

Abstract
Drug-induced pulmonary arterial hypertension (D-PAH) is a form of World Health Organization Group 1 pulmonary hypertension (PH) defined by severe small vessel loss and obstructive vasculopathy, which leads to progressive right heart failure and death. To date, 16 different compounds have been associated with D-PAH, including anorexigens, recreational stimulants, and more recently, several Food and Drug Administration-approved medications. Although the clinical manifestation, pathology, and hemodynamic profile of D-PAH are indistinguishable from other forms of pulmonary arterial hypertension, its clinical course can be unpredictable and to some degree dependent on removal of the offending agent. Because only a subset of individuals develop D-PAH, it is probable that genetic susceptibilities play a role in the pathogenesis, but the characterization of the genetic factors responsible for these susceptibilities remains rudimentary. Besides aggressive treatment with PH-specific therapies, the major challenge in the management of D-PAH remains the early identification of compounds capable of injuring the pulmonary circulation in susceptible individuals. The implementation of pharmacovigilance, precision medicine strategies, and global warning systems will help facilitate the identification of high-risk drugs and incentivize regulatory strategies to prevent further outbreaks of D-PAH. The goal for this review is to inform clinicians and scientists of the prevalence of D-PAH and to highlight the growing number of common drugs that have been associated with the disease.
Keywords: drug-induced pulmonary hypertension, pathology, pharmacovigilance, prevention
FIFTY YEARS OF DRUG-INDUCED PULMONARY ARTERIAL HYPERTENSION: A HISTORICAL PERSPECTIVE
Until 1968, primary pulmonary hypertension (PPH) was a poorly characterized disease because of the challenges of studying pulmonary vascular physiology and the low prevalence of the disease. Despite the fact that the pathology had already been documented, the physiology of PH was unknown until the advent of right heart catheterization (RHC) in the 1950s (11). However, in 1968, international interest in further characterizing PH increased because of a rapid rise in the number of cases of PPH in Switzerland, Germany, and Austria (58, 59).
Three years prior, aminorex fumarate (sold under the name Menocil), an amphetamine-like appetite suppressant, had come on the market as a novel treatment for obesity (Fig. 1). In the years that followed, an increase in the incidence of PPH was noted. One or two in 1,000 aminorex users developed PH, a significantly higher incidence rate than primary pulmonary arterial hypertension (PAH, estimated at 1–2 cases per million per year). For almost 60% of patients diagnosed within this time frame, symptoms began ~6 mo after the initiation of aminorex intake, although some noted symptoms within weeks of starting to use the drug. While several patients had regression of their disease after discontinuing aminorex, the mortality rate was high, with nearly one-half of the aminorex-exposed PPH patients dying within 10 years of diagnosis from pulmonary hypertension (PH)-related complications (53). Aminorex was taken off the market in Europe in 1972 following these case reports but remained on the market in the United States (U.S.) until 1973.
Fig. 1.

Molecular structure of amphetamine-derived anorexinogens.
In response to the aminorex-associated PPH epidemic, the World Health Organization (WHO) organized the First WHO Pulmonary Hypertension Conference in 1973 to assess the current state of knowledge and define the nomenclature and classification system that would be used to describe PH in the future. The conference led to a commitment to better understand the nature of normal pulmonary circulation and to carefully study cases of PH (62). In the U.S., the National Heart, Lung, and Blood Institute of the National Institutes of Health created a national database of PH cases shortly after the First WHO Pulmonary Hypertension Conference. Within just a few years, nearly 200 patients had been added to the database, and these early patients’ data contributed significantly to the evolving understanding of the pathophysiology of PH (125).
Around the same time, cases of PH were associated with other drugs and substances, raising further concern that PH could be triggered by substance exposure. Toxic oil syndrome was a scleroderma-like disease with vascular endothelial swelling and proliferation associated with PH triggered by exposure to a toxic rapeseed oil anilin derivative (50). Approximately 20,000 cases of PH associated with toxic oil syndrome were identified, resulting in >300 deaths (80). In 1989, another outbreak of PH, described as eosinophilia-myalgia syndrome, occurred in Mexico in association with a toxic product of l-tryptophan (64). In many of these cases, PH developed in patients who seemed to have an underlying genetic predisposition that was unmasked by exposure to the substance (7). In addition, phenformin, one of the early biguanides, was associated with the development of PH in a few patients; the PH resolved upon cessation of the drug (41).
Most notable of the substances associated with PH during this time was fenfluramine, popularly prescribed in combination with phentermine as “Fen-phen,” which became available on the market in the late 1970s. Like aminorex, fenfluramine is an amphetamine-like drug with indirect serotonergic activity used for weight loss. In 1981, the first report of fenfluramine-associated PPH was published (34). The International Primary Pulmonary Hypertension Study (IPPHS), a multicenter case control study conducted between 1992 and 1994, subsequently demonstrated a definite risk of PH associated with anorexic agents like fenfluramine (2). Abenhaim et al. recruited patients diagnosed with primary PH at centers throughout France, Belgium, the United Kingdom, and the Netherlands. Patients with diagnoses associated with secondary PH, such as interstitial lung disease or myocardial disease, were excluded. Age- and gender-matched controls were identified from regional primary care practices. The exposure status was assessed via structured face-to-face interviews by trained blinded interviewers with additional information provided from review of the medical chart. The drugs of interest included fenfluramine and its relative dexfenfluramine, amphetamine-like anorexic agents, and other appetite suppressants. The study included 95 cases and 355 controls. Use of anorexic drugs, predominantly fenfluramine and dexfenfluramine, was associated with an odds ratio of 6.3 (95% confidence interval: 3.0–13.2) of having primary PH, and if those substances had been used for more than 3 mo, the association was even stronger with an odds ratio of 23.1 (6.9–77.7). Tragically, dexfenfluramine was approved in the U.S. for treatment of obesity in 1996. Of note, the editorial that accompanied the New England Journal of Medicine article by Abenhaim et al. concluded that the benefits of these agents outweighed the risks of PAH, thus allowing prescription of the medication to continue in the U.S. (92).
In the years following IPPHS, two additional case-control studies, the Surveillance of North American Pulmonary Hypertension study (126) and the Surveillance of Pulmonary Hypertension in America study (144), confirmed that there was a strong association between fenfluramine exposure and PH. During this same time, evidence mounted that fenfluramine and its relatives were associated with cardiac valvular lesions (23). It was hypothesized that this association was because of the serotonergic effects of fenfluramine and its relatives that led to heart lesions similar to those seen in carcinoid syndrome (96), which interestingly is not associated with pulmonary vascular disease. Shortly after the publication of these reports, in 1997, fenfluramine and dexfenfluramine were taken off the market.
Benfluorex hydrochloride (Mediator) was yet another derivative of fenfluramine marketed as a weight loss and diabetes drug. It was approved in Europe in the 1970s, and, because benfluorex was approved as a treatment for diabetes and metabolic syndrome, rather than as an anorexigen, it did not receive the same level of scrutiny as fenfluramine and dexfenfluramine did in the 1980s and 1990s. As a result, benfluorex was available on the market into the 2000s. However, in 2009 and 2012, two case series were published describing an association between benfluorex exposure and PH (14, 131). Both case series described a combination of PH and valvulopathy, as was seen with fenfluramine and dexfenfluramine, although not all patients with fluramine-induced PH had valve disease. After the first case series in 2009, the European Medicines Agency recommended withdrawal of benfluorex from the market.
RECENT OUTBREAKS OF DRUG-INDUCED PULMONARY ARTERIAL HYPERTENSION ASSOCIATED WITH MEDICATIONS AND TOXINS
Interferons
Interferons (IFN) are cytokines released by immunomodulatory cells in response to infections and malignancies. IFN have been developed as therapy for a variety of immunologic and malignant processes (51). Among these, PEGylated INF-α2a (Pegasys) and -β (Pegintron) are Food and Drug Administration (FDA) approved for the treatment of multiple sclerosis, hematological malignancies, and chronic hepatitis C virus infection. In the last 5 years, case reports have linked IFN use with the development of PAH and raised safety concerns regarding the use of these agents in clinical practice (45, 73, 84). The 2014 French PH registry study reported 53 patients with PAH, 48 of which developed PAH after starting IFN while the remainder had a history of PAH before the initiation of therapy (132). Of the 53 patients, 48 had been treated with IFN-α for a mean duration of 7.7 mo for chronic hepatitis C virus (HCV) (n = 47) and chronic myelogenous leukemia (CML, n = 1). The remaining five patients had been treated with IFN-β for multiple sclerosis, and the time between treatment initiation and diagnosis of PAH was 59–117 mo (i.e., 5–10 yr). Most of these patients developed severe PAH, and two of them died as a result of PAH-related complications. Also, these patients exposed to IFN demonstrated worsening of symptoms and decreased exercise capacity and hemodynamic variables, all of which were parameters reversible in most cases after IFN discontinuation and did not require further treatment with PAH-specific agents. Given the strength of the data accrued so far, IFNs are now recognized as “possible” risk factors for drug-induced pulmonary arterial hypertension (D-PAH) in the most recent 2015 European Society of Cardiology and European Respiratory Society PH guidelines (47).
In an effort to bypass the significant toxicity of IFN for the treatment of HCV, new direct-acting antiviral agents (DAAs) like sofosbuvir have recently become available with demonstrable rates of cure as high as 85% in some studies when combined with ribavirin (49, 83). Whereas use of sofosbuvir and other DAAs has increased in the last 3 years, it must be noted that three cases of severe PAH in HCV patients treated with sofosbuvir-based regimes have been reported (124). Whether PAH in these patients could also be explained by the presence of other risk factors such as HIV and portal hypertension is unknown, but physicians should be aware of this possible D-PAH risk and report cases to pharmacovigilance groups for further monitoring.
Dasatinib
Tyrosine kinase inhibitors (TKIs) are a key treatment for CML, a hematological malignancy caused by chromosomal translocation that leads to the pathogenic tyrosine kinase protein breakpoint cluster region/Abelson (BCR/ABL) (103). TKIs like imatinib (Gleevec) and dasatinib (Sprycel) are being used to treat CML since they can inhibit BCR/ABL kinase activity in CML cells and induce prolonged remission. However, up to 20% of CML patients do not achieve a complete cytogenic response with imatinib. For these patients that do not respond to imatinib, a more potent TKI like dasatinib can be used to induce CML remission. Despite the therapeutic benefit of dasatinib in patients with CML, numerous studies have now shown that dasatinib can lead to PAH development in a subset of these patients.
In 2012, cases were reported from the France PH registry where nine patients had previously been treated with imatinib and subsequently with dasatinib (103). Both drugs were used to treat CML, although dasatinib was prescribed when the disease was found to be nonresponsive to imatinib. At the time of PH diagnosis, the patients presented with progressive dyspnea as well as bilateral pleural effusions in six of nine patients. With the previous evidence of a potential relationship between dasatinib exposure and development of precapillary PH, dasatinib treatment was discontinued in all patients and substituted with the alternate potent TKI nilotinib. In six patients, specific PAH therapy was not necessary, and within 8–36 mo across patients, eight out of nine patients showed clinical and/or hemodynamic improvement. However, despite this improvement, no patient had complete recovery, and two patients died at the time of follow-up. Of these two patients, one did not show any hemodynamic improvement and had the most severe hemodynamic compromise of all nine patients, whereas the other patient experienced sudden death while on a long-distance airplane flight. It must be noted that none of the affected patients had mutations in bone morphogenic protein receptor 2 (BMPR2), that is, mutations of a gene associated with both familial and sporadic forms of PAH.
Since the publication of this report, a study using the Bristol-Myers-Squibb (BMS) international pharmacovigilance database has reported on the clinical course of 41 patients with a diagnosis of dasatinib-induced PAH confirmed by RHC between 2006 and 2013 (134). In this cohort, PAH appeared to develop across a wide timeframe that ranged from as little as 1 mo to almost 7 yr after the initial exposure. Furthermore, cessation of dasatinib led to improvement of PAH in 94% of cases and complete resolution in 58%. Because of the small sample size, the investigators were not able to identify patient characteristics or risk factors associated with dasatinib-induced PAH. Also, a repeat RHC was performed for only five patients, and follow up echocardiograms were performed for only seven patients. Subsequently, Weatherald et al. reported changes in hemodynamics and clinical variables over a period of 1–81 mo (median range 24 mo) in 21 RHC-proven dasatinib-PAH cases from the French Pulmonary Hypertension Registry (146). Similar to the BMS study, most patients demonstrated improvement in hemodynamics, functional class, and 6-min walk distance after cessation of dasatinib. However, more than one-third of patients had persistent PAH when reassessed by RHC during follow-up, and 43% remained symptomatic despite being off dasatinib. The results of this study highlight the possibility that dasatinib can injure the pulmonary vasculature. The authors conclude that patients exposed to dasatinib should receive regular follow-up and initiate therapy if there is clinical and/or hemodynamic evidence of PAH.
To date, no definite cases of PAH have been documented in association with imatinib or nilotinib. It is worth pointing out that, until recently, imatinib had been studied for treatment of PAH, but a phase 3 study failed to demonstrate consistent clinical benefits and raised safety concerns around the drug (67). Of note, cases of PAH associated with bosutinib and ponatinib have also been reported (66, 119), further stressing the need for continued pharmacovigilance over the TKIs.
Mitomycin C
Mitomycin C (MMC) is a bioreductive alkylating agent that is being used as chemotherapy in the management of various malignancies (120). In recent years, MMC has been identified as a possible risk factor for pulmonary veno-occlusive disease (PVOD) (46), a rare form of PAH with an incidence of <0.5 cases per million per year and associated with a very poor prognosis, since most patients die within 2 year of diagnosis (68, 104). Perros et al. described seven cases of MMC-induced PVOD that were identified in the French PH Registry between June 2012 and December 2014 (116). In each case, the primary malignancy was squamous anal cancer, and all patients received MMC, although five of the seven cases also received 5-fluoruracil. The authors note that, in France, treatment with MMC is almost exclusively used in the setting of anal cancer. PVOD was diagnosed in these cases based on clinical, functional, radiological, and hemodynamic changes; a histologic diagnosis was precluded by the tenuous overall clinical condition of patients. Known risk factors for PAH were identified in one patient, who tested positive for human immunodeficiency virus (HIV) infection. The median age of these cases was 53 yr with a predominance of females (6 of 7). At the time of diagnosis, four patients were within New York Heart Association functional class III and three cases in class IV. Even after specific PAH therapies (either as monotherapy or a combination of endothelin-receptor antagonists plus phosphodiesterase type 5 inhibitors or prostacyclins), four of seven patients died: two because of right heart failure, one from local tumor progression, and one patient from severe pneumonia. The authors conclude that unexplained dyspnea after MMC therapy should raise the suspicion of PVOD and pulmonary toxicity. Of note, MMC may not be the only chemotherapeutic agent linked to PVOD, since an analysis has also shown that alkylating agents may represent a drug class that may be linked to PVOD development in susceptible individuals (122).
