

ABSTRACT
Cross-linked polymer beads with different cross-linking agent loading were prepared by carrying out cross-linking suspension copolymerization of styrene-divinylbenzene (St- DVB) monomers using guar gum (GG) and xanthan gum (XG) from bioresources as eco-friendly suspension biopolymer stabilizers in the presence of non reactive diluents. The effects of GG and XG as suspension biostabilizers on the characteristics of the styrene copolymer beads were investigated regarding thermal properties, porosity characteristics, solvent swelling ratio, and surface morphologies using TGA, DSC, XRD, SEM, BET analyses. Spherical and regular beads with smooth surface were produced and the average particle size was in the range 170–290 μm (50–80 mesh size). The porosity characteristics of the produced beads including surface area and pore volume were in range 0.45 m2/g and 32–45 ml/g, respectively. Overall, the present article provided a novel route to prepare cross-linked polystyrene copolymer beads with tunable porosity suitable for catalyst support.
KEYWORDS: Cross-linked polystyrene copolymer, suspension stabilizer, Polymer support, microporous, suspension polymerization
1. Introduction
Polymer supports, also referred to as polymeric reagents and functional polymers, are beaded or microspherical polymer products with a particle size range of about 5–500 μm [1–3]. They have been used in an increasingly large number of applications, for example, separation and purification, catalysis, drug delivery, solid phase organic synthesis, microencapsulation of phase change materials and templates for nanoparticle growth [4–10]. Such applications, especially as support for catalyst immobilization frequently require large particle size area, which necessitates the formation of pores (of the required dimensions) in the bead structure. The major thrust behind the widespread use of polymer supports is the simplification achieved in separation and purification of catalysts, products, synthetic intermediates, or decontamination from impurities, side products or toxins.
A wide range of polymer supports are used for various applications. These polymers are produced by several different synthetic routes and polymerization techniques, and each polymer type is employed in a variety of morphological grades (i.e. gel type, microporous or macroporous). Organic polymer supports are usually produced by suspension polymerization or suspension cross-linking in finely beaded form (ca. 5–500 μm). The introduction of the anchoring points (functional groups) into the polymer beads is conventionally accomplished either by copolymerization of appropriate monomer mixtures, or by functionalization of preformed beaded polymers. Vinyl-type polymer supports can be produced by either of these two methods [11–16].
A fundamentally more important aspect of polymer supports is the chemical structure of the polymer backbone. Chemical structure determines polymer-solvent compatibility in bulk, as well as polymer-solvent substrate interactions at the molecular level. In this respect, it is particularly noteworthy that the most frequently used polymeric support is polystyrene due to its environmental stability and hydrophobic nature. Styrene-divinylbenzene (St-DVB) copolymers are extensively used as polymeric supports for polymer bonded catalysts and reagents [17,18]. The permeability limits according to the molecular size and the accessibility of reactive and catalytic sites depend on the porous structure and swelling properties of the copolymers [19]. It has been established that these properties are deeply influenced by the copolymer synthesis conditions: suspension stabilizers, crosslinking degree, porogenic agent or diluent nature, etc [20–26].
The suspension-like polymerization process, by which is a simple, cheap, robust and environmental friendly method for the preparation of suitable cross-linked polystyrene beads. The most important feature of suspension polymerization is the formation of stable droplets of monomer suspended in the nonsolvent phase (water). The nonsolvent phase may be thought of as providing millions of tiny ‘spherical molds’ in which the polymer beads are formed constrained by surface tension. Stabilizers, typically a mixture of simple inorganic salts together with large, polar organic species, such as poly(vinylpyrrolidone), are added to the mixture to help reduce the surface tension of the droplets. This stabilization of the droplets prevents aggregation of the ‘molds’ which leads to misshapen beads [27,28].
Styrene-based resins used for the preparation of ion exchange resins and polymer supports are also obtained by oil-in-water (O/W) method of styrene (St) and divinylbenzene (DVB) (and a functional monomer) [29,30]. In the preparation of the polymer beads, the dispersion of the organic phase in the continuous phase is the determining step in establishing the particle size distribution of the final polymer beads. This is influenced by the geometrical factors of the reactor, operating parameters and substance parameters (type and concentration of stabilizers, viscosities of continuous and dispersed phases, interfacial tension) [31,32].
The addition of suspension stabilizers plays a very important role in the stabilization of liquid-liquid dispersions. In suspension polymerization technique, suspending agents are used to form a film or skin around the droplet/particle surface in order to prevent coalescence and agglomeration by a mechanism analogous to steric stabilization [33]. The most commonly used suspension (droplet) stabilizers for (O/W) suspension polymerization are styrene-maleic anhydride copolymer [34–37], salts of acrylic acid polymers [38], sodium dodecyl sulfate [39], polyvinyl alcohol (PVA) [40,41], poly(vinylpyrrolidone) (PVP) [42,43]. A wide range of other water soluble polymers, including gelatin, cellulose ethers, and synthetic polymers are also used. Scarcely soluble inorganic salts such as calcium carbonate (talc), phosphates, and sulfates may also be employed, either alone or in combination with organic stabilizers [25,26,44,45].
In the present study we have focused on the possibility of the use of natural water-dispersible biodegradable polymer for the preparation of polystyrene beads due to its different chemical nature relative to expensive synthetic ones, for example, poly(vinylpyrrolidone) (PVP). Several authors have analyzed the effects of the agitation rate, the cross-linker content, the type and amount of porogen agent (diluent), solvent, the type and concentration of synthetic suspension stabilizer polymer, the type and concentration of initiator, and also the operation conditions (temperature and time of polymerization, etc.) on the preparation of cross-linked polystyrene beads using the suspension polymerization method [46–53]. However, the effect of natural biodegradable polymers, guar gum, xanthan and carboxymethyl cellulose as suspension stabilizers on the preparation of cross-linked polystyrene beads by means of suspension polymerization has not been previously reported.
In continuation of recent works on the use of polymeric catalysts in organic transformations [43,54–57] and continued seeking for suitable polymer supports to immobilize catalysts on them, herein, this contribution describes in detail the use of eco-friendly suspending agents from bioresouces, guar and xanthan gums as stabilizing agents in suspension polymerization reactions, as well as their influence on the size and surface area of the synthesized polymer particles. For the purposes of this work, the synthesis of poly(St-co-DVB) cross-linked beads was chosen as the main suspension polymerization system to be studied. Thus, cross-linked polymer beads based on St and DVB monomeric mixtures with different DVB content were prepared by suspension copolymerization and the effect of guar gum, xanthan gum, and carboxymethyl cellulose as natural water-dispersible polymers was investigated regarding the diverse properties of the resultant microporous beads (thermal properties, morphology, particle size, etc.) (Scheme 1).
Scheme 1.

Preparation of cross-linked polystyrene copolymer.
