

Abstract
Bradykinin-induced activation of the pulmonary endothelium triggers a rise in intracellular Ca2+ that activates nitric oxide (NO)-dependent vasorelaxation. Chronic hypoxia is commonly associated with increased pulmonary vascular tone, which can cause pulmonary hypertension in responsive individuals. In the present study, we tested the hypothesis that long-term high-altitude hypoxia (LTH) diminishes bradykinin-induced Ca2+ signals and inhibits endothelial nitric oxide synthase (eNOS), prostacyclin (PGI2), and large-conductance K+ (BKCa) channels in sheep, which are moderately responsive to LTH, resulting in decreased pulmonary arterial vasorelaxation. Pulmonary arteries were isolated from ewes kept near sea level (720 m) or at high altitude (3,801 m) for >100 days. Vessel force was measured with wire myography and endothelial intracellular Ca2+ with confocal microscopy. eNOS was inhibited with 100 μM NG-nitro-l-arginine methyl ester (l-NAME), PGI2 production was inhibited with 10 µM indomethacin that inhibits cyclooxygenase, and BKCa channels were blocked with 1 mM tetraethylammonium. Bradykinin-induced endothelial Ca2+ signals increased following LTH, but bradykinin relaxation decreased. Furthermore, some vessels contracted in response to bradykinin after LTH. l-NAME sensitivity decreased, suggesting that eNOS dysfunction played a role in uncoupling Ca2+ signals and bradykinin relaxation. The Ca2+ ionophore A-23187 (10 µM) elicited an enhanced Ca2+ response following LTH while relaxation was unchanged although l-NAME sensitivity increased. Additionally, BKCa function decreased during bradykinin relaxation following LTH. Western analysis showed that BKCa α-subunit expression was increased by LTH while that for the β1 subunit was unchanged. Overall, these results suggest that those even moderately responsive to LTH can have impaired endothelial function.
Keywords: calcium ion, confocal microscopy, endothelial nitric oxide synthase, long-term high-altitude hypoxia, nitric oxide, potassium channels, sheep
INTRODUCTION
The regulation of pulmonary vascular smooth muscle tone requires a delicate balance between the activation of vasoconstrictive and vasorelaxant pathways. Disruption of this balance toward vasoconstriction can cause pulmonary hypertension. Pulmonary hypertension commonly leads to right heart failure and systemic vascular dysfunction and contributes to the pathogenesis of pulmonary and systemic edema, all of which induce significant morbidity and mortality (63). The risk of developing pulmonary hypertension because of chronic hypoxia is especially high among populations living permanently at high altitude (>2,500 m) (36), which currently amounts to ~140 million people worldwide (43). Numerous studies have indicated that endothelium-derived relaxing factors associated with nitric oxide (NO) is crucial in the modifications associated with loss of vasodilatory capacity and development of pulmonary hypertension (1, 20, 38).
Endothelial cells play a key role in determining the overall level of vessel relaxation (1, 48), and endothelium-dependent relaxation is partially mediated through bradykinin stimulation (22). Bradykinin is a potent vasodilatory and inflammatory substance that is important to normal physiology and pathology. As such, the roles of bradykinin in the pulmonary circulation and relationship to pulmonary hypertension have been explored across various age groups by our laboratory and others (4, 6, 22, 38). Bradykinin activation of the pulmonary endothelium stimulates relaxation through a number of pathways that are largely dependent on a rise in the intracellular Ca2+ concentration (48). The most widely studied of these pathways is bradykinin-induced activation of endothelial NO synthase (eNOS), which catalyzes the generation of NO (45). NO diffuses from the endothelium into smooth muscle cells and causes downstream stimulation of soluble guanylate cyclase (sGC), which activates pathways that lead to vasorelaxation (2, 32). However, bradykinin stimulation of the endothelium also activates other pathways, particularly phospholipase A2, which causes relaxation through prostacyclin (PGI2)-dependent activation of the smooth muscle cells (12).
