

Abstract
The buildup of fat in the liver (hepatic steatosis) is the first step in a series of incidents that may drive hepatic disease. Obesity is the leading cause of nonalcoholic fatty liver disease (NAFLD), in which hepatic steatosis progresses to liver disease. Chronic alcohol exposure also induces fat accumulation in the liver and shares numerous similarities to obesity-induced NAFLD. Regardless of whether hepatic steatosis is due to obesity or long-term alcohol use, it still may lead to hepatic fibrosis, cirrhosis, or possibly hepatocellular carcinoma. The antioxidant bilirubin and the enzyme that generates it, biliverdin reductase A (BVRA), are components of the heme catabolic pathway that have been shown to reduce hepatic steatosis. This review discusses the roles for bilirubin and BVRA in the prevention of steatosis, their functions in the later stages of liver disease, and their potential therapeutic application.
Keywords: ALD, bilirubin, BVRA, fatty liver disease, fibrosis, NAFLD, PPARα
INTRODUCTION
Obesity has grown to epidemic proportions throughout the world (36, 72). Obesity causes an increase in the release of adipokines and free fatty acids from adipose tissue, which leads to a systemic chronic inflammatory state (71). The inflammation along with hepatic lipid accumulation results in the development of nonalcoholic fatty liver disease (NAFLD), which can progress to nonalcoholic steatohepatitis (NASH; 68, 71). The incidence of NAFLD has paralleled the high rates of obesity worldwide and has become the most common cause of chronic liver disease (11, 21).
NAFLD is diagnosed by lipid accumulation within hepatocytes that accounts for >5% of the total liver weight (63). The prevalence of NAFLD in the United States is estimated at 30%, and a majority of these patients were asymptomatic (56, 81). However, 10.3% of patients with NAFLD eventually progress to a more serious hepatic disease and exhibit fibrosis (56). Hepatic steatosis is also seen in alcoholic liver disease (ALD) resulting from long-term alcohol abuse (8, 105). NAFLD and ALD have similar pathological progressions from simple hepatic steatosis to steatohepatitis, fibrosis, and then cirrhosis or possibly hepatocellular carcinoma (105). However, NAFLD usually has a greater degree of hepatic fatty degeneration whereas ALD has more pronounced inflammation and venous fibrosis (105).
Recent research has demonstrated an important function of bilirubin and the enzyme that produces it, biliverdin reductase A (BVRA), in protecting the liver against lipid accumulation and hepatic disease (54, 61, 83). Interestingly, alcoholic hepatitis, a type of ALD, is associated with higher serum conjugated bilirubin (74), whereas conversely, obese patients have lower total bilirubin serum levels (5). These findings suggest that hepatocyte damage may play a role in the development of hepatic steatosis and alterations of serum bilirubin levels in alcoholic hepatitis and obesity. This review discusses the recent findings of the protective roles of bilirubin and BVRA in hepatic diseases.
BILIRUBIN AND HEPATIC STEATOSIS
Bilirubin is generated from the breakdown of heme present in hemoproteins (e.g., hemoglobin and myoglobin) that is released from the catabolism of red blood cells. The heme ring is broken open by heme oxygenase forming biliverdin, which is reduced to bilirubin by biliverdin reductase (BVR; Fig. 1; 104). Snyder and colleagues showed that bilirubin was capable of protecting cells from the oxidative stress of a 10,000-fold increase in hydrogen peroxide (3, 91). Furthermore, they proposed a biliverdin-bilirubin redox cycle in which bilirubin, the antioxidant, reduces reactive oxygen species (ROS) and is consequently reoxidized to biliverdin. This proposed redox cycle would be capable of managing the oxidative stress perpetuated by a 10,000-fold surge in hydrogen peroxide concentration (91). However, it is possible that this redox recycling of bilirubin to biliverdin was an artifact of the cell culture conditions utilized by Sedlak and Snyder (91) as Maghzal et al. showed that the redox cycle might play an insufficient part in BVR’s role as a cellular antioxidant, at least in HeLa human cervical cancer cells (64). These authors argue that bilirubin may regenerate itself through hydrogen abstraction from the central methylene group, which would generate a resonance-stabilized tetrapyrrole radical lowering its lipophilicity (64). In cells that are more acceptable to lipid peroxidation, such as adipocytes and hepatocytes, it is plausible that the BVR-bilirubin cycle may be more pronounced. Gonzalez-Sanchez et al. showed in HepG2, Alexander, and HuH-7 human liver cell lines that BLVRA mRNA was inversely proportional to the level of ROS production (31). Indeed, a global Blvra knockout (KO) mouse had severe oxidative stress due to low plasma bilirubin levels (14). Thus, there is a need for more in vivo studies, particularly in humans, to reveal the nature of the BVR-bilirubin cycle.
Fig. 1.
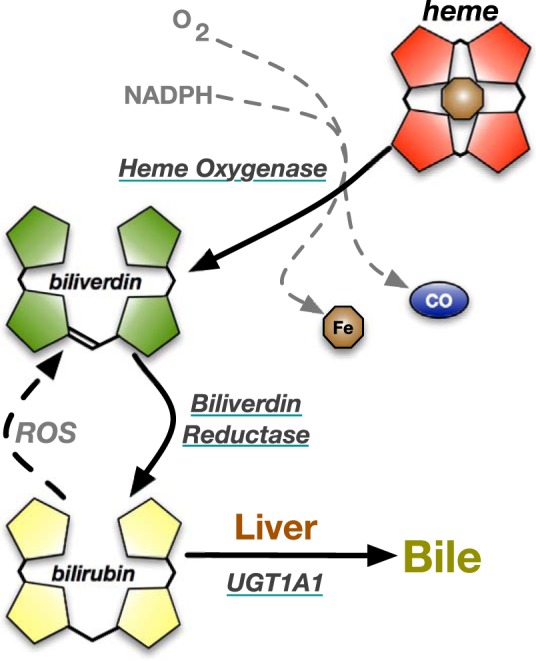
Heme catabolic pathway. Heme contains a porphyrin ring and a central iron atom. In heme degradation, heme oxygenase breaks open the porphyrin ring with NADPH and oxygen (O2) resulting in the release of iron (Fe), carbon monoxide (CO), and biliverdin. Biliverdin is reduced by biliverdin reductase to bilirubin. Reactive oxygen species (ROS) can recycle bilirubin back to biliverdin. In the liver, bilirubin is conjugated with glucuronic acid by UDP-glucuronosyltransferase 1A1 (UGT1A1) and excreted into the bile.
If bilirubin is not reoxidized to biliverdin, it is conjugated to glucuronic acid in hepatocytes by UDP-glucuronosyltransferase 1A1 (UGT1A1) to make it more soluble for biliary excretion (34, 114). The conjugated bilirubin is transported into the bile via the ATP-dependent multidrug-resistant protein transporter MRP2 (34, 114). Once in the gut, the conjugated bilirubin is reduced by the microflora to urobilinogen and further metabolized into stercobilin or urobilin, which are excreted in the feces and urine, respectively (34, 92, 102, 104). However, bilirubin is not simply a waste product of heme catabolism (109). It is also a potent antioxidant particularly when it comes to protecting lipids from oxidation (90, 109, 124). Within the last decade, bilirubin’s role has expanded as a protector against inflammation (87, 111, 121), diabetes (1, 13, 122), cardiovascular disease (47, 49, 124), metabolic syndrome (33, 55), obesity (48), and, in particular, chronic liver disease (41, 46, 53). Elevated total serum bilirubin levels have been reported to be negatively associated with NAFLD and NASH in patients undergoing routine health screening (54). In obese children, those diagnosed with NAFLD by liver ultrasonography have lower levels of total serum bilirubin than those without NAFLD (61). Puri et al. investigated children diagnosed with NAFLD and observed that total serum bilirubin levels were significantly lower in patients with NASH (83). In addition, total serum bilirubin levels were negatively correlated with the degree of steatosis and NAFLD activity score (83). These studies support an additional role of bilirubin as a protective factor against the progression and development of chronic liver disease.
Traditionally, elevated serum bilirubin levels have been considered a biomarker for liver disease and jaundice. However, it is important to differentiate between elevated serum conjugated and unconjugated bilirubin levels. Unconjugated bilirubin levels typically range from 2 to 8 µM, and for conjugated they range from 0.06 to 0.48 µM or 3–5% of total bilirubin (23, 24, 34). Patients with jaundice and hepatobiliary damage have higher concentrations of conjugated bilirubin (79 µM; 24, 34, 85), which indicates irreversible damage to hepatocytes. Meanwhile, several studies have shown a negative correlation between a patient’s serum unconjugated bilirubin levels and chronic liver disease, particularly in the severity of NAFLD and progression to NASH (41, 54, 83, 87). Salomone et al. performed a retrospective study to assess hepatic steatosis, inflammation, and fibrosis in NAFLD patients (87), in which they showed that unconjugated bilirubin levels were lower in patients with more severe inflammation and fibrosis, suggesting that unconjugated hyperbilirubinemia is protective (87). Therefore, it is imperative to report the serum conjugated and unconjugated bilirubin levels to clarify which form of bilirubin is eliciting the hepatic protection.
