

Abstract
While accounting for only 2% of the body's weight, the brain utilizes up to 20% of the body's total energy. Not surprisingly, metabolic dysfunction and energy supply-and-demand mismatch have been implicated in a variety of neurological and psychiatric disorders. Mitochondria are responsible for providing the brain with most of its energetic demands, and the brain uses glucose as its exclusive energy source. Exploring the role of mitochondrial dysfunction in the etiology of psychiatric disease is a promising avenue to investigate further. Genetic analysis of mitochondrial activity is a cornerstone in understanding disease pathogenesis related to metabolic dysfunction. In concert with neuroimaging and pathological study, genetics provides an important bridge between biochemical findings and clinical correlates in psychiatric disease. Mitochondrial genetics has several unique aspects to its analysis, and corresponding special considerations. Here, we review the components of mitochondrial genetic analysis – nuclear DNA, mitochondrial DNA, mitochondrial pathways, pseudogenes, nuclear-mitochondrial mismatch, and microRNAs – that could contribute to an observable clinical phenotype. Throughout, we highlight psychiatric diseases that can arise due to dysfunction in these processes, with a focus on schizophrenia and bipolar disorder.
Keywords: Mitochondria, Mitochondrial DNA, Oxidative phosphorylation, Mitonuclear crosstalk, Mitochondrial dynamics, Gene set analysis, Schizophrenia, Bipolar disorder
Introduction
Mitochondria are intracellular organelles that have coexisted with eukaryotic cells as endosymbionts for nearly 2 billion years. Mitochondria are the metabolic powerhouse of the cell, primarily responsible for the conversion of energy-carrying molecules into ATP through the process of oxidative phosphorylation (OXPHOS). In addition to their metabolic function, mitochondria regulate other cellular processes such as reactive oxygen species (ROS) generation, calcium flux, and apoptosis [1].
Mitochondrial function within the cell is finely tuned, and physiologic control is more complex than in other organelles. Unlike other organelles in animal cells, mitochondria carry their own genetic information, termed mitochondrial DNA (mtDNA). In humans, 37 genes are encoded by the mtDNA genome. The vast majority of genes involved in mitochondrial function, however, are located in the nucleus. Indeed, proteins encoded by over 1,100 genes operate within mitochondria [2], many of which have been implicated in neurological and psychiatric diseases.
The human brain has a high energy demand, and it is not surprising that metabolic dysfunction has been linked to a host of neurological and psychiatric disease states. Mitochondria are the main source of energy for neurons and they play an important role in many neuronal processes such as neurodevelopment, neurotransmission, and synaptic plasticity [3, 4, 5]. Due to its fundamental importance, it is reasonable to hypothesize that dysfunction of the mitochondrial organelle in the brain may play a major role in the pathophysiology of mental illness [6, 7, 8, 9]. This is particularly true for those dysfunctions involving a disturbance of energy metabolism, as proposed for schizophrenia (SCZ) and bipolar disorder (BD) [10, 11, 12, 13].
Variation in mitochondrial genetics includes SNPs, deletions, intracellular variation in mtDNA copies (heteroplasmy), disrupted pathways, epigenetics, and chemical changes, which may influence mitochondrial gene expression and function [14, 15]. Certain inherited mutations in mtDNA cause congenital, multisystem disorders with neuropsychiatric manifestations, such as MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes) [16] and Leigh syndrome [17]. mtDNA impairment also contributes to multifactorial conditions such as cancer, metabolic disease, and aging [15]. Furthermore, loss of mtDNA integrity leads to dysfunction in mitochondrial activity, potentially leading to neurodegenerative diseases such as Parkinson and Alzheimer disease [18, 19].
In this paper, we will explore the various cellular and metabolic pathways in which mitochondria participate, with a focus on mitochondrial genetics and pathways (Fig. 1). We will look at genetic pathways involved in mitochondrial function – including OXPHOS, mitonuclear crosstalk, and mitochondrial dynamics – and provide an overview of the techniques used to investigate mitochondrial pathways, such as gene set analysis. These pathways are specifically related to mitochondrial activity with respect to energy generation. We will touch on the analysis of pseudogenes, microRNA (miRNA), and nuclear-mitochondrial mismatch in the pathogenesis of psychiatric conditions. Throughout, we will highlight psychiatric diseases that can arise due to dysfunction in these organelles.
Fig. 1.
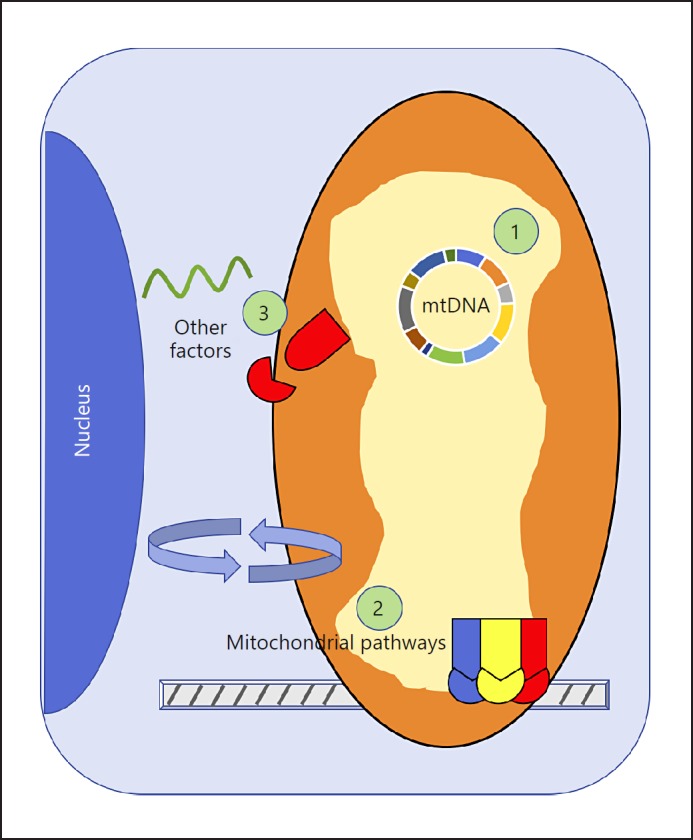
Mitochondrial genetics and pathways that contribute to psychiatric disease. On a genetic level, mitochondrial dysfunction can be caused by several mechanisms. (1) Mitochondrial DNA (mtDNA) – Variation within the mtDNA can contribute to mitochondrial dysfunction. Homoplasmic variants arise via maternally inherited mutations within the mtDNA, and are distributed equally across organs and cells. Heteroplasmic variants can be either inherited or acquired and generally increase over time, leading to heterogeneity in mtDNA sequences. Haplogroups are linked sets of mtDNA genetic variants shared between individuals as a consequence of common ancestry. (2) Mitochondrial pathways – Biological pathways under nuclear genetic control are necessary to ensure proper mitochondrial function. Oxidative phosphorylation facilitates aerobic respiration and provides ATP for the cell. Mitonuclear crosstalk describes the communication between the mitochondria and the nucleus. Mitochondrial dynamics includes fusion, fission, transport, and mitophagy, and ensures an appropriate number of mitochondria in the right location within the cell. (3) Other mitochondrial factors – MicroRNAs add an additional level of control over gene expression. The nuclear-mitochondrial mismatch phenomenon describes the coevolution of two distinct genomes (nDNA and mtDNA), and the hypothetical incompatibility of combining evolutionarily divergent genomes (nuclear and mitochondrial). Mitochondrial pseudogenes are sequences of mtDNA that have been incorporated into the nuclear genome, but are themselves noncoding.
Mitochondrial Genetics
Mitochondrial Genes
Genes coding for proteins involved in cellular metabolism and mitochondrial function are located both in the nucleus and in the mitochondria themselves. Even genes that ultimately contribute to the same protein complex may be from both mtDNA and nuclear DNA (nDNA). For example, of the five protein complexes that make up the OXPHOS pathway, complexes I, III, IV, and V are composed of a combination of genes found within the mitochondria and the nucleus (genes for complex II are encoded exclusively by the nucleus) [20]. Thus, when assessing for a mitochondrial genetic contribution to a specific phenotype or disease, both nDNA and mtDNA must be analyzed.
The mitochondrial genome is approximately 16.5 kb in length and encodes 37 genes in humans: 13 genes code for peptides involved in the OXPHOS protein complexes, 22 genes code for transfer RNAs (tRNAs), and 2 genes code for ribosomal RNAs (rRNAs) [21, 22], as shown in Figure 2. Specifically, the mtDNA genes include 7 genes of complex I (ND1–6), 1 gene of complex III (Cyt b), 3 genes of complex IV (COI–III), and 2 genes of complex V (ATP synthase; ATP6 and ATP8). In genetic analysis, nucleus-encoded mitochondrial genes can be treated like other nuclear genes and are amenable to genome-wide association studies (GWAS), described elsewhere [23]. In this section we will focus on some of the unique attributes of genetic analysis involving mtDNA. These include SNP variation, haplogroup stratification, and heteroplasmy.
Fig. 2.

Annotated map of mitochondrial genome (mtDNA) genome for schizophrenia (SCZ) and bipolar disorder (BD). The human mtDNA is 16,569 bp in length and codes for 13 oxidative phosphrylation genes, 22 tRNAs, and 2 rRNAs. SNPs conferring a risk for developing SCZ and BD are shown.
It is estimated that in humans the rate of mutation is roughly 10–20 times greater in mtDNA than in nDNA [15]. Consequently, there is more variation per unit length in mtDNA than there is in nDNA. As SNP variants in the mitochondrial genome are common within the general population, especially in the noncoding, poorly conserved mtDNA regions, researchers have developed a series of criteria to support the authenticity of postulated pathogenic variants [24]. These “canonical criteria” are as follows: (1) the mutation is not a known neutral polymorphism; (2) the mutation must be in an evolutionarily conserved site, suggesting a functional role for the SNP; (3) the mutation is usually heteroplasmic; (4) for heteroplasmic variants, the degree of heteroplasmy should correlate clinically with a phenotype; and (5) the mutation can be corroborated biochemically using tissue samples (initially described with muscle biopsy). With respect to determining whether a particular variant is novel or not, several databases exist, including dbSNP [25], MITOMAP [26], mtDB [27], PhyloTree [28] and MSeqDR [29]. We currently refer to mtDB and MSeqDR for most applications.
Heteroplasmy refers to heterogeneity within the mtDNA sequences of a single organism, or even within a single cell. Most of these heteroplasmic variants are completely benign, occurring within the noncoding mtDNA D-loop region, and one-quarter of all humans inherit a mixture of these genomes at fertilization through complete maternal inheritance [30]. Notwithstanding, pathogenic heteroplasmic variants can be found as the source of mitochondrial genetic disorders, and the clinical symptomatology usually correlates with the degree of heteroplasmy [31]. Furthermore, these variants usually accumulate with age, and disorders engendered by heteroplasmic variants are often acquired via accumulation with age [8].
