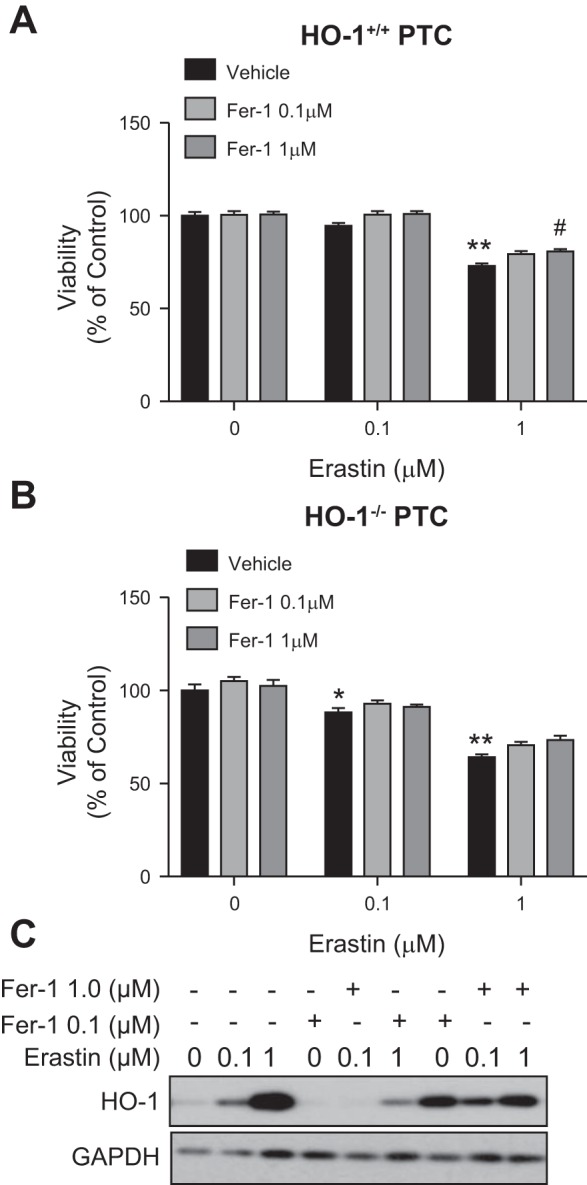
Ferrostatin-1 (Fer-1) attenuates erastin-induced ferroptosis in HO-1+/+ and HO-1−/− PTCs. Cell viability in HO-1+/+ (A) and HO-1−/− (B) PTCs treated with either 0.1 or 1 μM erastin with or without Fer-1 (0.1 or 1 μM) for 16 h. C: representative Western blot showing cells treated with erastin in the presence or absence of Fer-1. Data represented as means ± SE of four to six independent experiments with four to eight replicates in each experiment; *P < 0.05 and **P < 0.01 compared with vehicle-treated cells; #P < 0.01 compared with erastin-only treatments.
DISCUSSION
The results of this study demonstrate that renal PTCs undergo cell death in response to treatment with the ferroptosis inducers erastin or RSL3. Ferroptosis is an iron-dependent process, and erastin treatment appears to induce ferroptosis through oxidative stress and inhibition of the system xc- (cystine/glutamate) antiporter, whereas RSL3 induces ferroptosis via inhibition of GPX4 activity (9, 14, 54). Our results highlight several key findings: 1) both erastin and RSL3 induce HO-1 expression in PTCs; 2) HO-1-deficient PTCs are highly sensitive to erastin- and RSL3-induced ferroptosis; 3) erastin-induced cell death in PTCs is iron dependent—iron supplementation results in enhanced cell death, whereas iron chelation reduces cell death and HO-1 expression; 4) replenishment of glutathione in the presence of erastin inhibits cell death and reduces erastin-induced HO-1 expression; and 5) ferrostatin-1, a ferroptosis inhibitor, attenuates cellular stress and death in PTCs undergoing erastin-induced ferroptosis.