Although Perros et al. reported that a majority of MMC-induced PVOD patients died (4 of 7) in the French PH Registry despite PAH-specific therapies, there are conflicting case reports of MMC-induced PAH that improved with standard treatment. For example, Botros et al. report a case of a 60-yr-old woman diagnosed with anal carcinoma and then treated with MMC, 5-flurouracil, and local radiotherapy. Within 3 mo, diagnosis of PAH was established upon workup for symptoms of shortness of breath and ankle edema. PVOD was initially suspected; nonetheless, bosentan was added to diuretics and oral anticoagulation, and the patient recovered rapidly, no longer dependent on supplemental oxygen after 3 mo. Follow-up after 1 year confirmed that she had become symptom free while on the continued regimen that included bosentan therapy (13).
Selective Serotonin Reuptake Inhibitors
Selective serotonin reuptake inhibitors (SSRIs) are a recent addition to the list of PAH-associated drugs and toxins for their involvement in persistent pulmonary hypertension of the newborn (PPHN) (136). Multiple studies have been published with discordant results on the association of SSRIs with PPHN (5, 18, 19, 75, 147, 150, 151), but one population-based cohort study looking at over 1.6 million infants found that infants born to mothers who had taken SSRIs in late pregnancy were more than two times as likely to develop PPHN (79). Based on this large study, and the fact that serotonin signaling likely plays a role in pulmonary smooth muscle cell function (more on this in Aminorex, fenfluramine, and serotonin signaling), SSRIs are classified as having a “definite” association with PAH. However, PAH in adults exposed to SSRIs has not been thoroughly investigated.
Methamphetamine
Methamphetamine (METH) is a highly addictive and potent neurostimulant with systemic effects that can result in severe organ dysfunction and premature death (52). The molecular structure of methamphetamine is similar to that of aminorex fumarate and fenfluramine, stimulants known to cause D-PAH (2, 43) (Fig. 1). As of 2010, the worldwide prevalence of METH use was estimated at 14.3–52.5 million, second only to cannabis as the most common illicit substance of use. In California, METH use was associated with an incidence of 12–18 cases/10,000 hospital admissions between 2006 and 2013 (54). These epidemiological data stress the growing threat of METH to our society and the likelihood that practitioners will encounter patients afflicted with acute and chronic complications associated with METH use.
The link between METH and PAH has been reported by Schaiberger et al. and Chin et al. (21, 133); furthermore, PET studies have shown that intravenous d-[11C]METH accumulates primarily in the lung and could serve as an active insult to the pulmonary circulation (143). Currently, METH use is considered a “likely” risk factor for the development of D-PAH (48), but it is evident that much more work needs to be done to clarify the clinical and pathological features of METH-induced PAH (METH-PAH).
Zamanian et al. recently reported our decade-long experience with METH-PAH and queried the Healthcare Cost and Utilization Project California database for admission related to METH abuse and PAH (156). METH-PAH was defined as PAH in the setting of significant METH exposure following documentation of more than three episodes of use per week for >3 mo. In our study population, most patients reported inhalation/smoking as the preferred administration route; the self-reported median METH exposure was 60 mo, which was similar across both genders. Most importantly, Kaplan-Meier analysis showed a 5- and 10-yr event-free survival of 47.2 and 25%, respectively, in METH-PAH vs. 64.5 and 45.7% in idiopathic pulmonary hypertension (IPAH) (50).
Using explanted lung tissue, we were able to document that lungs from METH-PAH patients demonstrated characteristic vascular changes similar to those of IPAH, including angiomatoid plexiform lesions with slit-like vascular channels within the artery (156). However, analysis of hemodynamic parameters showed that, compared with IPAH, METH-PAH is characterized by more severe pulmonary vascular disease at baseline, as evidenced by higher right atrial pressure, lower stroke volume index, and a more dilated and dysfunctional right ventricle. The severity of hemodynamic and right ventricle (RV) impairment in METH-PAH is worse than that described in dasatinib-PAH, further underscoring the harmful impact of this toxin on pulmonary vascular health.
HIV and Drug Abuse
Although individuals infected with HIV are at increased risk for developing PAH, incidence is more common in HIV-infected drug-abusing individuals. Multiple reports document increased risk of PAH development in HIV-infected individuals abusing illicit drugs such as cocaine, opioids, methamphetamine, and marijuana (22, 61, 69, 95, 102, 128, 154). Intravenous drug use (IVDU) accounts for one-third of all new HIV infection in the U.S. (8) and has been identified as the major risk factor of HIV infection in the individuals diagnosed with HIV-PAH. Various systematic reviews and reports on HIV-PH have reported IVDU to be a risk factor in 42–78% of HIV-infected patients diagnosed with PH (6, 72, 97, 99, 111). A report published by Quezada et al. (118) reported a 9.9% prevalence of PAH related to HIV among which 58.6% patients were intravenous drug users. In addition, another recent study (123) reported a PAH prevalence of 3% in a HIV-infected patient cohort that involved 51% intravenous drug users.
Enhanced pulmonary vascular remodeling in HIV-infected lung tissues from intravenous cocaine and/or opioids abusers (31) indicates that IVDU and HIV-1 potentially act in concert to cause a pulmonary arteriopathy. Studies demonstrate that the direct action of HIV proteins released by the infected lymphocytes and macrophages plays a major role in the development of HIV-PAH (71, 142). Corroborating these findings, it has been shown that cocaine exposure triggers severe pulmonary vascular remodeling and PH in a noninfectious HIV-transgenic (Tg) rat model expressing HIV-1 proteins (24, 90, 98). HIV-Tg rats exposed to cocaine (40 mg/kg) for 21 days had higher mean pulmonary arterial pressure and right ventricle systolic pressure (RVSP) compared with age-matched saline-treated HIV-Tg rats or wild-type rats given either cocaine or saline. Importantly, the combination of cocaine and viral proteins had a synergistic effect in augmenting pulmonary smooth muscle cell proliferation in the vasculature (24, 31). The lung tissues from simian immunodeficiency virus-infected rhesus macaques exposed chronically to morphine (opioid) also demonstrated a severe pulmonary vascular arteriopathy with obliteration of vessels resulting from enhanced proliferation of endothelial cells (137). Oxidative stress-mediated enhanced proliferation of endothelial cells was not only observed upon combined treatment of viral proteins with morphine but also with cocaine and methamphetamine when compared with either treatment alone (137).
Taken together, these findings suggest that illicit use of intravenous drugs by HIV patients acts as a second hit that modifies and potentiates pulmonary vascular remodeling, resulting in increased incidence and severity of HIV-PAH in HIV-infected drug abusers. Complicating the clinical scenario further, other variables often coexist in HIV-infected intravenous drug users such as opportunistic lung infections, hepatitis-related liver disease, or cigarette smoking, which may also contribute to the milieu conducive to pulmonary vascular injury.
Tobacco Smoke
The impact of tobacco smoking on the lung circulation remains a controversial topic in the field of D-PAH, yet the link between the development of PH secondary to small vessel loss from emphysema and destruction of lung parenchyma is not in doubt. Studies have shown that exposure to cigarette smoke extract can induce endothelial cell apoptosis (109) and also can trigger the production of antiendothelial antibodies in susceptible individuals (139). Animal studies have also shown that acute smoke exposure induces cell proliferation in pulmonary arterioles, ultimately leading to muscularization with chronic exposure and thus development of PAH (153). This may be in part the result of a significant increase in gene expression of endothelin and vascular endothelial growth factor, while exposed to cigarette smoke, with a disproportionately low expression of vasodilator-related proteins such as endothelial nitric oxide synthase (152). In 2009, Ferrer et al. investigated the contribution of cigarette smoking to PH in COPD at early disease stages (42). Nineteen male Hartley guinea pigs were exposed to daily cigarette smoke for either 3 or 6 mo and were compared with controls exposed to sham smoke during the same periods of time. When the animals were killed and pulmonary vessels were examined, it was found that endothelial dysfunction was seen at 3 mo of exposure and did not increase further after 6 mo of cigarette smoke exposure. At 3 mo, smoke-exposed samples showed poorly differentiated smooth muscle cells proliferating in small vessels. Furthermore, prolonged smoke exposure led to full muscularization of small pulmonary vessels, wall thickening, and increased contractility of the main pulmonary artery. Finally, lung expression of endothelial nitric oxide synthase was decreased in the group exposed to cigarette smoke.
In 2013, Keusch et al. performed an international, multicenter, observational, case-control study from 2008 to 2012 in which 472 patients with PAH and chronic thromboembolic pulmonary hypertension (CTEPH) were asked about their smoking exposure and habits (78). Included patients were from Switzerland, Germany, and Austria who met inclusion criteria of a mean pulmonary artery pressure ≥25 mmHg and pulmonary artery wedge pressure ≤15 mmHg. Those with concomitant chronic lung disease (WHO group III) and other confounding comorbidities (WHO group V) were excluded from the analysis; 233 of the included 472 patients (49%) were smokers and predominantly men (71%). The included cases were then compared against a group of healthy controls, and it was determined that significantly more PAH and CTEPH men were active smokers (71 vs. 57% of the healthy controls, P = 0.0007). When combining rates of active smoking and exposure to second-hand smoke, PAH and CTEPH patients had an overall higher active and passive tobacco smoke exposure compared with healthy controls, 68 vs. 51% in women (P < 0.0001) and 82 vs. 69% in men (P = 0.0002). Furthermore, active smokers among PAH and CTEPH women and men were significantly younger compared with nonsmokers (women: age 52 ± 13 vs. 58 ± 18 yr, P = 0.0051; men: age 57 ± 15 vs. 64 ± 17 yr, P = 0.0288). In all, the authors concluded that the increased prevalence of active and passive cigarette smoke exposure among the PAH and CTEPH population, along with a lower age at PH diagnosis in smokers, may indicate a pathogenic role of smoking. Finally, Trip et al. reported that a considerable number of patients in their Dutch cohort of IPAH patients were smokers that had a surprisingly low diffusion capacity for carbon dioxide, suggesting that in these patients there was a diminished pulmonary capillary bed (140).
Chemical Solvents
Occupational chemical exposure has been reported as a risk factor for PVOD and systemic sclerosis, a disease also associated with obstructive arterial and venous remodeling and PAH (33). A case-control study conducted at the National Reference Centre for Severe Pulmonary Hypertension in France showed that a significant occupational history of exposure was evident in 72.7% of PVOD patients vs. 27.7% of IPAH patients (116). Among PVOD patients, the most common occupational exposures included trichloroethylene (58.3%) with a median latency between first exposure and PVOD diagnosis of 48 yr and median exposure duration of 17 yr, suggesting that chronic and repeated exposure is probably necessary to induce PVOD. Compared with nonexposed patients, those with trichloroethylene exposure were older (54.8 ± 21.4 vs. 67.9 ± 8.1) and did not harbor any eukaryotic translation initiation factor 2-α kinase 4 (EIF2AK4) mutations, a risk factor for familial and sporadic PVOD. The authors suggest that individual susceptibility must be important and raises the possibility that polymorphisms in cytochrome P-450 2E1 (CYP2E1) could be linked to altered metabolism of trichloroethylene and could affect the susceptibility to trichloroethylene-induced toxicity in exposed patients (86).
Mechanisms of D-PAH Pathogenesis: Data From In Vivo and In Vitro Models
The last two decades have seen major advances in the understanding of the pathobiology of PAH. At present, severe PAH is thought to require the combination of genetic and environmental factors (i.e., the 2-hit hypothesis) that precipitate a phenotypic switch in the cellular milieu of the pulmonary vascular wall, resulting in increased pulmonary vasoconstriction, abnormal vascular remodeling, and right heart hypertrophy. D-PAH provides a unique window allowing the study of how environmental events such as exposure to certain drugs and/or toxins may precipitate such a cell phenotype switch in susceptible individuals. However, as we shall see in the following section, our understanding of D-PAH has been significantly impaired by the absence of animal models that recapitulate the clinical and pathological features of D-PAH upon exposure to known offending agents. For example, Byrne-Quinn and Grover administered aminorex or amphetamine to calves and assessed pulmonary hemodynamics after 4 wk of drug treatment; the animals did not develop PH (17). Mlczoch et al. treated pigs with fenfluramine alone or in combination with aminorex and found no evidence for PH or alterations of the pulmonary vascular tone (101).
PAH is a very complex and heterogeneous group of diseases in which multiple cell types, tissues, and even distant organs are thought to contribute to its development. The group of diseases is also characterized by several clinical features, not all of which may be present in each individual patient. Rodent models of PAH often do not recapitulate the prominent features of human pathobiology. Animal models of PAH use either chronic hypoxia or monocrotaline (MCT) to induce pulmonary endothelial injury, but a “second-hit” seems to be necessary to achieve an obliterative pulmonary vascular histopathology that resembles that observed in the human disease. For example, MCT combined with pneumonectomy or a shunt causes severe PAH with neointimal lesions and right ventricular hypertrophy, but MCT alone only caused moderate RVH and limited arterial muscularization (112, 149). More than 15 years ago, a new model of D-PAH had been developed. This model is built on the combination of a subcutaneous injection of the vascular endothelial growth factor (VEGF) receptor blocker Sugen 5614 (the first hit) with chronic hypoxia or pneumonectomy (the second hit) (1). In either case the rats reproducibly develop severe PAH because of a vasoobliterative microangiopathy, and they die from right heart failure. The drug Sugen 5416 causes apoptosis of lung vascular endothelial cells, hypothetically because the endothelial cells organ-specifically require intact VEGF signaling for their maintenance (3). Like the monocrotaline rat model, the Sugen 5416 models are models of drug-induced pulmonary endothelial cell injury. It is worth mentioning that cancer therapeutics that target VEGF are currently in use to treat various malignancies; despite one report on PH complicating severe cardiotoxicity with bevacizumab (i.e., Avastin) (20), no isolated cases of PAH have been associated with this drug.