An important area of study with respect to heteroplasmy is how these mutations are propagated from mother to daughter and subsequent maternal progeny. The mechanism could be through vegetative segregation, relaxed replication, replicative advantage, maintenance of wild types, or a combination of the four [31]. In vegetative segregation, there is unequal partitioning of mitochondria from the parent to the daughter cell, resulting in a skewed distribution of the heteroplasmic variants in the offspring. Alternatively, heteroplasmy levels can also change in otherwise nondividing cells, as mitochondria and mtDNA are continually recycled in a process called relaxed replication. Interestingly, the level of heteroplasmy within a given cell can increase, decrease, or remain stable over time, and this effect is often tissue specific. The underlying mechanisms are not yet worked out, but it is likely that nuclear genes influence this regulation. Another way that heteroplasmy levels can change is through a process called replicative advantage [32]. In replicative advantage, the mutation itself confers either an advantage or a disadvantage relative to the wild-type copy with respect to mtDNA replication. For example, a heteroplasmic change with a large deletion may produce an mtDNA molecule that requires less time to replicate, allowing for favored replication of the mutated version relative to the wild type [33]. In this way, heteroplasmy levels set within the cell. Lastly, in maintenance of the wild type hypothesis, mtDNA replication is upregulated as a whole in the setting of heteroplasmic mutations in order to maintain a steady-state concentration of wild-type mtDNA [34]. The net result of this process is that, in an attempt to maintain normal wild type levels, the mutated mtDNA is also increased due to the nonselective mitochondrial biogenesis. In other words, the presence of nonfunctional heteroplasmic variants forces the cell to increase the number of mitochondria in the cell in order to keep up with the metabolic demand, which nonselectively increases both wild-type and heteroplasmic forms.
On the other hand, various internal control mechanisms may restrict the propagation of damaged or heteroplasmic mitochondria. On an organismal level, mutated mtDNAs that result in nonfunctional OXPHOS complexes could cause impaired respiratory chain function, leading to an altered mitochondrial membrane potential, which feeds back to the nucleus to restrict the transport of essential nucleus-encoded mitochondrial proteins to those specific mitochondria [31]. Additionally, the level of heteroplasmy passed down from mother to offspring during oogenesis is lower than the somatic heteroplasmy of the mother. This is thought to be controlled through the hypothetical “genetic bottleneck,” whereby the amount of mtDNA is deliberately kept low in primordial germ cells so as to limit inherited heteroplasmy [35], although the mechanism remains unclear.
There is a high correlation between ancestry and specific mtDNA variants. These linked sets of mtDNA genetic variants shared between individuals as a consequence of common ancestry are called haplogroups. Several studies have found haplogroup-specific susceptibility to a host of phenotypes ranging from neuropsychiatric disorders to aging profiles [36, 37]. Haplogroups are traditionally assigned an alphabetical letter corresponding to the particular set of gene variants harbored by their mtDNA. The standard haplogroup phylogenetic tree was established by van Oven and Kayser [28] and is updated online at http://www.phylotree.org. Interestingly, biochemical differences in OXPHOS activity have been observed between haplogroups, which may explain why individuals from certain haplogroups reach a pathological bioenergetic threshold more readily than others [38].
In summary, when analyzing mtDNA, one must consider SNP variants, heteroplasmy, and haplogroups. Publicly available pipelines, such as mtDNA-Server [39], can be used to analyze mtDNA sequences to detect each of these genetic components for further investigations.
Mitochondrial Genetics in Disease
For over a decade, there has been much interest in exploring the relationship between mitochondrial genetic variation and psychiatric disease, especially for SCZ and BD. One way to test whether mitochondria play a causal role in major psychoses is to search for inherited variants in mitochondrial genes that influence the risk of the disease.
For SCZ, positive findings were reported regarding a relationship between haplogroup HV and SCZ in an Israeli-Arab sample [40], and between haplogroup J-T and onset of SCZ in an Italian sample [41]. In another example, Hudson et al. [42] found nominally significant associations between SCZ and SNPs defining haplogroup U. However, other studies do not support these findings [43, 44, 45, 46], although the approaches used to assign individuals to haplogroups varied between the studies. For example, the Israeli-Arab sample study found a significant association when dividing the HV lineage into the H haplogroup and the non-H HV lineage haplogroups, while Mosquera-Miguel et al. [45] found no association with SCZ by including four distinct haplogroups (H, H1, H3, and HV). As for specific loci, a small study identified the 12096A heteroplasmic variant, which codes for ND4, in 5 of 6 mother/child pairs with SCZ, and in no controls [47].
Recently, our group performed the largest study thus far to investigate the role of mtDNA SNPs in SCZ risk (n = 4,778 cases; 5,819 controls). Using data from the Illumina HumanExome array chip, we tested for genetic associations of common and rare SNPs, and haplogroups, with SCZ susceptibility. We found nominally significant associations for a SNP in the cytochrome B gene (p = 0.007) and for haplogroup J (p = 0.02), with the p value threshold set at 0.001. Rare variants were also nominally significantly associated with SCZ (burden p = 0.007) [48].
For BD, Kato et al. [49] reported that the mtDNA mutations 5178C and 10398A were statistically associated with a lower brain intracellular pH in BD subjects in comparison with healthy controls. In another study, mutation 3644C was suggested to increase the risk for BD, and the finding was replicated in an independent sample [50]. In addition, the 3243G variant was shown to be associated with both SCZ and BD [51]. Kazuno et al. [52] showed that haplogroup N9a was overrepresented among BD subjects. They also reported that mutation 12358G was associated with BD, but they could not replicate this association in another sample. Haplogroup U has also been identified as predisposing patients with BD to symptoms of psychosis [53]. Figure 2 outlines the mtDNA mutations that have been implicated in SCZ and BD.
Integrity of the mitochondrial genome is crucial for normal neuronal function. Impaired function can lead to an increase in ROS production. mtDNA mutations can lead to greater electron leakage along the electron transport chain (ETC), exceeding the cell's antioxidant potential. This mechanism is thought to increase the risk of developing psychotic disorders such as SCZ and BD [54]. Evolutionary pressures such as ambient temperatures influenced the development of OXPHOS efficiency. For example, in haplogroup N, following northward migration from Africa to Europe, the 10398G>A variant encoding the A114T substitution reduced the coupling efficiency of the ETC complexes, resulting in greater heat production per generation of ATP [55]. This phenomenon may explain the predilection of certain haplogroup associations with mental illness. Either oxidative damage to proteins, lipids, or DNA within neurons or the resultant accumulation of toxic metabolites can explain the pathogenesis of psychiatric conditions as a result of ROS generation [54].
Mutations in the mitochondrial genome may also lead to an underproduction in ATP available for neuronal activity. As with congenital mitochondrial disorders such as MELAS, the precise mechanism by which decreased energy production contributes to neuropsychiatric symptoms is unknown. Association studies have found a decrease in pH in BD, presumably secondary to increased lactic acid production via glycolysis, as will be described later in the section devoted to the OXPHOS pathway.
We have focused thus far on the mitochondrial genome, and how perturbations can impact function. Next, we will expand our consideration of mitochondrial dysfunction to encompass the nuclear genome, focusing on pathways coordinated by the mitochondria.
Mitochondrial Pathways
Genetic Analysis of Mitochondrial Pathways
While shown to be heritable, most psychiatric disorders such as SCZ and BD are polygenic, with multiple affected loci conferring a risk for developing the disease [56]. As such, GWAS looking for single-gene variants may struggle to detect small contributions from multiple genes. To address this limitation, a relatively new technique of analysis of highly complex diseases with a polygenic etiology is gene set analysis [57, 58]. Instead of looking for variation in particular SNPs or genes, in gene set analysis geneticists look for changes in biological pathway activity. This is accomplished by selecting a group of genes to be analyzed together, usually related to each other by virtue of a common biological pathway such as OXPHOS – or mitochondrial dynamics in the case of mitochondrial genetics.
Gene set analysis is typically used as a secondary tool for genetic analysis and can complement prior GWAS [59]. In selecting a gene set, a combination of gene pathway databases and manual curation from a literature search is advisable. Examples of these gene sets can be found in Tables 1, 2, 3, described in greater detail below. Ideally, a gene set should contain between 10 and 200 genes [60]. Next, the statistical method and analytical tool can be selected.
Table 1.
OXPHOS pathway gene set genes
| Complex I | Complex II | Complex III | Complex IV | Complex V | Other | |
|---|---|---|---|---|---|---|
| NDUFA1 | NDUFS7 | SDHA | CYC1 | COX4I1 | ATP5A1 | AARS2 |
| NDUFA2 | NDUFS8 | SDHB | CYCS | COX4I2 | ATP5B | BID |
| NDUFA3 | NDUFV1 | SDHC | UQCR10 | COX5A | ATP5C1 | COQ9 |
| NDUFA4 | NDUFV2 | SDHD | UQCRB | COX5B | ATP5D | DLD |
| NDUFA5 | NDUFV3 | SDHAF1 | UQCRC1 | COX6A1 | ATP5E | IMMP1L |
| NDUFA6 | MT-ND1 | SDHAF2 | UQCRC2 | COX6A2 | ATP5F1 | IMMP2L |
| NDUFA7 | MT-ND2 | SDHAF3 | UQCRFS1 | COX7B | ATP5G1 | SAMM50 |
| NDUFA8 | MT-ND3 | UQCRH | COX7C | ATP5G2 | ||
| NDUFA9 | MT-ND4 | UQCRQ | COX8A | ATP5G3 | ||
| NDUFA10 | MT-ND4L | MT-CYB | COX8C | ATP5H | ||
| NDUFA11 | MT-ND5 | BCS1L | MT-CO1 | ATP5I | ||
| NDUFA12 | MT-ND6 | LYRM7 | MT-CO2 | ATP5J | ||
| NDUFA13 | ACAD9 | PMPCB | MT-CO3 | ATP5J2 | ||
| NDUFAB1 | ATP5SL | SLC25A33 | BCS1L | ATP5L | ||
| NDUFAF1 | BCS1L | TTC19 | CHCHD5 | ATP5O | ||
| NDUFB1 | COA1 | UQCC1 | COA1 | ATP5S | ||
| NDUFB2 | ECSIT | UQCC2 | COA3 | ATPIF1 | ||
| NDUFB3 | FOXRED1 | UQCR10 | COA4 | MT-ATP6 | ||
| NDUFB4 | NDUFAF2 | UQCR11 | COA5 | MT-ATP8 | ||
| NDUFB5 | NDUFAF3 | COX10 | ATPAF1 | |||
| NDUFB6 | NDUFAF4 | COX14 | ATPAF2 | |||
| NDUFB7 | NDUFAF5 | COX15 | OXA1L | |||
| NDUFB8 | NDUFAF6 | COX17 | PPIF | |||
| NDUFB9 | NDUFAF7 | COX19 | TMEM70 | |||
| NDUFB10 | NDUFAF8 | COX20 | USMG5 | |||
| NDUFB11 | NUBPL | COX6B1 | ||||
| NDUFC1 | OXA1L | COX6C | ||||
| NDUFC2 | PARK7 | COX7A2L | ||||
| NDUFS1 | TIMM21 | PET100 | ||||
| NDUFS2 | TIMMDC1 | PET117 | ||||
| NDUFS3 | TMEM126B | SCO1 | ||||
| NDUFS4 | SCO2 | |||||
| NDUFS5 | SURF1 | |||||
| NDUFS6 | TIMM21 |
Functionally related gene sets were selected from the Gene Ontology Consortium [152]. Genes that are categorized under the terms describing the structure or assembly of the respiratory complexes were included in the oxidative phosphorylation (OXPHOS) pathway. The genes were reconciled with MitoCarta and the literature where necessary.
Table 2.