Our initial hypothesis was that HO-1, because of its enzymatic activity, generates iron and therefore has the potential to enhance ferroptosis in PTCs. However, we found that PTCs proficient in HO-1 exhibit protection from ferroptosis. It is possible that elevated intracellular heme levels in HO-1-deficient cells render them increasingly susceptible to ferroptosis. HO-1 is a cytoprotective enzyme that is important for maintaining redox homeostasis and also provides a potent antioxidant defense mechanism in response to cell stress (24, 36) by breaking down toxic heme into carbon monoxide, biliverdin (further reduced by biliverdin reductase to bilirubin), and iron (24, 31, 32, 44). Numerous reports in animal models of AKI indicate that expression of HO-1 is key in protection against kidney injury. For example, in the rhabdomyolysis model of AKI, induction of HO before injury resulted in a significant attenuation of structural damage, prevented kidney failure, and reduced mortality (33). This protective role of HO-1 has also been demonstrated in several animal models of AKI (reviewed in Ref. 31). In addition, recent studies have linked genetic polymorphisms in the human HO-1 gene to the development of AKI in cardiac surgery patients (6, 23). Similarly, differential expression of genes, which modulate cellular iron content and iron homeostasis, has been demonstrated to regulate ferroptosis.
Induction of HO-1 in response to cellular stress is coupled with coexpression of the iron sequestering protein ferritin. Iron bound in the ferritin shell is in a mineralized, nontoxic state which cannot be used by the cell (2). Such a coupled response when HO-1 is induced may underlie the protective effects of HO-1 against ferroptosis, the latter presumably associated with an increase in the free labile cellular iron pool due to cellular oxidant stress. Sun et al. (42) demonstrated increased resistance to ferroptotic cell death in hepatocellular carcinoma cells through Nrf2 expression and activation as well as upregulation of Nrf2 target genes (quinone oxidoreductase 1, heme oxygenase-1, and ferritin heavy chain 1) involved in iron and ROS metabolism. More recently, increased mitochondrial ferritin expression in neuroblastoma cells and in drosophila was shown to significantly inhibit erastin-induced ferroptosis (48). Other proteins which modulate iron metabolism have been reported to regulate ferroptosis. The iron response element binding protein (IREB2) has been demonstrated to contribute to the accumulation of lipid peroxides during erastin-induced ferroptotic death in both HT-1080 and Calu-1 cells (8). Heat shock protein (HSP1) on the other hand decreases intracellular iron concentrations by inhibition of transferrin receptor (Trf1) recycling. Thus carcinoma cells depleted of HSPB1 were shown to be more sensitive to erastin-induced ferroptosis, whereas overexpression of HSPB1 resulted in inhibition of ferroptosis (2). Therefore, further studies are necessary to fully assess the role of iron regulatory proteins in modulating ferroptosis in the context of HO-1 depletion.
Even though the expression of HO-1 has been shown to be upregulated during ferroptosis, the role it plays in this form of regulated cell death has not been clearly elucidated. Kwon et al. (22) previously demonstrated that the presence of HO-1 in freshly isolated lung fibroblasts from HO-1+/+ mice accelerates erastin-induced ferroptosis and that lack of HO-1 attenuates this decrease in cell viability. On the other hand, Sun et al. (42) demonstrated that the induction of HO-1 in erastin-treated hepatocellular carcinoma cells is protective against ferroptosis. Our results demonstrate that HO-1-deficient PTCs are more susceptible to erastin-induced ferroptosis, thus indicating that HO-1 plays a protective role in mitigating cell death in this model. These differing results regarding HO-1 expression observed during ferroptotic cell death may indicate that the role of HO-1 in ferroptosis is context dependent and cell-type dependent.