The benefits of unconjugated bilirubin are also evident in patients with Gilbert’s syndrome (GS). These patients have unconjugated hyperbilirubinemia without hepatic damage or hemolysis due to a genetic polymorphism in the UGT1A1 gene (25). The UGT1A1 gene encodes the enzyme UDP-glucuronosyltransferase, which is responsible for the conjugation of bilirubin in the liver. UGT1A1*28 is the most common UGT1A1 polymorphism in patients with GS and contains an additional TA repeat in the UGT1A1 TATA promoter sequence. Patients with GS have a lower risk of cardiovascular disease and NAFLD (60, 89, 110). GS patients have been shown to have lower levels of serum cholesterol, triglycerides, and proinflammatory cytokines including interleukin-6 (IL-6; 112). In addition, Lin et al. found that obese children with the UGT1A1*6 variant in the coding region had slightly elevated plasma bilirubin levels and were less likely to be diagnosed with NAFLD (61). Another study determined that patients with unconjugated hyperbilirubinemia were less likely to develop NASH and had a less severe form of liver fibrosis based on the smaller degree of liver stiffness measured in these patients (53). Overall, studies continue to show that patients with UGT1A1 polymorphisms acquire health benefits from unconjugated hyperbilirubinemia. These benefits could be a result of bilirubin’s protection against lipid accumulation and chronic liver disease.
Several clinical studies on the effects of hyperbilirubinemia, particularly unconjugated hyperbilirubinemia, have demonstrated bilirubin’s protection against cardiovascular diseases (43, 97) and metabolic dysfunction (50). It is unlikely that these health benefits could be obtained solely from bilirubin’s antioxidant properties. Recently, a novel function for bilirubin has been uncovered as an activator of peroxisome proliferator-activated receptor-α (PPARα; 99). PPARα is a transcription factor that regulates genes involved in lipid metabolism, particularly in hepatic lipid homeostasis (106, 107). Bilirubin has been identified as an agonist of PPARα resulting in an increase in β-oxidation fat-burning genes resulting in a decrease in hepatic lipid accumulation (99). A humanized mouse model for GS that contains the human UGT1A1*28 polymorphism had unconjugated hyperbilirubinemia (38) and, on a high-fat diet, had decreased lipid accumulation and increased PPARα activity compared with controls (38). Mölzer et al. conducted a study with GS patients and reported similar increases in PPARα expression (73). However, the role of PPARα transcription in regard to the protective metabolic effects of bilirubin on the liver has not been directly evaluated in humans.
Bilirubin has also been shown to increase the p450 enzyme Cyp1a1 in hepatocytes, which was suggested to occur by possible activation of the aryl hydrocarbon receptor (AhR; 95). However, the Cyp1a1 promoter has two PPAR response elements that are known to be regulated by PPARα (94). The AhR is a ligand-activated transcription factor that is involved in cell differentiation and carcinogenesis, including the development of several cancers (27, 88), and inhibiting AhR levels reduces gastric tumors in mice (101, 119). Furthermore, obesity and fatty liver are prevented by inhibition of the AhR in both female and male mice (75). The fatty liver and lipid-lowering effect of bilirubin has been shown to be mediated by direct binding to PPARα (99). However, there may be other targets that bilirubin activates that also need to be investigated.
Furthermore, higher plasma bilirubin levels reduce the risks of hepatocellular carcinoma recurrence (35). In addition to PPARα, bilirubin has been shown to activate other cellular targets, but not by direct binding, which include the ERK1/2 pathway, which may mediate bilirubin’s antioncogenic effect. One study performed treated BALB/c nude mice injected with human colorectal carcinoma cells with bilirubin and found a dramatic reduction in tumor growth compared with vehicle-treated mice (78). Bilirubin inhibited tumor cell growth through the phosphorylation and activation of ERK1/2 and its upstream kinase, MEK (78). It has been well documented that hepatocellular carcinoma has an upregulation of the ERK1/2 pathway via phosphorylation of the ERK1/2 pathway components leading to cell survival and growth (59). Further studies are needed to understand the impact of bilirubin on the ERK1/2 pathway and to clarify its protective role in liver disease progression and the development of hepatocellular carcinoma.
THE ROLE OF BILIVERDIN REDUCTASE IN HEPATIC STEATOSIS
BVRA reduces biliverdin IXα to bilirubin IXα (76), and it can also function as a signaling molecule and transcription factor (2, 65, 76). There is also a BVRB isoform that generates bilirubin IXβ, but this form of bilirubin is only produced the first few weeks after birth (7, 76, 82). A benefit of the fetal bilirubin IXβ is that it is more soluble than bilirubin IXα, which does not require conjugation for its excretion, which is required for excretion of bilirubin IXα (16). Very little is known about BVRB, and its function in adulthood has not been studied, even though it is still expressed (76). BVRA has been the most studied and is a cell surface protein that has been shown to mediate anti-inflammatory effects via the phosphatidylinositol 3-kinase (PI3K) and Akt pathway (116). The protective actions of BVRA were recently demonstrated by a global BVRA-null mouse (14). BVRA has also been shown to prevent hepatic lipid accumulation and disease progression (37). Hinds et al. developed a liver-specific BVRA KO mouse and found significantly higher liver weight, hepatic triglycerides, and Oil Red O staining in the liver after high-fat diet feeding in the KO compared with the wild-type mice (37). In addition, the liver-specific BVRA KO mice had upregulated de novo lipogenesis enzymes including fatty acid synthase, an enzyme involved in synthesizing fatty acids (37). Moreover, liver-specific BVRA KO mice had a decrease in phospho-AMPK, a downstream inhibitor of enzymes involved in fatty acid synthesis, and an increase in the active form of acetyl-CoA carboxylase, the rate-limiting enzyme in fatty acid synthesis (37). These results imply that BVRA or bilirubin in the liver is an essential factor that mediates fat accumulation.
Glycogen synthase kinase-3β (GSK3β) breaks down glycogen and promotes lipid synthesis in the liver through its inhibition of lipid metabolic enzyme transcription (79). GSK3β activity is regulated by phosphorylation at serine 9 (pSer9), which inactivates the enzyme (98). BVRA has been shown to control the Ser9 phosphorylation of GSK3β (29). GSK3β is a target of Akt, and its activity is most likely regulated by the BVR-PI3K-Akt pathway [reviewed in more detail by O’Brien et al. (76) and Rochette et al. (86)], as was shown in liver-specific BVRA KO mice (37) and by others (69). Phosphatase and tensin homolog (PTEN) negatively regulates the PI3K-Akt pathway through its phosphatase activity (67, 100, 116), but the impact of BVRA on PTEN has yet to be investigated. In liver-specific BVRA KO mice, there was an increase in GSK3β activity as demonstrated by a significant decrease in the levels of pSer9 GSK3β in the liver (37). PPARα is also a phosphoprotein that is regulated by phosphorylation at several different residues (9). One such residue is serine 73 (pSer73), which marks the protein for degradation via the ubiquitin pathway (Fig. 2; 37). Phosphorylation of PPARα at pSer73 is mediated by GSK3β, and BVRA KO mice exhibited an increase in pSer73 PPARα in the liver as well as a decrease in the total levels of PPARα (37). In addition, liver-specific BVRA KO mice exhibited significantly decreased levels of known PPARα target genes such as fibroblast growth factor 21 (FGF21; 37). Through PPARα activation, FGF21 has been shown to regulate hepatic lipid metabolism and attenuate hepatic steatosis (10, 18, 37, 39, 42, 66). PPARα KO mice exhibit an enhanced fatty liver and lower serum FGF21 levels (15). Most likely, BVRA plays a pivotal role in preventing the inhibition of PPARα by phosphorylating and inhibiting GSK3β, which allows for the upregulation of β-oxidation and the downregulation of de novo lipogenesis genes to promote lipid metabolism and glycogen storage in the liver (37, 107).
Fig. 2.