2. Experimental
2.1. Starting materials and instruments
The polymerizable monomers, styrene (St, Industrial grade) and divinylbenzene (DVB, containing 65% DVB isomers, the remainder mainly being 3- and 4-ethylvinylbenzene) were supplied by TPC (Tabriz Petrochemical Complex, Iran). DVB was extracted with sodium hydroxide solution (1%, 2˟ 10 ml) to remove the inhibitor, and then were washed by water (3x 30 ml) until neutralization. The oil-soluble initiator, 2,2ʹ- azobisisobutyronitrile (AIBN) was supplied by TPC and used as received. Guar gum (GG), xanthan gum (XG, petroleum grade), and carboxymethyl cellulose (CMC) were provided by National Iranian South Oil Company (NISOC) and were used without further purification. Poly(vinyl alcohol) (PVA), having a weight-average molecular weight of 86–87,000 and a degree of hydrolysis of 88%, was supplied by Aldrich.Thermogravimetric (TGA) measurements were carried out using a TGA/DTA Bahar: STA 503 in the range from 25 to 600°C under air atmosphere; the heating rate was 10 °C min−1. The DSC measurements were performed with a DSC Pyris 6, Perkin Elmer, at the heating rate of 10°Cmin−1 in nitrogen atmosphere, in the range from 25 to 400°C. FTIR spectra of the studied copolymers were obtained employing a Unicam Matteson 1000 spectrometer, using KBr pallets, the contents of the sample in the pallet was about 3%. The surface morphology of the beads was examined by a Hitachi scanning electron microscopy (SEM). The texture of the beads in dry state, i.e., surface area (S) and pore volume (Vp), were analyzed by nitrogen adsorption measurements using BET and BJH methods, respectively. The average size and size distribution of copolymer beads were determined by sieving (screen) analyses performed by using a Shimadzu shaker sieving unit, with Endcotte stainless steel standard sieves. All the solvents were of synthesis grade and were used as obtained. Nitrogen was of high-purity grade (99.99%). Distilled water was used as the continuous phase in all reactions and water washings.
2.2. Preparation of cross-linked polystyrene copolymer beads
The suspension polymerization (shown in Tables 1 and 2) was carried out in a 250 ml three-necked round-bottomed flask equipped with a mechanical stirrer, nitrogen gas inlet, and reflux condenser that is placed in a thermostated water bath. All reactions were carried out at 80°C. A mixture of distilled water (500 parts) containing GG/or XG and XG/PVA (wt% in relation to the monomers) and NaCl (2% w/v in relation to water volume) was first introduced into the flask and stirred at room temperature for 1 h. NaCl, water soluble salt was used to promote the salting-out of the organic phase. The suspending medium was then heated to the reaction temperature. While stirring nitrogen was purged from the mixture. A mixture of the monomers (styrene and several weight ratios of DVB cross-linker, 4, 6, 8, 10%) and the diluents (toluene/n-heptane (3:2) mixture, 100 parts), containing AIBN as the initiator (1 mol% in relation to the monomers) was added dropwise, through a dropping funnel to the flask in about 30 min. The mixture was agitated, until the organic components were dispersed as fine droplets, and then heated at 80°C.The stirring rate was kept at 350–400 rpm and the reaction was allowed to proceed for 18 h. After the polymerization, the obtained copolymer beads were hot filtered out on a Buchner funnel under reduced pressure, and treated in 1N HCl at boiling temperature for 15 min, washed twice with hot water, and then vacuum filtered to remove the stabilizer. The copolymer beads were extracted with acetone for a few hours in a soxhlet apparatus to remove porogen and residual monomers, and then were washed twice with acetone, three times with methanol to ensure complete removal of impurities. Finally, the samples were dried in a vacuum oven at 50°C for at least 8 h.
Table 1.
Experiments performed to evaluate the potential of GG as a stabilizing agent. Basic recipe and reaction conditions of the investigated suspension polymerization reactions*.
| Ingredient | GG, % |
Crosslinking agent, % |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| G0.5 | G1 | G1.5 | G2 | D4 | D6 | D8 | D10 | ||
| Discontinuous (dispersed) phase | |||||||||
| St, % | 92 | 92 | 92 | 92 | 96 | 94 | 92 | 90 | |
| DVB, % | 8 | 8 | 8 | 8 | 4 | 6 | 8 | 10 | |
| Toluene, % | 60 | 60 | 60 | 60 | |||||
| n-Heptane, % | 40 | 40 | 40 | 40 | |||||
| Continuous phase Guar gum, % | AIBN, % | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 |
| Water, % | (500 parts) | ||||||||
| NaCl, % | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| 0.5 | 1 | 1.5 | 2 | 2 | 2 | 2 | 2 | ||
*For all reactions, aqueous phase r to organic phase (O/W) ratio = 5/1; reaction temperature = 80°C;
polymerization time = 18 h; agitation rate = 250 rpm
Table 2.
Experiments performed to evaluate the potential of XG as a stabilizing agent. Basic recipe and reaction conditions of the investigated suspension polymerization reactions*.
| Ingredient | XG, % |
Crosslinking agent, % |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| X0.5 | X1 | X1.5 | X2 | D4 | D6 | D8 | D10 | ||
| Discontinuous (dispersed) phase | St, % | 92 | 92 | 92 | 92 | 96 | 94 | 92 | 90 |
| DVB, % | 8 | 8 | 8 | 8 | 4 | 6 | 8 | 10 | |
| Toluene, % | 60 | 60 | 60 | 60 | |||||
| n-Heptane, % | 40 | 40 | 40 | 40 | |||||
| AIBN, % | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | |
| Continuous phase | Water, % | (500 parts) | |||||||
| NaCl, % | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| Xanthan gum, % | 0.5 | 1 | 1.5 | 2 | 2 | 2 | 2 | 2 | |
*For all reactions, aqueous phase to organic phase (O/W) ratio = 5/1; reaction temperature = 80°C;
polymerization time = 18 h; agitation rate = 250 rpm
The monomer dilution degree was fixed at 100% in relation to the volume of the monomers mixture.The volume ratio between the two phases (organic/aqueous) was kept constant at 1/5 for all reactions.The overall conversion of the monomers to solid copolymer was determined gravimetrically.
The same procedure was used to prepare cross-linked polystyrene beads in the presence of different weight percentages of DVB cross-linker and suspension stabilizers.
2.3. Resin swelling measurements
To a stoppered test tube containing the dried copolymer sample (2 g), 10 ml of toluene was added and the sample was soaked (swollen) in toluene at ambient temperature for overnight. Toluene was filtered and beads were padded dried with a filter paper and finally dried and weighed.