Previous studies have shown that glutathione depletion is a potent stimulus for HO-1 induction (15, 17, 30, 59), providing a potential mechanism for erastin-induced HO-1 expression. We observed complete attenuation of erastin-induced cell death in both HO-1+/+ and HO-1−/− cells with glutathione supplementation by using NAC. In addition, in cells that were cotreated with NAC, we also observed significant reduction in HO-1 levels. Our findings indicate that regardless of the presence or absence of HO-1, depletion of glutathione by erastin is necessary for its ability to induce ferroptosis. Other investigators have also shown that cellular glutathione plays a key protective role in the suppression of ferroptosis (55). For example, several studies have demonstrated that GPX4 is a key regulator of ferroptotic cell death. GPX4 is a phospholipid hydroperoxidase enzyme, which protects against lipid peroxidation in lipid bilayers of cell membranes (7, 39, 50). Genetic inactivation or pharmacological inhibition of GPX4 promotes ferroptotic cell death in a variety of in vitro models (11, 14, 38, 51). Moreover, genetic deletion of GPX4 in animal models has been demonstrated to cause increased accumulation of oxidized phospholipid products in kidneys, induction of AKI and death in mice (14). Whether HO-1 and GPX4 expression synergistically protect from ferroptosis is an interesting question for further investigation.
We propose that a few explanations are plausible for the role of HO-1 in ferroptosis. First, HO-1 induction is a protective antioxidant response combating ferroptosis. Second, HO-1 promotes ferroptosis via release of its enzymatic product, iron. Third, HO-1 induction is simply associated with the cellular stress caused by lipid ROS and glutathione depletion. More recent studies, however, have demonstrated that ferroptosis is not a nonspecific ROS-induced cell death process, but rather involves a well-defined arachidonate lipoxygenase-mediated lipid peroxidation signature that predominantly oxidizes phosphatidylethanolamine (PE) and phosphatidylinositol 4,5-bisphosphate (PIP2) in the plasma membrane (18, 19, 57). In addition, lipid peroxidation may stimulate the immune system and potentiate a process termed necroinflammation (26). Given the established role of HO-1 in inflammation, it would be interesting to evaluate the involvement of HO-1 in necroinflammation.
In addition to the well-known classical pathways of cell death such as apoptosis and necrosis, more recent studies have identified novel and distinct cell death pathways such as necroptosis, mitochondrial permeability transition-dependent regulated necrosis, parthanatos, and ferroptosis in AKI (13, 14, 21, 25, 27, 29, 41, 47). Our studies demonstrate ferroptosis can drive cell death in renal PTCs, reinforcing the supposition that it is an important mechanism by which PTC death may occur in AKI. Furthermore, HO-1 expression demonstrates an antiferroptotic effect, which may partially explain the aggravated damage proximal tubules experience secondary to AKI in the setting of HO-1 deficiency (16, 34).
GRANTS
This work is supported by an American Heart Association predoctoral fellowship 17PRE33370121 to J. Lever, and the National Institutes of Health Grants T32-DK-7545 and 5K12GM088010 to O. Adedoyin (appointee), K01 DK103931 to S. Bolisetty, R01-DK59600 to A. Agarwal and J. George; and the core resource of the UAB-UCSD O’Brien Center (P30-DK079337) to A. Agarwal.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
O.A., J.G., and A.A. conceived and designed research; O.A., R.B., A.M.T., J.M.L., and S.B. performed experiments; O.A., R.B., S.B., J.G., and A.A. analyzed data; O.A., R.B., J.M.L., J.G., and A.A. interpreted results of experiments; O.A. and R.B. prepared figures; O.A. drafted manuscript; O.A., J.M.L., J.G., and A.A. edited and revised manuscript; O.A., R.B., A.M.T., J.M.L., S.B., J.G., and A.A. approved final version of manuscript.