Mitonuclear crosstalk gene set genes
| Anterograde crosstalk | Retrograde crosstalk | Quality control | Integrated cellular stress response |
|---|---|---|---|
| CAMK4 | ATF2 | AFG3L2 | ATF3 |
| CAMKK2 | CAMK4 | AKTl | ATF4 |
| CREB | CEBPG | CEBPG | DDIT3 |
| ESRRA | CREB | CLPP | EIF2AK4 |
| ESRRB | DDIT3 | CLPX | EIF2S1 |
| ESRRG | EGRI | COX5B | TRIB3 |
| GABPA | KEAP1 | DDIT3 | |
| MEF2A | MAPK1 | DNAJA3 | |
| NCOR1 | MAPK8 | FOXO3 | |
| NRF1 | MAPK9 | HSF1 | |
| NRIP1 | MAPK10 | HSP90AA1 | |
| PARP1 | MTOR | HSPA4 | |
| PARP2 | NFATC1 | HSPD1 | |
| PPARA | NFE2L2 | HSPE1 | |
| PPARG | NFKB1 | HTRA2 | |
| PPARGC1A | NFKB2 | JUN | |
| PPARGC1B | PPARGC1A | LONP1 | |
| PPRC1 | PPP3CC | MAPK9 | |
| PRKAA1 | PRKAA1 | NFKB2 | |
| PRKACA | NRF1 | ||
| PRKACB | PMPCB | ||
| SIRTl | SIRT3 | ||
| TP53 | SIRT7 | ||
| SMARCA4 | |||
| SPG7 | |||
| SSBP1 | |||
| STAR | |||
| TIMM17A | |||
| TXN2 | |||
| YME1L1 |
Table 3.
Mitochondrial dynamics gene set genes
| Fusion | Fission | Transport | Mitophagy |
|---|---|---|---|
| AFG3L2 | BNIP3 | CLUH | ATPIF1 |
| BAK1 | COX10 | DNM1L | BNIP3 |
| BAX | DHODH | KIF1B | BNIP3L |
| BNIP3 | DNM1L | LRPPRC | CISD2 |
| CHCHD3 | DNM2 | LRRK2 | DNM1L |
| DNM1L | DNM3 | MAIP1 | FIS1 |
| FIS1 | FIS1 | MAP1S | FUNDC1 |
| GDAP1 | GDAP1 | MFN1 | FUNDC2 |
| MFF | LRRK2 | MFN2 | HK2 |
| MFN1 | MARCH5 | MGARP | HTRA2 |
| MFN2 | MFF | MST01 | MFN2 |
| MIEF1 | MIEF1 | MULI | MULI |
| MIEF2 | MIEF2 | OPA1 | PARK7 |
| MUL1 | MTFP1 | RHOT1 | PINK1 |
| OMA1 | MTFR1 | RHOT2 | SQSTM1 |
| OPA1 | MTFR1L | SYNJ2BP | TOMM7 |
| PLD6 | MTFR2 | TRAK1 | TSPO |
| PRKN | MUL1 | TRAK2 | VDAC1 |
| STOML2 | MY019 | UBB | |
| USP30 | OPA1 | ||
| PINK1 | |||
| PRKN | |||
| SLC25A46 |
Functionally related gene sets were selected from the Gene Ontology Consortium [152]. Genes that are categorized under the terms describing mitochondrial fission, fusion, axonal transport, membrane organization, localization, and mitophagy were included in the mitochondrial dynamics pathway. The genes were reconciled with MitoCarta and the literature where necessary.
MAGMA and INRICH are two platforms that can be used to conduct gene set analyses. MAGMA uses competitive gene set analysis to detect genetic pathways enriched for a particular phenotype. In MAGMA, SNPs are combined and assigned to a gene, and an “association score” is calculated for each gene included in the analysis with respect to the phenotype in question. The group of association scores for the genes included in the gene set of interest is compared to the remaining genes using a one-tailed two-sample t test and is assessed for significance [61]. INRICH is also a competitive gene set analysis tool that assigns chromosomal intervals to a set of variants with high significance and then determines whether genes falling within these intervals belong to certain pathways. The results are compared to multiple null sets consisting of random intervals, and considered significant if the true test data show a greater correlation to the pathway genes [62]. Additional information regarding existing tools for this kind of analysis, as well as a performance comparison between them, was described elsewhere [61]. Guidance regarding the gene set analysis procedure is provided by Mooney and Wilmot [60].
Although a powerful tool, gene set analysis has several limitations and warrants special considerations. These include generating a list of genes from a series of SNPs, dealing with overlapping genes involved in multiple gene sets, and inconsistent gene set sizes, among other factors. These considerations are discussed at length in a review by Wang et al. [57].
In considering a genetic mitochondrial basis for psychiatric disease, it is valuable to address the physiologic role of mitochondria within a cell. Mitochondria are part of different cellular circuits, and the mitochondrial gene sets or pathways might differ between diseases. Since there is evidence that the underlying pathophysiology of SCZ is related to impaired brain metabolism, and since mitochondrial dysfunction has been correlated with SCZ in several studies, an aberrant mitochondrial pathway is an attractive theory for SCZ pathogenesis.
Evidence has suggested that nucleus-encoded mitochondrial genes may play a role in SCZ [63, 64], although the mitochondrion-related pathways were not significantly associated with the disease (n = 9,379 cases; 7,736 controls) [65]. Further evidence includes an enrichment of expression from quantitative trait locus variants in nucleus-encoded mitochondrial genes [66], and an excess of large (> 500-kb deletions) and rare copy number variants in the set of nucleus-encoded mitochondrial genes in SCZ patients [67]. Our group recently conducted a hypothesis-driven mitochondrial gene-based analysis using the Psychiatric Genomics Consortium (PGC) summary statistic results for SCZ to test whether variants in nucleus-encoded mitochondrial genes of pathways influence susceptibility to SCZ [154]. Our findings identified 19 genes that were significantly associated with SCZ, the associations of 9 of which were replicated in an independent sample. For BD, there are fewer studies including mitochondrial nuclear genes than for SCZ, but the Psychiatric GWAS Consortium Bipolar Disorder Working Group has failed to identify mitochondrial nuclear loci to be associated with the disease [68].
Oxidative Phosphorylation
OXPHOS within mitochondria is the main source of aerobic energy for neuronal functioning. OXPHOS converts high energy reducing agents to usable energy by transferring electrons along the ETC and then coupling the generated electrochemical gradient to produce an electromotive force, which is harnessed to convert ADP into ATP. First, NADH is oxidized by complex I, and succinate is oxidized by complex II, after which the electrons are transferred to coenzyme Q, then to complex III, then to complex IV, and lastly to the final electron acceptor, oxygen, to generate water. The passage of these electrons through the ETC generates energy, which is used to pump protons from the mitochondrial matrix into the mitochondrial inner membrane. The resulting electrochemical gradient draws protons through complex V, also called ATP synthase, which converts this kinetic energy into chemical energy as a high-energy phosphate in the form of ATP [55].
Complex I, also called NADH:ubiquinone oxidoreductase, is the largest member of the OXPHOS system, comprising 45 subunits. Complex I is responsible for transferring electrons from NADH to coenzyme Q. It contributes nearly 40% of the electromotive force and generates significant ROS in the process [69]. Complex I and complex III contain Fe-S clusters, and are therefore the most susceptible to oxidative damage [70]. Although contributing less to the overall energy production, complex IV has traditionally been used as a metric for neuronal energy utilization [71]. Both complex I and complex IV have been investigated most extensively in the context of psychiatric disorders, as will be described later. In addition to the subunits themselves, genes involved in the assembly of the ETC complexes have been implicated in disease. These assembly genes, as well as the remaining OXPHOS genes encoded by both the mitochondrial and the nuclear genomes, are listed in Table 1.
Numerous imaging studies have shown that the brain's metabolic activity is disrupted in SCZ. Biochemical studies investigating OXPHOS integrity and specific ETC complex function in SCZ have returned inconsistent results. Ben-Shachar [20] reported increased activity of complex I in SCZ patients with active (acute or chronic) psychosis, with decreased complex I activity in patients with residual SCZ, as compared to healthy controls. Additionally, dopamine binds to and reversibly inhibits complex I, further suggesting an association between complex I and SCZ. In contrast, earlier studies reported reduced complex IV activity in SCZ with unaffected complex I [72, 73]. Since many studies have been conducted on either postmortem samples or extracted blood products, it is suggested that these differing experimental conditions may have contributed to the conflicting findings. In support of OXPHOS dysfunction in SCZ, a small parallel transcriptomic, proteomic, and metabolomic study found OXPHOS to be the most significantly downregulated pathway in patients compared to controls [74]. More details on the role of the OXPHOS pathway in the context of SCZ pathophysiology can be found in a recent review [75].
With respect to other psychiatric disorders, Andreazza et al. [76] found that complex I activity is decreased in the prefrontal cortex of BD patients compared to controls. Parenthetically, this study did not detect a significant difference in complex I activity between SCZ patients and controls. Lending further support to OXPHOS impairment in BD, numerous studies have described an association between BD and lower intracellular pH [77, 78]. This biochemical alteration may derive from deficient OXPHOS with a concomitant increase in oxygen-independent metabolism (glycolysis), resulting in increased lactic acid levels [79]. Washizuka et al. [80] found that NDUFV2 haplotype frequencies were statistically different between BD cases and healthy controls. The association was replicated in another study, but in the opposite direction [81].
Mitonuclear Crosstalk
As described above, the majority of mitochondrial genes are encoded by the nucleus, with several of the OXPHOS components present in the mtDNA. Thus, mitochondria and the nucleus finely coordinate the transcription, translation, and import of mitochondrial proteins to ensure the proper stoichiometric relationship between the various OXPHOS components. Furthermore, the nucleus provides tight control over mitochondrial function due to the importance of mitochondria in many cellular processes.
Communication between mitochondria and the nucleus is crucial in regulating the metabolic needs of the cell [82]. This communication, termed “mitonuclear crosstalk,” occurs bidirectionally: the nucleus can influence mitochondrial structure, replication, biogenesis, fission, and fusion, the number of mitochondria, and the mitophagy turnover rate [83]. On the other hand, mitochondria influence the replication and expression of nuclear genes through production of ATP and other high-energy intermediates that are produced with the involvement of mitochondria, such as acetyl-CoA, and S-aden osyl methionine [84]. Therefore, disturbances of this crosstalk may lead to deficiencies in the mitochondrial energy-generating system, causing or contributing to disease.
The pathways that coordinate mitochondrion-nucleus communication are categorized into anterograde and retrograde crosstalk. Signals sent from the nucleus to mitochondria constitute anterograde regulation, while those sent from mitochondria to the nucleus constitute retrograde regulation (Fig. 3a) [85]. Anterograde crosstalk culminates in the expression of nucleus-encoded mitochondrial genes which induce mtDNA gene expression, ultimately upregulating mitochondrial OXPHOS activity. In this way, mitochondrial activity can be directed by nuclear events. Two nuclear transcription factors primarily control the expression of the nucleus-encoded mitochondrial genes: nuclear respiratory factor 1 (NRF1) and GA-binding protein-α (GABPα). Additionally, peroxisome proliferator-activated receptors (PPARs), estrogen-related receptors (ERRs), and myocyte-specific enhancer factor 2 (MEF2) likewise control the expression of these mitochondrial genes, but to a lesser extent.
Fig. 3.