Swelling (%) = [(increase in the weight of the beads)/(original weight of the beads)] × 100
3. Results and discussion
3.1. Preparation of cross-linked polystyrene copolymer beads
In our previous works, different lightly cross-linked polymer beads based on St/DVB monomers were successfully prepared by means of aqueous suspension radical polymerization technique using conventional synthetic suspension stabilizers (PVP) in the presence of non reactive diluents and utilized as polymer supports to immobilize catalysts in one step [43,54,55]. The cross-linked polystyrene functionalization is largely dependent on the bead diameter. Beads with diameter greater than 150 μm possess optimal handling properties. Larger diameter resins are greatly reducing the problem of static often associated with smaller diameter resins, which makes these smaller resins more difficult to manipulate. Although smaller diameter beads often have increased chemical reaction rates, this needs to be balanced by the improved handling and increased functionality per bead associated with larger beads. Thus, a new procedure for cross-linking suspension polymerization using natural water-dispersible polymers is desirable.
This work is a first attempt to elucidate the influence of GG, XG used as stabilizing agents in suspension polymerization reactions. In the first experiments carried out in this research the concentration of GG, XG in the aqueous phase of the polymerization reactions were varied to evaluate their effect on the average particle size of the resulting polymer beads. These experiments were performed according to the recipe and experimental conditions presented in Tables 1 and 2. In the present study, a relatively low monomer phase weight fraction of 0.1 was used to obtain pearl-like particles. Higher monomer phase fraction could not allow the copolymerization to form beads, but gave aggregates or big lump of polymer mass. However, It may increase as a normal commercial practice (> 0.5) by increasing the amount of droplet stabilizing agent or the stirring rate. Two series of copolymer beads were prepared using two biopolymer suspending agents, GG, XG, at a fixed diluent-monomer volume ratio, but with various amounts of DVB (Tables 1 and 2).
3.2. The influence of different biopolymer suspension stabilizers on the bead formation process
The important feature of suspension agents is their amphipathic character, which explains their ability to lower the interfacial tension and to locate at the monomer/water interface [38]. It is well known that the suspension stabilizer form a film or skin around the droplet/particle surface, and this layer prevents coalescence and agglomeration by a mechanism analogous to steric stabilization, which is known to protect particle against flocculation [33,46,58]. This behavior could affect the morphology, the particle size and the amount of surface area and pore volume of the microparticles. To investigate this dependence, different experiments using natural biopolymers (GG and XG), on the basis of experimental conditions described in Tables 3 and 4 were done. Guar gum (GG) is a nonionic edible plant polysaccharide with extensive applications in food, paper, textile, and petroleum industry [59,60]. It is a polysaccharide with one of the highest molecular weights of all naturally occurring water soluble polymers and that has an approximate 1:2 ratio of D-galactose to D-mannose. The main advantages for using guar are its low cost, easy availability and capacity to form viscous solutions and gels at low concentration. Xanthan gum (XG) is an extracellular heteropolysaccharide and its structural unit consists of D-glucopyranose glucan backbone with side chains of D-mannopyranose-D- glucuronid acid-D-mannopyranose on alternating residue, It can be used in food and pharmaceutical industry and oil recovery [61,62]. Guar gum is in fact the powdered endosperm of the seeds of the Cyamopsis tetragonolobus which contains a complex polysaccharide called galactomannan. This hydroxyl group is rich high molecular weight polymer consisting of d-mannose backbone on which d-galactose units are attached as side-chains. These functional groups form hydrogen bonding with water to impart viscosity and thickening to the solution [63].
Table 3.
Effect of the concentration of GG on bead properties*.
| Run | G0.5 | G1 | G1.5 | G2 |
|---|---|---|---|---|
| GG concentration, wt% | 0.5 | 1 | 1.5 | 2 |
| Yield, % | 10 | 23 | 53 | 65 |
| Bead size distribution, wt% | ||||
| ≤ 0.177 mm, ≥ 80 mesh | Fused | 9.15 | 13 | 19.45 |
| 0.177–0.297 mm, 80 mesh | Fused | 41.85 | 53.25 | 58.45 |
| 0.297–0.707 mm, 50 mesh | Fused | 29.80 | 22.30 | 15.77 |
| ≥ 0.707 mm, 25 mesh | Fused | 19.21 | 11.50 | 6.37 |
| % Swelling | 65.40 | 68.50 | 70.58 | |
| Glass transition temperature, °C | 110 | 111 | 115 | |
| Surface area, m2/g | 26 | 29 | 35 | |
| Pore volume, cm3/g | 0.16 | 0.18 | 0.195 |
*Diluent composition (Toluene/Heptane: 3/2); DVB concentration = 8%
Table 4.
Effect of the concentration of XG on bead properties*.
| Run | X0.5 | X1 | X1.5 | X2 | X3 |
|---|---|---|---|---|---|
| XG concentration, wt% | 0.5 | 1 | 1.5 | 2 | 3 |
| Yield, % | 9 | 18 | 49 | 68 | 69 |
| Bead size distribution, wt% | |||||
| ≤ 0.177 mm, ≥ 80 mesh | Fused | 6.11 | 9.12 | 12.26 | 11.90 |
| 0.177–0.297 mm, 80 mesh | Fused | 24.40 | 31.20 | 37.32 | 36.20 |
| 0.297–0.707 mm, 50 mesh | Fused | 35.32 | 30.65 | 26.02 | 27.30 |
| ≥ 0.707 mm, 25 mesh | Fused | 34.20 | 29.08 | 24.16 | 24.20 |
| % Swelling | 52. 40 | 56.20 | 60.63 | 61.0 | |
| Glass transition temperature, °C | 109 | 111 | 113 | 113 | |
| Surface area, m2/g | 25 | 28 | 34 | 40 | |
| Pore volume, cm3/g | 0.155 | 0.165 | 0.19 | 0.215 |
*Diluent composition (Toluene/Heptane: 3/2); DVB concentration = 8%
The concentration of the polymeric dispersant is one of the most important factors controlling nucleation, i.e., the number of particles increases and their size decreases as the dispersant content is raised. The efficiency of stabilizing function of guar gum at different concentrations on bead formation process and properties shown in Table 3. Different dispersant concentrations gave various copolymer particle sizes. Due to heterogeneity of particle size obtained for GG, copolymer beads were sieved (screen mesh size) in order to quantify their average particle size. The polymer particle size distribution determined by screen analysis is given by screen mesh size and by the millimeters (mm) or microns (µ) corresponding to the screen mesh size. At a GG concentration 2 wt%, 19.45 wt% of particles have a particle size higher than 80 mesh, 74.25 wt% between 50–80 mesh, and only 6.37 wt% lower than 50 mesh. The highest overall conversion (expressed as % yield) was at a GG concentration 2 wt%. The bead size distributions measured for various GG concentrations are summarized in Table 3. The effect of the increasing GG concentration, and hence, viscosity of the continuous phase, on the particle size distribution (PSD) is depicted in Figure 1, which shows that the increasing concentration of GG leads to smaller particle sizes and causes the PSD to shift towards smaller diameters and to narrower slightly. Table 3, shows a remarkable product distribution in that almost 76 percent of the particles fall in the 50 mesh and 80 mesh size. As the amount of GG present is increased the polymer particle size decreases; but the product particle size distribution maintains a relatively narrow range.