REFERENCES
- 1.Adedoyin OO, Loftin CD. Microsomal prostaglandin E synthase-1 expression by aortic smooth muscle cells attenuates the differentiated phenotype. J Cardiovasc Pharmacol 68: 127–142, 2016. doi: 10.1097/FJC.0000000000000395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem Sci 41: 274–286, 2016. doi: 10.1016/j.tibs.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bolisetty S, Traylor A, Joseph R, Zarjou A, Agarwal A. Proximal tubule-targeted heme oxygenase-1 in cisplatin-induced acute kidney injury. Am J Physiol Renal Physiol 310: F385–F394, 2016. doi: 10.1152/ajprenal.00335.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolisetty S, Traylor AM, Kim J, Joseph R, Ricart K, Landar A, Agarwal A. Heme oxygenase-1 inhibits renal tubular macroautophagy in acute kidney injury. J Am Soc Nephrol 21: 1702–1712, 2010. doi: 10.1681/ASN.2010030238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 15: 348–366, 2016. doi: 10.1038/nrd.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Curtis LM, Agarwal A. Heme oxygenase-1 gene polymorphisms—toward precision medicine for AKI. J Am Soc Nephrol 27: 3229–3231, 2016. doi: 10.1681/ASN.2016060699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dächert J, Schoeneberger H, Rohde K, Fulda S. RSL3 and Erastin differentially regulate redox signaling to promote Smac mimetic-induced cell death. Oncotarget 7: 63779–63792, 2016. doi: 10.18632/oncotarget.11687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B III, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149: 1060–1072, 2012. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, Stockwell BR. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 3: e02523, 2014. doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol 10: 9–17, 2014. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- 11.Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, Prokisch H, Trümbach D, Mao G, Qu F, Bayir H, Füllekrug J, Scheel CH, Wurst W, Schick JA, Kagan VE, Angeli JP, Conrad M. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13: 91–98, 2017. doi: 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feldkamp T, Park JS, Pasupulati R, Amora D, Roeser NF, Venkatachalam MA, Weinberg JM. Regulation of the mitochondrial permeability transition in kidney proximal tubules and its alteration during hypoxia-reoxygenation. Am J Physiol Renal Physiol 297: F1632–F1646, 2009. doi: 10.1152/ajprenal.00422.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, Basavarajappa D, Rådmark O, Kobayashi S, Seibt T, Beck H, Neff F, Esposito I, Wanke R, Förster H, Yefremova O, Heinrichmeyer M, Bornkamm GW, Geissler EK, Thomas SB, Stockwell BR, O’Donnell VB, Kagan VE, Schick JA, Conrad M. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16: 1180–1191, 2014. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gallorini M, Petzel C, Bolay C, Hiller KA, Cataldi A, Buchalla W, Krifka S, Schweikl H. Activation of the Nrf2-regulated antioxidant cell response inhibits HEMA-induced oxidative stress and supports cell viability. Biomaterials 56: 114–128, 2015. doi: 10.1016/j.biomaterials.2015.03.047. [DOI] [PubMed] [Google Scholar]
- 16.Hull TD, Kamal AI, Boddu R, Bolisetty S, Guo L, Tisher CC, Rangarajan S, Chen B, Curtis LM, George JF, Agarwal A. Heme oxygenase-1 regulates myeloid cell trafficking in AKI. J Am Soc Nephrol 26: 2139–2151, 2015. doi: 10.1681/ASN.2014080770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin XY, Lee SH, Park PH, Hur J, Kim SA, Kim HS, Sohn DH. 2′-Methoxy-4‘6’-bis(methoxymethoxy)chalcone inhibits nitric oxide production in lipopolysaccharide-stimulated RAW 264.7 macrophages. Basic Clin Pharmacol Toxicol 106: 454–460, 2010. doi: 10.1111/j.1742-7843.2009.00524.x. [DOI] [PubMed] [Google Scholar]