Sketch of mitonuclear crosstalk mechanism. a Anterograde and retrograde components of the crosstalk are shown in response to cellular stressors. b Quality control, integrated stress response, and extracellular communication components of the crosstalk are shown in response to mitochondrial stressors. ROS, reactive oxygen species; mtDNA, mitochondrial DNA; OXPHOS, oxidative phosphorylation; UPRmt, mitochondrial unfolded protein response.
NRFs, PPARs, and ERRs are activated by a similar common pathway, with PGC1α (also called PPARGC1A) acting as the master regulator [86]. MEF2, on the other hand, is controlled by a separate pathway. For the common PGC1α-NRF/PPAR/ERR pathway, nucleus-encoded mitochondrial genes are induced by exercise or caloric restriction (via AMPK and SIRT1-mediated activation) [87], increased calcium (via CAMKIV and others) [88], or cold temperature (via cAMP response element-binding protein [CREB] and others). DNA damage and telomere dysfunction inhibit the activation of mitochondrial genes through PARP1 and p53, respectively, presumably to prevent further damage from increased ROS production [89, 90]. Calcium elevation due to exercise can also stimulate calcineurin, which activates MEF2, the last transcription factor controlling mitochondrial gene expression [91]. In summary, NRF, PPAR, ERR, and MEF all serve to exert nuclear control on upregulated mitochondrial activity.
Retrograde crosstalk is initiated in response to OXPHOS dysfunction or mtDNA mutation [85]. These conditions manifest in the form of decreased ATP synthesis, increased ROS generation, or calcium release from a leaky mitochondrial membrane. Low ATP leads to a high AMP/ATP ratio, thus activating the AMPK pathway to stimulate mitochondrial biogenesis through activation of PGC1α, as described above [92]. An elevated ROS concentration can also activate AMPK, as well as activating JNK and inhibiting KEAP1, with a concomitant antioxidant response via NFE2L2 translocation to the nucleus [93]. Moreover, both ROS and increased calcium activate NF-κB, which promotes the expression of nuclear genes involved in calcium metabolism [94]. Increased calcium levels can also activate calcium metabolism genes in an NF-κB-independent manner. Through these methods, mitochondria can exert control over nuclear gene expression.
Mitonuclear crosstalk also includes mitochondrial quality control and proteostasis [95], the integrated cellular stress response, and extracellular signaling (Fig. 3b). The mitochondrial quality control system includes chaperones and proteases encoded by the nuclear genome which are important to maintain mitochondrial proteostasis. There are three different proteostasis responses involved in mitochondrion-nucleus crosstalk. First, the mitochondrial unfolded protein response acts by activating the expression of proteases, chaperones, and other stress response genes. Second, proteolytic stress induces the expression of some mitochondrial proteases. Third, the heat shock response activates mitochondrial chaperones. In total, these mechanisms serve to protect the cell from mitochondrial dysfunction by responding to disrupted mitochondrial proteostasis.
For mitochondrial damage not related to disrupted proteostasis, the integrated stress response regulates the cellular response to mitochondrial dysfunction [96]. The main initiation factor for the integrated stress response is eukaryotic translation initiation factor 2 subunit alpha (EIF2α; also called EIF2S1). The net result of the integrated stress response is to inhibit protein synthesis globally, with activation of expression for stress response genes. These include ATF4, CHOP (also called DDIT3), and TRIB3 [97].
Lastly, mitonuclear crosstalk includes extracellular signaling, whereby mitokines (mitochondrion-derived signaling molecules) are released from the cell in response to mitochondrial stress, and subsequently interact with the nucleus of a neighboring cell. The mechanisms underlying this process are not well understood, and even the identities of the mitokines themselves are not fully elucidated. Nevertheless, mitokines are involved in systemic metabolism [98] and have been implicated in a wide spectrum of neuronal processes [99]. The pathways described here underlying mitonuclear crosstalk and regulation were recently reviewed by Quiroós et al. [85]; the associated genes for mitonuclear crosstalk gene set analysis are listed in Table 2.
Although not investigated explicitly, several of the players involved in mitonuclear crosstalk have been implicated in the pathophysiology of SCZ and BD. In one study, levels of the mitochondrial activator PPAR-γ were reduced in patients with SCZ compared to healthy controls [100]. This was attributed to a proinflammatory state, but the authors also noted the possible connection with mitochondrial function.
Mitonuclear crosstalk is a compelling pathway to consider in the etiology of BD. BD is characterized by biphasic states of high energy production (mania) and low energy production (euthymia or depression). This suggests inappropriate mitochondrial cell signaling. Levels of CREB were disrupted in patients with BD [101], and mRNA transcripts encoding various components of the ETC were reduced [102, 103]. SIRT1, SIRT2, and SIRT6 were all decreased in depressive patients with BD [104]. The signaling components CREB and SIRT1 outlined here are involved in diverse biological processes. It is unclear whether changes in these components are causative of pathology, or rather a response to an increased calcium concentration, inflammation, or oxidative stress [105]. It is further confounding when considering how cellular metabolic signaling is related to the different manic/ depressive states of disease, especially in BD. Additional studies are required to untangle the role of mitonuclear crosstalk in both SCZ and BD.
Mitochondrial Dynamics
Central to mitochondrial function is mitochondrial dynamics. Mitochondrial dynamics comprises such processes as mitochondrial fission, fusion, transport within the cell, and mitophagy [106, 107]. Intact mitochondrial dynamics has been linked to normal brain development, while impaired dynamics leads to neuronal injury and developmental dysfunction [108]. Mitochondria regularly undergo fission and fusion as part of mitochondrial dynamics. Fusion is facilitated by the mitofusins Mfn1 and Mfn2, as well as optic atrophy 1 (OPA1), while fission is facilitated by dynamin-1-like protein (DNM1L; also called DRP1) and dynamin 2 (DNM2). Loss of the mitofusin proteins prevents fusion from occurring and is associated with changes in mitochondrial shape and structure, loss of the mitochondrial membrane potential, and loss of mtDNA stability [109, 110]. Conversely, fusion is associated with increased mtDNA quality and fidelity [106]. Disruption of normal mitochondrial fission and fusion leads to neurodegeneration in the CNS and elsewhere in the body as well [111, 112]. Indeed, mutations affecting these proteins cause clinical neurological conditions such as Charcot-Marie-Tooth disease type IIA (Mfn2 mutation) and dominant optic atrophy (Opa1 mutation).
Coordinated mitochondrial transport is especially crucial in neurons, as the site of mitochondrial biogenesis (cell soma) may be located far from the site of energy demand (e.g., axons). Mitochondrial transport is largely controlled via sensing of the calcium concentration by the GTPase Miro, located on the outer mitochondrial membrane. Specifically, Miro serves to stop mitochondrial transit, acting as a “traffic cop” when sensing calcium [113]. Miro is part of the mitochondrial rho GTPase (RHOT) family. Mitochondria move along microtubules; the motor protein kinesin complex mediates anterograde transport, while the dyneins mediate retrograde transport [114]. The kinesin-related protein 5 family (KIF5A–C) comprises specific motor proteins that are thought to play a vital role in the transport of mitochondria in the nervous system. Mutations in the genes encoding KIF5A–C have been implicated in a host of neurodevelopmental disorders [115, 116, 117, 118, 119, 120, 121, 122], as summarized by Flippo and Strack [107]; however, it is unclear whether these derive from impaired mitochondrial transport or from impaired transport of other organelles via KIF5. Interestingly, KIF5A interacts with DISC1 (disrupted in schizophrenia 1), a protein whose namesake highlights its relation to SCZ and other psychiatric disorders [123, 124]. It is also possible that Mfn2 participates in mitochondrial transport in addition to fission/fusion, as Mfn2 dysfunction (or deletion in model organisms) is associated with impaired mitochondrial localization.
Lastly, autophagy, or mitophagy, refers to the controlled destruction of mitochondria within the cell. Several biochemical stressors can trigger mitophagy, including hypoxia, energy stress, increased OXPHOS activity, and a disrupted mitochondrial membrane potential [106, 125]. The most studied mechanism of mitophagy is the Pink1/parkin-mediated mitophagy pathway [126]. Under physiologic conditions, the kinase Pink1 is localized inside mitochondria. With a disrupted membrane potential, Pink1 is released and associates with proteins on the outer mitochondrial membrane, causing them to become phosphorylated. This phosphorylation recruits parkin, which is followed by ubiquitination of several outer mitochondrial membrane proteins. This ubiquitination signals p62, proteasomal degradation, and autophagic membranes to complete the destruction of the mitochondrion. The genes involved in mitochondrial dynamics are listed in Table 3.
There is evidence to suggest an impairment in mitochondrial dynamics in the etiology of SCZ and BD. Rosenfeld et al. [127] reported decreased levels of Opa1 (profusion protein) in SCZ patients, but not in BD patients. Furthermore, two candidate genes for SCZ – G72 and DISC1 – participate in mitochondrial dynamics. Specifically, G72 (also called DAOA) has been shown to regulate mitochondrial fission [128], and DISC1 interacts with many of the transport proteins [124], as discussed. A recent paper by Flippo and Strack [107] reviewed mitochondrial dynamics in the context of SCZ.
Also for mitochondrial dynamics, more mitochondria of a smaller area were found in the prefrontal cortex of postmortem BD samples compared to controls [129]. In the same study, peripheral cells from patients with BD were found to have a greater density of mitochondria closer to the nucleus with a steeper decline at greater distances, and this finding was replicated in an independent sample of antemortem BD patients and controls. The morphology (shape) of mitochondria also differed for BD patients, showing a greater number of atypical cup- and disc-shaped mitochondria. These findings provide pathological support for impaired mitochondrial biogenesis, mitophagy, and/or transport in patients with BD. That being said, although there are many reports showing an association with impaired mitochondrial dynamics, there is no direct evidence for the involvement of mitochondrial trafficking or mitophagy in the pathogenesis of BD or SCZ.
We have explored three mitochondrial pathways in the context of SCZ and BD. It should be noted that different disorders may be conducive to analysis using different pathways. The pathways of choice should be based on strong biological hypotheses to minimize the number of gene sets required for analysis. In the next section, we will touch briefly on some developing topics in mitochondrial genetics.
Other Topics in Mitochondrial Genetics
Pseudogenes
Pseudogenes are recognizable copies of nucleus-encoded genes that have subsequently lost their function (i.e., they are noncoding) [130]. While the majority of mitochondrial genes migrated to the nucleus millions of years ago, the process of nuclear shuttling of mitochondrial genes into the nuclear genome still occurs today. Mitochondrial pseudogenes are sequences of mtDNA that have been incorporated into the nuclear genome. These nuclear mitochondrial pseudogenes (NUMTs) retain a similar genetic sequence to that of the true mtDNA, but they are not expressed by the nuclear transcriptomic machinery, and are thus nonfunctional. Nevertheless, they pose a challenge to the amplification and further analysis of mtDNA, because PCR primers that are designed to amplify mtDNA specifically may inadvertently hybridize to NUMTs as well [131], sometimes preferentially [132].
Currently, NUMTs are largely a technical nuisance that have led to false-positive results in mtDNA analysis [133]. However, several studies have correlated nonmitochondrial pseudogenes with a risk propensity for several diseases such as cancer [134]. It is likely that NUMTs will play a role in the future of mitochondrial genetic analysis.