Figure 1.
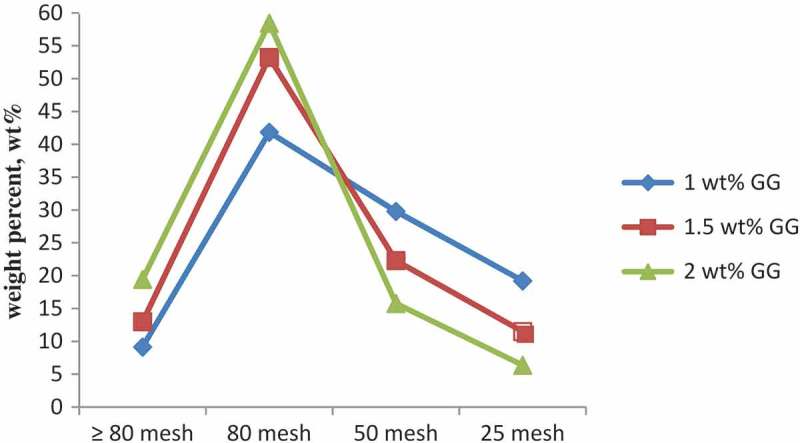
Size distribution of the cross-linked beads at the different GG concentrations following suspension polymerization.
Also, it was found that increase in the amount of guar gum used results in still further decrease in the copolymer product size but usage much in excess of the 3 percent shown ceases to afford significant size decrease. Very large amounts of guar gum tend to decrease the yield of polymer product somewhat.
At a GG concentration 0.5 wt% (Run G0.5), the polymeric beads formed were clustered and fused, no bead formation could be observed, because the suspension stabilizer concentration was too low to make enough droplet stability to keep them from coalescing during polymerization and resulted in a thinner layer of interfacial polymer and a less stable suspension. Thus, this amount of the suspending agent GG is not enough to help the bead formation. When the GG concentration at a fixed DVB content was increased from 1 to 2 wt% (G1 to G2), the overall yield and average particle size were increased, and the particle shape improved. Furthermore, increasing the GG concentration could result in an increase in droplet stability by decreasing the droplet collision frequency.
Suspension copolymerization was also performed with four different XG concentrations (0.5,1,2,3wt%). Table 4 shows the effect of XG concentration on dispersion copolymerization of St, DVB. The presence of 0.5–0.75 weight percent XG, or less, based on (St+ DVB) charge, in the polymerization zone resulted in polymer product in the form of one solid mass around the stirring magnet. The effect of the increasing XG concentration on the (PSD) is depicted in Figure 2, which shows that the increasing concentration of XG leads to smaller particle sizes and causes the PSD to shift towards smaller diameters and to narrower slightly, but less than the equivalent amount of GG, and the product particle size distribution maintains a relatively narrow range. In general, St/DVB particles obtained using XG were opaque with spherical shape, although some agglomerates were also observed (Figure 7(b)).
Figure 2.

Size distribution of the cross-linked beads at the different XG concentrations following suspension polymerization.
Figure 7.

SEM micrographs of the polymer beads synthesized by different stabilizer. (a) GG, (b) XG, (c) CMC.
It can be seen that these copolymer particles, controlled by the dispersant, have a nearly micro-spherical shape with a smooth surface. Different XG concentrations also gave various copolymer particle sizes. The copolymer particle sizes obtained were in the range of 25–80 mesh with bead size distribution (wt%) shown in Table 4.
Also, it was found that, similar to GG, increase in the amount of XG used results in still further decrease in the copolymer product size but usage much in excess of the 4 percent shown ceases to afford significant size decrease. Very large amounts of XG tend to decrease the yield of polymer product somewhat.
The copolymerization using (XG + PVA) suspension agents at a weight ratio of XG to PVA of 2/1 and a range of XG usage ranging from 0.1 to 3 percent at a fixed DVB content (8 wt%) were carried out. It was found that at 4 percent of PVA alone, St and DVB did not copolymerized to a particulate polymer in the procedure used herein. However, using the combination agent particulate polymer product is produced in high yield at a usage of only 0.1 percent XG plus 0.05 percent PVA and this production continues at high yield even at 4.0 percent XG plus 2.0 percent PVA. At a weight ratio XG to PVA (2/1) and XG concentrations based on monomer, 0.1, 0.2, 0.5, 1, 1.5, 2; the yields (wt), were 79, 78, 75, 73, 71, 70%, respectively, and the porosities were 30, 31, 36, 41, 46, 51%, respectively. At equivalent xanthan gum usage, the XG alone process gives product of larger size than the combination agent process; however, both processes give a relatively narrow range of particle sizes. The combination process gives a definitely more porous product than the xanthan gum alone process at equivalent agent usage.
When the combination of XG/PVA (2/1) with an overall concentration 2 wt% at a fixed DVB content (8 wt%) was used as stabilizer, almost 93 wt% of particles have a particle size 80 mesh and higher, and the particle shape improved (Table 5). The copolymer particles have a spherical shape and are stabilized in a dispersion medium because of repulsive forces generated by barriers of the soluble mixed dispersant. When two surfaces, each covered by a layer of adsorbed soluble polymer chains, approach each other within a distance less than the combined thickness of the adsorbed layers, an interaction between the polymer layers will occur. This is the source of steric stabilization generating a repulsive force between the opposing surfaces.When the system was polymerized without the dispersant, agglomerated particles were formed. It is explained that the polymerizing system without a stabilizer brings about the attractive force which operates between two adjacent particles, usually called Vander Waals force, originating in the interactions between the atoms and molecules of which the particles are composed [64].
Table 5.
Effect of the concentration of crosslinking agent (DVB) on bead properties*.
| Run | D4 | D6 | D8 | D10 | D15 |
|---|---|---|---|---|---|
| Crosslinking agent concentration, wt% | 4 | 6 | 8 | 10 | 15 |
| Yield, % | 11 | 22 | 46 | 69 | 70.50 |
| Bead size distribution, wt% | |||||
| ≤ 0.177 mm, ≥ 80 mesh | 46 | 20.75 | 15.6 | 26.53 | 33.59 |
| 0.177–0.297 mm, 80 mesh | 28 | 64.15 | 78.12 | 63.26 | 58.77 |
| 0.297–0.707 mm, 50 mesh | 8 | 11.32 | 3.1 | 8.16 | 3.55 |
| ≥ 0.707 mm, 25 mesh | 18 | 3.7 | 3.1 | 2.0 | 4.08 |
| % Swelling | 84.52 | 83.07 | 60.63 | 58.16 | 52.38 |
| Glass transition temperature, °C | 104 | 113.7 | 116 | 125 | 132 |
*Diluent composition (Toluene/n-Heptane: 3/2); XG/PVA (2/1), overall concentration 2%wt
The possibility of using CMC as a stabilizer in the procedure used herein with five different CMC concentrations (0.5, 1, 1.5, 2, 3, 4 wt%) at a fixed DVB content (8 wt%) was studied. It was observed with a range of CMC usage ranging from 0.5 to 3 percent, the polymerizable monomers, St and DVB, did not copolymerized to a particulate polymer in the procedure used herein. However, at a CMC concentration 4 wt% alone, randomly irregular aggregated particles were formed.