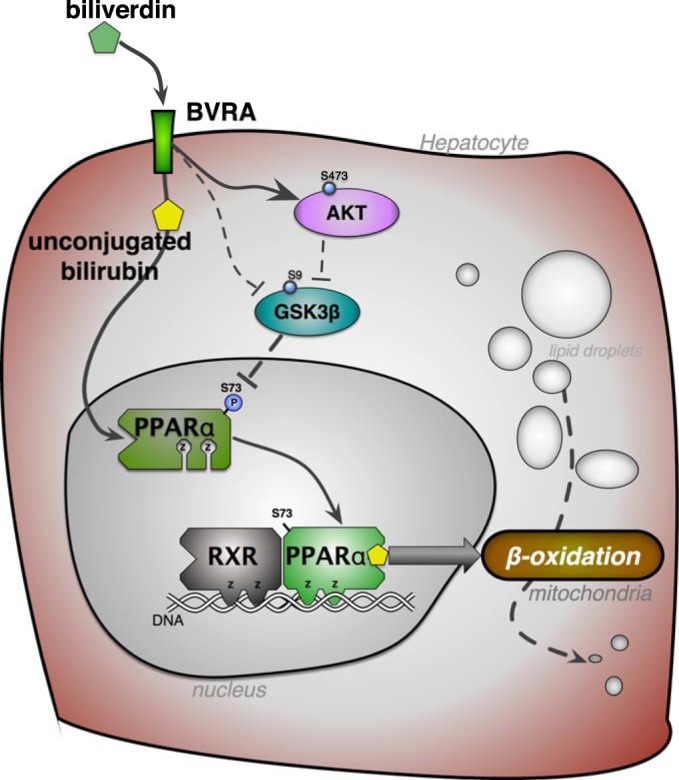
Biliverdin reductase signaling reduces hepatic fat accumulation. Biliverdin reductase A (BVRA) generation of unconjugated bilirubin from biliverdin activates peroxisome proliferator-activated receptor-α (PPARα; 99), which is a transcription factor that interacts with the retinoid X receptor (RXR) to increase genes in the β-oxidation pathway that burn fatty acids in the mitochondria. BVRA activates Akt, which inhibits glycogen synthase kinase-3β (GSK3β) through increased phosphorylation at serine 9 (S9) to prevent hepatic steatosis (37). BVRA can also directly interact with GSK3β to regulate activity (69). Inhibition of GSK3β results in increased activity of PPARα via decreased phosphorylation (P) at serine 73 (37). Z, zinc finger DNA binding domains. The signaling functions of BVRA and bilirubin have been reviewed further by Hamoud et al. (34), O’Brien et al. (76), and Rochette et al. (86).
BILIRUBIN, BILIVERDIN REDUCTASE, AND INFLAMMATION
Inflammation is a key component in the progression of simple hepatic steatosis to steatohepatitis. Inflammation is regulated by a multitude of factors including the toll-like receptor 4 (TLR4) signaling pathway. Experiments performed by Seki et al. on TLR4 mutant mice demonstrated that 3 wk post-bile duct ligation, the TLR4 mutant mice had reduced hepatic fibrosis, decreased expression of fibrotic marker α-smooth muscle actin (α-SMA), and decreased Sirius red staining for collagen (93). In comparison, the wild-type mice showed overt hepatic fibrosis in this same time frame (93). Five days post-bile duct ligation the TLR4 mutant mice showed an active suppression of hepatic fibrogenesis markers including collagen-α1(I), α-SMA, TGF-β1, and tissue inhibitor of metalloproteinases 1 (TIMP-1), which are known to be upregulated during hepatic stellate cell (HSC) activation and fibrosis (26, 93). Recently, TLR5 knockout mice were shown to develop hepatic lipogenesis and the metabolic syndrome (96), but whether BVRA or bilirubin impacts this receptor is unknown. Thus, these experiments suggest that the TLR4, and possibly TLR5, signaling pathway is a key player in HSC activation and fibrogenesis (93)
Lipopolysaccharide (LPS) is secreted by the gut microbiota and is a ligand for TLR4 (80). When mice were treated with antibiotics to reduce LPS secretion by the gut microbiota, TLR4 activation was reduced resulting in a downregulation of HSC activation as measured by a significant reduction in collagen and α-SMA expression (93). This study also found that quiescent HSCs are the cells most influenced by TLR4 activation in the liver (93). In addition, LPS, the TLR4 ligand, also increases HSC sensitivity to TGF-β1, a profibrogenic cytokine and proposed activator of HSCs (93). Also, BVRA may regulate the TLR4 signaling pathway in HSCs. Wegiel et al. identified BVRA as an inhibitor of TLR4 in liver macrophages (117). Biliverdin induces the nitrosylation of BVRA by nitric oxide, which results in transport to the nucleus, where it binds to the TLR4 promoter to inhibit its expression (117). Thus, BVRA may prevent fibrogenesis by antagonizing the TLR4 signaling pathway not only in macrophages but also in HSCs (Fig. 3). However, more work is needed to determine the role of BVRA and bilirubin in TLR4- or TLR5-mediated hepatic steatosis and fibrosis.
Fig. 3.
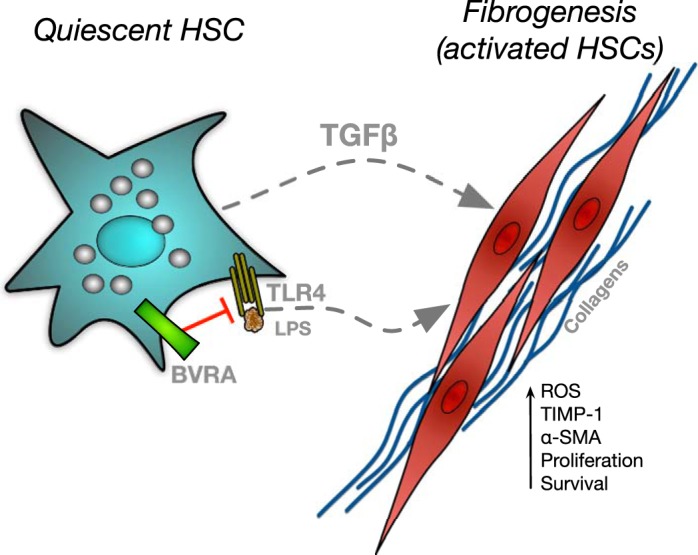
Proposed inhibitory role of biliverdin reductase A (BVRA) in fibrogenesis. Hepatic stellate cells (HSCs) are activated by lipopolysaccharide (LPS) binding to toll-like receptor 4 (TLR4) or stimulation by transforming growth factor-β (TGF-β). Activated HSCs increase collagen production as well as several other factors to promote liver fibrogenesis. BVRA can attenuate fibrogenesis through antagonism of TLR4. α-SMA, α-smooth muscle actin; ROS, reactive oxygen species; TIMP-1, tissue inhibitor of metalloproteinases 1.
Macrophages are an important component of the inflammatory cascade in the liver and can be divided into two distinct classes: M1, proinflammatory, and M2, anti-inflammatory macrophages (66). M1 macrophages release inflammatory cytokines such as interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α; 66), and IL-12 and promote the inflammatory process. M2 macrophages, however, are reparative in nature and produce high levels of IL-10 and TGF-β and low levels of IL-12. Recently, Hu et al. reported the critical role of BVRA in the polarization of M2 macrophages in vitro and in response to renal ischemia-reperfusion injury in vivo (44). In a series of elegant experiments, the authors established that overexpression of BVRA in cultured macrophages promoted the M2 phenotype as indexed by expression of IL-10 levels whereas knockdown of BVRA promoted an M1-like phenotype (44). There is little known about the role of BVRA in the liver, especially on inflammation and hepatic immune cells. Bilirubin, a marker for liver disease, may have another protective role as an antioxidant to reduce inflammation.
Bilirubin has a complex effect on the inflammatory response in the periphery versus the central nervous system. Both induction of heme oxygenase as well as bilirubin treatment have been demonstrated to have anti-inflammatory actions in many inflammatory conditions such as colitis and organ transplantation (113, 118, 120, 123). Bilirubin treatment in these conditions has been shown to decrease the levels of proinflammatory markers including myeloperoxidase, TNF-α, and IL-1β (120). Bilirubin treatment can also lower the levels of proinflammatory and proapoptotic genes including monocyte chemoattractant protein-1, caspase-3, and caspase-8 to improve allograft function and survival (113). Despite these reported anti-inflammatory effects, bilirubin has also been reported to promote inflammation in microglia and nerve cells (22, 32, 62), albeit at extremely high levels. This is consistent with the toxic effects of bilirubin on the central nervous system in infants with hyperbilirubinemia (77) and highlights the dichotomous relationship between bilirubin and inflammation. Further investigations are needed to determine whether the hyperbilirubinemia consists of unconjugated or conjugated bilirubin and to investigate whether high levels of bilirubin IXβ cause similar complications for infants.