As the list of offending agents continues to grow, it is imperative that we understand animal models better and develop additional models of D-PAH to gain a better understanding of the differences in metabolic pathways and detoxification responses compared with humans.
Aminorex, Fenfluramine, and Serotonin Signaling
The serotonin pathway has been identified by both experimental and clinical studies as a major contributor to disease severity and progression. Early studies that led to “The Serotonin Hypothesis” in PAH discovered a significant correlation between the consumption of a class of diet pills that augment serotonin signaling and the development of PAH in humans (2, 76). Treatment with one of these drugs, fenfluramine, has been shown to increase the level of circulating serotonin, or 5-hydroxytryramine (5-HT), by interacting with the 5-HT transporter (5-HTT) in platelets to cause the release of 5-HT (44, 94). PAH patients that have not been exposed to fenfluramine may also have elevated plasma 5-HT levels and increased expression of 5-HTT (37, 38, 65), placing serotonin signaling as a possible disease trigger. Serotonin can also signal through 5-HT receptors, including 5-HT1b and 5-HT2a. Studies in humans and rodents have identified 5-HT1b to play a role in mediating vasoconstriction of small pulmonary arteries (77, 105). The contributions of 5-HTT and 5-HT1b to the development of PH have been shown to be synergistic, since inhibiting both with chemical antagonists is effective in preventing 5-HT-induced PH (106). The effect of excessive serotonin signaling in the lung appears to occur through smooth muscle cells (SMCs), which respond to 5-HT with increased proliferation (85, 117), a hallmark of PAH. SMCs have also been shown to be resistant to apoptosis, with 5-HT signaling through 5-HTT and 5-HT1b to activate the ERK pathway (88). Hypoxia is also found to increase circulating 5-HT levels in the airways and in cultured SMCs (36, 74), as well as to increase the expression of 5-HTT in SMCs located in the media of distal remodeled vessels of chronic hypoxic rats (36). SSRIs such as fluoxetine can competitively inhibit 5-HTT (4) and prevent the growth response of PA-SMC to 5-HT in vitro (36). Treatment with fluoxetine protects mice from chronic hypoxic-induced PH (93) and reverses MCT-induced PH in rats (55, 57). Moreover, fluoxetine has a suppressive effect on the chronic methamphetamine-induced pulmonary vascular remodeling in rats via inhibition of serotonin transporter 5-HTT (87). Although these studies support a therapeutic role for 5-HTT inhibition, a recent study by de Raaf and colleagues documented no difference in hemodynamics and lung angioproliferative changes in 5-HTT knockout and wild-type rats exposed to Sugen 5416 (29), questioning the role of serotonin in neointima formation and angioproliferative vascular remodeling.
The most important finding from animal studies investigating the roles of fenfluramine and dexfenfluramine is that these anorexigens indeed have an impact on lung homeostasis and vascular dynamics. Unfortunately, however, while in humans these drugs are strongly linked to disease onset, the results from fenfluramine and dexfenfluramine rodent studies have not produced a detailed understanding of the mechanisms of how these drugs contribute to the development of PAH. In fact, in one study, dexfenfluramine appeared to protect the rat lung vasculature against MCT-induced vascular disease (100). Similarly, Rochefort et al. found that the hypoxia-induced PH in rats was attenuated with continuous dexfenfluramine treatment (127). Other studies find that dexfenfluramine treatment does not affect the hypoxia-induced PH seen in rats after 4 wk, but discontinuation of treatment thereafter with continued hypoxia leads to severe PH with marked increases in 5-HTT expression and right ventricular hypertrophy (30, 35, 39). However, an earlier pair of studies from one laboratory found that in hypoxic dogs there was an increased vascular tone when the animals had been treated with dexfenfluramine. The conclusion of the authors was that the drug treatment was associated with a change in pulmonary vascular resistance that was unrelated to the vasoreactivity in response to hypoxia (107, 108). Chronic fenfluramine treatment in dogs was also found to lead to pulmonary vasoconstriction, but only in the presence of a second hit like endothelin-1 exposure (10). Collectively, these studies suggest that dexfenfluramine plays an active role in hypoxia-induced pulmonary vascular changes and suggest that the drug may strike a sensitive balance in how the lung tissue deals with oxygen availability.
Fenfluramine and dexfenfluramine are known to augment serotonin signaling by either increasing circulating 5-HT, 5-HTT expression, and/or 5-HT receptors with a notable effect in pulmonary smooth muscle cells. The Dempsie laboratory has described how anorexigens could have off target affects in the lung. This laboratory employed transgenic mice, either lacking tryptophan hydroxylase-1 or overexpressing 5-HTT, to ask how dexfenfluramine and the serotonin pathway cooperate to cause lung pathology. The results clearly showed that circulating 5-HT is necessary for dexfenfluramine treatment to cause vascular changes and increased pulmonary pressure, but the results also indicate that dexfenfluramine is protective against hypoxia-induced PH, and that this latter effect is likewise serotonin dependent (28). The serotonin pathway is also the subject of a study investigating whether METH generates lung disease in rats (145). Epidemiological studies place METH as a likely contributor to PAH, and the drug was also shown to cause pulmonary toxicity in mice (148). Wang et al. demonstrated that METH exposure in rats resulted in pulmonary toxicity with increased inflammation and arterial remodeling, and the authors argue that augmented serotonin signaling causes the observed phenotype. However, as is the case with other animal models of PAH, the rats do not present all of the clinical characteristics of PAH, since they do not develop increased pulmonary arterial pressure or right heart hypertrophy. Last, and of considerable clinical importance, severe PAH is not a hallmark of syndromes caused by serotonin-producing tumors.
BMPR2 Mutations and Serotonin
BMPR2 mutations are the most common mutations associated with heritable PAH, accounting for 80% of the cases; in addition, BMPR2 mutations can also be found in ~25% of cases of IPAH, supporting a key role in the pathogenesis (40). Interestingly, only ~20% of BMPR2 mutation carriers develop PAH, strongly suggesting that a “second hit,” perhaps also in the form of an environmental stressor, is required for disease development. One study has revealed that 9% of patients with fenfluramine-associated PAH are carriers of BMPR2 mutations (70). A pathological link between BMPR2 haploinsufficiency and serotonin exposure has been uncovered using BMPR2+/− mice treated with a serotonin infusion. Compared with wild-type animals, infusion of serotonin resulted in increased pulmonary artery systolic pressure, right ventricular hypertrophy, and pulmonary artery remodeling in BMPR2+/− mice, an effect that was further exaggerated under hypoxic conditions (an environmental stimulus known to trigger pulmonary vascular remodeling and vasoconstriction). Exposure to serotonin also resulted in greater suppression of BMPR2 signaling activity, as evidence by reduced phosphorylation (i.e., activation) of Smad1/5, the downstream signaling mediators of BMPR2, upon stimulation with the ligand BMP-2 (89). This suggests that serotonin may be a second hit required to uncover PAH in the face of BMPR2 haploinsufficiency (91).
Dasatinib
To date, there are no effective biomarkers nor any tailored therapies to treat individuals with dasatinib-induced PAH (103). Recently, Guignabert et al. used a combination of cell and animal models to elucidate the molecular mechanisms by which dasatinib provokes damage to the pulmonary circulation (56). Using clinically relevant doses (1 mg·kg−1·day−1) of dasatinib, the investigators pretreated 4-wk-old male Wistar rats before exposing them to MCT or hypoxia, stimuli known to trigger vascular remodeling and PH in rats. Compared with vehicle- or imatinib-pretreated controls, dasatinib-pretreated rats demonstrated significantly higher pulmonary arterial pressures, right ventricular hypertrophy, lower cardiac output, and vascular remodeling. Moreover, prominent infiltration of inflammatory cells (CD3+ lymphocytes, macrophages, and leukocytes) was observed around the remodeled vessels of the dasatinib-pretreated animals, suggesting a role for immune dysregulation in the pathogenesis of dasatinib-induced PAH. In addition, there was evidence for endothelial dysfunction as demonstrated by 1) an increase in the expression and release of markers of endothelial injury (intercellular adhesion molecule, vascular cell adhesion molecule-1, and E-selectin) in both animals and patients, 2) activation of the unfolded protein response (an endoplasmic reticulum stress response not seen with imatinib), and 3) reactive oxygen species (ROS)-dependent in vitro endothelial cell apoptosis that could be reduced by treatment with the antioxidant NAC. Taken together, these findings suggest that dasatinib could induce PAH in susceptible individuals by triggering ROS-dependent endothelial apoptosis, small vessel loss, and vascular remodeling.
MMC and PVOD
Following the description of their clinical cohort of PVOD patients with a history of MMC exposure, Perros et al. used rats to study the effect of MMC on the pulmonary circulation (116). A single dose of MMC (4 mg/kg single dose) was sufficient to produce PH with pulmonary arterial and venous remodeling that histologically resembled pulmonary capillary hemangiomatosis and PVOD. Interestingly, female rats were more prone to develop severe disease as evidenced by reduced survival and more severely altered hemodynamics. Mechanistically, it was proposed that MMC depletes levels of general control nonderepressible 2 (GCN2) protein in the lungs and mimics PVOD induced by mutations in the EIF2AK4 gene, which has been identified as the predisposing gene related to heritable PVOD. Of note, although there were no changes in BMPR2 levels, there was evidence of disruption of Smad1/5/8 signaling, suggesting a possible cross talk between BMPR2 and GCN2. In an effort to test therapeutic approaches, rats were given amifostine, a cytoprotective adjuvant commonly used to prevent nephrotoxicity by alkylating chemotherapy agents, simultaneously with MMC, resulting in significant improvement in survival, RV hypertrophy, and pulmonary hemodynamics compared with rats treated with MMC alone (116). This improvement was attributed to less muscularized distal microvessels and more low-resistance nonmuscularized distal microvessels, ultimately with a smaller percentage of occluded distal microvessels compared with rats treated with MMC alone. The authors conclude that simultaneous administration of amifostine may reduce the risk of developing PVOD.
Amphetamine and Methamphetamine
Amphetamine derivatives are metabolized by two major liver enzymes [CYP2D6 and carboxylesterase 1 (CES1)]. Although CYP2D6 is required for phase 1 metabolism of many drugs, CES1 is mainly involved in the detoxification of cocaine and heroin as well as FDA-approved stimulants (e.g., methylphenidate) (63, 157). Polymorphisms in these enzymes can contribute to chronic organ injury by affecting rate of metabolism (16, 26, 110, 138, 159) (81), but whether polymorphisms are linked to METH-PAH has not been explored.
To assess CES1 and CYP2D6 protein expression in pulmonary arteries, we performed immunofluorescence studies using lung tissue sections from two healthy donors and four METH-PAH patients obtained at the time of autopsy or transplant (113). Our studies demonstrated that CYP2D6 is expressed in the endothelium of small pulmonary microvessels; however, we found no difference in CYP2D6 expression between healthy donor and METH-PAH lungs (see The issue of drug metabolism by pulmonary cells regarding the role of CYP in pulmonary drug metabolism). In contrast, CES1 expression was found to be reduced or absent in remodeled vessels of all four METH-PAH samples compared with healthy patient samples (Fig. 2). To assess whether healthy human pulmonary microvascular endothelial cells (PMVECs) have the capacity to metabolize METH, we analyzed cell media and lysates of METH-treated PMVECs for metabolites using LC/MS. Analysis of the cell media did not identify significant amounts of METH or any known metabolites in either the treated or nontreated group, suggesting internalization and/or degradation of METH. However, analysis of cell lysates from METH-treated PMVECs demonstrated at least six hydroxyl isomers of METH produced via metabolic detoxification.
Fig. 2.

Carboxylesterase 1 (CES1) expression is reduced in vascular lesions of methamphetamine-induced pulmonary arterial hypertension (METH-PAH). A: representative immunofluorescence studies of lung sections stained for cytochrome P-450 (CYP) 2D6 (red) obtained from healthy donor and METH-PAH patients. No difference was seen among our 4 METH-PAH patients. B: representative immunofluorescence studies of lung sections stained for CES1 (red) obtained from healthy donor (top) and 4 METH-PAH patients. CD31 (green) stains for endothelial cells. Scale bar = 25 μm.
Further analysis of CES1 polymorphisms in the METH-PAH patient population led to the discovery of rs115629050. Found exclusively in METH-PAH samples, this missense single nucleotide variant is located within the active site of the enzyme, which could impair its detoxifying properties. Using a combination of genetic and mutation analysis, we found that reduction of CES1 via siRNA or introduction of a mutant form of CES1 carrying the single nucleotide variant led to greater in vitro PMVEC apoptosis in the setting of METH exposure. We next proceeded to explore the mechanism by which CES1 regulates PMVEC survival in the setting of METH exposure. Studies in neurons and in brain endothelial cells have shown that METH triggers production of ROS (25, 130) that results in activation of autophagy (12, 115, 121, 158). Autophagy is a process that can protect cells under stress until homeostasis is restored; in the setting of catastrophic injury, autophagy can facilitate apoptosis. Based on our findings, CES1 deficiency reduces the buffering capacity of autophagy in favor of apoptosis in response to METH exposure (Fig. 3).
Fig. 3.