Previous studies from our group and others showed that exposure to chronic hypoxia altered the regulation of vessel relaxation by various receptor-signaling systems (6, 16, 21). It is well known that chronic hypoxia is an important risk factor in the development of pulmonary hypertension, since it enhances pulmonary vasoconstriction and reduces vasodilatory capacity, resulting in increased pulmonary pressures and pulmonary hypertension (48). Although the effects of chronic hypoxia on endothelium-dependent relaxation have been widely studied, the mechanisms by which they occur are still not completely understood, and there is great variation depending on the individuals or animal species examined (48, 69). Thus, we sought to design a series of studies to examine how chronic hypoxia affects bradykinin-induced vasorelaxation in pulmonary arteries. We tested the specific hypothesis that chronic hypoxia impairs normal bradykinin-mediated relaxation through loss of pathways that are coupled to eNOS and phospholipase A2 signaling. This hypothesis was tested on arteries isolated from female adult sheep housed near sea level (normoxic) or exposed to long-term high-altitude hypoxia (LTH) for 110 days, since sheep generally respond similar to humans, even though they are hyporesponsive relative to commonly used large animal models, including cattle or pigs (48, 69).
METHODS
Experimental animals.
Experiments were performed on sheep pulmonary arteries using procedures detailed in our previous reports (5, 6, 26). Sheep were chosen largely because chronic hypoxia affects the ovine pulmonary vasculature to a similar extent as that of humans (48). The studies were performed within the regulations of the Animal Welfare Act, the National Institutes of Health Guide for the Care and Use of Laboratory Animals, “The Guiding Principles in the Care and Use of Animals” approved by the Council of the American Physiological Society and approved by the Animal Care and Use Committee of Loma Linda University (LLU). The tissue preparation, wire myography, and imaging experimental procedures and protocols are based on previously published methodology (5, 26). Pulmonary arteries of fourth- to fifth-branch order with internal diameters of ~500–700 μm were isolated from two groups of adult ewes placed in either normoxic or LTH experimental groups. Smaller vessels such as these are generally more involved in contributing to elevated pulmonary vascular resistance from LTH. The normoxic animals were from Nebeker Ranch (720 m; Lancaster, CA) and were brought to LLU (353 m; arterial = 95 ± 5 mmHg) for experimental study. Animals in the LTH experimental group, also from Nebeker Ranch, were acclimatized to high altitude (3,801 m, = 60 ± 5 mmHg) at the Barcroft Laboratory, White Mountain Research Station (Bishop, CA) for ~110 days (26). We have used this model for previous examinations of the influence of LTH on pulmonary arterial function (21, 26, 49). The hypoxic animals were then transported to LLU for experimental study. To maintain hypoxic conditions, a tracheal catheter was placed in the ewe shortly after arrival. The tracheal catheter was used to administer N2 to the inspired air at a rate that was adjusted to maintain at ~60 mmHg, which is equivalent to the at the White Mountain Research Station (34). This was maintained until the time of the experimental study. Within 1 to 5 days of arriving at LLU, animals were anesthetized with thiamylal (10 mg/kg iv) followed by inhalation of 1.0–2.0% isoflurane. Lungs were removed and placed in ice-cold phosphate-free balanced salt solution (BSS) of the following composition (in mM): 126 NaCl, 5 KCl, 10 HEPES, 1 MgCl2, 2 CaCl2, and 10 glucose, with pH adjusted to 7.4 with NaOH.
Tissue preparation.
Pulmonary arteries were isolated and used immediately for contractility and imaging experiments at LLU. Care was taken during arterial isolation and wire mounting to ensure the endothelium was not damaged. Endothelial integrity was examined in confocal microscopy studies and by vasodilatory response to bradykinin as we have done previously (6). The arteries were cut into 5-mm-long rings in ice-cold BSS. Arteries were treated with the NO donor N-nitrosoproline (ProliNO) in BSS, which releases NO in a 1:2 ProliNO-to-NO molar ratio with a dissociation half-life of ~2 s at physiological pH and temperature (60). All other contraction studies were performed with a modified Krebs-Henseleit (K-H) solution containing (in mM) 120 NaCl, 4.8 KCl, 1.2 K2HPO4, 25 NaHCO3, 1.2 MgCl2, 2.5 CaCl2, and 10 glucose. To depolarize the arteries, NaCl was omitted from the BSS or K-H solution and replaced with equimolar KCl.