BILIRUBIN AND BILIVERDIN REDUCTASE IN FIBROSIS AND LIVER DISEASE
The HSC has been identified as the primary contributor to the development of hepatic fibrosis in liver disease (26). Upon liver injury, the HSCs become activated causing them to transform into contractile myofibroblast-like cells that have an increase in α-SMA expression, a protein involved in fibroblast contractility (Fig. 3; 4, 26, 40). Washington et al. analyzed liver biopsies of NAFLD and NASH patients and found that 74/76 of the NAFLD and NASH liver biopsies had an increase in α-SMA, the strongest marker for HSC activation, compared with controls (115). Reeves et al. examined liver biopsies from ALD patients and observed that the patients’ biopsies contained more activated HSCs than controls, demonstrated by a higher amount of α-SMA (84). The HSC activation was also positively correlated with the degree of hepatic steatosis (84). HSCs are spindle-shaped cells located in the space of Disse between the basolateral side of the hepatocytes and the sinusoidal endothelial cells (26). Quiescent HSCs have characteristic vitamin A droplets. The activated HSCs are characterized by a loss of vitamin A droplets, proliferation, and increased extracellular matrix (ECM) production causing a dramatic alteration in the liver parenchyma (26).
Another important pathological feature of both NAFLD and ALD is the oxidative stress mediated by the increase in ROS production (6). In ALD, oxidative stress results from the CYP2E1-catalyzed breakdown of ethanol to acetaldehyde to acetate (12). This reaction generates potent amounts of ROS leading to lipid peroxidation and oxidative liver damage through the formation of DNA adducts (12). NAFLD has also been shown to have increased levels of ROS and damaging lipid peroxidation products (6). Elevated serum bilirubin is a biomarker for liver disease and is often associated with jaundice (74). However, Tang et al. showed that bilirubin is most likely preventive in HSC activation (103), which may occur as an antioxidant or signaling through PPARα. However, this function still remains unknown.
Studies have shown that bilirubin attenuates the activation of HSCs and reduces their harmful effects on the liver. As a known antioxidant, bilirubin reduces ROS production by HSCs, which is an important feature of HSC activation. Increased ROS production reacts with lipids of the cell membrane, DNA bases, and mitochondria resulting in cellular oxidative stress and damage (45, 103). Tang et al. showed that when bilirubin levels are increased, there was a decrease in α-SMA expression and therefore a decrease in HSC activation (103). In a clinical study of pediatric biliary atresia, 19 patients demonstrated a negative correlation between α-SMA expression and serum bilirubin (20). These studies suggest that bilirubin could play a role in preventing HSC activation and subsequent hepatic fibrosis. Bilirubin also attenuates the ECM accumulation and HSC proliferation upon hepatic injury. TIMPs inhibit matrix metalloproteinases, or MMPs, preventing ECM degradation and increasing net accumulation. An increase in ECM accumulation is an important sign of liver fibrosis (103). Bilirubin decreases the TIMP-1-to-MMP-2 ratio in the liver signifying higher ECM degradation and lower accumulation (103). In addition, bilirubin has been shown to play a role in inhibiting HSC proliferation and increased apoptosis. Proliferation and survival of activated HSCs are observed in liver disease (Fig. 3; 103). Since HSCs are the main contributors to fibrosis, it is likely that bilirubin is exerting its effects on HSCs by preventing their activation and profibrotic activity.
POTENTIAL THERAPEUTIC APPLICATIONS OF BILIRUBIN AND BILIVERDIN REDUCTASE
Although several preclinical studies have demonstrated protective effects of bilirubin on the liver, one challenge is translating these findings to patients suffering from liver diseases. One issue with bilirubin is its insolubility in aqueous solutions making it difficult to deliver by traditional mechanisms. Recently, the solubility of bilirubin in aqueous solutions was markedly improved by covalent attachment of polyethylene glycol (PEG) resulting in PEGylated bilirubin nanoparticles (58). These PEGylated bilirubin nanoparticles were found to be effective against hepatocellular injury by reducing oxidative stress, proinflammatory cytokine production, and recruitment of neutrophils in a mouse model of ischemia-reperfusion injury (52). Bilirubin levels can also be increased in patients via antagonism of hepatic UGT1A1 to create a moderate hyperbilirubinemia. Antagonism of UGT1A1 can be achieved by several methods, including protease inhibitor drugs such as indinavir and atazanavir, antisense morpholinos, or a natural antagonist such as silymarin (17, 19, 108). All of these approaches have their limitations, and none have been specifically demonstrated to be protective against NAFLD.
Delivery of BVRA is another potential therapeutic avenue against NAFLD. Proteins such as BVRA when fused with a protein transduction domain, such as Tat, possess the ability to traverse the lipid bilayer membrane of mammalian cells. Recent studies have utilized this strategy to deliver BVRA both in vitro and in vivo to protect against inflammation (51, 57). Another encouraging area is the delivery of peptide sequences of BVRA. Several studies have demonstrated that cell-signaling activities of BVRA can be replicated via delivery of specific peptide sequences of the protein (28, 70). BVR peptides when combined with nanospheres were found to attenuate the development of hyperglycemia in obese, diabetic ob/ob mice (30). Although very promising in the potential regulation of blood glucose levels, the effects of BVR peptides on NAFLD have yet to be reported.
CONCLUSIONS
As NAFLD continues to be problematic worldwide, it is essential to uncover the mechanism involved and prevent the progression of hepatic disease. Some studies have shown a link between bilirubin and BVRA in the prevention of hepatic steatosis. Bilirubin possibly inhibits hepatocellular carcinomas, but more studies are needed to be conclusive. Suppression of UGT1A1 to increase unconjugated bilirubin or delivery of BVRA nanoparticles may serve as a useful therapeutic target for liver diseases such as NAFLD and NASH. Furthermore, bilirubin and BVRA may have potential treatment value in preventing the development and progression of chronic liver diseases and cancer. More studies are needed to elucidate their therapeutic potential and roles in the different stages of liver disease.
GRANTS
This work was supported by National Institutes of Health Grants L32-MD-009154 (T. D. Hinds, Jr.), K01-HL-125445 (T. D. Hinds, Jr.), P01-HL-051971 (D. E. Stec), P01-HL-088421 (D. E. Stec), and P20-GM-104357 (D. E. Stec).
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.W. and T.D.H. conceived and designed research; L.W. and T.D.H. prepared figures; L.W., A.-R.H., D.E.S., and T.D.H. drafted manuscript; L.W., A.-R.H., D.E.S., and T.D.H. edited and revised manuscript; L.W., A.-R.H., D.E.S., and T.D.H. approved final version of manuscript.