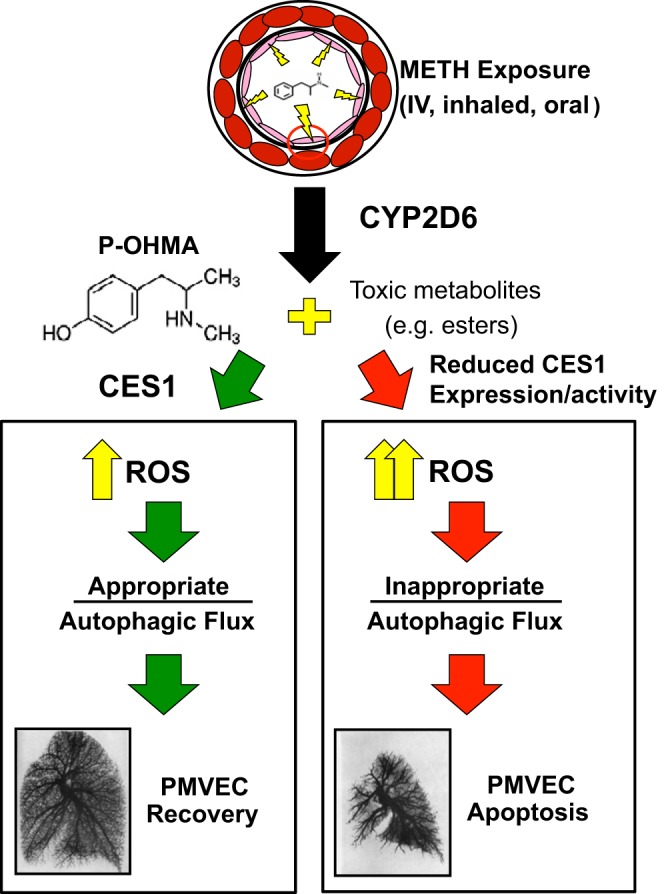
Proposed model of the role of CES1 in METH-PAH.
Amphetamines and DNA Damage
Hypoxia is one of the major environmental triggers of PH and is known to increase the severity of hemodynamic impairment and vascular remodeling in the context of BMPR2 mutations, serotonin stimulation, and VEGF receptor blockade (32, 89). Interestingly, hypoxia can also facilitate genomic instability by suppressing DNA repair responses and increasing rates of mutagenesis. Based on this fact, Chen et al. postulated that the combination of hypoxia and amphetamine (or METH) exposure would increase the risk of PAH by promoting the accumulation of damaged DNA and more severe pulmonary vascular remodeling. Compared with healthy donor cells, PMVECs purified from lungs of METH-PAH patients displayed greater evidence of DNA damage as determined by comet assay and γH2AX foci. Furthermore, whereas treatment of cells with amphetamine alone did not induce DNA damage, costimulation with doxorubicin (a compound that induces DNA breaks) resulted in greater DNA damage and endothelial apoptosis. The investigators show that amphetamine exposure results in the destabilization of hypoxia-inducible factor (HIF)-1α and an increase in mitochondrial oxidation, caspase activation, and DNA damage. Complementary to the in vitro studies, the authors used a chronic hypoxia mouse model to test whether METH administration could lead to more severe PH and vascular remodeling compared with hypoxia alone. They found no difference in the level of RVSP or degree of RV hypertrophy, but treatment with METH resulted in more severe pulmonary vascular remodeling, as demonstrated by an increase in muscularized distal precapillary arteries. The authors speculate that exposure to amphetamine or METH can dysregulate the normal cellular response to hypoxia and facilitate the accumulation of pathological mutations in PMVECs raising the risk for PAH, the effect of which could be further aggravated in the presence of other PAH-associated mutations.
The Issue of Drug Metabolism by Pulmonary Cells
Because D-PAH is caused by drugs in genetically susceptible individuals and likely also genetically susceptible animals, it is worthwhile to review here some of the known aspects of lung drug metabolism and detoxification that is largely accomplished by two different lung cells (alveolar type II cells and lung microvascular endothelial cells). While there is presently no good reason to link PAH with normal or abnormal alveolar type II cell function, there is an overwhelming amount of data that describe metabolic functions of the PMVECs, which include quantitative uptake of drugs (for example, propranolol), mediators of inflammation such as serotonin, and precursor lipids like arachidonic acid, the angiotensin-converting enzyme-dependent conversion of angiotensin, and secretion of endothelin (9, 135). In addition to active uptake in PMVEC and secretion of factors, the PMVEC are also home to drug-metabolizing enzymes of the CYP450 group. It has been recognized that next to the liver the lung is the second important drug-metabolizing organ. Drugs can: 1) damage the lung endothelium directly, 2) induce the high expression of one or more CYP450 enzymes, which can generate cytotoxic drug metabolites, and 3) generate endothelial cell toxic or immunotoxic compounds. The drug-metabolizing CYPP450 isozymes are linked to the aryl hydrocarbon receptor (Ahr). The prototypical activating ligand of this receptor is tetrachlorodibenzodioxin; other ligands are kynurenine and components of cigarette smoke. Binding of the Ahr to the cytosolic nuclear transporter aryl hydrocarbon receptor nuclear translocator (ARNT, which happens to be the transcription factor HIF1-β) generates a transcriptionally active heterodimer that can bind to consensus regulatory sequences (xenobiotic response elements) located upstream in the promoter of target genes, which are CYP450 enzyme-encoding genes. For example, epidermal growth factor, TNF-α, and TGF-β are downstream targets of Ahr activation (60). Activation of Ahr also causes immunosuppression by modulating dendritic cell behavior (15). This Ahr-ARNT-CYP450 axis thus links drug metabolism with inflammation and immune responses (Fig. 4). It is of interest for the purpose of this review that dexfenfluramine induces CYP1B1 (27) and that Sugen 5416 is a powerful inducer of CYP1B1 (27-fold) and of CYP1A1 (260-fold). Table 1 lists a number of drugs and agents and their metabolism by various CYP450 isozymes or induction of CYP450 isozymes by the drugs; this list also includes trichloroethylene (82, 155), which is hypothesized to cause PVOD (see Chemical solvents).
Fig. 4.

The aryl hydrocarbon receptor (Ahr)-aryl hydrocarbon receptor nuclear translocator (ARNT)-CYP450 signaling pathway and its proposed role in drug-induced pulmonary arterial hypertension (D-PAH).
Table 1.
List of CYPs associated with different drugs known to produce D-PAH
| Drugs | Cytochromes |
|---|---|
| Monocrotaline | Cyt3A4 |
| Fenfluramine | Cyt1B1 |
| Metamphetamine | Cyt 3A4, 2D6 |
| Dasatinib | Cyt 3A4, 1A1,1B1 |
| Interferon | Cyt 3A4, 1A2 |
| Sugen 5416 | Cyt 1A1, 1B1 |
| Trichloroethylene | Cyt 4A10 |
| Cigarette smoke components | Cyt 1A1, 2A6, 2E1, 3A |
CYP, cytochrome P-450; D-PAH, drug-induced pulmonary arterial hypertension.
Finally, there are direct and indirect toxic effects of inhaled cigarette smoke components on lung cells, including lung vascular endothelial cells (141). Cigarette smoke components induce several CYP450 enzymes (114); they can alter estrogen metabolism and destroy lung capillaries following induction of endothelial cell apoptosis. Whether cigarette smoking facilitates the development of PAH in BMPR2 mutation carriers is unknown.
PREVENTING THE NEXT D-PAH EPIDEMIC
Physician Awareness
Because agents associated with drug-induced PH are newly identified, it becomes necessary to evaluate the current state of identification, communication, and education regarding culprit drugs and potential prevention of their toxic effects. From the point of view of the drug-prescribing physician, the importance of clinical screening and drug-taking history remains a crucial first step to identifying potential drugs that may inadvertently lead to PH. Thus, awareness of commonly reported associative agents, recognition of the pattern of injury (e.g., PAH vs. PVOD), and knowledge of diagnostic resources remain the current mainstay of initial suspicion for D-PAH.
Pharmacovigilance
In the U.S., the FDA is tasked with protecting public health by assuring the safety of human drug products. In 2006, to specifically address the process of identifying drug safety and toxicity, the FDA requested an evaluation by the Institute of Medicine. The resulting report identified three primary areas of focus to support a robust drug product safety program (drug safety science, operations and management, and safety communications). Within the FDA, the Center for Drug Evaluation and Research (CDER) is responsible for implementing these areas of focus. The CDER evaluates new drugs before marketing and performs postmarketing safety surveillance, including monitoring use of marketed drugs for unexpected health risks such as PH. The premarket review includes animal testing for toxicity across multiple species. Next, a phase 1 study is performed, with an emphasis in safety, and the most frequent side effects and drug pharmacokinetics are identified. Typically, this phase involves 20–80 subjects. After a phase 2 study emphasizes the desired effectiveness of the drug, a phase 3 study gathers more data about both safety and effectiveness, typically involving several hundred to 3,000 subjects. Finally, a new drug application is reviewed, and manufacturing facilities are scrutinized for safety purposes.
In 2008, the FDA launched its Safety-First Initiative, the mission of which was to provide equal attention to postmarketing drug safety as that given for premarketing review. Within the FDA, there are two major systems for postmarketing drug surveillance. First, the FDA Adverse Events Reporting System constitutes the “passive” system for identifying potential associations between a particular drug and toxicities. It comprises an electronic database of spontaneously submitted adverse events, also known as the MedWatch program, in which the public and drug sponsors or manufacturers voluntarily report cases of unintended side effects of marketed drugs. The intent is to identify serious, rare, or unexpected “safety signals” that may indicate new or unanticipated drug safety concerns. CDER then uses data-mining techniques to identify reports of greatest value. The second major system for postmarketing drug surveillance is the Sentinel System, the “active” system in which national integrated electronic databases are shared and integrated to monitor drug safety. The aim of this system is to proactively assess medical product safety in the real world, using collaboration between academic medical centers, healthcare systems, health insurers, and patient advocacy groups. Patient identifiers are removed, and large real-world data sets are then analyzed to draw associations between drugs and potential adverse events.
Limitations to the current FDA model for pharmacovigilance rest in the frequency and quality of reporting an adverse event. In the current passive system for adverse event reporting, MedWatch, 5% of reports annually are by the public, whereas 95% are by drug sponsors or manufacturers. This in turn brings to question the likelihood of reporting such events, dependent on the amount of time a product has been on the market or the degree of publicity around a particular adverse event. In addition, accuracy of reports is confounded by polypharmacy or comorbid patient conditions. Last, the CDER does not require a causal relationship to be proven before reporting, which may lead to inaccurate reporting of drug toxicity.
Drug Discovery
A major determinant of successful drug discovery is selection of agents with minimal toxicity. Monitoring for drug toxicity should be implemented early in the discovery process and maintained beyond drug approval, since some toxicities may not be evident in short-term studies. This is the focus of medicinal chemistry, a discipline devoted to improving the therapeutic and safety profiles of agents and whose participation in the drug development process can improve the quality of agents tested in clinical studies. Drug modifications that can improve the therapeutic index include chemical modifications to the molecular structure to improve solubility and biodistribution as well as designing novel delivery vehicles (e.g., nanocarriers, biogels) to target the drug to the pulmonary vasculature. In addition to the methods described above, there are numerous in silico tools (e.g., DEREK, TOPKAT, and MCASE) that can predict possible toxicity of a given candidate compound by analyzing its molecular structure and comparing it against a database of compounds with similar molecular structures and known toxicity profile. A major benefit of this approach is that structural analysis can help chemists modify the original molecule to improve target selectivity and pharmacological profile while reducing its toxicity.
Another approach to screening for drug toxicity is the use of patient-derived inducible pluripotent stem cells to generate differentiated vascular (endothelial, smooth muscle, fibroblasts) cells and cardiomyocytes. In addition to screening for efficacy of novel compounds, these cells provide a platform to screen the toxicity profile of multiple candidate compounds simultaneously with high-throughput technology in cells derived from either circulating blood or skin fibroblasts. Induced pluripotent stem cell-derived endothelial cells from PAH patients have become available and will likely help accelerate development of such platforms for the future (129).
Last, it is worth mentioning that animal models of PAH used for preclinical testing of compounds are rarely screened for evidence of possible systemic toxicity for the test compound. Animal models can be used to document and quantify in vivo toxicity of candidate compounds by searching for changes in clinical parameters (e.g., behavior, vital signs, EKG) and organ dysfunction through the study of explanted tissues or analysis of biomarkers in body fluids. One must also consider the toxicity of compounds such as Sugen 5416 and MCT, which are used routinely to induce PAH in rodents when it comes to assess potential treatment effects of drugs developed for the treatment of PAH.
Conclusions
In the last 50 years, numerous drugs and substances have been implicated in the development of PH (Table 2). One challenge in identifying drugs associated with PH is the relative rarity of PH and the frequent latency between starting a drug and the development of clinical symptoms. Another challenge is that animal models may not reproduce the most salient features of human forms of severe PAH, perhaps in part because the animals lack a required and human-specific genetic makeup. As a result, there continues to be a need for pharmacovigilance and the anticipation that there will be new drugs discovered that cause PAH in susceptible individuals.
Table 2.
Updated clinical classification of PAH
| Definite | Possible | Likely | Unlikely |
|---|---|---|---|
| Aminorex | Cocaine | Amphetamines | Oral contraceptives |
| Fenfluramine | Phenylpropanolamine | l-Tryptophan | Estrogen |
| Dexfenfluramine | St. John's wort | Methamphetamines | Cigarette smoking |
| Toxic rapeseed oil | Chemotherapeutic agents | Dasatanib | |
| Benfluorex | Interferon-α and -β | ||
| SSRIs | Amphetamine-like drugs |
SSRI, selective serotonin reuptake inhibitors.
The drugs and agents highlighted here cover ~50 yr since the first epidemic of drug-induced PAH, and it is highly likely that more drugs will be identified as risk factors for PAH. In the past year, reports of PAH associated with other FDA-approved drugs, such as IFN and mitomycin, have been published, further illustrating the expanding role of prescription drugs as a cause of PH. As our understanding of the PAH pathobiology improves, we must pay attention to pathways and gene modifiers that participate in drug metabolism. Precision medicine tools such as gene sequencing, exosome analysis, and bioinformatics can serve in detecting genetic susceptibility factors that increase the risk of PAH in subsets of patients, but await validation in future studies.