Contraction studies.
Wire-mounted pulmonary arterial rings were suspended in organ baths (Radnoti Glass Instruments, Monrovia, CA) containing 5 or 10 ml of modified K-H solution or BSS maintained at 37°C. Arteries in modified K-H were aerated with 95% O2-5% CO2 (pH 7.4), whereas unaerated HEPES-buffered BSS was used for the ProliNO studies to prevent sparge-induced loss of liberated NO in the atmosphere. Each ring was suspended between two tungsten triangles passed through the lumen: one triangle was anchored to the glass hook at the bottom of the organ chamber while the other was connected to a tissue hook attached to a low-compliance force transducer (Radnoti Glass Instruments) that measured isometric force (5, 6, 26). The transducers were connected to an analog-to-digital data interface (Powerlab 16/30; A/D Instruments, Colorado Springs, CO, or MP100; Biopac, Goleta, CA) attached to a computer. The changes in tension were recorded using Chart 5.5 or 7.0 (AD Instruments), or AcqKnowledge 3.9 (Biopac), and the data were stored on digital media for later analysis.
At the beginning of each experiment, vessels were equilibrated without tension for ≥30 min. Each vessel was tensioned to ~0.75 g, and the vessel tension was allowed to stabilize, as previously described (5, 6, 26). To allow for comparative evaluation of smooth muscle contraction and relaxation, isolated pulmonary arterial rings bathed in modified K-H were stimulated with 125 mM KCl (high K) to cause membrane depolarization and subsequent L-type Ca2+ channel activation (49). In some experiments, the tension was normalized to a control response obtained with high K. To evaluate dose-response relaxation characteristics, bradykinin was used to relax the arteries following precontraction with 10 µM phenylephrine (PE) or serotonin (5-HT). In these experiments, the tension was normalized to the maximum 10 µM PE-precontracted tension or serotonin. Care was taken to add bradykinin shortly after the maximum response to each dose to avoid bradykinin type 2 receptor (B2R) desensitization.
Confocal microscopy studies.
Changes in the intracellular Ca2+ were measured in pulmonary arterial endothelial cells in situ, with the Ca2+-sensitive dye fluo 4-AM (Invitrogen, Carlsbad, CA) using a Zeiss 710 NLO laser scanning confocal imaging workstation (Thornwood, NY) with an inverted microscope (Zeiss Axio Observer) using procedures based on those previously described (6, 26). Fluo 4-AM was dissolved in DMSO and added from a 1 mM stock solution to the arterial suspension at a final fluo 4 concentration of 10 μM, along with 0.1% Pluronic F-127 for 1 h at room temperature in the dark in BSS. Arterial segments then were washed with BSS for 30 min to allow for dye esterification and cut into linear strips. The arterial segments were pinned to Sylgard (Ellsworth Adhesives, Germantown, WI) and placed in an open-bath imaging chamber (Warner Instruments, Hamden, CT) mounted on the confocal imaging stage. Cells were illuminated at 488 nm with a krypton argon laser, and the emitted light was collected using a photomultiplier tube with a band-limited spectral grating of range 493–622 nm with full-frame images made every 700 ms. To ensure that the endothelial intracellular Ca2+ concentration was recorded, the pinhole was adjusted to provide an imaging depth of 5.4 μm. The sample was then focused above the internal elastic lamina layer, which has significant autofluorescence when excited at 488 nm in this preparation. This imaging depth is substantially deeper than an individual endothelial cell based on morphological examination of fixed and live preparations (data not shown). This depth of imaging accounts for sample ruffling and allows for the examination of many more endothelial cells than otherwise would have been achieved. The influence of Ca2+ signals from smooth muscle was mitigated because myocytes are on the opposite side of the internal elastic lamina, and not in the primary imaging plane. Images were acquired at a 12-bit sampling depth. Recordings were made using an immersion ×40 Plan Apochromat, 1.0-numeric aperture objective mounted on an inverterscope objective inverter (LSM Tech, Wellsville, PA), which allowed for imaging in an upright configuration. To prevent arterial movement during recordings, arteries were pretreated for 1 h with a cocktail including 10 μM Y-27632 to inhibit Rho kinase, 10 μM cytochalasin D to inhibit actin polymerization, and 10 μM ML-7 to inhibit myosin light chain kinase, as previously described (5, 6). The concentrations of these drugs were chosen based on the outcomes of wire myography studies where the influence of the drugs on vessel reactivity was examined individually (data not shown). Regions of interest were detected automatically post hoc using the LC Pro plug-in for ImageJ (14). For presentation and quantification purposes, the fractional fluorescence intensity was automatically calculated using LC Pro. False positives were excised from the final dataset by their relationship to the timing of bradykinin application and by visual analysis of the data.