REFERENCES
- 1.Abbasi A, Deetman PE, Corpeleijn E, Gansevoort RT, Gans RO, Hillege HL, van der Harst P, Stolk RP, Navis G, Alizadeh BZ, Bakker SJ. Bilirubin as a potential causal factor in type 2 diabetes risk: a Mendelian randomization study. Diabetes 64: 1459–1469, 2015. doi: 10.2337/db14-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmad Z, Salim M, Maines MD. Human biliverdin reductase is a leucine zipper-like DNA-binding protein and functions in transcriptional activation of heme oxygenase-1 by oxidative stress. J Biol Chem 277: 9226–9232, 2002. doi: 10.1074/jbc.M108239200. [DOI] [PubMed] [Google Scholar]
- 3.Baranano DE, Rao M, Ferris CD, Snyder SH. Biliverdin reductase: a major physiologic cytoprotectant. Proc Natl Acad Sci USA 99: 16093–16098, 2002. doi: 10.1073/pnas.252626999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 115: 209–218, 2005. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belo L, Nascimento H, Kohlova M, Bronze-da-Rocha E, Fernandes J, Costa E, Catarino C, Aires L, Mansilha HF, Rocha-Pereira P, Quintanilha A, Rêgo C, Santos-Silva A. Body fat percentage is a major determinant of total bilirubin independently of UGT1A1*28 polymorphism in young obese. PLoS One 9: e98467, 2014. doi: 10.1371/journal.pone.0098467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhardwaj P, Madan K, Thareja S, Joshi YK, Saraya A. Comparative redox status in alcoholic liver disease and nonalcoholic fatty liver disease. Hepatol Int 2: 202–208, 2008. doi: 10.1007/s12072-008-9060-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blumenthal SG, Stucker T, Rasmussen RD, Ikeda RM, Ruebner BH, Bergstrom DE, Hanson FW. Changes in bilirubins in human prenatal development. Biochem J 186: 693–700, 1980. doi: 10.1042/bj1860693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bruha R, Dvorak K, Petrtyl J. Alcoholic liver disease. World J Hepatol 4: 81–90, 2012. doi: 10.4254/wjh.v4.i3.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burns KA, Vanden Heuvel JP. Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta 1771: 952–960, 2007. doi: 10.1016/j.bbalip.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Camporez JP, Asrih M, Zhang D, Kahn M, Samuel VT, Jurczak MJ, Jornayvaz FR. Hepatic insulin resistance and increased hepatic glucose production in mice lacking Fgf21. J Endocrinol 226: 207–217, 2015. doi: 10.1530/JOE-15-0136. [DOI] [PubMed] [Google Scholar]
- 11.Cazzo E, Pareja JC, Chaim EA. Nonalcoholic fatty liver disease and bariatric surgery: a comprehensive review. Sao Paulo Med J 135: 277–295, 2017. doi: 10.1590/1516-3180.2016.0306311216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ceni E, Mello T, Galli A. Pathogenesis of alcoholic liver disease: role of oxidative metabolism. World J Gastroenterol 20: 17756–17772, 2014. doi: 10.3748/wjg.v20.i47.17756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen J, Wang J, Zhang X, Zhu H. Inverse relationship between serum bilirubin levels and diabetic foot in Chinese patients with type 2 diabetes mellitus. Med Sci Monit 23: 5916–5923, 2017. doi: 10.12659/MSM.907248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen W, Maghzal GJ, Ayer A, Suarna C, Dunn LL, Stocker R. Absence of the biliverdin reductase-a gene is associated with increased endogenous oxidative stress. Free Radic Biol Med 115: 156–165, 2018. doi: 10.1016/j.freeradbiomed.2017.11.020. [DOI] [PubMed] [Google Scholar]
- 15.Chen X, Ward SC, Cederbaum AI, Xiong H, Lu Y. Alcoholic fatty liver is enhanced in CYP2A5 knockout mice: the role of the PPARα-FGF21 axis. Toxicology 379: 12–21, 2017. doi: 10.1016/j.tox.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cunningham O, Gore MG, Mantle TJ. Initial-rate kinetics of the flavin reductase reaction catalysed by human biliverdin-IXβ reductase (BVR-B). Biochem J 345: 393–399, 2000. doi: 10.1042/bj3450393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.D’Andrea V, Pérez LM, Sánchez Pozzi EJ. Inhibition of rat liver UDP-glucuronosyltransferase by silymarin and the metabolite silibinin-glucuronide. Life Sci 77: 683–692, 2005. doi: 10.1016/j.lfs.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 18.De Sousa-Coelho AL, Relat J, Hondares E, Pérez-Martí A, Ribas F, Villarroya F, Marrero PF, Haro D. FGF21 mediates the lipid metabolism response to amino acid starvation. J Lipid Res 54: 1786–1797, 2013. doi: 10.1194/jlr.M033415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dekker D, Dorresteijn MJ, Pijnenburg M, Heemskerk S, Rasing-Hoogveld A, Burger DM, Wagener FA, Smits P. The bilirubin-increasing drug atazanavir improves endothelial function in patients with type 2 diabetes mellitus. Arterioscler Thromb Vasc Biol 31: 458–463, 2011. doi: 10.1161/ATVBAHA.110.211789. [DOI] [PubMed] [Google Scholar]
- 20.Dong R, Luo Y, Zheng S. α-SMA overexpression associated with increased liver fibrosis in infants with biliary atresia. J Pediatr Gastroenterol Nutr 55: 653–656, 2012. doi: 10.1097/MPG.0b013e3182680be3. [DOI] [PubMed] [Google Scholar]
- 21.Dowman JK, Tomlinson JW, Newsome PN. Pathogenesis of non-alcoholic fatty liver disease. QJM 103: 71–83, 2010. doi: 10.1093/qjmed/hcp158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Falcão AS, Fernandes A, Brito MA, Silva RF, Brites D. Bilirubin-induced immunostimulant effects and toxicity vary with neural cell type and maturation state. Acta Neuropathol 112: 95–105, 2006. doi: 10.1007/s00401-006-0078-4. [DOI] [PubMed] [Google Scholar]
- 23.Fevery J, Muraca M, Mesa V, Van Steenbergen W, Blanckaert N. Plasma bilirubin pigments in health and disease. Mol Aspects Med 9: 391–404, 1987. doi: 10.1016/0098-2997(87)90005-7. [DOI] [PubMed] [Google Scholar]
- 24.Fevery J, Vanstapel F, Blanckaert N. Bile pigment metabolism. Baillieres Clin Gastroenterol 3: 283–312, 1989. doi: 10.1016/0950-3528(89)90002-X. [DOI] [PubMed] [Google Scholar]
- 25.Fretzayas A, Moustaki M, Liapi O, Karpathios T. Gilbert syndrome. Eur J Pediatr 171: 11–15, 2012. doi: 10.1007/s00431-011-1641-0. [DOI] [PubMed] [Google Scholar]
- 26.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 88: 125–172, 2008. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fritz WA, Lin TM, Safe S, Moore RW, Peterson RE. The selective aryl hydrocarbon receptor modulator 6-methyl-1,3,8-trichlorodibenzofuran inhibits prostate tumor metastasis in TRAMP mice. Biochem Pharmacol 77: 1151–1160, 2009. doi: 10.1016/j.bcp.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gibbs PE, Lerner-Marmarosh N, Poulin A, Farah E, Maines MD. Human biliverdin reductase-based peptides activate and inhibit glucose uptake through direct interaction with the kinase domain of insulin receptor. FASEB J 28: 2478–2491, 2014. doi: 10.1096/fj.13-247015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gibbs PE, Miralem T, Lerner-Marmarosh N, Maines MD. Nanoparticle delivered human biliverdin reductase-based peptide increases glucose uptake by activating IRK/Akt/GSK3 axis: the peptide is effective in the cell and wild-type and diabetic Ob/Ob mice. J Diabetes Res 2016: 4712053, 2016. doi: 10.1155/2016/4712053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gibbs PE, Miralem T, Maines MD. Biliverdin reductase: a target for cancer therapy? Front Pharmacol 6: 119, 2015. doi: 10.3389/fphar.2015.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez-Sanchez E, Perez MJ, Nytofte NS, Briz O, Monte MJ, Lozano E, Serrano MA, Marin JJG. Protective role of biliverdin against bile acid-induced oxidative stress in liver cells. Free Radic Biol Med 97: 466–477, 2016. doi: 10.1016/j.freeradbiomed.2016.06.016. [DOI] [PubMed] [Google Scholar]
- 32.Gordo AC, Falcão AS, Fernandes A, Brito MA, Silva RF, Brites D. Unconjugated bilirubin activates and damages microglia. J Neurosci Res 84: 194–201, 2006. doi: 10.1002/jnr.20857. [DOI] [PubMed] [Google Scholar]
- 33.Guzek M, Jakubowski Z, Bandosz P, Wyrzykowski B, Smoczyński M, Jabloiska A, Zdrojewski T. Inverse association of serum bilirubin with metabolic syndrome and insulin resistance in Polish population. Przegl Epidemiol 66: 495–501, 2012. [PubMed] [Google Scholar]