- 18.Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, Kapralov AA, Amoscato AA, Jiang J, Anthonymuthu T, Mohammadyani D, Yang Q, Proneth B, Klein-Seetharaman J, Watkins S, Bahar I, Greenberger J, Mallampalli RK, Stockwell BR, Tyurina YY, Conrad M, Bayır H. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13: 81–90, 2017. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kan CFK, Singh AB, Stafforini DM, Azhar S, Liu J. Arachidonic acid downregulates acyl-CoA synthetase 4 expression by promoting its ubiquitination and proteasomal degradation. J Lipid Res 55: 1657–1667, 2014. doi: 10.1194/jlr.M045971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kie JH, Kapturczak MH, Traylor A, Agarwal A, Hill-Kapturczak N. Heme oxygenase-1 deficiency promotes epithelial-mesenchymal transition and renal fibrosis. J Am Soc Nephrol 19: 1681–1691, 2008. doi: 10.1681/ASN.2007101099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim J, Long KE, Tang K, Padanilam BJ. Poly(ADP-ribose) polymerase 1 activation is required for cisplatin nephrotoxicity. Kidney Int 82: 193–203, 2012. doi: 10.1038/ki.2012.64. [DOI] [PubMed] [Google Scholar]
- 22.Kwon MY, Park E, Lee SJ, Chung SW. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 6: 24393–24403, 2015. doi: 10.18632/oncotarget.5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leaf DE, Body SC, Muehlschlegel JD, McMahon GM, Lichtner P, Collard CD, Shernan SK, Fox AA, Waikar SS. Length polymorphisms in heme oxygenase-1 and AKI after cardiac surgery. J Am Soc Nephrol 27: 3291–3297, 2016. doi: 10.1681/ASN.2016010038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lever JM, Boddu R, George JF, Agarwal A. Heme oxygenase-1 in kidney health and disease. Antioxid Redox Signal 25: 165–183, 2016. doi: 10.1089/ars.2016.6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z. Regulated cell death in AKI. J Am Soc Nephrol 25: 2689–2701, 2014. doi: 10.1681/ASN.2014030262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linkermann A, Stockwell BR, Krautwald S, Anders HJ. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol 14: 759–767, 2014. doi: 10.1038/nri3743. [DOI] [PubMed] [Google Scholar]
- 27.Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz PS, Weinlich R, Vanden Berghe T, Vandenabeele P, Pasparakis M, Bleich M, Weinberg JM, Reichel CA, Bräsen JH, Kunzendorf U, Anders HJ, Stockwell BR, Green DR, Krautwald S. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci USA 111: 16836–16841, 2014. doi: 10.1073/pnas.1415518111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25: 402–408, 2001. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 29.Martin-Sanchez D, Ruiz-Andres O, Poveda J, Carrasco S, Cannata-Ortiz P, Sanchez-Nino MD, Ruiz Ortega M, Egido J, Linkermann A, Ortiz A, Sanz AB. Ferroptosis, but not necroptosis, is important in nephrotoxic folic acid-induced AKI. J Am Soc Nephrol, 28: 218–229, 2017. doi: 10.1681/ASN.2015121376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mukhopadhyay D, Srivastava R, Chattopadhyay A. Sodium fluoride generates ROS and alters transcription of genes for xenobiotic metabolizing enzymes in adult zebrafish (Danio rerio) liver: expression pattern of Nrf2/Keap1 (INrf2). Toxicol Mech Methods 25: 364–373, 2015. doi: 10.3109/15376516.2015.1025348. [DOI] [PubMed] [Google Scholar]
- 31.Nath KA. Heme oxygenase-1 and acute kidney injury. Curr Opin Nephrol Hypertens 23: 17–24, 2014. doi: 10.1097/01.mnh.0000437613.88158.d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nath KA. Heme oxygenase-1: a redoubtable response that limits reperfusion injury in the transplanted adipose liver. J Clin Invest 104: 1485–1486, 1999. doi: 10.1172/JCI8827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nath KA, Balla G, Vercellotti GM, Balla J, Jacob HS, Levitt MD, Rosenberg ME. Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J Clin Invest 90: 267–270, 1992. doi: 10.1172/JCI115847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nath KA, Haggard JJ, Croatt AJ, Grande JP, Poss KD, Alam J. The indispensability of heme oxygenase-1 in protecting against acute heme protein-induced toxicity in vivo. Am J Pathol 156: 1527–1535, 2000. doi: 10.1016/S0002-9440(10)65024-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ooko E, Saeed ME, Kadioglu O, Sarvi S, Colak M, Elmasaoudi K, Janah R, Greten HJ, Efferth T. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine 22: 1045–1054, 2015. doi: 10.1016/j.phymed.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 36.Poss KD, Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci USA 94: 10919–10924, 1997. doi: 10.1073/pnas.94.20.10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rewa O, Bagshaw SM. Acute kidney injury-epidemiology, outcomes and economics. Nat Rev Nephrol 10: 193–207, 2014. doi: 10.1038/nrneph.2013.282. [DOI] [PubMed] [Google Scholar]