The next step of our studies involved the preparation of a series of polystyrene-divinyl benzene cross-linked resins from 2 to 10% DVB by traditional suspension radical polymerization method. There are a number of variables which must be considered when altering the cross-linking ratios; thus the relative amounts of St and DVB needed to be taken into account during the synthesis. In our cases, the incorporation of DVB used in the polymerization mixtures was such as to give a lightly cross-linked copolymer bead (Tables 1 and 2). It was observed that the crude yield of the polymerization was dependent on the amount of cross-linking agent added to the reaction (Figure 3). The yields of synthesis increased with increasing cross-linking ratio, with a 2% resin giving a yield of only 32% whereas the 6–8, 8–10 resins consistently yielded almost 60% and 70%, respectively. Since increasing cross-linking confers upon a growing polymer increasing insolubility in the medium, it follows that a higher proportion of soluble polymer will remain in the cases of the lowest cross-linked resins, and given the fact that these will be washed out of the resins by the workup procedure, the yields of reaction will increase with increasing resin cross-linking. In all cases, the size distribution of the obtained beads was carefully controlled by stirring, and the beads were sieved to give the distribution shown in Figure 4. Of these beads the 50–80 mesh size (or 170–290 μm) beads were used in the subsequent studies.
Figure 3.

Relationship between the crude yield of the polymerization reaction and resin cross-linking.
Figure 4.

Size distribution of the 4–10% cross-linked beads following suspension polymerization using the GG, XG, XG/PVA as stabilizers.
Figure 5 shows the relation between yield of copolymers prepared with GG as a function of the crosslinker DVB concentration.When the DVB content increases, the polymerization rate and the final monomer conversion increase. The yield of the copolymers increased from about 52 to 75%. DVB has a higher reactivity than styrene. It helps to decrease the activation energy of polymerization and to increase the reaction rate. As the polymerization proceeds, the polymerization rate slows down. In the last stage, the number of nuclei does not rise any more, and the amount of monomers and cross linking agent decreases. At the same time, the crosslinking density of the particles becomes higher.
Figure 5.

The yield of the copolymers with changing crosslinker DVB concentration (Yield of copolymer in weight percent of St + DVB charge).
The FTIR spectrum of copolymers exhibited an absorption at 3025 cm−1 attributed to aromatic C-H stretching (Figure 6). In addition, bands at 1600, 1487, 1447 cm−1 confirmed the presence of carbon- carbon double bond phenyl stretching. The strong absorption at 696, 751 cm−1 due to out of plane bending of monosubstituted benzenes and 2912, 2840 cm−1 due to aliphatic C-H stretching. The characteristic absorption bands of the prepared copolymers were in agreement with the reported literature [65].
Figure 6.

FTIR spectrum of St-DVB copolymer obtained using GG as stabilizer.
The surface morphology and particle size of some of the synthesized polymer beads were also investigated by SEM to gain an insight into the formation of pores. Images of the morphologies obtained are shown in Figure 7, which shows, when GG was used as stabilizer, all particles are perfectly regular, spherical shape and almost uniform in their size and that the surface is even and smooth (Figure 7(a)). The diameters of the beads were about 0.17–0.707 mm. The even and smooth surface of the particles also demonstrate that the monomer diluent is a good solvent for the polymer and the beads are microporous in nature. It was also observed that when XG used as stabilizer, all particles are perfectly regular, spherical and almost heterogeneous in their size, a heterogeneous surface morphology and that the surface of particles is even and smooth (Figure 7(b)).The average particle size was approximately 0.298 mm. However, with CMC as stabilizer, randomly irregular aggregated particles with rough surface were observed (Figure 7(c)). It could be hypothesized that GG, XG (especially XG/PVA) preferred to adsorb on the surface of the polymer than phase change materials, resulting in lower interfacial tension between the polymer and the aqueous phase, and leading to a smooth surface. The repeating structure of guar/xanthan gums, i.e. D-galactose to D-mannose (1/2 ratio) and D-glucopyranose glucan backbone with side chains of D-mannopyranose-D- glucuronid acid-D-mannopyranose on alternating residue, could result in greater their density enabling cooperative effect among hydroxyl interacting sites that give rise to enhanced stabilizing function. These functional groups form hydrogen bonding with water to impart viscosity and thickening to the solution.
The percentage of volume swelling (% swelling) of the copolymer beads in toluene as solvent were measured and the data are shown in Figure 8. At a fixed pore former ratio, a higher swelling percentage was achieved in copolymers synthesized with GG, XG as stabilizer agents. In both cases, higher DVB content resulted in lower swelling percentage at an equal DVB content. Figure 8 shows the relation between The percentage of volume swelling of copolymers prepared with GG, XG as a function of the cross-linking degree. The % swelling in toluene of the beads decreases with an increasing cross-linking density of the copolymer chains. The copolymer beads are therefore less flexible, leading to a decrease in the % swelling and these results in turn led to a decrease in the diffusion coefficient of the copolymer.
Figure 8.
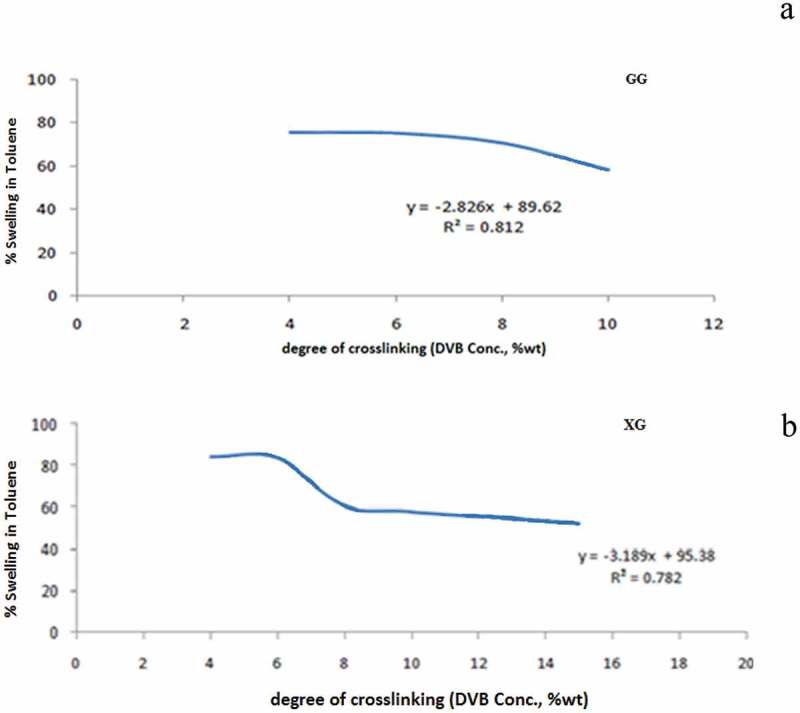
Swelling (%) of 4–10% DVB cross-linked polystyrene beads in toluene solvent.