MicroRNAs
miRNAs are small, noncoding RNAs that can bind to and inhibit or degrade endogenous mRNA transcripts, adding an additional layer of control to gene expression [135]. Several key mitochondrial processes are controlled by specific miRNAs, including OXPHOS (e.g., miR-338 and COXIV), cell signaling (e.g., miR-696 and PGC1α), fission (e.g., miR-30 and p53/Drp1), and mitophagy (e.g., miR-21 and PTEN), among others [136]. miRNAs have been linked to function in the brain, and they play a role in neurodevelopment, plasticity, metabolism, apoptosis, and disease [137].
The study of miRNA is complicated by the fact that different tissues express different miRNAs, leading to varying control depending on the analyzed sample. The DIANA-TarBase v8.0 database [138] compiles publications that demonstrate miRNA-gene interactions through laboratory experiments on various tissues. With respect to mitochondrial pathways, 17 out of 65 genes in OXPHOS complex I, 28 out of 49 genes in mitonuclear crosstalk, 12 out of 23 genes in mitochondrial fission, and 11 out of 20 genes in mitophagy are controlled by one or more miRNAs with validated activity in the human brain. The field of miRNA is still growing, and it is expected that our understanding of miRNA regulation will develop further as more miRNAs are identified. Nevertheless, miRNAs have already been studied in the context of psychiatric disease, although mitochondrial miRNAs have not been identified as contributing to disease. A review by Beveridge and Cairns [139] can be found on the subject of miRNA in SCZ.
Mitochondrial-Nuclear DNA Mismatch
Nuclear-mitochondrial mismatch is related to the mitonuclear crosstalk pathway, whereby the nuclear genome is required to interact with the mitochondrial genome to maintain proper physiologic function [140]. As discussed, mitochondrial genetics is a bigenomic system consisting of more than 1,000 genes in the autosomal chromosomes (nucleus) and 37 genes encoded by mtDNA. However, these two genomes have different modes of inheritance. For nDNA, the genetic information is inherited equally from both parents and, due to recombination during meiosis, a novel combination of genetic information can be introduced into the offspring. On the other hand, mtDNA is passed on by maternal inheritance exclusively, with no (or little [141]) recombination.
Haplogroup-defining variants in the mtDNA are ancient; they arose during the out-of-Africa human migration, adjusting mitochondrial performance to allow humans to adapt to new environmental conditions. These haplogroup-defining mtDNA variants may be neutral in a native population where the nuclear genome coevolved with the mitochondrial genome, but they might be harmful in the context of another ancestral genetic background, potentially altering the cellular physiology and leading to diseases. This scenario is named the mitochondrial-nuclear DNA mismatch (or misalignment) hypothesis.
Consequences of this hypothesis have been shown in animal models. For example, congenic mice (mtDNA transferred from one mice strain to another by mating) showed altered cognitive functioning (learning, exploration) when compared to wild-type strains [142]. Similarly, an increased susceptibility to experimental autoimmune encephalomyelitis and anxiety-like behavior was reported for murine conplastic strains [143]. Another study in conplastic mouse models reported that the interplay between mtDNA and nDNA alters mitochondrial function, triggering cellular adaptive response, with a resulting impact on an organism's metabolism and ageing [144]. The importance of nuclear-mitochondrial mismatch gained traction with the recent legislation in the UK to permit mitochondrial replacement therapy for women with known mitochondrial disorders [145]. Recent studies did not find any deleterious effects from mitochondrial-nuclear DNA mismatch, despite the theoretical risk [37]. However, this is still a developing field requiring considerable research.
Discussion
Mitochondrial genetics comes with unique challenges and considerations in comparison to nuclear GWAS analysis. We have addressed some of the specifics of analyzing mtDNA sequences such as SNPs, heteroplasmy, and haplogroups. We also explored three pathways related to mitochondrial function – OXPHOS, mitonuclear crosstalk, and mitochondrial dynamics – all of which show evidence of involvement in the pathophysiology of psychiatric diseases. Future areas of consideration such as NUMTs, miRNAs, and nuclear-mitochondrial mismatch were presented. Considering the numerous interactions of mitochondria in the cellular physiology and their vital role in energy generation and metabolism, mitochondrial genetics is a promising avenue to explore. However, there are challenges and limitations to mitochondrial genetic analysis.
In standard genetic studies, mtDNA SNPs are excluded as noise due to variable mtDNA copy numbers and heteroplasmy. As such, they are removed from data samples prior to GWAS analysis. Furthermore, mtDNA requires its own quality control which is distinct from that of the nuclear genome, and there is no standard analysis protocol for analyzing mtDNA from genotyping arrays. Lastly, because of these limitations, there is a dearth of comprehensive data sets that include mtDNA, further hindering the study of this important field. Indeed, a consortium of mitochondrial genetics researchers with data sets containing mtDNA sequencing would benefit the field.
Notwithstanding the technical challenges, mitochondrial genetics may elucidate essential mechanisms underlying various diseases. This is especially true for multifactorial diseases, where geneticists have struggled to identify significant loci. There are roughly 1,100 nucleus- encoded mitochondrial genes that encode proteins functioning within the mitochondria. Moreover, these genes should be considered in the context of the numerous physiological pathways in which they are involved, such as electrolyte and ROS homeostasis, cell survival, and energy regulation. Indeed, the effects of mitochondrial control on nuclear transcription are complex and extensive. Whole-transcriptome analysis through RNA sequencing showed that the expression of over 66% of the genes in the human genome are controlled by mitochondria to some degree [6]. As such, the role of this organelle in disease susceptibility and pathophysiology cannot be overstated.
Here we focused primarily on two psychiatric conditions, SCZ and BD, but mitochondria play a role in other psychiatric diseases as well. Mitochondrial genetics has been employed to explore eating disorders, depression, substance abuse, and antipsychotic side effect profiles [9, 146, 147, 148, 149, 150]. Elucidating a role for mitochondrial pathways implicated in the pathophysiology of psychiatric diseases could have profound implications for drug development and treatment. The mitochondria possess numerous druggable targets for novel therapeutics. In particular, ROS has been shown to engender psychiatric diseases, and controlling ROS can be achieved by targeting mitochondria.
In this paper, we reviewed mitochondrial genetics in the context of psychiatric disease. However, these principles can be applied to other diseases. Mitochondria have evolved to adapt to caloric availability, ambient oxygen levels, and environmental conditions. They regulate diverse activities such as tissue maintenance, work expenditure, and fighting infection [55]. Considerable research has investigated the role of the mitochondrial physiology in endocrine, metabolic and cardiovascular, inflammatory, and neurodegenerative diseases [6] with an increasing emphasis on mitochondrial genetics [151]. The tools and research strategies outlined here will contribute towards enhancing the field of mitochondrial genetics across the scope of this organelle's involvement in human disease.
Disclosure Statement
J.L.K. has been funded by Genome Canada, The Ontario Ministry of Research and Innovation, CIHR, a BBRF/NARSAD Distinguished Investigator Award, and the Judy and Larry Tanenbaum Foundation. J.L.K. has also received speaker honoraria and expenses from Eli Lilly, Novartis, and Shire, and consultant honoraria and expenses from Roche. A.B.C., V.F.G., and Z.L.Z. declare no conflict of interest.
Acknowledgments
We are grateful for the discussions with and input from Dr. Ana Andreazza and Dr. Marquis Vawter. We would like to acknowledge the Judy and Larry Tanenbaum Family Foundation for their support of this study.
References
- 1.Verge B, Alonso Y, Valero J, Miralles C, Vi lella E, Martorell L. Mitochondrial DNA (mtDNA) and schizophrenia. Eur Psychiatry. 2011;26:45–56. doi: 10.1016/j.eurpsy.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 2.Calvo SE, Clauser KR, Mootha VK. Mito Carta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2015;44:D1251–D1257. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ly CV, Verstreken P. Mitochondria at the synapse. Neuroscientist. 2006;12:291–299. doi: 10.1177/1073858406287661. [DOI] [PubMed] [Google Scholar]
- 4.Cheng A, Hou Y, Mattson MP. Mitochondria and neuroplasticity. ASN Neuro. 2010;2 doi: 10.1042/AN20100019. e00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martins-de-Souza D, Harris LW, Guest PC, Bahn S. The role of energy metabolism dysfunction and oxidative stress in schizophrenia revealed by proteomics. Antioxid Redox Signal. 2011;15:2067–2079. doi: 10.1089/ars.2010.3459. [DOI] [PubMed] [Google Scholar]
- 6.Picard M, Wallace DC, Burelle Y. The rise of mitochondria in medicine. Mitochondrion. 2016;30:105–116. doi: 10.1016/j.mito.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rollins B, Martin MV, Sequeira PA, Moon EA, Morgan LZ, Watson SJ, Schatzberg A, Akil H, Myers RM, Jones EG, et al. Mitochondrial variants in schizophrenia, bipolar disorder, and major depressive disorder. PLoS One. 2009;4 doi: 10.1371/journal.pone.0004913. e4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sequeira A, Martin MV, Rollins B, Moon EA, Bunney WE, Macciardi F, Lupoli S, Smith EN, Kelsoe J, Magnan CN, et al. Mitochondrial mutations and polymorphisms in psychiatric disorders. Front Genet. 2012;3:103. doi: 10.3389/fgene.2012.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manji H, Kato T, Di Prospero NA, Ness S, Beal MF, Krams M, Chen G. Impaired mitochondrial function in psychiatric disorders. Nat Rev Neurosci. 2012;13:293–307. doi: 10.1038/nrn3229. [DOI] [PubMed] [Google Scholar]
- 10.Gonçalves VF, Andreazza AC, Kennedy JL. Mitochondrial dysfunction in schizophrenia: an evolutionary perspective. Hum Genet. 2015;134:13–21. doi: 10.1007/s00439-014-1491-8. [DOI] [PubMed] [Google Scholar]