- 34.Hamoud AR, Weaver L, Stec DE, Hinds TD Jr. Bilirubin in the liver-gut signaling axis. Trends Endocrinol Metab 29: 140–150, 2018. doi: 10.1016/j.tem.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han S, Yang JD, Sinn DH, Ko JS, Kim JM, Shin JC, Son HJ, Gwak MS, Joh JW, Kim GS. Higher bilirubin levels of healthy living liver donors are associated with lower posttransplant hepatocellular carcinoma recurrence. Transplantation 100: 1933–1938, 2016. doi: 10.1097/TP.0000000000001293. [DOI] [PubMed] [Google Scholar]
- 36.Hassan K, Bhalla V, El Regal ME, A-Kader HH. Nonalcoholic fatty liver disease: a comprehensive review of a growing epidemic. World J Gastroenterol 20: 12082–12101, 2014. doi: 10.3748/wjg.v20.i34.12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hinds TD Jr, Burns KA, Hosick PA, McBeth L, Nestor-Kalinoski A, Drummond HA, AlAmodi AA, Hankins MW, Vanden Heuvel JP, Stec DE. Biliverdin reductase A attenuates hepatic steatosis by inhibition of glycogen synthase kinase (GSK) 3β phosphorylation of serine 73 of peroxisome proliferator-activated receptor (PPAR) α. J Biol Chem 291: 25179–25191, 2016. doi: 10.1074/jbc.M116.731703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hinds TD Jr, Hosick PA, Chen S, Tukey RH, Hankins MW, Nestor-Kalinoski A, Stec DE. Mice with hyperbilirubinemia due to Gilbert’s syndrome polymorphism are resistant to hepatic steatosis by decreased serine 73 phosphorylation of PPARα. Am J Physiol Endocrinol Metab 312: E244–E252, 2017. doi: 10.1152/ajpendo.00396.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hinds TD Jr, Sodhi K, Meadows C, Fedorova L, Puri N, Kim DH, Peterson SJ, Shapiro J, Abraham NG, Kappas A. Increased HO-1 levels ameliorate fatty liver development through a reduction of heme and recruitment of FGF21. Obesity (Silver Spring) 22: 705–712, 2014. doi: 10.1002/oby.20559. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Hinz B, Celetta G, Tomasek JJ, Gabbiani G, Chaponnier C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell 12: 2730–2741, 2001. doi: 10.1091/mbc.12.9.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hjelkrem M, Morales A, Williams CD, Harrison SA. Unconjugated hyperbilirubinemia is inversely associated with non-alcoholic steatohepatitis (NASH). Aliment Pharmacol Ther 35: 1416–1423, 2012. doi: 10.1111/j.1365-2036.2012.05114.x. [DOI] [PubMed] [Google Scholar]
- 42.Hondares E, Rosell M, Gonzalez FJ, Giralt M, Iglesias R, Villarroya F. Hepatic FGF21 expression is induced at birth via PPARα in response to milk intake and contributes to thermogenic activation of neonatal brown fat. Cell Metab 11: 206–212, 2010. doi: 10.1016/j.cmet.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hopkins PN, Wu LL, Hunt SC, James BC, Vincent GM, Williams RR. Higher serum bilirubin is associated with decreased risk for early familial coronary artery disease. Arterioscler Thromb Vasc Biol 16: 250–255, 1996. doi: 10.1161/01.ATV.16.2.250. [DOI] [PubMed] [Google Scholar]
- 44.Hu Z, Pei G, Wang P, Yang J, Zhu F, Guo Y, Wang M, Yao Y, Zeng R, Liao W, Xu G. Biliverdin reductase A (BVRA) mediates macrophage expression of interleukin-10 in injured kidney. Int J Mol Sci 16: 22621–22635, 2015. doi: 10.3390/ijms160922621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jaeschke H, Ramachandran A. Reactive oxygen species in the normal and acutely injured liver. J Hepatol 55: 227–228, 2011. doi: 10.1016/j.jhep.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jang BK. Elevated serum bilirubin levels are inversely associated with nonalcoholic fatty liver disease. Clin Mol Hepatol 18: 357–359, 2012. doi: 10.3350/cmh.2012.18.4.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jansen T, Daiber A. Direct antioxidant properties of bilirubin and biliverdin. Is there a role for biliverdin reductase? Front Pharmacol 3: 30, 2012. doi: 10.3389/fphar.2012.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jenko-Pražnikar Z, Petelin A, Jurdana M, Žiberna L. Serum bilirubin levels are lower in overweight asymptomatic middle-aged adults: an early indicator of metabolic syndrome? Metabolism 62: 976–985, 2013. doi: 10.1016/j.metabol.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 49.Kang SJ, Kim D, Park HE, Chung GE, Choi SH, Choi SY, Lee W, Kim JS, Cho SH. Elevated serum bilirubin levels are inversely associated with coronary artery atherosclerosis. Atherosclerosis 230: 242–248, 2013. doi: 10.1016/j.atherosclerosis.2013.06.021. [DOI] [PubMed] [Google Scholar]
- 50.Karadag F, Sengul CB, Enli Y, Karakulah K, Alacam H, Kaptanoglu B, Kalkanci O, Herken H. Relationship between serum bilirubin levels and metabolic syndrome in patients with schizophrenia spectrum disorders. Clin Psychopharmacol Neurosci 15: 153–162, 2017. doi: 10.9758/cpn.2017.15.2.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim HR, Kim DW, Jo HS, Cho SB, Park JH, Lee CH, Choi YJ, Yeo EJ, Park SY, Kim ST, Yu YH, Kim DS, Kim HA, Cho SW, Han KH, Park J, Eum WS, Choi SY. Tat-biliverdin reductase A inhibits inflammatory response by regulation of MAPK and NF-κB pathways in Raw 264.7 cells and edema mouse model. Mol Immunol 63: 355–366, 2015. doi: 10.1016/j.molimm.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 52.Kim JY, Lee DY, Kang S, Miao W, Kim H, Lee Y, Jon S. Bilirubin nanoparticle preconditioning protects against hepatic ischemia-reperfusion injury. Biomaterials 133: 1–10, 2017. doi: 10.1016/j.biomaterials.2017.04.011. [DOI] [PubMed] [Google Scholar]
- 53.Kumar R, Rastogi A, Maras JS, Sarin SK. Unconjugated hyperbilirubinemia in patients with non-alcoholic fatty liver disease: a favorable endogenous response. Clin Biochem 45: 272–274, 2012. doi: 10.1016/j.clinbiochem.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 54.Kwak MS, Kim D, Chung GE, Kang SJ, Park MJ, Kim YJ, Yoon JH, Lee HS. Serum bilirubin levels are inversely associated with nonalcoholic fatty liver disease. Clin Mol Hepatol 18: 383–390, 2012. doi: 10.3350/cmh.2012.18.4.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kwon KM, Kam JH, Kim MY, Kim MY, Chung CH, Kim JK, Linton JA, Eom A, Koh SB, Kang HT. Inverse association between total bilirubin and metabolic syndrome in rural Korean women. J Womens Health (Larchmt) 20: 963–969, 2011. doi: 10.1089/jwh.2010.2453. [DOI] [PubMed] [Google Scholar]
- 56.Le MH, Devaki P, Ha NB, Jun DW, Te HS, Cheung RC, Nguyen MH. Prevalence of non-alcoholic fatty liver disease and risk factors for advanced fibrosis and mortality in the United States. PLoS One 12: e0173499, 2017. doi: 10.1371/journal.pone.0173499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee SJ, Kang HK, Eum WS, Park J, Choi SY, Kwon HY. Tat-biliverdin reductase A protects INS-1 cells from human islet amyloid polypeptide-induced cytotoxicity by alleviating oxidative stress and ER stress. Cell Biol Int 41: 514–524, 2017. doi: 10.1002/cbin.10750. [DOI] [PubMed] [Google Scholar]
- 58.Lee Y, Kim H, Kang S, Lee J, Park J, Jon S. Bilirubin nanoparticles as a nanomedicine for anti-inflammation therapy. Angew Chem Int Ed Engl 55: 7460–7463, 2016. doi: 10.1002/anie.201602525. [DOI] [PubMed] [Google Scholar]
- 59.Li L, Zhao GD, Shi Z, Qi LL, Zhou LY, Fu ZX. The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC. Oncol Lett 12: 3045–3050, 2016. doi: 10.3892/ol.2016.5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lin JP, O’Donnell CJ, Schwaiger JP, Cupples LA, Lingenhel A, Hunt SC, Yang S, Kronenberg F. Association between the UGT1A1*28 allele, bilirubin levels, and coronary heart disease in the Framingham Heart Study. Circulation 114: 1476–1481, 2006. doi: 10.1161/CIRCULATIONAHA.106.633206. [DOI] [PubMed] [Google Scholar]
- 61.Lin YC, Chang PF, Hu FC, Chang MH, Ni YH. Variants in the UGT1A1 gene and the risk of pediatric nonalcoholic fatty liver disease. Pediatrics 124: e1221–e1227, 2009. doi: 10.1542/peds.2008-3087. [DOI] [PubMed] [Google Scholar]