TGA can accurately track the in situ weight variations of the copolymer bead samples during heating process to investigate their thermal degradation. Figure 9(a) shows TGA plots of copolymer beads prepared with GG, XG at an equal DVB content. It was obviously seen that both samples showed two-step degradations and similar patterns, and a little weight loss (about 4%) at lower heating temperature (215°C) was resulted from the residual monomer and physically adsorbed water. The main weight loss stage was the random bond scission or the polymer chains. Two copolymer bead samples prepared using GG and XG as stabilizing agents (8.0 wt.% DVB) had 50% weight loss at 410 and 445°C, and 89% weight loss at 441 and 459°C, respectively. The two samples completely degraded at about 620 oC. It is also worth noticing that these copolymer beads yield thermal degradation with very little or none ash residue. Figure 9(b) shows TGA plots of the copolymer beads prepared using GG at different DVB content. The thermal stabilization of the copolymer beads was enhanced with increasing DVB content. No obvious weight loss was observed between 50 and 320°C for all samples. Comparatively, at 4 wt.% DVB loading, the copolymer bead had 50 and 89% weight loss at 402 and 490°C, respectively, and completely degraded at 615°C. At 8 wt.% DVB, the copolymer bead had 50 and 89% weight loss at 412 and 502°C, respectively, and completely degraded at 625°C. The results strongly show that the thermal stability property of copolymer beads increased as the cross-linker DVB loading increased.
Figure 9.

Thermograms of (a) Poly(St-DVB) copolymer prepared using GG, XG,(b) Poly(St-DVB) copolymers prepared using GG at different DVB content.
The DSC analysis provide analogous information (Figure 11). The maximum decomposition temperature (50 wt.% weight loss) for the copolymer bead prepared using GG is 410°C while for XG 425°C. No exothermic peak in the range 200–250 oC was observed. For this peak, polymer cross-linking is responsible. Absence of such peaks in the case of copolymer beads prepared using GG and XG indicates that all double bonds of monomers were consumed during copolymerization. The Tg values of the synthetic copolymer beads (with XG) for various contents of the DVB cross-linking agent (4–15 wt.%) are shown in Table 5. The Tg values were in the range of 107–133°C. It can be seen that the DVB content in the copolymer exerts a direct impact on the glass transition temperature of the resulting copolymer. The relation between Tg and the DVB content is presented in Figure 10.
Figure 11.

DSC thermograph of the copolymer bead at 6 wt.% DVB using XG as suspension stabilizer.
Figure 10.

Effect of the crosslinking agent concentration on glass transition temperature (Tg) of the copolymer beads.
The higher the DVB content, the higher the observed Tg values. Theoretically, the limited macromolecular relaxation by cross-linking site in the network reduces chain flexibility and elevates the Tg of the beads [66] as well as decreasing the solvent swelling Table 6. Furthermore, the Tg values of the synthesized copolymer beads for various toluene/heptane ratios of 100/0, 80/20, 60/40, 80/20 as diluents for the monomers are shown in Table 7. At a fixed cross-linking agent concentration, the Tg range of these polymers is about 107–109°C, which indicates that the variation of the toluene/heptanes volume ratio has an insignificant effect on the Tg values of the beads, because the composition of the monomer system is almost unchanged for cross-linking copolymerization.
Table 6.
Effect of the concentration of crosslinking agent on bead properties*.
| Run | D4 | D6 | D8 | D10 | D15 | ||
|---|---|---|---|---|---|---|---|
| Crosslinking agent concentration, wt% | 4 | 6 | 8 | 10 | 15 | ||
| Yield, % | 54 | 59 | 64 | 69 | 71 | ||
| Bead size distribution, wt% | |||||||
| ≤ 0.177 mm, > 80 mesh | 10.16 | 20 | 19.4 | 26.19 | 29.36 | ||
| 0.177–0.297 mm, 80 mesh | 57.62 | 50 | 58.45 | 52.38 | 49.19 | ||
| 0.297–0.707 mm, 50 mesh | 28.81 | 22.5 | 15.77 | 19.04 | 14.40 | ||
| ≥ 0.707mm, 25 mesh | 3.38 | 7.50 | 6.37 | 2.3 | 4.31 | ||
| % Swelling | 79.49 | 75.12 | 70.58 | 58.16 | 49.12 | ||
| Glass transition temperature, °C | 105 | 111 | 116 | 126 | 131 |
*Diluent composition (Toluene/Heptane: 3/2); Guar gum concentration, 2% wt
Table 7.
Effect of the Toluene/n-Heptane diluent composition on bead properties*.
| Run | H00 | H20 | H40 | H60 | H80 | H100 |
|---|---|---|---|---|---|---|
| Diluent composition (Tol./Hep), wt.% | 100/0 | 80/20 | 60/40 | 40/60 | 20/80 | |
| Yield, % | 58.5 | 60.75 | 64.7 | 59.6 | 58.78 | 56.5 |
| Bead size distribution, wt% | ||||||
| ≤ 0. mm, ≥ 80 mesh | 55.05 | 30.02 | 15.6 | 27.02 | 41.78 | 47.1 |
| 0.177–0.297 mm, 80 mesh | 25.41 | 59.66 | 78.12 | 61.08 | 50 | 28.26 |
| 0.297–0.707 mm, 50 mesh | 9.7 | 6.20 | 3.1 | 5.28 | 3.4 | 12.3 |
| ≥ 0.707mm, 25 mesh | 9.7 | 4.09 | 3.1 | 5.9 | 4.79 | 12.3 |
| Average bead size, mm | 0.275 | 0.255 | 0.25 | 0.26 | 0.243 | 0.29 |
| % Swelling | 65.20 | 67.12 | 76.20 | 64.40 | 55.40 | 50.10 |
| Glass transition temperature, °C | 107 | 107 | 109 | 108 | 109 | 109 |
| Surface area, m2/g | 18.445 | 24.246 | 31.24 | 38.214 | 43.21 | 45.20 |
| Pore volume, cm3/g | 0.102 | 0.115 | 0.155 | 0.162 | 0.172 | 0.218 |
*DVB concentration = 8 wt.%; Guar gum concentration, 2 wt.%
Differences between the results obtained from the TG and DSC analyses are caused by the oven atmosphere. In the case of DSC it is dynamic atmosphere of nitrogen while the TG measurements are carried out under static air atmosphere.