- 11.Hjelm BE, Rollins B, Mamdani F, Lauterborn JC, Kirov G, Lynch G, Gall CM, Sequeira A, Vawter MP. Evidence of mitochondrial dysfunction within the complex genetic etiology of schizophrenia. Mol Neuropsychiatry. 2015;1:201–219. doi: 10.1159/000441252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rajasekaran A, Venkatasubramanian G, Berk M, Debnath M. Mitochondrial dysfunction in schizophrenia: pathways, mechanisms and implications. Neurosci Biobehav Rev. 2015;48:10–21. doi: 10.1016/j.neubiorev.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 13.Gonul AS, Coburn K, Kula M. Cerebral blood flow, metabolic, receptor, and transporter changes in bipolar disorder: the role of PET and SPECT studies. Int Rev Psychiatry. 2009;21:323–335. doi: 10.1080/09540260902962131. [DOI] [PubMed] [Google Scholar]
- 14.Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wallace DC, Chalkia D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a021220. a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ciafaloni E, Ricci E, Shanske S, Moraes C, Silvestri G, Hirano M, Simonetti S, Angelini C, Donati M, Garcia C, et al. MELAS: clinical features, biochemistry, and molecular genetics. Ann Neurol. 1992;31:391–398. doi: 10.1002/ana.410310408. [DOI] [PubMed] [Google Scholar]
- 17.Rahman S, Blok R, Dahl HH, Danks D, Kirby D, Chow C, Christodoulou J, Thorburn D. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol. 1996;39:343–351. doi: 10.1002/ana.410390311. [DOI] [PubMed] [Google Scholar]
- 18.Santos RX, Correia SC, Zhu X, Smith MA, Moreira PI, Castellani RJ, Nunomura A, Perry G. Mitochondrial DNA oxidative damage and repair in aging and Alzheimer's disease. Antioxid Redox Signal. 2013;18:2444–2457. doi: 10.1089/ars.2012.5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cieri D, Brini M, Calì T. Emerging (and converging) pathways in Parkinson's disease: keeping mitochondrial wellness. Biochem Biophys Res Commun. 2017;483:1020–1030. doi: 10.1016/j.bbrc.2016.08.153. [DOI] [PubMed] [Google Scholar]
- 20.Ben-Shachar D. The interplay between mitochondrial complex I, dopamine and Sp1 in schizophrenia. J Neural Transm (Vienna) 2009;116:1383–1396. doi: 10.1007/s00702-009-0319-5. [DOI] [PubMed] [Google Scholar]
- 21.Taanman J-W. The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta. 1999;1410:103–123. doi: 10.1016/s0005-2728(98)00161-3. [DOI] [PubMed] [Google Scholar]
- 22.Vafai SB, Mootha VK. Mitochondrial disorders as windows into an ancient organelle. Nature. 2012;491:374–383. doi: 10.1038/nature11707. [DOI] [PubMed] [Google Scholar]
- 23.Clarke GM, Anderson CA, Pettersson FH, Cardon LR, Morris AP, Zondervan KT. Basic statistical analysis in genetic case-control studies. Nat Protoc. 2011;6:121–133. doi: 10.1038/nprot.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DiMauro S, Schon EA. Mitochondrial DNA mutations in human disease. Am J Med Ge net. 2001;106:18–26. doi: 10.1002/ajmg.1392. [DOI] [PubMed] [Google Scholar]
- 25.Sherry ST, Ward M-H, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brandon MC, Lott MT, Nguyen KC, Spolim S, Navathe SB, Baldi P, Wallace DC. MITOMAP: a human mitochondrial genome database - 2004 update. Nucleic Acids Res. 2005;33:D611–D613. doi: 10.1093/nar/gki079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ingman M, Gyllensten U. mtDB: Human Mitochondrial Genome Database, a resource for population genetics and medical sciences. Nucleic Acids Res. 2006;34:D749–D751. doi: 10.1093/nar/gkj010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Oven M, Kayser M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat. 2009;30:E386–E394. doi: 10.1002/humu.20921. [DOI] [PubMed] [Google Scholar]
- 29.Falk MJ, Shen L, Gonzalez M, Leipzig J, Lott MT, Stassen AP, Diroma MA, Navarro-Gomez D, Yeske P, Bai R, et al. Mitochondrial Disease Sequence Data Resource (MSeqDR): a global grass-roots consortium to facilitate deposition, curation, annotation, and integrated analysis of genomic data for the mitochondrial disease clinical and research communities. Mol Genet Metab. 2015;114:388–396. doi: 10.1016/j.ymgme.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Payne BA, Wilson IJ, Yu-Wai-Man P, Coxhead J, Deehan D, Horvath R, Taylor RW, Samuels DC, Santibanez-Koref M, Chinnery PF. Universal heteroplasmy of human mitochondrial DNA. Hum Mol Genet. 2012;22:384–390. doi: 10.1093/hmg/dds435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet. 2015;16:530–542. doi: 10.1038/nrg3966. [DOI] [PubMed] [Google Scholar]
- 32.Takeda K, Takahashi S, Onishi A, Hanada H, Imai H. Replicative advantage and tissue-specific segregation of RR mitochondrial DNA between C57BL/6 and RR heteroplasmic mice. Genetics. 2000;155:777–783. doi: 10.1093/genetics/155.2.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clark KA, Howe DK, Gafner K, Kusuma D, Ping S, Estes S, Denver DR. Selfish little circles: transmission bias and evolution of large deletion-bearing mitochondrial DNA in Caenorhabditis briggsae nematodes. PLoS One. 2012;7 doi: 10.1371/journal.pone.0041433. e41433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chinnery PF, Samuels DC, Elson J, Turnbull DM. Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: is there a common mechanism? Lancet. 2002;360:1323–1325. doi: 10.1016/S0140-6736(02)11310-9. [DOI] [PubMed] [Google Scholar]
- 35.Cree LM, Samuels DC, de Sousa Lopes SC, Rajasimha HK, Wonnapinij P, Mann JR, Dahl H-HM, Chinnery PF. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat Genet. 2008;40:249–254. doi: 10.1038/ng.2007.63. [DOI] [PubMed] [Google Scholar]
- 36.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rishishwar L, Jordan IK. Implications of human evolution and admixture for mitochondrial replacement therapy. BMC Genomics. 2017;18:140. doi: 10.1186/s12864-017-3539-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gómez-Durán A, Pacheu-Grau D, López-Gallardo E, Díez-Sánchez C, Montoya J, López-Pérez MJ, Ruiz-Pesini E. Unmasking the causes of multifactorial disorders: OXPHOS differences between mitochondrial haplogroups. Hum Mol Genet. 2010;19:3343–3353. doi: 10.1093/hmg/ddq246. [DOI] [PubMed] [Google Scholar]
- 39.Weissensteiner H, Forer L, Fuchsberger C, Schöpf B, Kloss-Brandstätter A, Specht G, Kronenberg F, Schönherr S. mtDNA-Server: next-generation sequencing data analysis of human mitochondrial DNA in the cloud. Nucleic Acids Res. 2016;44:W64–W69. doi: 10.1093/nar/gkw247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amar S, Shamir A, Ovadia O, Blanaru M, Reshef A, Kremer I, Rietschel M, Schulze TG, Maier W, Belmaker R, et al. Mitochondrial DNA HV lineage increases the susceptibility to schizophrenia among Israeli Arabs. Schizophr Res. 2007;94:354–358. doi: 10.1016/j.schres.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 41.Magri C, Gardella R, Barlati SD, Valsecchi P, Sacchetti E, Barlati S. Mitochondrial DNA haplogroups and age at onset of schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:496–501. doi: 10.1002/ajmg.b.30496. [DOI] [PubMed] [Google Scholar]
- 42.Hudson G, Gomez-Duran A, Wilson IJ, Chinnery PF. Recent mitochondrial DNA mutations increase the risk of developing common late-onset human diseases. PLoS Genet. 2014;10 doi: 10.1371/journal.pgen.1004369. e1004369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ueno H, Nishigaki Y, Kong Q-P, Fuku N, Kojima S, Iwata N, Ozaki N, Tanaka M. Analysis of mitochondrial DNA variants in Japanese patients with schizophrenia. Mitochondrion. 2009;9:385–393. doi: 10.1016/j.mito.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 44.Bertolin C, Magri C, Barlati S, Vettori A, Perini GI, Peruzzi P, Mostacciuolo ML, Vazza G. Analysis of complete mitochondrial genomes of patients with schizophrenia and bipolar disorder. J Hum Genet. 2011;56:869–872. doi: 10.1038/jhg.2011.111. [DOI] [PubMed] [Google Scholar]
- 45.Mosquera-Miguel A, Torrell H, Abasolo N, Arrojo M, Paz E, Ramos-Ríos R, Agra S, Páramo M, Brenlla J, Martínez S, et al. No evidence that major mtDNA European haplogroups confer risk to schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2012;159B:414–421. doi: 10.1002/ajmg.b.32044. [DOI] [PubMed] [Google Scholar]
- 46.Torrell H, Salas A, Abasolo N, Morén C, Garrabou G, Valero J, Alonso Y, Vilella E, Costas J, Martorell L. Mitochondrial DNA (mtDNA) variants in the European haplogroups HV, JT, and U do not have a major role in schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2014;165B:607–617. doi: 10.1002/ajmg.b.32264. [DOI] [PubMed] [Google Scholar]
- 47.Martorell L, Segués T, Folch G, Valero J, Joven J, Labad A, Vilella E. New variants in the mitochondrial genomes of schizophrenic patients. Eur J Hum Genet. 2006;14:520. doi: 10.1038/sj.ejhg.5201606. [DOI] [PubMed] [Google Scholar]
- 48.Gonçalves VF, Giamberardino SN, Crowley JJ, Vawter MP, Saxena R, Bulik CM, Yilmaz Z, Hultman CM, Sklar P, Kennedy JL, Sullivan PF, Knight J. Examining the role of common and rare mitochondrial variants in schizophrenia. PLoS One. 2018;13 doi: 10.1371/journal.pone.0191153. e0191153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kato T, Kunugi H, Nanko S, Kato N. Mitochondrial DNA polymorphisms in bipolar disorder. J Affect Disord. 2001;62:151–164. doi: 10.1016/s0165-0327(99)00173-1. [DOI] [PubMed] [Google Scholar]
- 50.Munakata K, Tanaka M, Mori K, Washizuka S, Yoneda M, Tajima O, Akiyama T, Nanko S, Kunugi H, Tadokoro K, et al. Mitochondrial DNA 3644T→C mutation associated with bipolar disorder. Genomics. 2004;84:1041–1050. doi: 10.1016/j.ygeno.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 51.Munakata K, Iwamoto K, Bundo M, Kato T. Mitochondrial DNA 3243A>G mutation and increased expression of LARS2 gene in the brains of patients with bipolar disorder and schizophrenia. Biol Psychiatry. 2005;57:525–532. doi: 10.1016/j.biopsych.2004.11.041. [DOI] [PubMed] [Google Scholar]
- 52.Kazuno A, Munakata K, Mori K, Nanko S, Kunugi H, Nakamura K, Mori N, Yamada K, Yoshikawa T, Kato N, et al. Mitochondrial DNA haplogroup analysis in patients with bipolar disorder. Am J Med Genet B Neuropsychiatr Genet. 2009;150:243–247. doi: 10.1002/ajmg.b.30804. [DOI] [PubMed] [Google Scholar]
- 53.Frye MA, Ryu E, Nassan M, Jenkins GD, Andreazza AC, Evans JM, McElroy SL, Oglesbee D, Highsmith WE, Biernacka JM. Mitochondrial DNA sequence data reveals association of haplogroup U with psychosis in bipolar disorder. J Psychiatr Res. 2017;84:221–226. doi: 10.1016/j.jpsychires.2016.09.027. [DOI] [PubMed] [Google Scholar]