- 62.Loftspring MC, Johnson HL, Feng R, Johnson AJ, Clark JF. Unconjugated bilirubin contributes to early inflammation and edema after intracerebral hemorrhage. J Cereb Blood Flow Metab 31: 1133–1142, 2011. doi: 10.1038/jcbfm.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.López-Velázquez JA, Silva-Vidal KV, Ponciano-Rodríguez G, Chávez-Tapia NC, Arrese M, Uribe M, Méndez-Sánchez N. The prevalence of nonalcoholic fatty liver disease in the Americas. Ann Hepatol 13: 166–178, 2014. [PubMed] [Google Scholar]
- 64.Maghzal GJ, Leck MC, Collinson E, Li C, Stocker R. Limited role for the bilirubin-biliverdin redox amplification cycle in the cellular antioxidant protection by biliverdin reductase. J Biol Chem 284: 29251–29259, 2009. doi: 10.1074/jbc.M109.037119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maines MD. Biliverdin reductase: PKC interaction at the cross-talk of MAPK and PI3K signaling pathways. Antioxid Redox Signal 9: 2187–2195, 2007. doi: 10.1089/ars.2007.1805. [DOI] [PubMed] [Google Scholar]
- 66.Marino JS, Stechschulte LA, Stec DE, Nestor-Kalinoski A, Coleman S, Hinds TD Jr. Glucocorticoid receptor β induces hepatic steatosis by augmenting inflammation and inhibition of the peroxisome proliferator-activated receptor (PPAR) α. J Biol Chem 291: 25776–25788, 2016. doi: 10.1074/jbc.M116.752311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matsuda S, Kobayashi M, Kitagishi Y. Roles for PI3K/AKT/PTEN pathway in cell signaling of nonalcoholic fatty liver disease. ISRN Endocrinol 2013: 472432, 2013. doi: 10.1155/2013/472432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Milić S, Lulić D, Štimac D. Non-alcoholic fatty liver disease and obesity: biochemical, metabolic and clinical presentations. World J Gastroenterol 20: 9330–9337, 2014. doi: 10.3748/wjg.v20.i28.9330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miralem T, Lerner-Marmarosh N, Gibbs PE, Jenkins JL, Heimiller C, Maines MD. Interaction of human biliverdin reductase with Akt/protein kinase B and phosphatidylinositol-dependent kinase 1 regulates glycogen synthase kinase 3 activity: a novel mechanism of Akt activation. FASEB J 30: 2926–2944, 2016. doi: 10.1096/fj.201600330RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miralem T, Lerner-Marmarosh N, Gibbs PE, Tudor C, Hagen FK, Maines MD. The human biliverdin reductase-based peptide fragments and biliverdin regulate protein kinase Cδ activity: the peptides are inhibitors or substrate for the protein kinase C. J Biol Chem 287: 24698–24712, 2012. doi: 10.1074/jbc.M111.326504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mirza MS. Obesity, visceral fat, and NAFLD: querying the role of adipokines in the progression of nonalcoholic fatty liver disease. ISRN Gastroenterol 2011: 592404, 2011. doi: 10.5402/2011/592404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mitchell NS, Catenacci VA, Wyatt HR, Hill JO. Obesity: overview of an epidemic. Psychiatr Clin North Am 34: 717–732, 2011. doi: 10.1016/j.psc.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mölzer C, Wallner M, Kern C, Tosevska A, Schwarz U, Zadnikar R, Doberer D, Marculescu R, Wagner KH. Features of an altered AMPK metabolic pathway in Gilbert’s syndrome, and its role in metabolic health. Sci Rep 6: 30051, 2016. doi: 10.1038/srep30051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morgan TR. Management of alcoholic hepatitis. Gastroenterol Hepatol (NY) 3: 97–99, 2007. [PMC free article] [PubMed] [Google Scholar]
- 75.Moyer BJ, Rojas IY, Kerley-Hamilton JS, Nemani KV, Trask HW, Ringelberg CS, Gimi B, Demidenko E, Tomlinson CR. Obesity and fatty liver are prevented by inhibition of the aryl hydrocarbon receptor in both female and male mice. Nutr Res 44: 38–50, 2017. doi: 10.1016/j.nutres.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.O’Brien L, Hosick PA, John K, Stec DE, Hinds TD Jr. Biliverdin reductase isozymes in metabolism. Trends Endocrinol Metab 26: 212–220, 2015. doi: 10.1016/j.tem.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Odell GB. Studies in kernicterus. I. The protein binding of bilirubin. J Clin Invest 38: 823–833, 1959. doi: 10.1172/JCI103864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ollinger R, Kogler P, Troppmair J, Hermann M, Wurm M, Drasche A, Königsrainer I, Amberger A, Weiss H, Ofner D, Bach FH, Margreiter R. Bilirubin inhibits tumor cell growth via activation of ERK. Cell Cycle 6: 3078–3085, 2007. doi: 10.4161/cc.6.24.5022. [DOI] [PubMed] [Google Scholar]
- 79.Oreña SJ, Torchia AJ, Garofalo RS. Inhibition of glycogen-synthase kinase 3 stimulates glycogen synthase and glucose transport by distinct mechanisms in 3T3-L1 adipocytes. J Biol Chem 275: 15765–15772, 2000. doi: 10.1074/jbc.M910002199. [DOI] [PubMed] [Google Scholar]
- 80.Park BS, Lee JO. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med 45: e66, 2013. doi: 10.1038/emm.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Paschos P, Paletas K. Non alcoholic fatty liver disease and metabolic syndrome. Hippokratia 13: 9–19, 2009. [PMC free article] [PubMed] [Google Scholar]
- 82.Pereira PJ, Macedo-Ribeiro S, Párraga A, Pérez-Luque R, Cunningham O, Darcy K, Mantle TJ, Coll M. Structure of human biliverdin IXβ reductase, an early fetal bilirubin IXβ producing enzyme. Nat Struct Biol 8: 215–220, 2001. doi: 10.1038/84948. [DOI] [PubMed] [Google Scholar]
- 83.Puri K, Nobili V, Melville K, Corte CD, Sartorelli MR, Lopez R, Feldstein AE, Alkhouri N. Serum bilirubin level is inversely associated with nonalcoholic steatohepatitis in children. J Pediatr Gastroenterol Nutr 57: 114–118, 2013. doi: 10.1097/MPG.0b013e318291fefe. [DOI] [PubMed] [Google Scholar]
- 84.Reeves HL, Burt AD, Wood S, Day CP. Hepatic stellate cell activation occurs in the absence of hepatitis in alcoholic liver disease and correlates with the severity of steatosis. J Hepatol 25: 677–683, 1996. doi: 10.1016/S0168-8278(96)80238-8. [DOI] [PubMed] [Google Scholar]
- 85.Roche SP, Kobos R. Jaundice in the adult patient. Am Fam Physician 69: 299–304, 2004. [PubMed] [Google Scholar]
- 86.Rochette L, Zeller M, Cottin Y, Vergely C. Redox functions of heme oxygenase-1 and biliverdin reductase in diabetes. Trends Endocrinol Metab 29: 74–85, 2018. doi: 10.1016/j.tem.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 87.Salomone F, Li Volti G, Rosso C, Grosso G, Bugianesi E. Unconjugated bilirubin, a potent endogenous antioxidant, is decreased in patients with non-alcoholic steatohepatitis and advanced fibrosis. J Gastroenterol Hepatol 28: 1202–1208, 2013. doi: 10.1111/jgh.12155. [DOI] [PubMed] [Google Scholar]
- 88.Schlezinger JJ, Liu D, Farago M, Seldin DC, Belguise K, Sonenshein GE, Sherr DH. A role for the aryl hydrocarbon receptor in mammary gland tumorigenesis. Biol Chem 387: 1175–1187, 2006. doi: 10.1515/BC.2006.145. [DOI] [PubMed] [Google Scholar]
- 89.Schwertner HA, Vítek L. Gilbert syndrome, UGT1A1*28 allele, and cardiovascular disease risk: possible protective effects and therapeutic applications of bilirubin. Atherosclerosis 198: 1–11, 2008. doi: 10.1016/j.atherosclerosis.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 90.Sedlak TW, Saleh M, Higginson DS, Paul BD, Juluri KR, Snyder SH. Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc Natl Acad Sci USA 106: 5171–5176, 2009. doi: 10.1073/pnas.0813132106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sedlak TW, Snyder SH. Bilirubin benefits: cellular protection by a biliverdin reductase antioxidant cycle. Pediatrics 113: 1776–1782, 2004. doi: 10.1542/peds.113.6.1776. [DOI] [PubMed] [Google Scholar]
- 92.Sekera ER, Rudolph HL, Carro SD, Morales MJ, Bett GCL, Rasmusson RL, Wood TD. Depletion of stercobilin in fecal matter from a mouse model of autism spectrum disorders. Metabolomics 13: 132, 2017. doi: 10.1007/s11306-017-1277-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat Med 13: 1324–1332, 2007. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 94.Sérée E, Villard PH, Pascussi JM, Pineau T, Maurel P, Nguyen QB, Fallone F, Martin PM, Champion S, Lacarelle B, Savouret JF, Barra Y. Evidence for a new human CYP1A1 regulation pathway involving PPAR-α and 2 PPRE sites. Gastroenterology 127: 1436–1445, 2004. doi: 10.1053/j.gastro.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 95.Sinal CJ, Bend JR. Aryl hydrocarbon receptor-dependent induction of Cyp1a1 by bilirubin in mouse hepatoma Hepa 1c1c7 cells. Mol Pharmacol 52: 590–599, 1997. doi: 10.1124/mol.52.4.590. [DOI] [PubMed] [Google Scholar]