In the meantime, physicians must remain vigilant of their patient population and establish close interactions between national drug regulatory agencies, national PH networks, and PAH patient associations to pursue leads and spread the word across the globe. We encourage clinicians to collect information regarding relevant drug exposure as part of the medical history and send out blood or urine for toxicology if there is suspicion of active stimulant use. Also, current guidelines recommend the use of FDA-approved vasodilators to treat patients with D-PAH as aggressively as those afflicted with other forms of WHO 1 PAH. Ultimately, prevention may prove to be the best strategy to deal with D-PAH, and it will help elevate awareness of this severe and often fatal disease.
GRANTS
V. A. de Jesus Perez is supported by National Institutes of Health (NIH) Grants R01-HL-134776 and R01-HL-134776-02, the Pulmonary Fibrosis Foundation, and the Pulmonary Hypertension Association. R. T. Zamanian is supported by NIH Grants NHLBI-HV-10-05, 1U01-HL-107393-01, PAR-09-185, and N01-HV-00242 and the Vera Moulton Wall Center. K. Yuan is supported by an American Heart Association Scientist Development Award and a Parker B. Francis Fellowship Award. N. K. Dhillon is supported by NIH Grants R01-DA-034542, R01-DA-042715 02, and 5 R01-HL-129875-02.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.E.O., K.Y., N.F.V., and V.A.d.J.P. prepared figures; M.E.O., K.Y., C.R., H.T., E.A.S., N.K.D., N.F.V., R.T.Z., and V.A.d.J.P. drafted manuscript; M.E.O., K.Y., C.R., H.T., E.A.S., N.K.D., N.F.V., R.T.Z., and V.A.d.J.P. edited and revised manuscript; M.E.O., K.Y., H.T., E.A.S., N.K.D., N.F.V., R.T.Z., and V.A.d.J.P. approved final version of manuscript.
REFERENCES
- 1.Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF, Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 121: 2747–2754, 2010. doi: 10.1161/CIRCULATIONAHA.109.927681. [DOI] [PubMed] [Google Scholar]
- 2.Abenhaim L, Moride Y, Brenot F, Rich S, Benichou J, Kurz X, Higenbottam T, Oakley C, Wouters E, Aubier M, Simonneau G, Bégaud B; International Primary Pulmonary Hypertension Study Group . Appetite-suppressant drugs and the risk of primary pulmonary hypertension. N Engl J Med 335: 609–616, 1996. doi: 10.1056/NEJM199608293350901. [DOI] [PubMed] [Google Scholar]
- 3.Al-Husseini A, Kraskauskas D, Mezzaroma E, Nordio A, Farkas D, Drake JI, Abbate A, Felty Q, Voelkel NF. Vascular endothelial growth factor receptor 3 signaling contributes to angioobliterative pulmonary hypertension. Pulm Circ 5: 101–116, 2015. doi: 10.1086/679704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson IM, Tomenson BM. The efficacy of selective serotonin re-uptake inhibitors in depression: a meta-analysis of studies against tricyclic antidepressants. J Psychopharmacol 8: 238–249, 1994. doi: 10.1177/026988119400800407. [DOI] [PubMed] [Google Scholar]
- 5.Andrade SE, McPhillips H, Loren D, Raebel MA, Lane K, Livingston J, Boudreau DM, Smith DH, Davis RL, Willy ME, Platt R. Antidepressant medication use and risk of persistent pulmonary hypertension of the newborn. Pharmacoepidemiol Drug Saf 18: 246–252, 2009. doi: 10.1002/pds.1710. [DOI] [PubMed] [Google Scholar]
- 6.Araújo I, Enjuanes-Grau C, Lopez-Guarch CJ, Narankiewicz D, Ruiz-Cano MJ, Velazquez-Martin T, Delgado J, Escribano P. Pulmonary arterial hypertension related to human immunodeficiency virus infection: A case series. World J Cardiol 6: 495–501, 2014. doi: 10.4330/wjc.v6.i6.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arnaiz-Villena A, Martinez-Laso J, Corell A, Allende L, Rosal M, Gomez-Reino JJ, Vicario JL. Frequencies of HLA-A24 and HLA-DR4-DQ8 are increased and that of HLA-B blank is decreased in chronic toxic oil syndrome. Eur J Immunogenet 23: 211–219, 1996. doi: 10.1111/j.1744-313X.1996.tb00116.x. [DOI] [PubMed] [Google Scholar]
- 8.Banerjee A, Strazza M, Wigdahl B, Pirrone V, Meucci O, Nonnemacher MR. Role of mu-opioids as cofactors in human immunodeficiency virus type 1 disease progression and neuropathogenesis. J Neurovirol 17: 291–302, 2011. doi: 10.1007/s13365-011-0037-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baretella O, Vanhoutte PM. Endothelium-dependent contractions: prostacyclin and endothelin-1, partners in crime? Adv Pharmacol 77: 177–208, 2016. doi: 10.1016/bs.apha.2016.04.006. [DOI] [PubMed] [Google Scholar]
- 10.Barman SA, Isales CM. Fenfluramine potentiates canine pulmonary vasoreactivity to endothelin-1. Pulm Pharmacol Ther 11: 183–187, 1998. doi: 10.1006/pupt.1998.0135. [DOI] [PubMed] [Google Scholar]
- 11.Barst RJ. Pulmonary hypertension: past, present and future. Ann Thorac Med 3: 1–4, 2008. doi: 10.4103/1817-1737.37832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beauvais G, Atwell K, Jayanthi S, Ladenheim B, Cadet JL. Involvement of dopamine receptors in binge methamphetamine-induced activation of endoplasmic reticulum and mitochondrial stress pathways. PLoS One 6: e28946, 2011. doi: 10.1371/journal.pone.0028946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Botros L, Van Nieuw Amerongen GP, Vonk Noordegraaf A, Bogaard HJ. Recovery from mitomycin-induced pulmonary arterial hypertension. Ann Am Thorac Soc 11: 468–470, 2014. doi: 10.1513/AnnalsATS.201312-426LE. [DOI] [PubMed] [Google Scholar]
- 14.Boutet K, Frachon I, Jobic Y, Gut-Gobert C, Leroyer C, Carlhant-Kowalski D, Sitbon O, Simonneau G, Humbert M. Fenfluramine-like cardiovascular side-effects of benfluorex. Eur Respir J 33: 684–688, 2009. doi: 10.1183/09031936.00086308. [DOI] [PubMed] [Google Scholar]
- 15.Bruhs A, Haarmann-Stemmann T, Frauenstein K, Krutmann J, Schwarz T, Schwarz A. Activation of the arylhydrocarbon receptor causes immunosuppression primarily by modulating dendritic cells. J Invest Dermatol 135: 435–444, 2015. doi: 10.1038/jid.2014.419. [DOI] [PubMed] [Google Scholar]
- 16.Bruxel EM, Salatino-Oliveira A, Genro JP, Zeni CP, Polanczyk GV, Chazan R, Rohde LA, Hutz MH. Association of a carboxylesterase 1 polymorphism with appetite reduction in children and adolescents with attention-deficit/hyperactivity disorder treated with methylphenidate. Pharmacogenomics J 13: 476–480, 2013. doi: 10.1038/tpj.2012.25. [DOI] [PubMed] [Google Scholar]
- 17.Byrne-Quinn E, Grover RF. Aminorex (Menocil) and amphetamine: acute and chronic effects on pulmonary and systemic haemodynamics in the calf. Thorax 27: 127–131, 1972. doi: 10.1136/thx.27.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chambers CD, Hernandez-Diaz S, Van Marter LJ, Werler MM, Louik C, Jones KL, Mitchell AA. Selective serotonin-reuptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N Engl J Med 354: 579–587, 2006. doi: 10.1056/NEJMoa052744. [DOI] [PubMed] [Google Scholar]
- 19.Chambers CD, Johnson KA, Dick LM, Felix RJ, Jones KL. Birth outcomes in pregnant women taking fluoxetine. N Engl J Med 335: 1010–1015, 1996. doi: 10.1056/NEJM199610033351402. [DOI] [PubMed] [Google Scholar]
- 20.Chen J, Du F, Hu B, Chi C, Chu H, Jiang L, Li P, Gong Z. Severe cardiotoxicity in a patient with colorectal cancer treated with bevacizumab. Anticancer Res 37: 4557–4561, 2017. [DOI] [PubMed] [Google Scholar]
- 21.Chin KM, Channick RN, Rubin LJ. Is methamphetamine use associated with idiopathic pulmonary arterial hypertension? Chest 130: 1657–1663, 2006. doi: 10.1378/chest.130.6.1657. [DOI] [PubMed] [Google Scholar]
- 22.Collazos J, Martínez E, Fernández A, Mayo J. Acute, reversible pulmonary hypertension associated with cocaine use. Respir Med 90: 171–174, 1996. doi: 10.1016/S0954-6111(96)90160-2. [DOI] [PubMed] [Google Scholar]
- 23.Connolly HM, Crary JL, McGoon MD, Hensrud DD, Edwards BS, Edwards WD, Schaff HV. Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med 337: 581–588, 1997. doi: 10.1056/NEJM199708283370901. [DOI] [PubMed] [Google Scholar]
- 24.Dalvi P, Spikes L, Allen J, Gupta VG, Sharma H, Gillcrist M, Montes de Oca J, O’Brien-Ladner A, Dhillon NK. Effect of cocaine on pulmonary vascular remodeling and hemodynamics in human immunodeficiency virus-transgenic rats. Am J Respir Cell Mol Biol 55: 201–212, 2016. doi: 10.1165/rcmb.2015-0264OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.da Silva DD, Silva E, Carmo H. Combination effects of amphetamines under hyperthermia - the role played by oxidative stress. J Appl Toxicol 34: 637–650, 2014. doi: 10.1002/jat.2889. [DOI] [PubMed] [Google Scholar]
- 26.de la Torre R, Yubero-Lahoz S, Pardo-Lozano R, Farré M. MDMA, methamphetamine, and CYP2D6 pharmacogenetics: what is clinically relevant? Front Genet 3: 235, 2012. doi: 10.3389/fgene.2012.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dempsie Y, MacRitchie NA, White K, Morecroft I, Wright AF, Nilsen M, Loughlin L, Mair KM, MacLean MR. Dexfenfluramine and the oestrogen-metabolizing enzyme CYP1B1 in the development of pulmonary arterial hypertension. Cardiovasc Res 99: 24–34, 2013. doi: 10.1093/cvr/cvt064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dempsie Y, Morecroft I, Welsh DJ, MacRitchie NA, Herold N, Loughlin L, Nilsen M, Peacock AJ, Harmar A, Bader M, MacLean MR. Converging evidence in support of the serotonin hypothesis of dexfenfluramine-induced pulmonary hypertension with novel transgenic mice. Circulation 117: 2928–2937, 2008. doi: 10.1161/CIRCULATIONAHA.108.767558. [DOI] [PubMed] [Google Scholar]
- 29.de Raaf MA, Kroeze Y, Middelman A, de Man FS, de Jong H, Vonk-Noordegraaf A, de Korte C, Voelkel NF, Homberg J, Bogaard HJ. Serotonin transporter is not required for the development of severe pulmonary hypertension in the Sugen hypoxia rat model. Am J Physiol Lung Cell Mol Physiol 309: L1164–L1173, 2015. doi: 10.1152/ajplung.00127.2015. [DOI] [PubMed] [Google Scholar]
- 30.Desbuards N, Antier D, Rochefort GY, Apfeldorfer CS, Schenck E, Hanton G, Hyvelin JM. Dexfenfluramine discontinuous treatment does not worsen hypoxia-induced pulmonary vascular remodeling but activates RhoA/ROCK pathway: consequences on pulmonary hypertension. Eur J Pharmacol 602: 355–363, 2009. doi: 10.1016/j.ejphar.2008.11.025. [DOI] [PubMed] [Google Scholar]
- 31.Dhillon NK, Li F, Xue B, Tawfik O, Morgello S, Buch S, Ladner AO. Effect of cocaine on human immunodeficiency virus-mediated pulmonary endothelial and smooth muscle dysfunction. Am J Respir Cell Mol Biol 45: 40–52, 2011. doi: 10.1165/rcmb.2010-0097OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diebold I, Hennigs JK, Miyagawa K, Li CG, Nickel NP, Kaschwich M, Cao A, Wang L, Reddy S, Chen PI, Nakahira K, Alcazar MA, Hopper RK, Ji L, Feldman BJ, Rabinovitch M. BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab 21: 596–608, 2015. doi: 10.1016/j.cmet.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dorfmüller P, Humbert M, Perros F, Sanchez O, Simonneau G, Müller KM, Capron F. Fibrous remodeling of the pulmonary venous system in pulmonary arterial hypertension associated with connective tissue diseases. Hum Pathol 38: 893–902, 2007. doi: 10.1016/j.humpath.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 34.Douglas JG, Munro JF, Kitchin AH, Muir AL, Proudfoot AT. Pulmonary hypertension and fenfluramine. Br Med J (Clin Res Ed) 283: 881–883, 1981. doi: 10.1136/bmj.283.6296.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eddahibi S, Adnot S, Frisdal E, Levame M, Hamon M, Raffestin B. Dexfenfluramine-associated changes in 5-hydroxytryptamine transporter expression and development of hypoxic pulmonary hypertension in rats. J Pharmacol Exp Ther 297: 148–154, 2001. [PubMed] [Google Scholar]
- 36.Eddahibi S, Fabre V, Boni C, Martres MP, Raffestin B, Hamon M, Adnot S. Induction of serotonin transporter by hypoxia in pulmonary vascular smooth muscle cells. Relationship with the mitogenic action of serotonin. Circ Res 84: 329–336, 1999. doi: 10.1161/01.RES.84.3.329. [DOI] [PubMed] [Google Scholar]