- 54.Machado AK, Pan AY, da Silva TM, Duong A, Andreazza AC. Upstream pathways controlling mitochondrial function in major psychosis: a focus on bipolar disorder. Can J Psychiatry. 2016;61:446–456. doi: 10.1177/0706743716648297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wallace DC. Bioenergetics in human evolution and disease: implications for the origins of biological complexity and the missing genetic variation of common diseases. Phil Trans R Soc B. 2013;368:20120267. doi: 10.1098/rstb.2012.0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gandal MJ, Leppa V, Won H, Parikshak NN, Geschwind DH. The road to precision psychiatry: translating genetics into disease mechanisms. Nat Neurosci. 2016;19:1397–1407. doi: 10.1038/nn.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang L, Jia P, Wolfinger RD, Chen X, Zhao Z. Gene set analysis of genome-wide association studies: methodological issues and perspectives. Genomics. 2011;98:1–8. doi: 10.1016/j.ygeno.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramanan VK, Shen L, Moore JH, Saykin AJ. Pathway analysis of genomic data: concepts, methods, and prospects for future development. Trends Genet. 2012;28:323–332. doi: 10.1016/j.tig.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mooney MA, Nigg JT, McWeeney SK, Wilmot B. Functional and genomic context in pathway analysis of GWAS data. Trends Genet. 2014;30:390–400. doi: 10.1016/j.tig.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mooney MA, Wilmot B. Gene set analysis: A step-by-step guide. Am J Med Genet B Neuropsychiatr Genet. 2015;168:517–527. doi: 10.1002/ajmg.b.32328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11 doi: 10.1371/journal.pcbi.1004219. e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee PH, O'Dushlaine C, Thomas B, Purcell SM. INRICH: interval-based enrichment analysis for genome-wide association studies. Bioinformatics. 2012;28:1797–1799. doi: 10.1093/bioinformatics/bts191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schizophrenia Working Group of the Psychiatric Genomics Consortium Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ripke S, O'Dushlaine C, Chambert K, Moran JL, Kähler AK, Akterin S, Bergen SE, Collins AL, Crowley JJ, Fromer M, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 2013;45:1150–1159. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci. 2015;18:199–209. doi: 10.1038/nn.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim Y, Xia K, Tao R, Giusti-Rodriguez P, Vladimirov V, van den Oord E, Sullivan P. A meta-analysis of gene expression quantitative trait loci in brain. Transl Psychiatry. 2014;4 doi: 10.1038/tp.2014.96. e459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Szatkiewicz JP, Neale BM, O'Dushlaine C, Fromer M, Goldstein JI, Moran JL, Chambert K, Kähler A, Magnusson PK, Hultman CM, et al. Detecting large copy number variants using exome genotyping arrays in a large Swedish schizophrenia sample. Mol Psychiatry. 2013;18:1178–1184. doi: 10.1038/mp.2013.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Psychiatric GWAS Consortium Bipolar Disorder Working Group Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43:977–983. doi: 10.1038/ng.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ben-Shachar D. Mitochondrial multifaceted dysfunction in schizophrenia; complex I as a possible pathological target. Schizophr Res. 2017;187:3–10. doi: 10.1016/j.schres.2016.10.022. [DOI] [PubMed] [Google Scholar]
- 70.Clay HB, Sillivan S, Konradi C. Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int J Dev Neurosci. 2011;29:311–324. doi: 10.1016/j.ijdevneu.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wong-Riley MT. Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989;12:94–101. doi: 10.1016/0166-2236(89)90165-3. [DOI] [PubMed] [Google Scholar]
- 72.Cavelier L, Jazin EE, Eriksson I, Prince J, Båve U, Oreland L, Gyllensten U. Decreased cytochrome-c oxidase activity and lack of age- related accumulation of mitochondrial DNA deletions in the brains of schizophrenics. Genomics. 1995;29:217–224. doi: 10.1006/geno.1995.1234. [DOI] [PubMed] [Google Scholar]
- 73.Maurer I, Zierz S, Möller H-J. Evidence for a mitochondrial oxidative phosphorylation defect in brains from patients with schizophrenia. Schizophr Res. 2001;48:125–136. doi: 10.1016/s0920-9964(00)00075-x. [DOI] [PubMed] [Google Scholar]
- 74.Prabakaran S, Swatton J, Ryan M, Huffaker S, Huang J-J, Griffin J, Wayland M, Freeman T, Dudbridge F, Lilley K, et al. Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol Psychiatry. 2004;9:684–697, 643. doi: 10.1038/sj.mp.4001511. [DOI] [PubMed] [Google Scholar]
- 75.Bergman O, Ben-Shachar D. Mitochondrial oxidative phosphorylation system (OXPHOS) deficits in schizophrenia: possible interactions with cellular processes. Can J Psychiatry. 2016;61:457–469. doi: 10.1177/0706743716648290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Andreazza AC, Shao L, Wang J-F, Young LT. Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Arch Gen Psychiatry. 2010;67:360–368. doi: 10.1001/archgenpsychiatry.2010.22. [DOI] [PubMed] [Google Scholar]
- 77.Hamakawa H, Murashita J, Yamada N, Inubushi T, Kato N, Kato T. Reduced intracellular pH in the basal ganglia and whole brain measured by 31P-MRS in bipolar disorder. Psychiatry Clin Neurosci. 2004;58:82–88. doi: 10.1111/j.1440-1819.2004.01197.x. [DOI] [PubMed] [Google Scholar]
- 78.Stork C, Renshaw PF. Mitochondrial dysfunction in bipolar disorder: evidence from magnetic resonance spectroscopy research. Mol Psychiatry. 2005;10:900–919. doi: 10.1038/sj.mp.4001711. [DOI] [PubMed] [Google Scholar]
- 79.Regenold WT, Phatak P, Marano CM, Sassan A, Conley RR, Kling MA. Elevated cerebrospinal fluid lactate concentrations in patients with bipolar disorder and schizophrenia: implications for the mitochondrial dysfunction hypothesis. Biol Psychiatry. 2009;65:489–494. doi: 10.1016/j.biopsych.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Washizuka S, Kakiuchi C, Mori K, Kunugi H, Tajima O, Akiyama T, Nanko S, Kato T. Association of mitochondrial complex I subunit gene NDUFV2 at 18p11 with bipolar disorder. Am J Med Genet B Neuropsychiatr Genet. 2003;120:72–78. doi: 10.1002/ajmg.b.20041. [DOI] [PubMed] [Google Scholar]
- 81.Xu C, Li PP, Kennedy JL, Green M, Hughes B, Cooke RG, Parikh SV, Warsh JJ. Further support for association of the mitochondrial complex I subunit gene NDUFV2 with bipolar disorder. Bipolar Disord. 2008;10:105–110. doi: 10.1111/j.1399-5618.2008.00535.x. [DOI] [PubMed] [Google Scholar]
- 82.Woodson JD, Chory J. Coordination of gene expression between organellar and nuclear genomes. Nat Rev Genet. 2008;9:383–395. doi: 10.1038/nrg2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wallace DC. The epigenome and the mitochondrion: bioenergetics and the environment [corrected] Genes Dev. 2010;24:1571–1573. doi: 10.1101/gad.1960210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wallace DC, Fan W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion. 2010;10:12–31. doi: 10.1016/j.mito.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Quirós PM, Mottis A, Auwerx J. Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol. 2016;17:213–226. doi: 10.1038/nrm.2016.23. [DOI] [PubMed] [Google Scholar]
- 86.Cagin U, Enriquez JA. The complex crosstalk between mitochondria and the nucleus: what goes in between? Int J Biochem Cell Biol. 2015;63:10–15. doi: 10.1016/j.biocel.2015.01.026. [DOI] [PubMed] [Google Scholar]
- 87.Reznick RM, Shulman GI. The role of AMP-activated protein kinase in mitochondrial biogenesis. J Physiol. 2006;574:33–39. doi: 10.1113/jphysiol.2006.109512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wu H, Kanatous SB, Thurmond FA, Gallardo T, Isotani E, Bassel-Duby R, Williams RS. Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science. 2002;296:349–352. doi: 10.1126/science.1071163. [DOI] [PubMed] [Google Scholar]
- 89.Luo X, Kraus WL. On PAR with PARP: cellular stress signaling through poly (ADP- ribose) and PARP-1. Genes Dev. 2012;26:417–432. doi: 10.1101/gad.183509.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sahin E, Colla S, Liesa M, Moslehi J, Müller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wu H, Rothermel B, Kanatous S, Rosenberg P, Naya FJ, Shelton JM, Hutcheson KA, DiMaio JM, Olson EN, Bassel-Duby R, et al. Activation of MEF2 by muscle activity is mediated through a calcineurin-dependent pathway. EMBO J. 2001;20:6414–6423. doi: 10.1093/emboj/20.22.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Garcia-Roves PM, Osler ME, Holmström MH, Zierath JR. Gain-of-function R225Q mutation in AMP-activated protein kinase γ3 subunit increases mitochondrial biogenesis in glycolytic skeletal muscle. J Biol Chem. 2008;283:35724–35734. doi: 10.1074/jbc.M805078200. [DOI] [PubMed] [Google Scholar]
- 93.Palikaras K, Lionaki E, Tavernarakis N. Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ. 2015;22:1399–1401. doi: 10.1038/cdd.2015.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Biswas G, Anandatheerthavarada HK, Zaidi M, Avadhani NG. Mitochondria to nucleus stress signaling. J Cell Biol. 2003;161:507–519. doi: 10.1083/jcb.200211104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mottis A, Jovaisaite V, Auwerx J. The mitochondrial unfolded protein response in mammalian physiology. Mamm Genome. 2014;25:424–433. doi: 10.1007/s00335-014-9525-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 97.Michel S, Canonne M, Arnould T, Renard P. Inhibition of mitochondrial genome expression triggers the activation of CHOP-10 by a cell signaling dependent on the integrated stress response but not the mitochondrial unfolded protein response. Mitochondrion. 2015;21:58–68. doi: 10.1016/j.mito.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 98.Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim Y-N, Kim SS, Kim DH, Hur KY, Kim HK, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. 2013;19:83–92. doi: 10.1038/nm.3014. [DOI] [PubMed] [Google Scholar]
- 99.Deng P, Haynes CM. The mitokine quest(ion) Cell Res. 2016;26:1265–1266. doi: 10.1038/cr.2016.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Martínez-Gras I, Pérez-Nievas BG, García-Bueno B, Madrigal JL, Andrés-Esteban E, Rodríguez-Jiménez R, Hoenicka J, Palomo T, Rubio G, Leza JC. The anti-inflammatory prostaglandin 15d-PGJ2 and its nuclear receptor PPARγ are decreased in schizophrenia. Schizophr Res. 2011;128:15–22. doi: 10.1016/j.schres.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 101.Ren X, Rizavi HS, Khan MA, Bhaumik R, Dwivedi Y, Pandey GN. Alteration of cyclic-AMP response element binding protein in the postmortem brain of subjects with bipolar disorder and schizophrenia. J Affect Disord. 2014;152:326–333. doi: 10.1016/j.jad.2013.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sun X, Wang J-F, Tseng M, Young LT. Downregulation in components of the mitochondrial electron transport chain in the postmortem frontal cortex of subjects with bipolar disorder. J Psychiatry Neurosci. 2006;31:189–196. [PMC free article] [PubMed] [Google Scholar]
- 103.Konradi C, Eaton M, MacDonald ML, Walsh J, Benes FM, Heckers S. Molecular evidence for mitochondrial dysfunction in bipolar disorder. Arch Gen Psychiatry. 2004;61:300–308. doi: 10.1001/archpsyc.61.3.300. [DOI] [PubMed] [Google Scholar]
- 104.Abe N, Uchida S, Otsuki K, Hobara T, Yamagata H, Higuchi F, Shibata T, Watanabe Y. Altered sirtuin deacetylase gene expression in patients with a mood disorder. J Psychiatr Res. 2011;45:1106–1112. doi: 10.1016/j.jpsychires.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 105.Morris G, Walder K, McGee SL, Dean OM, Tye SJ, Maes M, Berk M. A model of the mitochondrial basis of bipolar disorder. Neurosci Biobehav Rev. 2017;74((pt A)):1–20. doi: 10.1016/j.neubiorev.2017.01.014. [DOI] [PubMed] [Google Scholar]