4. Conclusion
This contribution shows, for the first time, that guar and xanthan gums have potential as stabilizing agents in the synthesis of polymers by suspension polymerization as a result of their surface active properties. This approach allows tuning of the average particle size of the synthesized polymer beads by adjusting the concentration of the investigated natural polymers in the aqueous phase of the suspension. The effect of two new natural and biodegradable suspension stabilizers on the process of poly(St-DVB) bead formation was investigated. The concentration window of the stabilizers in toluene/heptane for formation of regular microspheres with smooth surface and nearly narrow size distribution was 2 wt%.The obtained porous copolymer beads may be useful for catalyst immobilization due to their high thermal stability and swelling ratio, and appropriate average diameter in number. This approach using environmentally benign suspension stabilizers could make a valuable contribution to the existing processes in the field of porous copolymer bead synthesis. Efforts to develop other biopolymer based systems and their combinations with synthetic ones in low amounts as suspending agents for applications in suspension and emulsion polymerizations are underway in our laboratory.
Funding Statement
This work was supported by the this work was partially supported by Shahid Beheshti University Research Council. ;
Acknowledgments
The authors gratefully acknowledge the partial support of this work by Shahid Beheshti University Research Council. We are also grateful from TPC, NISOC for providing chemicals.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Lu J, Toy Patrick H.. Chem Rev. 2009;109:815. [DOI] [PubMed] [Google Scholar]
- [2].Arshady R. Adv Mater. 1991;3(4):182. [Google Scholar]
- [3].Arshady R. Suspension, emulsion, and dispersion polymerization: A methodological survey. Colloid Polym Sci. 1992;270:717–732. [Google Scholar]
- [4].Huck CW, Bonn GK. Recent developments in polymer-based sorbents for solid-phase extraction. J Chromatogr. 2005;885(1–2):51. [DOI] [PubMed] [Google Scholar]
- [5].Sharma MM. Some novel aspects of cationic ion-exchange resins as catalysts. Funct Polym. 1995;26(1–3):3. [Google Scholar]
- [6].Walsh D, Wu D, Chang Y-T. Current Opinion in Chemical Biology. 2003;7:353. [DOI] [PubMed] [Google Scholar]
- [7].Davis ME. New vistas in zeolite and molecular sieve catalysis. Acc Chem Res. 1993;26(3):111. [Google Scholar]
- [8].Li J-R, Kuppler RJ, Zhou H-C. Selective gas adsorption and separation in metal–organic frameworks. Chem Soc Rev. 2009;38(5):1477. [DOI] [PubMed] [Google Scholar]
- [9].Lee J, Farha OK, Roberts J, et al. Metal–organic framework materials as catalysts. Chem Soc Rev. 2009;38(5):1450. [DOI] [PubMed] [Google Scholar]
- [10].Kaur P, Hupp Joseph T, Nguyen SonBinh T. Am Chem Soc Catal. 2011;1:819. [Google Scholar]
- [11].Arshady R, Ledwith A. Reactive Polym. 1983;1:159. [Google Scholar]
- [12].Arshady R. Preparation of polymer nano- and microspheres by vinyl polymerization techniques. J Microencapsulation. 1988;5:101. [DOI] [PubMed] [Google Scholar]
- [13].Arshady R. Microspheres and microcapsules: A survey of manufacturing techniques. Part 1: suspension cross-linking. Polym Eng Sci. 1989;29:1746. [Google Scholar]
- [14].Arshady R. Makromol Chem. 1988;189:1295–1303. [Google Scholar]
- [15].Arshady R. Angew Makromol Chem. 1989;170:43. [Google Scholar]
- [16].Arshady R, Basato M, Corain B, et al. Preparation of isocyano polymer supports and their complexes with catalytically relevant transition metal centers. J Mol Catal. 1989;53:111. [Google Scholar]
- [17].Sherrington DC. Chem. Commun. 1998;21:2275. [Google Scholar]
- [18].Yuan HG, Kalfas G, Ray WH. SUSPENSION POLYMERIZATION. J Macromol Sci., Rev Macromol Chem Phys. 1991;C31(2–3):215. [Google Scholar]
- [19].Vivaldo-Lima E, Wood PE, Hamielec AE, et al. An Updated Review on Suspension Polymerization. Ind Eng Chem Res. 1997;36(4):939. [Google Scholar]
- [20].Hamielec AE, Tobita H. Polymerization process. In ullmann’s encyclopedia of industrial chemistry. Weinheim, Germany: Wiley-VCH; 2002. [Google Scholar]
- [21].Sherrington DC In: Sherrington DC, Hodge P, editors. Polymer supported reactions in organic synthesis. Chichester: Wiley; Chapter 1; 1980. [Google Scholar]
- [22].Howdle SM, Jerabek K, Leocorbo V, et al. Sherrington DC. Polymer. 2000;41(19):7273–7277. [Google Scholar]
- [23].Erbay E, Okay O. Macroporous styrene-divinylbenzene copolymers: formation of stable porous structures during the copolymerization. Polym Bull. 1998;41(3):379. [Google Scholar]
- [24].Poinescu IC, Vlad CD. Effect of polymeric porogens on the properties of poly(styrene-co-divinylbenzene). Eur Polym J. 1997;33(9):1515. [Google Scholar]
- [25].Arshady R. Beaded polymer supports and gels. J Chromatogr. 1991;586:181. [Google Scholar]
- [26].Arshady R, Ledwith A. React Polym. 1983;1:159. [Google Scholar]
- [27].Brooks Brian W. Suspension polymerization processes. Chem Eng Technol. 2010;33(11):1737. [Google Scholar]
- [28].Kun KA, Kunin R. Macroreticular resins. III. Formation of macroreticular styrene–divinylbenzene copolymers. J Polym Sci Polym Chem. 1968;6:2689. [Google Scholar]
- [29].Millar JR, Smith DG, Kressman TRE. J Chem Soc. 1965;304. [Google Scholar]
- [30].Coutinho FMB, Neves MAFS, Dias ML. Porous structure and swelling properties of styrene-divinylbenzene copolymers for size exclusion chromatography. JAppl Polym Sci. 1997;65(7):1257. [Google Scholar]
- [31].Garcia-Diego C, Cuellar J. Synthesis of macroporous poly(styrene-co-divinylbenzene) microparticles using n-heptane as the porogen: quantitative effects of the DVB concentration and the monomeric fraction on their structural characteristics. Ind Eng Chem Res. 2005;44:8237–8247. [Google Scholar]