- 38.Sakai O, Yasuzawa T, Sumikawa Y, Ueta T, Imai H, Sawabe A, Ueshima S. Role of GPx4 in human vascular endothelial cells, and the compensatory activity of brown rice on GPx4 ablation condition. Pathophysiology, 24: 9–15, 2016. doi: 10.1016/j.pathophys.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 39.Sakamoto K, Sogabe S, Kamada Y, Matsumoto SI, Kadotani A, Sakamoto JI, Tani A. Discovery of GPX4 inhibitory peptides from random peptide T7 phage display and subsequent structural analysis. Biochem Biophys Res Commun 482: 195–201, 2017. doi: 10.1016/j.bbrc.2016.11.035. [DOI] [PubMed] [Google Scholar]
- 40.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675, 2012. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A, Stockwell BR. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc 136: 4551–4556, 2014. doi: 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63: 173–184, 2016. doi: 10.1002/hep.28251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tonnus W, Linkermann A. “Death is my Heir”—ferroptosis connects cancer pharmacogenomics and ischemia-reperfusion injury. Cell Chem Biol 23: 202–203, 2016. doi: 10.1016/j.chembiol.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 44.Tracz MJ, Alam J, Nath KA. Physiology and pathophysiology of heme: implications for kidney disease. J Am Soc Nephrol 18: 414–420, 2007. doi: 10.1681/ASN.2006080894. [DOI] [PubMed] [Google Scholar]
- 45.Venkatachalam MA, Bernard DB, Donohoe JF, Levinsky NG. Ischemic damage and repair in the rat proximal tubule: differences among the S1, S2, and S3 segments. Kidney Int 14: 31–49, 1978. doi: 10.1038/ki.1978.87. [DOI] [PubMed] [Google Scholar]
- 46.Wang HE, Muntner P, Chertow GM, Warnock DG. Acute kidney injury and mortality in hospitalized patients. Am J Nephrol 35: 349–355, 2012. doi: 10.1159/000337487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang S, Zhang C, Hu L, Yang C. Necroptosis in acute kidney injury: a shedding light. Cell Death Dis 7: e2125, 2016. doi: 10.1038/cddis.2016.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang YQ, Chang SY, Wu Q, Gou YJ, Jia L, Cui YM, Yu P, Shi ZH, Wu WS, Gao G, Chang YZ. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front Aging Neurosci 8: 308, 2016. doi: 10.3389/fnagi.2016.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ 23: 369–379, 2016. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA 113: E4966–E4975, 2016. doi: 10.1073/pnas.1603244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, Stockwell BR. Regulation of ferroptotic cancer cell death by GPX4. Cell 156: 317–331, 2014. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol 26: 165–176, 2016. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 15: 234–245, 2008. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu H, Guo P, Xie X, Wang Y, Chen G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J Cell Mol Med 21: 648–657, 2017. doi: 10.1111/jcmm.13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu X, Long YC. Crosstalk between cystine and glutathione is critical for the regulation of amino acid signaling pathways and ferroptosis. Sci Rep 6: 30033, 2016. doi: 10.1038/srep30033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, Zeh HJ, Kang R, Tang D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol 2: e1054549, 2015. doi: 10.1080/23723556.2015.1054549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun 478: 1338–1343, 2016. doi: 10.1016/j.bbrc.2016.08.124. [DOI] [PubMed] [Google Scholar]
- 58.Zarjou A, Bolisetty S, Joseph R, Traylor A, Apostolov EO, Arosio P, Balla J, Verlander J, Darshan D, Kuhn LC, Agarwal A. Proximal tubule H-ferritin mediates iron trafficking in acute kidney injury. J Clin Invest 123: 4423–4434, 2013. doi: 10.1172/JCI67867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang D, Shen J, Wang C, Zhang X, Chen J. GSH-dependent iNOS and HO-1 mediated apoptosis of human Jurkat cells induced by nickel(II). Environ Toxicol 24: 404–414, 2009. doi: 10.1002/tox.20440. [DOI] [PubMed] [Google Scholar]