- 37.Eddahibi S, Hanoun N, Lanfumey L, Lesch KP, Raffestin B, Hamon M, Adnot S. Attenuated hypoxic pulmonary hypertension in mice lacking the 5-hydroxytryptamine transporter gene. J Clin Invest 105: 1555–1562, 2000. doi: 10.1172/JCI8678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eddahibi S, Humbert M, Fadel E, Raffestin B, Darmon M, Capron F, Simonneau G, Dartevelle P, Hamon M, Adnot S. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J Clin Invest 108: 1141–1150, 2001. doi: 10.1172/JCI200112805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eddahibi S, Raffestin B, Launay JM, Sitbon M, Adnot S. Effect of dexfenfluramine treatment in rats exposed to acute and chronic hypoxia. Am J Respir Crit Care Med 157: 1111–1119, 1998. doi: 10.1164/ajrccm.157.4.9704095. [DOI] [PubMed] [Google Scholar]
- 40.Evans JD, Girerd B, Montani D, Wang XJ, Galiè N, Austin ED, Elliott G, Asano K, Grünig E, Yan Y, Jing ZC, Manes A, Palazzini M, Wheeler LA, Nakayama I, Satoh T, Eichstaedt C, Hinderhofer K, Wolf M, Rosenzweig EB, Chung WK, Soubrier F, Simonneau G, Sitbon O, Gräf S, Kaptoge S, Di Angelantonio E, Humbert M, Morrell NW. BMPR2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta-analysis. Lancet Respir Med 4: 129–137, 2016. doi: 10.1016/S2213-2600(15)00544-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fahlén M, Bergman H, Helder G, Rydén L, Wallentin I, Zettergren L. Phenformin and pulmonary hypertension. Br Heart J 35: 824–828, 1973. doi: 10.1136/hrt.35.8.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferrer E, Peinado VI, Díez M, Carrasco JL, Musri MM, Martínez A, Rodríguez-Roisin R, Barberà JA. Effects of cigarette smoke on endothelial function of pulmonary arteries in the guinea pig. Respir Res 10: 76, 2009. doi: 10.1186/1465-9921-10-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fishman AP. Aminorex to fen/phen: an epidemic foretold. Circulation 99: 156–161, 1999. doi: 10.1161/01.CIR.99.1.156. [DOI] [PubMed] [Google Scholar]
- 44.Friström S, Airaksinen MM, Halmekoski J. Release of platelet 5-hydroxytryptamine by some anorexic and other sympathomimetics and their acetyl derivatives. Acta Pharmacol Toxicol (Copenh) 41: 218–224, 1977. doi: 10.1111/j.1600-0773.1977.tb02142.x. [DOI] [PubMed] [Google Scholar]
- 45.Fruehauf S, Steiger S, Topaly J, Ho AD. Pulmonary artery hypertension during interferon-alpha therapy for chronic myelogenous leukemia. Ann Hematol 80: 308–310, 2001. doi: 10.1007/s002770100298. [DOI] [PubMed] [Google Scholar]
- 46.Gagnadoux F, Capron F, Lebeau B. Pulmonary veno-occlusive disease after neoadjuvant mitomycin chemotherapy and surgery for lung carcinoma. Lung Cancer 36: 213–215, 2002. doi: 10.1016/S0169-5002(01)00479-2. [DOI] [PubMed] [Google Scholar]
- 47.Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M, Aboyans V, Vaz Carneiro A, Achenbach S, Agewall S, Allanore Y, Asteggiano R, Paolo Badano L, Albert Barbera J, Bouvaist H, Bueno H, Byrne RA, Carerj S, Castro G, Erol C, Falk V, Funck-Brentano C, Gorenflo M, Granton J, Iung B, Kiely DG, Kirchhof P, Kjellstrom B, Landmesser U, Lekakis J, Lionis C, Lip GY, Orfanos SE, Park MH, Piepoli MF, Ponikowski P, Revel MP, Rigau D, Rosenkranz S, Voller H, Luis Zamorano J; ESC Scientific Document Group . 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 37: 67–119, 2016. doi: 10.1093/eurheartj/ehv317. [DOI] [PubMed] [Google Scholar]
- 48.Galiè N, Simonneau G. The Fifth World Symposium on Pulmonary Hypertension. J Am Coll Cardiol Suppl 62: D1–D3, 2013. doi: 10.1016/j.jacc.2013.10.030. [DOI] [PubMed] [Google Scholar]
- 49.Gane EJ, Stedman CA, Hyland RH, Ding X, Svarovskaia E, Symonds WT, Hindes RG, Berrey MM. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med 368: 34–44, 2013. doi: 10.1056/NEJMoa1208953. [DOI] [PubMed] [Google Scholar]
- 50.Garcia-Dorado D, Miller DD, Garcia EJ, Delcan JL, Maroto E, Chaitman BR. An epidemic of pulmonary hypertension after toxic rapeseed oil ingestion in Spain. J Am Coll Cardiol 1: 1216–1222, 1983. doi: 10.1016/S0735-1097(83)80133-8. [DOI] [PubMed] [Google Scholar]
- 51.George PM, Badiger R, Alazawi W, Foster GR, Mitchell JA. Pharmacology and therapeutic potential of interferons. Pharmacol Ther 135: 44–53, 2012. doi: 10.1016/j.pharmthera.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 52.Gotway MB, Marder SR, Hanks DK, Leung JW, Dawn SK, Gean AD, Reddy GP, Araoz PA, Webb WR. Thoracic complications of illicit drug use: an organ system approach. Radiographics 22: S119–S135, 2002. doi: 10.1148/radiographics.22.suppl_1.g02oc01s119. [DOI] [PubMed] [Google Scholar]
- 53.Greiser E. Epidemiologic studies on the relation between use of appetite depressants and primary vascular pulmonary hypertension. Internist (Berl) 14: 437–442, 1973. [PubMed] [Google Scholar]
- 54.Gruenewald PJ, Ponicki WR, Remer LG, Waller LA, Zhu L, Gorman DM. Mapping the spread of methamphetamine abuse in California from 1995 to 2008. Am J Public Health 103: 1262–1270, 2013. doi: 10.2105/AJPH.2012.300779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guignabert C, Izikki M, Tu LI, Li Z, Zadigue P, Barlier-Mur AM, Hanoun N, Rodman D, Hamon M, Adnot S, Eddahibi S. Transgenic mice overexpressing the 5-hydroxytryptamine transporter gene in smooth muscle develop pulmonary hypertension. Circ Res 98: 1323–1330, 2006. doi: 10.1161/01.RES.0000222546.45372.a0. [DOI] [PubMed] [Google Scholar]
- 56.Guignabert C, Phan C, Seferian A, Huertas A, Tu L, Thuillet R, Sattler C, Le Hiress M, Tamura Y, Jutant EM, Chaumais MC, Bouchet S, Manéglier B, Molimard M, Rousselot P, Sitbon O, Simonneau G, Montani D, Humbert M. Dasatinib induces lung vascular toxicity and predisposes to pulmonary hypertension. J Clin Invest 126: 3207–3218, 2016. doi: 10.1172/JCI86249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guignabert C, Raffestin B, Benferhat R, Raoul W, Zadigue P, Rideau D, Hamon M, Adnot S, Eddahibi S. Serotonin transporter inhibition prevents and reverses monocrotaline-induced pulmonary hypertension in rats. Circulation 111: 2812–2819, 2005. doi: 10.1161/CIRCULATIONAHA.104.524926. [DOI] [PubMed] [Google Scholar]
- 58.Gurtner HP. Pulmonary hypertension, “plexogenic pulmonary arteriopathy” and the appetite depressant drug aminorex: post or propter? Bull Eur Physiopathol Respir 15: 897–923, 1979. [PubMed] [Google Scholar]
- 59.Gurtner HP, Gertsch M, Salzmann C, Scherrer M, Stucki P, Wyss F. Are the primary vascular forms of chronic pulmonary heart disease becoming more common?. Schweiz Med Wochenschr 98: 1579–1589, 1968. [PubMed] [Google Scholar]
- 60.Haarmann-Stemmann T, Bothe H, Abel J. Growth factors, cytokines and their receptors as downstream targets of arylhydrocarbon receptor (AhR) signaling pathways. Biochem Pharmacol 77: 508–520, 2009. doi: 10.1016/j.bcp.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 61.Haim DY, Lippmann ML, Goldberg SK, Walkenstein MD. The pulmonary complications of crack cocaine. A comprehensive review. Chest 107: 233–240, 1995. doi: 10.1378/chest.107.1.233. [DOI] [PubMed] [Google Scholar]
- 62.Hatano S and Strasser T. Primary Pulmonary Hypertension: Report on a WHO Meeting, Geneva, 15–17 October 1973. Geneva: World Health Organization, 1976. [Google Scholar]
- 63.Hatfield MJ, Umans RA, Hyatt JL, Edwards CC, Wierdl M, Tsurkan L, Taylor MR, Potter PM. Carboxylesterases: general detoxifying enzymes. Chem Biol Interact B 259: 327–331, 2016. doi: 10.1016/j.cbi.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hertzman PA, Falk H, Kilbourne EM, Page S, Shulman LE. The eosinophilia-myalgia syndrome: the Los Alamos Conference. J Rheumatol 18: 867–873, 1991. [PubMed] [Google Scholar]
- 65.Hervé P, Launay JM, Scrobohaci ML, Brenot F, Simonneau G, Petitpretz P, Poubeau P, Cerrina J, Duroux P, Drouet L. Increased plasma serotonin in primary pulmonary hypertension. Am J Med 99: 249–254, 1995. doi: 10.1016/S0002-9343(99)80156-9. [DOI] [PubMed] [Google Scholar]
- 66.Hickey PM, Thompson AA, Charalampopoulos A, Elliot CA, Hamilton N, Kiely DG, Lawrie A, Sabroe I, Condliffe R. Bosutinib therapy resulting in severe deterioration of pre-existing pulmonary arterial hypertension. Eur Respir J 48: 1514–1516, 2016. doi: 10.1183/13993003.01004-2016. [DOI] [PubMed] [Google Scholar]
- 67.Hoeper MM, Barst RJ, Bourge RC, Feldman J, Frost AE, Galié N, Gómez-Sánchez MA, Grimminger F, Grünig E, Hassoun PM, Morrell NW, Peacock AJ, Satoh T, Simonneau G, Tapson VF, Torres F, Lawrence D, Quinn DA, Ghofrani HA. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation 127: 1128–1138, 2013. doi: 10.1161/CIRCULATIONAHA.112.000765. [DOI] [PubMed] [Google Scholar]
- 68.Huertas A, Girerd B, Dorfmuller P, O'Callaghan D, Humbert M, Montani D. Pulmonary veno-occlusive disease: advances in clinical management and treatments. Expert Rev Respir Med 5: 217–229, 2011. doi: 10.1586/ers.11.15. [DOI] [PubMed] [Google Scholar]
- 69.Hughes A, Sathe N, Spagnola K. State Estimates of Substance Use from the 2005–2006 National Surveys on Drug Use and Health. DHHS Publication No. SMA 08–4311, NSDUH Series H-33. Rockville, MD: Dept Health Human Services, 2008. [Google Scholar]
- 70.Humbert M, Deng Z, Simonneau G, Barst RJ, Sitbon O, Wolf M, Cuervo N, Moore KJ, Hodge SE, Knowles JA, Morse JH. BMPR2 germline mutations in pulmonary hypertension associated with fenfluramine derivatives. Eur Respir J 20: 518–523, 2002. doi: 10.1183/09031936.02.01762002. [DOI] [PubMed] [Google Scholar]
- 71.Humbert M, Montani D, Perros F, Dorfmüller P, Adnot S, Eddahibi S. Endothelial cell dysfunction and cross talk between endothelium and smooth muscle cells in pulmonary arterial hypertension. Vascul Pharmacol 49: 113–118, 2008. doi: 10.1016/j.vph.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 72.Janda S, Quon BS, Swiston J. HIV and pulmonary arterial hypertension: a systematic review. HIV Med 11: 620–634, 2010. doi: 10.1111/j.1468-1293.2010.00829.x. [DOI] [PubMed] [Google Scholar]
- 73.Jochmann N, Kiecker F, Borges AC, Hofmann MA, Eddicks S, Sterry W, Baumann G, Trefzer U. Long-term therapy of interferon-alpha induced pulmonary arterial hypertension with different PDE-5 inhibitors: a case report. Cardiovasc Ultrasound 3: 26, 2005. doi: 10.1186/1476-7120-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Johnson DE, Georgieff MK. Pulmonary neuroendocrine cells. Their secretory products and their potential roles in health and chronic lung disease in infancy. Am Rev Respir Dis 140: 1807–1812, 1989. doi: 10.1164/ajrccm/140.6.1807. [DOI] [PubMed] [Google Scholar]
- 75.Källén B, Olausson PO. Maternal use of selective serotonin re-uptake inhibitors and persistent pulmonary hypertension of the newborn. Pharmacoepidemiol Drug Saf 17: 801–806, 2008. doi: 10.1002/pds.1570. [DOI] [PubMed] [Google Scholar]
- 76.Kay JM, Smith P, Heath D. Aminorex and the pulmonary circulation. Thorax 26: 262–270, 1971. doi: 10.1136/thx.26.3.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Keegan A, Morecroft I, Smillie D, Hicks MN, MacLean MR. Contribution of the 5-HT(1B) receptor to hypoxia-induced pulmonary hypertension: converging evidence using 5-HT(1B)-receptor knockout mice and the 5-HT(1B/1D)-receptor antagonist GR127935. Circ Res 89: 1231–1239, 2001. doi: 10.1161/hh2401.100426. [DOI] [PubMed] [Google Scholar]
- 78.Keusch S, Hildenbrand FF, Bollmann T, Halank M, Held M, Kaiser R, Kovacs G, Lange TJ, Seyfarth HJ, Speich R, Ulrich S. Tobacco smoke exposure in pulmonary arterial and thromboembolic pulmonary hypertension. Respiration 88: 38–45, 2014. doi: 10.1159/000359972. [DOI] [PubMed] [Google Scholar]
- 79.Kieler H, Artama M, Engeland A, Ericsson O, Furu K, Gissler M, Nielsen RB, Nørgaard M, Stephansson O, Valdimarsdottir U, Zoega H, Haglund B. Selective serotonin reuptake inhibitors during pregnancy and risk of persistent pulmonary hypertension in the newborn: population based cohort study from the five Nordic countries. Br Med J 344: d8012, 2012. doi: 10.1136/bmj.d8012. [DOI] [PubMed] [Google Scholar]