- [32].Tank R, Beldar A, Saxena A, et al. Effects of crosslinking agents and inert diluents on the characteristics of crosslinked polystyrene copolymers. J Appl Polym Sci. 2008;108:1531. [Google Scholar]
- [33].Vivaldo-Lima E, Wood PE, Hamielec AE. An updated review on suspensionpolymerization. Ind Eng Chem Res. 1997;36:939. [Google Scholar]
- [34].Zhang H, Wang X. Fabrication and performances of microencapsulated phase change materials based on n-octadecane core and resorcinol-modified melamine–formaldehyde shell. Eng Aspects. 2009;332:129. [Google Scholar]
- [35].Yu F, Chen Z, Zeng X. Colloid. Polym Sci. 2009;287:549. [Google Scholar]
- [36].Su JF, Wang LX, Ren L, et al. Mechanical properties and thermal stability of double-shell thermal-energy-storage microcapsules. J Appl Polym Sci. 2006;103:1295. [Google Scholar]
- [37].Jacobelli H, Bartholin M, Guyot A. Styrene divinyl benzene copolymers. I. Texture of macroporous copolymers with ethyl-2-hexanoic acid in diluent. J Appl Polym Sci. 1979;23:927. [Google Scholar]
- [38].Dowding PJ, Vincent B. Suspension polymerisation to form polymer beads. Colloids Surf, A. 2000;161:259. [Google Scholar]
- [39].Fan X, Zhang Z, Li G, et al. Attachment of solid particles to air bubbles in surfactant-free aqueous solutions. Chem Eng Sci. 2004;59:2639. [Google Scholar]
- [40].Bachtsi AR, Boutris CJ, Kiparissides C. Production of oil-containing crosslinked poly(vinyl alcohol) microcapsules by phase separation: effect of process parameters on the capsule size distribution. J Appl Polym Sci. 1996;60:9. [Google Scholar]
- [41].Hong K, Nakayama K, Park S. Effects of protective colloids on the preparation of poly(l-lactide)/poly(butylene succinate) microcapsules. Eur Polym J. 2002;38:305. [Google Scholar]
- [42].Sánchez L, Sánchez P, De Lucas A, et al. Microencapsulation of PCMs with a polystyrene shell. Colloid Polym Sci. 2007;285:1377. [Google Scholar]
- [43].Rahmatpour A. Polystyrene-supported GaCl3: A new, highly efficient and recyclable heterogeneous Lewis acid catalyst for tetrahydropyranylation of alcohols and phenols. Polyhedron. 2012;44:66. [Google Scholar]
- [44].Grulke EA. Encyl Polym Sci Eng. 1989;19:443. [Google Scholar]
- [45].Warson H. Polym Paint Color J. 1984;10:541 Grulke EA 1. [Google Scholar]
- [46].Konno M, Arai K. The effect of stabilizer on coalescence of dispersed drops in suspension polymerization of styrene. J Chem Eng Jpn. 1982;15:131. [Google Scholar]
- [47].Horak D, Pelzbauer Z, Svec F, et al. Reactive polymers. XXXIII. The influence of the suspension stabilizer on the morphology of a suspension polymer. J Appl Polym Sci. 1981;26:3205. [Google Scholar]
- [48].Coutinho FMB, Cid RCA. Styrene-divinylbenzene copolymers-formation of porous structure by using precipitants as diluents in suspension polymerization. Eur Polym J. 1990;26(11):1185. [Google Scholar]
- [49].Zhang HT, Huang JX, Jiang BB. Kinetics of polymerization and particle stabilization mechanism on dispersion copolymerization of styrene and divinylbenzene. J Appl Polym Sci. 2002;85:2230. [Google Scholar]
- [50].Rabelo D, Coutinho FMB. Porous structure formation and swelling properties of styrene-divinylbenzene copolymers. Eur Polym J. 1994;30(6):675. [Google Scholar]
- [51].Rabelo D, Coutinho FMB. Free-radical decay in a polymer blend PMMA/PEO studied by the EPR method. Eur Polym J. 1992;28(12):553. [Google Scholar]
- [52].Okay O, Angew D. Makromol. Chemie. 1988;157:15. [Google Scholar]
- [53].Luz CTL, Coutinho FMB. The influence of the diluent system on the porous structure formation of copolymers based on 2-vinylpyridine and divinylbenzene. Diluent system. III. heptane/methylethylketone. J Appl Polym Sci. 2004;91:666. [Google Scholar]
- [54].Rahmatpour A. Polystyrene-supported GaCl3 as a highly efficient and recyclable heterogeneous Lewis acid catalyst for one-pot synthesis of N-substituted pyrroles. J Organomet Chem. 2012;712:15. [Google Scholar]
- [55].Rahmatpour A. Polystyrene-supported GaCl3: A new, highly efficient and recyclable heterogeneous Lewis acid catalyst for acetylation and benzoylation of alcohols and phenols. Comptes Rendus Chem. 2012;15:1048. [Google Scholar]
- [56].Rahmatpour A. Polystyrene-supported GaCl3 as a highly efficient and reusable heterogeneous Lewis acid catalyst for the three-component synthesis of benzoxanthene derivatives. Monatsh Chem. 2013;144:1205. [Google Scholar]
- [57].Rahmatpour A, Vakili A, Azizian S. Polystyrene-Gallium Trichloride complex: a mild, highly efficient, and recyclable Polymeric Lewis Acid catalyst for chemoselective silylation of Alcohols and Phenols with Hexamethyldisilazane. Heteroat Chem. 2013;24(6):443. [Google Scholar]
- [58].Wolters D, Meyer-Zaika W, Bandermann F. Macromol. Mater Eng. 2001;286:94. [Google Scholar]
- [59].Izydorczyk M, Cui Steve W, Wang Q. Polysaccharide gums: structures, functional properties, and applications. Boca Raton: Taylor & Francis Group, LLC; 2015. Chapter 6. [Google Scholar]
- [60].Alsewailem Fares D. Applied researches in Polysaccharides. 2015;2:125–137. [Google Scholar]
- [61].GarcõÂa-Ochoaa F, Santosa VE, Casasb JA, et al. Xanthan gum: production, recovery, and properties. Biotech Adv. 2000;18:549–579. [DOI] [PubMed] [Google Scholar]
- [62].Dutta S. World J Pharma Life Sci. 2015;1(1):50–84. [Google Scholar]
- [63].Mudgil D, Barak S, Khatkar BS. Guar gum: processing, properties and food applications—a review. J Food Sci Tech. 2014;51(3):409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Barrett KE. Dispersion polymerization inorganic media. New York: John Wiley; 1974. [Google Scholar]
- [65].Mahdavian AR, Khoee S. A facile and efficient method for preparation of chiral supported poly(styrene–divinylbenzene) copolymers. React Func Polym. 2002;50:217. [Google Scholar]
- [66].Vaino Andrew R, Janda Kim D. Rapid identification of substrates for novel proteases using a combinatorial peptide library. J Comb Chem. 2000;2:579. [DOI] [PubMed] [Google Scholar]