- 96.Singh V, Chassaing B, Zhang L, San Yeoh B, Xiao X, Kumar M, Baker MT, Cai J, Walker R, Borkowski K, Harvatine KJ, Singh N, Shearer GC, Ntambi JM, Joe B, Patterson AD, Gewirtz AT, Vijay-Kumar M. Microbiota-dependent hepatic lipogenesis mediated by stearoyl CoA desaturase 1 (SCD1) promotes metabolic syndrome in TLR5-deficient mice. Cell Metab 22: 983–996, 2015. doi: 10.1016/j.cmet.2015.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Song YS, Koo BK, Cho NH, Moon MK. Effect of low serum total bilirubin levels (≤0.32 mg/dl) on risk of coronary artery disease in patients with metabolic syndrome. Am J Cardiol 114: 1695–1700, 2014. doi: 10.1016/j.amjcard.2014.08.043. [DOI] [PubMed] [Google Scholar]
- 98.Stambolic V, Woodgett JR. Mitogen inactivation of glycogen synthase kinase-3β in intact cells via serine 9 phosphorylation. Biochem J 303: 701–704, 1994. doi: 10.1042/bj3030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stec DE, John K, Trabbic CJ, Luniwal A, Hankins MW, Baum J, Hinds TD Jr. Bilirubin binding to PPARα inhibits lipid accumulation. PLoS One 11: e0153427, 2016. doi: 10.1371/journal.pone.0153427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stechschulte LA, Wuescher L, Marino JS, Hill JW, Eng C, Hinds TD Jr. Glucocorticoid receptor β stimulates Akt1 growth pathway by attenuation of PTEN. J Biol Chem 289: 17885–17894, 2014. doi: 10.1074/jbc.M113.544072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Su M, Qian C, Hu Y, Lu W, Huang R, Chen M, Chen J. Inhibitory effect of the low-toxic exogenous aryl hydrocarbon receptor modulator 3′3-diindolylmethane on gastric cancer in mice. Oncol Lett 14: 8100–8105, 2017. doi: 10.3892/ol.2017.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sundararaghavan VL, Sindhwani P, Hinds TD Jr. Glucuronidation and UGT isozymes in bladder: new targets for the treatment of uroepithelial carcinomas? Oncotarget 8: 3640–3648, 2017. doi: 10.18632/oncotarget.12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tang Y, Zhang Q, Zhu Y, Chen G, Yu F. Low concentrations of bilirubin inhibit activation of hepatic stellate cells in vitro. Mol Med Rep 15: 1647–1653, 2017. doi: 10.3892/mmr.2017.6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tomaro ML, Batlle AM. Bilirubin: its role in cytoprotection against oxidative stress. Int J Biochem Cell Biol 34: 216–220, 2002. doi: 10.1016/S1357-2725(01)00130-3. [DOI] [PubMed] [Google Scholar]
- 105.Toshikuni N, Tsutsumi M, Arisawa T. Clinical differences between alcoholic liver disease and nonalcoholic fatty liver disease. World J Gastroenterol 20: 8393–8406, 2014. doi: 10.3748/wjg.v20.i26.8393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.van Raalte DH, Li M, Pritchard PH, Wasan KM. Peroxisome proliferator-activated receptor (PPAR)-α: a pharmacological target with a promising future. Pharm Res 21: 1531–1538, 2004. doi: 10.1023/B:PHAM.0000041444.06122.8d. [DOI] [PubMed] [Google Scholar]
- 107.Varga T, Czimmerer Z, Nagy L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim Biophys Acta 1812: 1007–1022, 2011. doi: 10.1016/j.bbadis.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vera T, Granger JP, Stec DE. Inhibition of bilirubin metabolism induces moderate hyperbilirubinemia and attenuates ANG II-dependent hypertension in mice. Am J Physiol Regul Integr Comp Physiol 297: R738–R743, 2009. doi: 10.1152/ajpregu.90889.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vítek L. The role of bilirubin in diabetes, metabolic syndrome, and cardiovascular diseases. Front Pharmacol 3: 55, 2012. doi: 10.3389/fphar.2012.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vítek L, Jirsa M Jr, Brodanová M, Kalab M, Marecek Z, Danzig V, Novotný L, Kotal P. Gilbert syndrome and ischemic heart disease: a protective effect of elevated bilirubin levels. Atherosclerosis 160: 449–456, 2002. doi: 10.1016/S0021-9150(01)00601-3. [DOI] [PubMed] [Google Scholar]
- 111.Vogel ME, Idelman G, Konaniah ES, Zucker SD. Bilirubin prevents atherosclerotic lesion formation in low-density lipoprotein receptor-deficient mice by inhibiting endothelial VCAM-1 and ICAM-1 signaling. J Am Heart Assoc 6: e004820, 2017. doi: 10.1161/JAHA.116.004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wallner M, Marculescu R, Doberer D, Wolzt M, Wagner O, Vitek L, Bulmer AC, Wagner KH. Protection from age-related increase in lipid biomarkers and inflammation contributes to cardiovascular protection in Gilbert’s syndrome. Clin Sci (Lond) 125: 257–264, 2013. doi: 10.1042/CS20120661. [DOI] [PubMed] [Google Scholar]
- 113.Wang H, Lee SS, Dell’Agnello C, Tchipashvili V, d’Avila JC, Czismadia E, Chin BY, Bach FH. Bilirubin can induce tolerance to islet allografts. Endocrinology 147: 762–768, 2006. doi: 10.1210/en.2005-0632. [DOI] [PubMed] [Google Scholar]
- 114.Wang XC, Chowdhury JR, Chowdhury NR. Bilirubin metabolism: applied physiology. Curr Paediatr 16: 70–74, 2006. doi: 10.1016/j.cupe.2005.10.002. [DOI] [Google Scholar]
- 115.Washington K, Wright K, Shyr Y, Hunter EB, Olson S, Raiford DS. Hepatic stellate cell activation in nonalcoholic steatohepatitis and fatty liver. Hum Pathol 31: 822–828, 2000. doi: 10.1053/hupa.2000.8440. [DOI] [PubMed] [Google Scholar]
- 116.Wegiel B, Baty CJ, Gallo D, Csizmadia E, Scott JR, Akhavan A, Chin BY, Kaczmarek E, Alam J, Bach FH, Zuckerbraun BS, Otterbein LE. Cell surface biliverdin reductase mediates biliverdin-induced anti-inflammatory effects via phosphatidylinositol 3-kinase and Akt. J Biol Chem 284: 21369–21378, 2009. doi: 10.1074/jbc.M109.027433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wegiel B, Gallo D, Csizmadia E, Roger T, Kaczmarek E, Harris C, Zuckerbraun BS, Otterbein LE. Biliverdin inhibits Toll-like receptor-4 (TLR4) expression through nitric oxide-dependent nuclear translocation of biliverdin reductase. Proc Natl Acad Sci USA 108: 18849–18854, 2011. doi: 10.1073/pnas.1108571108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Willis D, Moore AR, Frederick R, Willoughby DA. Heme oxygenase: a novel target for the modulation of the inflammatory response. Nat Med 2: 87–90, 1996. doi: 10.1038/nm0196-87. [DOI] [PubMed] [Google Scholar]
- 119.Yin XF, Chen J, Mao W, Wang YH, Chen MH. A selective aryl hydrocarbon receptor modulator 3,3′-diindolylmethane inhibits gastric cancer cell growth. J Exp Clin Cancer Res 31: 46, 2012. doi: 10.1186/1756-9966-31-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhou JA, Jiang M, Yang X, Liu Y, Guo J, Zheng J, Qu Y, Song Y, Li R, Qin X, Wang X. Unconjugated bilirubin ameliorates the inflammation and digestive protease increase in TNBS-induced colitis. Mol Med Rep 16: 1779–1784, 2017. doi: 10.3892/mmr.2017.6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhou ZX, Chen JK, Hong YY, Zhou R, Zhou DM, Sun LY, Qin WL, Wang TC. Relationship between the serum total bilirubin and inflammation in patients with psoriasis vulgaris. J Clin Lab Anal 30: 768–775, 2016. doi: 10.1002/jcla.21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhu B, Wu X, Bi Y, Yang Y. Effect of bilirubin concentration on the risk of diabetic complications: a meta-analysis of epidemiologic studies. Sci Rep 7: 41681, 2017. doi: 10.1038/srep41681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhu H, Wang J, Jiang H, Ma Y, Pan S, Reddy S, Sun X. Bilirubin protects grafts against nonspecific inflammation-induced injury in syngeneic intraportal islet transplantation. Exp Mol Med 42: 739–748, 2010. doi: 10.3858/emm.2010.42.11.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ziberna L, Martelanc M, Franko M, Passamonti S. Bilirubin is an endogenous antioxidant in human vascular endothelial cells. Sci Rep 6: 29240, 2016. doi: 10.1038/srep29240. [DOI] [PMC free article] [PubMed] [Google Scholar]