- 106.Mishra P. Interfaces between mitochondrial dynamics and disease. Cell Calcium. 2016;60:190–198. doi: 10.1016/j.ceca.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 107.Flippo KH, Strack S. An emerging role for mitochondrial dynamics in schizophrenia. Schizophrenia Research. 2017;187:26–32. doi: 10.1016/j.schres.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Flippo KH, Strack S. Mitochondrial dynamics in neuronal injury, development and plasticity. J Cell Sci. 2017;130:671–681. doi: 10.1242/jcs.171017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- 110.Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 112.Chen Y, Liu Y, Dorn GW., 2nd Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. 2011;109:1327–1331. doi: 10.1161/CIRCRESAHA.111.258723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cai Q, Sheng Z-H. Moving or stopping mitochondria: Miro as a traffic cop by sensing calcium. Neuron. 2009;61:493–496. doi: 10.1016/j.neuron.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hirokawa N, Niwa S, Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68:610–638. doi: 10.1016/j.neuron.2010.09.039. [DOI] [PubMed] [Google Scholar]
- 115.Fink JK. Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013;126:307–328. doi: 10.1007/s00401-013-1115-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Goizet C, Boukhris A, Mundwiller E, Tallaksen C, Forlani S, Toutain A, Carriere N, Paquis V, Depienne C, Durr A, et al. Complicated forms of autosomal dominant hereditary spastic paraplegia are frequent in SPG10. Hum Mutat. 2009;30:E376–E385. doi: 10.1002/humu.20920. [DOI] [PubMed] [Google Scholar]
- 117.Jerath NU, Grider T, Shy ME. Progressive lower extremity weakness and axonal sensorimotor polyneuropathy from a mutation in KIF5A (c.611G>A;p.Arg204Gln) Case Rep Genet. 2015;2015:496053. doi: 10.1155/2015/496053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kaji S, Kawarai T, Miyamoto R, Nodera H, Pedace L, Orlacchio A, Izumi Y, Takahashi R, Kaji R. Late-onset spastic paraplegia type 10 (SPG10) family presenting with bulbar symptoms and fasciculations mimicking amyotrophic lateral sclerosis. J Neurol Sci. 2016;364:45–49. doi: 10.1016/j.jns.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 119.López E, Casasnovas C, Giménez J, Santamaría R, Terrazas JM, Volpini V. Identification of two novel KIF5A mutations in hereditary spastic paraplegia associated with mild peripheral neuropathy. J Neurol Sci. 2015;358:422–427. doi: 10.1016/j.jns.2015.08.1529. [DOI] [PubMed] [Google Scholar]
- 120.Poirier K, Lebrun N, Broix L, Tian G, Saillour Y, Boscheron C, Parrini E, Valence S, Saint Pierre B, Oger M, et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet. 2013;45:639–647. doi: 10.1038/ng.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rydzanicz M, Jagła M, Kosinska J, Tomasik T, Sobczak A, Pollak A, Herman-Sucharska I, Walczak A, Kwinta P, Płoski R. KIF5A de novo mutation associated with myoclonic seizures and neonatal onset progressive leukoencephalopathy. Clin Genet. 2017;91:769–773. doi: 10.1111/cge.12831. [DOI] [PubMed] [Google Scholar]
- 122.Willemsen MH, Ba W, Wissink-Lindhout WM, de Brouwer AP, Haas SA, Bienek M, Hu H, Vissers LE, van Bokhoven H, Kalscheuer V, et al. Involvement of the kinesin family members KIF4A and KIF5C in intellectual disability and synaptic function. J Med Genet. 2014;51:487–494. doi: 10.1136/jmedgenet-2013-102182. [DOI] [PubMed] [Google Scholar]
- 123.Lipina TV, Roder JC. Disrupted-in-schizophrenia-1 (DISC1) interactome and mental disorders: impact of mouse models. Neurosci Biobehav Rev. 2014;45:271–294. doi: 10.1016/j.neubiorev.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 124.Norkett R, Modi S, Kittler J. Mitochondrial roles of the psychiatric disease risk factor DISC1. Schizophr Res. 2017;187:47–54. doi: 10.1016/j.schres.2016.12.025. [DOI] [PubMed] [Google Scholar]
- 125.Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 126.Jin SM, Youle RJ. PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci. 2012;125:795–799. doi: 10.1242/jcs.093849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rosenfeld M, Brenner-Lavie H, Ari SG-B, Kavushansky A, Ben-Shachar D. Perturbation in mitochondrial network dynamics and in complex I dependent cellular respiration in schizophrenia. Biol Psychiatry. 2011;69:980–988. doi: 10.1016/j.biopsych.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 128.Kvajo M, Dhilla A, Swor D, Karayiorgou M, Gogos J. Evidence implicating the candidate schizophrenia/bipolar disorder susceptibility gene G72 in mitochondrial function. Mol Psychiatry. 2008;13:685–696. doi: 10.1038/sj.mp.4002052. [DOI] [PubMed] [Google Scholar]
- 129.Cataldo AM, McPhie DL, Lange NT, Punzell S, Elmiligy S, Ye NZ, Froimowitz MP, Hassinger LC, Menesale EB, Sargent LW, et al. Abnormalities in mitochondrial structure in cells from patients with bipolar disorder. Am J Pathol. 2010;177:575–585. doi: 10.2353/ajpath.2010.081068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bensasson D, Zhang D-X, Hartl DL, Hewitt GM. Mitochondrial pseudogenes: evolution's misplaced witnesses. Trends Ecol Evol. 2001;16:314–321. doi: 10.1016/s0169-5347(01)02151-6. [DOI] [PubMed] [Google Scholar]
- 131.Zhang D-X, Hewitt GM. Nuclear integrations: challenges for mitochondrial DNA markers. Trends Ecol Evol. 1996;11:247–251. doi: 10.1016/0169-5347(96)10031-8. [DOI] [PubMed] [Google Scholar]
- 132.Collura RV, Stewart C-B. Insertions and duplications of mtDNA in the nuclear genomes of Old World monkeys and hominoids. Nature. 1995;378:485–489. doi: 10.1038/378485a0. [DOI] [PubMed] [Google Scholar]
- 133.Wallace DC, Stugard C, Murdock D, Schurr T, Brown MD. Ancient mtDNA sequences in the human nuclear genome: a potential source of errors in identifying pathogenic mutations. Proc Natl Acad Sci. 1997;94:14900–14905. doi: 10.1073/pnas.94.26.14900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pink RC, Wicks K, Caley DP, Punch EK, Jacobs L, Carter DRF. Pseudogenes: pseudo-functional or key regulators in health and disease? RNA. 2011;17:792–798. doi: 10.1261/rna.2658311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 136.Li P, Jiao J, Gao G, Prabhakar BS. Control of mitochondrial activity by miRNAs. J Cell Biochem. 2012;113:1104–1110. doi: 10.1002/jcb.24004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Loohuis NO, Kos A, Martens G, Van Bokhoven H, Kasri NN, Aschrafi A. MicroRNA networks direct neuronal development and plasticity. Cell Mol Life Sci. 2012;69:89–102. doi: 10.1007/s00018-011-0788-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Karagkouni D, Paraskevopoulou MD, Chatzopoulos S, Vlachos IS, Tastsoglou S, Kanellos I, Papadimitriou D, Kavakiotis I, Maniou S, Skoufos G, et al. DIANA-TarBase v8: a decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2017;46:D239–D245. doi: 10.1093/nar/gkx1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Beveridge NJ, Cairns MJ. MicroRNA dysregulation in schizophrenia. Neurobiol Dis. 2012;46:263–271. doi: 10.1016/j.nbd.2011.12.029. [DOI] [PubMed] [Google Scholar]
- 140.Muir R, Diot A, Poulton J. Mitochondrial content is central to nuclear gene expression: profound implications for human health. Bioessays. 2016;38:150–156. doi: 10.1002/bies.201500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kraytsberg Y, Schwartz M, Brown TA, Ebralidse K, Kunz WS, Clayton DA, Vissing J, Khrapko K. Recombination of human mitochondrial DNA. Science. 2004;304:981. doi: 10.1126/science.1096342. [DOI] [PubMed] [Google Scholar]
- 142.Roubertoux PL, Sluyter F, Carlier M, Marcet B, Maarouf-Veray F, Chérif C, Marican C, Arrechi P, Godin F, Jamon M, et al. Mitochondrial DNA modifies cognition in interaction with the nuclear genome and age in mice. Nat Genet. 2003;35:65–69. doi: 10.1038/ng1230. [DOI] [PubMed] [Google Scholar]
- 143.Yu X, Gimsa U, Wester-Rosenlöf L, Kanitz E, Otten W, Kunz M, Ibrahim SM. Dissecting the effects of mtDNA variations on complex traits using mouse conplastic strains. Genome Res. 2009;19:159–165. doi: 10.1101/gr.078865.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Latorre-Pellicer A, Moreno-Loshuertos R, Lechuga-Vieco AV, Sánchez-Cabo F, Torroja C, Acín-Pérez R, Calvo E, Aix E, González-Guerra A, Logan A, et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature. 2016;535:561–565. doi: 10.1038/nature18618. [DOI] [PubMed] [Google Scholar]
- 145.Cohen IG, Savulescu J, Adashi EY. Transatlantic lessons in regulation of mitochondrial replacement therapy. Science. 2015;348:178–180. doi: 10.1126/science.aaa8153. [DOI] [PubMed] [Google Scholar]
- 146.Victor VM, Rovira-Llopis S, Saiz-Alarcon V, Sangüesa MC, Rojo-Bofill L, Bañuls C, Falcón R, Castelló R, Rojo L, Rocha M, et al. Altered mitochondrial function and oxidative stress in leukocytes of anorexia nervosa patients. PLoS One. 2014;9 doi: 10.1371/journal.pone.0106463. e106463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Eszlari N, Kovacs D, Petschner P, Pap D, Gonda X, Elliott R, Anderson IM, Deakin JFW, Bagdy G, Juhász G. Distinct effects of folate pathway genes MTHFR and MTHFD1L on ruminative response style: a potential risk mechanism for depression. Transl Psychiatry. 2016;6 doi: 10.1038/tp.2016.19. e745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sadakierska-Chudy A, Frankowska M, Filip M. Mitoepigenetics and drug addiction. Pharmacol Ther. 2014;144:226–233. doi: 10.1016/j.pharmthera.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 149.Gonçalves VF, Zai CC, Tiwari AK, Brandl EJ, Derkach A, Meltzer HY, Lieberman JA, Müller DJ, Sun L, Kennedy JL. A hypothesis-driven association study of 28 nuclear-encoded mitochondrial genes with antipsychotic-induced weight gain in schizophrenia. Neuropsychopharmacology. 2014;39:1347–1354. doi: 10.1038/npp.2013.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mittal K, Gonçalves VF, Harripaul R, Cuperfain AB, Rollins B, Tiwari AK, Zai CC, Maciukiewicz M, Müller DJ, Vawter MP, et al. A comprehensive analysis of mitochondrial genes variants and their association with antipsychotic-induced weight gain. Schizophr Res. 2017;187:67–73. doi: 10.1016/j.schres.2017.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron. 2011;70:1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gene Ontology Consortium Gene Ontology Consortium: going forward Nucleic Acids Res. 2015;43((database issue)) doi: 10.1093/nar/gku1179. D1049–D1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pellegrino MW, Nargund AM, Haynes CM. Signaling the mitochondrial unfolded protein response. Biochim Biophys Acta. 2013;1833:410–416. doi: 10.1016/j.bbamcr.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gonçalves VF, Cappi C, Hagen CM, Sequeira A, Vawter MP, Derkach A, Zai CC, Hedley PL, Bybjerg-Grauholm J, Pouget JG, et al. A comprehensive analysis of nuclear-encoded mitochondrial genes in schizophrenia. Biol Psychiatry. 2018;83:780–789. doi: 10.1016/j.biopsych.2018.02.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]