- 80.Kilbourne EM, Rigau-Perez JG, Heath CW Jr, Zack MM, Falk H, Martin-Marcos M, de Carlos A. Clinical epidemiology of toxic-oil syndrome. Manifestations of a New Illness. N Engl J Med 309: 1408–1414, 1983. doi: 10.1056/NEJM198312083092302. [DOI] [PubMed] [Google Scholar]
- 81.Laizure SC, Herring V, Hu Z, Witbrodt K, Parker RB. The role of human carboxylesterases in drug metabolism: have we overlooked their importance? Pharmacotherapy 33: 210–222, 2013. doi: 10.1002/phar.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lash LH, Chiu WA, Guyton KZ, Rusyn I. Trichloroethylene biotransformation and its role in mutagenicity, carcinogenicity and target organ toxicity. Mutat Res Rev Mutat Res 762: 22–36, 2014. doi: 10.1016/j.mrrev.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lawitz E, Mangia A, Wyles D, Rodriguez-Torres M, Hassanein T, Gordon SC, Schultz M, Davis MN, Kayali Z, Reddy KR, Jacobson IM, Kowdley KV, Nyberg L, Subramanian GM, Hyland RH, Arterburn S, Jiang D, McNally J, Brainard D, Symonds WT, McHutchison JG, Sheikh AM, Younossi Z, Gane EJ. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 368: 1878–1887, 2013. doi: 10.1056/NEJMoa1214853. [DOI] [PubMed] [Google Scholar]
- 84.Ledinek AH, Jazbec SS, Drinovec I, Rot U. Pulmonary arterial hypertension associated with interferon beta treatment for multiple sclerosis: a case report. Mult Scler 15: 885–886, 2009. doi: 10.1177/1352458509104593. [DOI] [PubMed] [Google Scholar]
- 85.Lee SL, Dunn J, Yu FS, Fanburg BL. Serotonin uptake and configurational change of bovine pulmonary artery smooth muscle cells in culture. J Cell Physiol 138: 145–153, 1989. doi: 10.1002/jcp.1041380120. [DOI] [PubMed] [Google Scholar]
- 86.Lipscomb JC, Garrett CM, Snawder JE. Cytochrome P450-dependent metabolism of trichloroethylene: interindividual differences in humans. Toxicol Appl Pharmacol 142: 311–318, 1997. doi: 10.1006/taap.1996.8040. [DOI] [PubMed] [Google Scholar]
- 87.Liu M, Wang Y, Wang HM, Bai Y, Zhang XH, Sun YX, Wang HL. Fluoxetine attenuates chronic methamphetamine-induced pulmonary arterial remodelling: possible involvement of serotonin transporter and serotonin 1B receptor. Basic Clin Pharmacol Toxicol 112: 77–82, 2013. doi: 10.1111/j.1742-7843.2012.00933.x. [DOI] [PubMed] [Google Scholar]
- 88.Liu Y, Tian HY, Yan XL, Fan FL, Wang WP, Han JL, Zhang JB, Ma Q, Meng Y, Wei F. Serotonin inhibits apoptosis of pulmonary artery smooth muscle cell by pERK1/2 and PDK through 5-HT1B receptors and 5-HT transporters. Cardiovasc Pathol 22: 451–457, 2013. doi: 10.1016/j.carpath.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 89.Long L, MacLean MR, Jeffery TK, Morecroft I, Yang X, Rudarakanchana N, Southwood M, James V, Trembath RC, Morrell NW. Serotonin increases susceptibility to pulmonary hypertension in BMPR2-deficient mice. Circ Res 98: 818–827, 2006. doi: 10.1161/01.RES.0000215809.47923.fd. [DOI] [PubMed] [Google Scholar]
- 90.Lund AK, Lucero J, Herbert L, Liu Y, Naik JS. Human immunodeficiency virus transgenic rats exhibit pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 301: L315–L326, 2011. doi: 10.1152/ajplung.00045.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.MacLean MR, Dempsie Y. The serotonin hypothesis of pulmonary hypertension revisited. Adv Exp Med Biol 661: 309–322, 2010. doi: 10.1007/978-1-60761-500-2_20. [DOI] [PubMed] [Google Scholar]
- 92.Manson JE, Faich GA. Pharmacotherapy for obesity – do the benefits outweigh the risks? N Engl J Med 335: 659–660, 1996. doi: 10.1056/NEJM199608293350910. [DOI] [PubMed] [Google Scholar]
- 93.Marcos E, Adnot S, Pham MH, Nosjean A, Raffestin B, Hamon M, Eddahibi S. Serotonin transporter inhibitors protect against hypoxic pulmonary hypertension. Am J Respir Crit Care Med 168: 487–493, 2003. doi: 10.1164/rccm.200210-1212OC. [DOI] [PubMed] [Google Scholar]
- 94.Martín F, Artigas F. Simultaneous effects of p-chloroamphetamine, d-fenfluramine, and reserpine on free and stored 5-hydroxytryptamine in brain and blood. J Neurochem 59: 1138–1144, 1992. doi: 10.1111/j.1471-4159.1992.tb08356.x. [DOI] [PubMed] [Google Scholar]
- 95.McCord J, Jneid H, Hollander JE, de Lemos JA, Cercek B, Hsue P, Gibler WB, Ohman EM, Drew B, Philippides G, Newby LK; American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology . Management of cocaine-associated chest pain and myocardial infarction: a scientific statement from the American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology. Circulation 117: 1897–1907, 2008. doi: 10.1161/CIRCULATIONAHA.107.188950. [DOI] [PubMed] [Google Scholar]
- 96.Mehendale HM, Morita T, Angevine LS. Effect of chlorphentermine on the pulmonary clearance of 5-hydroxytryptamine in rabbits in vivo. Pharmacology 26: 274–284, 1983. doi: 10.1159/000137811. [DOI] [PubMed] [Google Scholar]
- 97.Mehta NJ, Khan IA, Mehta RN, Sepkowitz DA. HIV-Related pulmonary hypertension: analytic review of 131 cases. Chest 118: 1133–1141, 2000. doi: 10.1378/chest.118.4.1133. [DOI] [PubMed] [Google Scholar]
- 98.Mermis J, Gu H, Xue B, Li F, Tawfik O, Buch S, Bartolome S, O’Brien-Ladner A, Dhillon NK. Hypoxia-inducible factor-1 α/platelet derived growth factor axis in HIV-associated pulmonary vascular remodeling. Respir Res 12: 103, 2011. doi: 10.1186/1465-9921-12-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mesa RA, Edell ES, Dunn WF, Edwards WD. Human immunodeficiency virus infection and pulmonary hypertension: two new cases and a review of 86 reported cases. Mayo Clin Proc 73: 37–45, 1998. doi: 10.1016/S0025-6196(11)63616-1. [DOI] [PubMed] [Google Scholar]
- 100.Mitani Y, Mutlu A, Russell JC, Brindley DN, DeAlmeida J, Rabinovitch M. Dexfenfluramine protects against pulmonary hypertension in rats. J Appl Physiol (1985) 93: 1770–1778, 2002. doi: 10.1152/japplphysiol.00500.2002. [DOI] [PubMed] [Google Scholar]
- 101.Mlczoch J, Weir EK, Reeves JT, Grover RF. Long term effects of the anorectic agent fenfluramine alone and in combination with aminorex on pulmonary and systemic circulation in the pig. Basic Res Cardiol 74: 313–320, 1979. doi: 10.1007/BF01907748. [DOI] [PubMed] [Google Scholar]
- 102.Mondy KE, Gottdiener J, Overton ET, Henry K, Bush T, Conley L, Hammer J, Carpenter CC, Kojic E, Patel P, Brooks JT, Investigators SUNS; SUN Study Investigators . High prevalence of echocardiographic abnormalities among hiv-infected persons in the era of highly active antiretroviral therapy. Clin Infect Dis 52: 378–386, 2011. doi: 10.1093/cid/ciq066. [DOI] [PubMed] [Google Scholar]
- 103.Montani D, Bergot E, Günther S, Savale L, Bergeron A, Bourdin A, Bouvaist H, Canuet M, Pison C, Macro M, Poubeau P, Girerd B, Natali D, Guignabert C, Perros F, O’Callaghan DS, Jaïs X, Tubert-Bitter P, Zalcman G, Sitbon O, Simonneau G, Humbert M. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation 125: 2128–2137, 2012. doi: 10.1161/CIRCULATIONAHA.111.079921. [DOI] [PubMed] [Google Scholar]
- 104.Montani D, Lau EM, Dorfmüller P, Girerd B, Jaïs X, Savale L, Perros F, Nossent E, Garcia G, Parent F, Fadel E, Soubrier F, Sitbon O, Simonneau G, Humbert M. Pulmonary veno-occlusive disease. Eur Respir J 47: 1518–1534, 2016. doi: 10.1183/13993003.00026-2016. [DOI] [PubMed] [Google Scholar]
- 105.Morecroft I, Heeley RP, Prentice HM, Kirk A, MacLean MR. 5-hydroxytryptamine receptors mediating contraction in human small muscular pulmonary arteries: importance of the 5-HT1B receptor. Br J Pharmacol 128: 730–734, 1999. doi: 10.1038/sj.bjp.0702841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Morecroft I, Loughlin L, Nilsen M, Colston J, Dempsie Y, Sheward J, Harmar A, MacLean MR. Functional interactions between 5-hydroxytryptamine receptors and the serotonin transporter in pulmonary arteries. J Pharmacol Exp Ther 313: 539–548, 2005. doi: 10.1124/jpet.104.081182. [DOI] [PubMed] [Google Scholar]
- 107.Naeije R, Maggiorini M, Delcroix M, Leeman M, Mélot C. Effects of chronic dexfenfluramine treatment on pulmonary hemodynamics in dogs. Am J Respir Crit Care Med 154: 1347–1350, 1996. doi: 10.1164/ajrccm.154.5.8912746. [DOI] [PubMed] [Google Scholar]
- 108.Naeije R, Wauthy P, Maggiorini M, Leeman M, Delcroix M. Effects of dexfenfluramine on hypoxic pulmonary vasoconstriction and embolic pulmonary hypertension in dogs. Am J Respir Crit Care Med 151: 692–697, 1995. doi: 10.1164/ajrccm/151.3_Pt_1.692. [DOI] [PubMed] [Google Scholar]
- 109.Nana-Sinkam SP, Lee JD, Sotto-Santiago S, Stearman RS, Keith RL, Choudhury Q, Cool C, Parr J, Moore MD, Bull TM, Voelkel NF, Geraci MW. Prostacyclin prevents pulmonary endothelial cell apoptosis induced by cigarette smoke. Am J Respir Crit Care Med 175: 676–685, 2007. doi: 10.1164/rccm.200605-724OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nemoda Z, Angyal N, Tarnok Z, Gadoros J, Sasvari-Szekely M. Carboxylesterase 1 gene polymorphism and methylphenidate response in ADHD. Neuropharmacology 57: 731–733, 2009. doi: 10.1016/j.neuropharm.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 111.Nunes H, Humbert M, Sitbon O, Morse JH, Deng Z, Knowles JA, Le Gall C, Parent F, Garcia G, Hervé P, Barst RJ, Simonneau G. Prognostic factors for survival in human immunodeficiency virus-associated pulmonary arterial hypertension. Am J Respir Crit Care Med 167: 1433–1439, 2003. doi: 10.1164/rccm.200204-330OC. [DOI] [PubMed] [Google Scholar]
- 112.Okada K, Tanaka Y, Bernstein M, Zhang W, Patterson GA, Botney MD. Pulmonary hemodynamics modify the rat pulmonary artery response to injury. A neointimal model of pulmonary hypertension. Am J Pathol 151: 1019–1025, 1997. [PMC free article] [PubMed] [Google Scholar]
- 113.Orcholski ME, Khurshudyan A, Shamskhou EA, Yuan K, Chen IY, Kodani SD, Morisseau C, Hammock BD, Hong EM, Alexandrova L, Alastalo TP, Berry G, Zamanian RT, de Jesus Perez VA. Reduced carboxylesterase 1 is associated with endothelial injury in methamphetamine-induced pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 313: L252–L266, 2017. doi: 10.1152/ajplung.00453.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Oyama T, Sugio K, Uramoto H, Kawamoto T, Kagawa N, Nadaf S, Carbone D, Yasumoto K. Cytochrome P450 expression (CYP) in non-small cell lung cancer. Front Biosci 12: 2299–2308, 2007. doi: 10.2741/2232. [DOI] [PubMed] [Google Scholar]
- 115.Park M, Hennig B, Toborek M. Methamphetamine alters occludin expression via NADPH oxidase-induced oxidative insult and intact caveolae. J Cell Mol Med 16: 362–375, 2012. doi: 10.1111/j.1582-4934.2011.01320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Perros F, Günther S, Ranchoux B, Godinas L, Antigny F, Chaumais MC, Dorfmüller P, Hautefort A, Raymond N, Savale L, Jaïs X, Girerd B, Cottin V, Sitbon O, Simonneau G, Humbert M, Montani D. Mitomycin-induced pulmonary veno-occlusive disease: evidence from human disease and animal models. Circulation 132: 834–847, 2015. doi: 10.1161/CIRCULATIONAHA.115.014207. [DOI] [PubMed] [Google Scholar]
- 117.Pitt BR, Weng W, Steve AR, Blakely RD, Reynolds I, Davies P. Serotonin increases DNA synthesis in rat proximal and distal pulmonary vascular smooth muscle cells in culture. Am J Physiol Lung Cell Mol Physiol 266: L178–L186, 1994. doi: 10.1152/ajplung.1994.266.2.L178. [DOI] [PubMed] [Google Scholar]
- 118.Quezada M, Martin-Carbonero L, Soriano V, Vispo E, Valencia E, Moreno V, de Isla LP, Lennie V, Almería C, Zamorano JL. Prevalence and risk factors associated with pulmonary hypertension in HIV-infected patients on regular follow-up. AIDS 26: 1387–1392, 2012. doi: 10.1097/QAD.0b013e328354f5a1. [DOI] [PubMed] [Google Scholar]
- 119.Quilot FM, Georges M, Favrolt N, Beltramo G, Foignot C, Grandvuillemin A, Montani D, Bonniaud P, Camus P. Pulmonary hypertension associated with ponatinib therapy. Eur Respir J 47: 676–679, 2016. doi: 10.1183/13993003.01110-2015. [DOI] [PubMed] [Google Scholar]
- 120.Ragonese M, Racioppi M, Bassi PF, Di Gianfrancesco L, Lenci N, Filianoti A, Recupero SM. Mitomycin C: new strategies to improve efficacy of a well-known therapy. Urologia 83, Suppl 2: 24–28, 2016. doi: 10.5301/uro.5000193. [DOI] [PubMed] [Google Scholar]
- 121.Ramirez SH, Potula R, Fan S, Eidem T, Papugani A, Reichenbach N, Dykstra H, Weksler BB, Romero IA, Couraud PO, Persidsky Y. Methamphetamine disrupts blood-brain barrier function by induction of oxidative stress in brain endothelial cells. J Cereb Blood Flow Metab 29: 1933–1945, 2009. doi: 10.1038/jcbfm.2009.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ranchoux B, Günther S, Quarck R, Chaumais MC, Dorfmüller P, Antigny F, Dumas SJ, Raymond N, Lau E, Savale L, Jaïs X, Sitbon O, Simonneau G, Stenmark K, Cohen-Kaminsky S, Humbert M, Montani D, Perros F. Chemotherapy-induced pulmonary hypertension: role of alkylating agents. Am J Pathol 185: 356–371, 2015. doi: 10.1016/j.ajpath.2014.10.021. [DOI] [PubMed] [Google Scholar]
- 123.Rasoulinejad M, Moradmand Badie S, Salehi MR, Seyed Alinaghi SA, Dehghan Manshadi SA, Zakerzadeh N, Foroughi M, Jahanjo Amin Abad F, Moradmand Badie B. Echocardiographic assessment of systolic pulmonary arterial pressure in HIV-positive patients. Acta Med Iran 52: 827–830, 2014. [PubMed] [Google Scholar]
- 124.Renard S, Borentain P, Salaun E, Benhaourech S, Maille B, Darque A, Bregigeon S, Colson P, Laugier D, Gaubert MR, Habib G. Severe Pulmonary Arterial Hypertension in Patients Treated for Hepatitis C With Sofosbuvir. Chest 149: e69–e73, 2016. doi: 10.1016/j.chest.2015.09.018. [DOI] [PubMed] [Google Scholar]
- 125.Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, Goldring RM, Groves BM, Koerner SK. Primary pulmonary hypertension. A national prospective study. Ann Intern Med 107: 216–223, 1987. doi: 10.7326/0003-4819-107-2-216. [DOI] [PubMed] [Google Scholar]
- 126.Rich S, Rubin L, Walker AM, Schneeweiss S, Abenhaim L. Anorexigens and pulmonary hypertension in the United States: results from the surveillance of North American pulmonary hypertension. Chest 117: 870–874, 2000. doi: 10.1378/chest.117.3.870. [DOI] [PubMed] [Google Scholar]
- 127.Rochefort GY, Lemaire MC, Eder V, Hanton G, Hyvelin JM, Bonnet P, Antier D. Dexfenfluramine does not worsen but moderates progression of chronic hypoxia-induced pulmonary hypertension. Eur J Pharmacol 550: 149–154, 2006. doi: 10.1016/j.ejphar.2006.08.050. [DOI] [PubMed] [Google Scholar]
- 128.Rosen MI, Black AC, Arnsten JH, Goggin K, Remien RH, Simoni JM, Golin CE, Bangsberg DR, Liu H. Association between use of specific drugs and antiretroviral adherence: findings from MACH 14. AIDS Behav 17: 142–147, 2013. doi: 10.1007/s10461-011-0124-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sa S, Gu M, Chappell J, Shao NY, Ameen M, Elliott KA, Li D, Grubert F, Li CG, Taylor S, Cao A, Ma Y, Fong R, Nguyen L, Wu JC, Snyder MP, Rabinovitch M. Induced pluripotent stem cell model of pulmonary arterial hypertension reveals novel gene expression and patient specificity. Am J Respir Crit Care Med 195: 930–941, 2017. doi: 10.1164/rccm.201606-1200OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sajja RK, Rahman S, Cucullo L. Drugs of abuse and blood-brain barrier endothelial dysfunction: A focus on the role of oxidative stress. J Cereb Blood Flow Metab 36: 539–554, 2016. doi: 10.1177/0271678X15616978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Savale L, Chaumais MC, Cottin V, Bergot E, Frachon I, Prevot G, Pison C, Dromer C, Poubeau P, Lamblin N, Habib G, Reynaud-Gaubert M, Bourdin A, Sanchez O, Tubert-Bitter P, Jaïs X, Montani D, Sitbon O, Simonneau G, Humbert M. Pulmonary hypertension associated with benfluorex exposure. Eur Respir J 40: 1164–1172, 2012. doi: 10.1183/09031936.00188611. [DOI] [PubMed] [Google Scholar]
- 132.Savale L, Sattler C, Günther S, Montani D, Chaumais M-C, Perrin S, Jaïs X, Seferian A, Jovan R, Bulifon S, Parent F, Simonneau G, Humbert M, Sitbon O. Pulmonary arterial hypertension in patients treated with interferon. Eur Respir J 44: 1627–1634, 2014. doi: 10.1183/09031936.00057914. [DOI] [PubMed] [Google Scholar]
- 133.Schaiberger PH, Kennedy TC, Miller FC, Gal J, Petty TL. Pulmonary hypertension associated with long-term inhalation of “crank” methamphetamine. Chest 104: 614–616, 1993. doi: 10.1378/chest.104.2.614. [DOI] [PubMed] [Google Scholar]
- 134.Shah NP, Wallis N, Farber HW, Mauro MJ, Wolf RA, Mattei D, Guha M, Rea D, Peacock A. Clinical features of pulmonary arterial hypertension in patients receiving dasatinib. Am J Hematol 90: 1060–1064, 2015. doi: 10.1002/ajh.24174. [DOI] [PubMed] [Google Scholar]
- 135.Shimoda LA, Sham JS, Sylvester JT. Altered pulmonary vasoreactivity in the chronically hypoxic lung. Physiol Res 49: 549–560, 2000. [PubMed] [Google Scholar]
- 136.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, Olschewski H, Robbins IM, Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol Suppl 62: D34–D41, 2013. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 137.Spikes L, Dalvi P, Tawfik O, Gu H, Voelkel NF, Cheney P, O’Brien-Ladner A, Dhillon NK. Enhanced pulmonary arteriopathy in simian immunodeficiency virus-infected macaques exposed to morphine. Am J Respir Crit Care Med 185: 1235–1243, 2012. doi: 10.1164/rccm.201110-1909OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sun Z, Murry DJ, Sanghani SP, Davis WI, Kedishvili NY, Zou Q, Hurley TD, Bosron WF. Methylphenidate is stereoselectively hydrolyzed by human carboxylesterase CES1A1. J Pharmacol Exp Ther 310: 469–476, 2004. doi: 10.1124/jpet.104.067116. [DOI] [PubMed] [Google Scholar]
- 139.Taraseviciene-Stewart L, Voelkel NF. Molecular pathogenesis of emphysema. J Clin Invest 118: 394–402, 2008. doi: 10.1172/JCI31811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Trip P, Nossent EJ, de Man FS, van den Berk IA, Boonstra A, Groepenhoff H, Leter EM, Westerhof N, Grünberg K, Bogaard HJ, Vonk-Noordegraaf A. Severely reduced diffusion capacity in idiopathic pulmonary arterial hypertension: patient characteristics and treatment responses. Eur Respir J 42: 1575–1585, 2013. doi: 10.1183/09031936.00184412. [DOI] [PubMed] [Google Scholar]
- 141.Voelkel NF. Cigarette Smoke is an Endothelial Cell Toxin. Am J Respir Crit Care Med 197: 274, 2018. doi: 10.1164/rccm.201706-1123LE. [DOI] [PubMed] [Google Scholar]
- 142.Voelkel NF, Tuder RM. Cellular and molecular mechanisms in the pathogenesis of severe pulmonary hypertension. Eur Respir J 8: 2129–2138, 1995. doi: 10.1183/09031936.95.08122129. [DOI] [PubMed] [Google Scholar]
- 143.Volkow ND, Fowler JS, Wang GJ, Shumay E, Telang F, Thanos PK, Alexoff D. Distribution and pharmacokinetics of methamphetamine in the human body: clinical implications. PLoS One 5: e15269, 2010. doi: 10.1371/journal.pone.0015269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Walker AM, Langleben D, Korelitz JJ, Rich S, Rubin LJ, Strom BL, Gonin R, Keast S, Badesch D, Barst RJ, Bourge RC, Channick R, Frost A, Gaine S, McGoon M, McLaughlin V, Murali S, Oudiz RJ, Robbins IM, Tapson V, Abenhaim L, Constantine G. Temporal trends and drug exposures in pulmonary hypertension: an American experience. Am Heart J 152: 521–526, 2006. doi: 10.1016/j.ahj.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 145.Wang Y, Liu M, Wang HM, Bai Y, Zhang XH, Sun YX, Wang HL. Involvement of serotonin mechanism in methamphetamine-induced chronic pulmonary toxicity in rats. Hum Exp Toxicol 32: 736–746, 2013. doi: 10.1177/0960327112468174. [DOI] [PubMed] [Google Scholar]
- 146.Weatherald J, Chaumais MC, Savale L, Jaïs X, Seferian A, Canuet M, Bouvaist H, Magro P, Bergeron A, Guignabert C, Sitbon O, Simonneau G, Humbert M, Montani D. Long-term outcomes of dasatinib-induced pulmonary arterial hypertension: a population-based study. Eur Respir J 50: 1700217, 2017. doi: 10.1183/13993003.00217-2017. [DOI] [PubMed] [Google Scholar]
- 147.Weir EK, Reeve HL, Huang JM, Michelakis E, Nelson DP, Hampl V, Archer SL. Anorexic agents aminorex, fenfluramine, and dexfenfluramine inhibit potassium current in rat pulmonary vascular smooth muscle and cause pulmonary vasoconstriction. Circulation 94: 2216–2220, 1996. doi: 10.1161/01.CIR.94.9.2216. [DOI] [PubMed] [Google Scholar]
- 148.Wells SM, Buford MC, Braseth SN, Hutchison JD, Holian A. Acute inhalation exposure to vaporized methamphetamine causes lung injury in mice. Inhal Toxicol 20: 829–838, 2008. doi: 10.1080/08958370801895121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.White RJ, Meoli DF, Swarthout RF, Kallop DY, Galaria II, Harvey JL, Miller CM, Blaxall BC, Hall CM, Pierce RA, Cool CD, Taubman MB. Plexiform-like lesions and increased tissue factor expression in a rat model of severe pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 293: L583–L590, 2007. doi: 10.1152/ajplung.00321.2006. [DOI] [PubMed] [Google Scholar]
- 150.Wichman CL, Moore KM, Lang TR, St Sauver JL, Heise RH Jr, Watson WJ. Congenital heart disease associated with selective serotonin reuptake inhibitor use during pregnancy. Mayo Clin Proc 84: 23–27, 2009. doi: 10.4065/84.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wilson KL, Zelig CM, Harvey JP, Cunningham BS, Dolinsky BM, Napolitano PG. Persistent pulmonary hypertension of the newborn is associated with mode of delivery and not with maternal use of selective serotonin reuptake inhibitors. Am J Perinatol 28: 19–24, 2011. doi: 10.1055/s-0030-1262507. [DOI] [PubMed] [Google Scholar]
- 152.Wright JL, Farmer SG, Churg A. A neutrophil elastase inhibitor reduces cigarette smoke-induced remodelling of lung vessels. Eur Respir J 22: 77–81, 2003. doi: 10.1183/09031936.03.00095703. [DOI] [PubMed] [Google Scholar]
- 153.Wright JL, Tai H, Churg A. Cigarette smoke induces persisting increases of vasoactive mediators in pulmonary arteries. Am J Respir Cell Mol Biol 31: 501–509, 2004. doi: 10.1165/rcmb.2004-0051OC. [DOI] [PubMed] [Google Scholar]
- 154.Yakel DL Jr, Eisenberg MJ. Pulmonary artery hypertension in chronic intravenous cocaine users. Am Heart J 130: 398–399, 1995. doi: 10.1016/0002-8703(95)90459-X. [DOI] [PubMed] [Google Scholar]
- 155.Yoo HS, Bradford BU, Kosyk O, Shymonyak S, Uehara T, Collins LB, Bodnar WM, Ball LM, Gold A, Rusyn I. Comparative analysis of the relationship between trichloroethylene metabolism and tissue-specific toxicity among inbred mouse strains: liver effects. J Toxicol Environ Health A 78: 15–31, 2015. doi: 10.1080/15287394.2015.958417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zamanian RT, Hedlin H, Greuenwald P, Wilson DM, Segal JI, Jorden M, Kudelko K, Liu J, Hsi A, Rupp A, Sweatt AJ, Tuder R, Berry GJ, Rabinovitch M, Doyle RL, De Jesus Perez V, Kawut SM. Features and outcomes of methamphetamine associated pulmonary arterial hypertension. Am J Respir Crit Care Med 197: 788–800, 2018. doi: 10.1164/rccm.201705-0943OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zanger UM, Raimundo S, Eichelbaum M. Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Naunyn Schmiedebergs Arch Pharmacol 369: 23–37, 2004. doi: 10.1007/s00210-003-0832-2. [DOI] [PubMed] [Google Scholar]
- 158.Zhang X, Banerjee A, Banks WA, Ercal N. N-Acetylcysteine amide protects against methamphetamine-induced oxidative stress and neurotoxicity in immortalized human brain endothelial cells. Brain Res 1275: 87–95, 2009. doi: 10.1016/j.brainres.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhu HJ, Patrick KS, Yuan HJ, Wang JS, Donovan JL, DeVane CL, Malcolm R, Johnson JA, Youngblood GL, Sweet DH, Langaee TY, Markowitz JS. Two CES1 gene mutations lead to dysfunctional carboxylesterase 1 activity in man: clinical significance and molecular basis. Am J Hum Genet 82: 1241–1248, 2008. doi: 10.1016/j.ajhg.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]