Western immunoblot assay.
To generate whole cell lysates, pulmonary arteries were harvested, cleaned, rapid-frozen in liquid nitrogen, and stored at −80°C. Tissues were then homogenized using glass-on-glass in a RIPA extraction buffer containing 10 mM DTT and a protease inhibitor cocktail (catalog no. M1745; Sigma-Aldrich, St. Louis, MO). Samples were centrifuged at 4°C for 20 min at 5,000 g, and supernatants were collected and analyzed using SDS-PAGE [10% large-conductance K+ (BKCa) channel α-subunit, 15% BKCa β-subunit] along with reference control samples. Separated proteins were transferred to nitrocellulose membranes at 300 mA for 1.5 h (BKCa α-subunit) or at 250 mA for 50 min (BKCa β1-subunit) in Towbin buffer (25 mM Tris, 192 mM glycine, and 20% methanol). Membranes were blocked using 5% milk in Tris-buffered saline at pH 7.45 for 1 h at room temperature with continuous shaking. Primary antibodies were incubated for 12 h at 4°C using the following dilutions: 300:1 for BK-α (catalog no. APC-021; Alomone, Jerusalem, Israel) and 400:1 for BK-β1 (catalog no. APC-036; Alomone) (6, 30). For visualization, membranes were incubated for 90 min with a secondary antibody conjugated to DyLight 800 (catalog no. 46422; Pierce Chemical, Rockford, IL). Subsequently, membranes were stripped and reprobed using antibodies against β-actin [monoclonal anti-β-actin produced in mouse, clone AC-74 (catalog no. A2228; Sigma-Aldrich)] as a loading control. Anti-β-actin was diluted 1:5,000 and incubated for 90 min in 5% milk in Tris-buffered saline with 0.1% Tween 20. Membranes were imaged on an infrared imaging system (CLx; LI-COR, Lincoln, NE), and individual protein bands were quantified using Image Studio version 5.2 software (LI-COR). Protein abundance was normalized using β-actin as a loading control and expressed as relative abundance compared with an arbitrary reference standard, which was pooled whole cell lysate obtained from pregnant adult sheep middle cerebral arteries.
Chemicals, drugs, and statistical methods.
Most reagents and chemicals were purchased from Sigma-Aldrich; bradykinin, Y-27632, and ML-7 were purchased from Tocris (Minneapolis, MN). Tetraethylammonium (TEA) was used to inhibit BKCa channels using dosing regimens and conditions as delineated in our previous studies of fetal and newborn pulmonary (6) and adult uterine (30) arteries from sheep. All time-series recordings were graphed with IGOR pro 6.0 (Wavemetrics, Lake Oswego, OR), and summarized data are presented as means ± SE. Statistical analyses were made using Prism 5.0 (Graphpad, La Jolla, CA). Data were evaluated for normality before comparative statistical analysis. Dose-response curves were fitted in Prism 5.0 using the Hill equation with nonlinear curve fit analysis (5). A total of 225 arterial segments from 43 sheep were tested for contractility. N values are displayed in the legends of the figures in the format “n = number of animals/number of arteries examined.” The Ca2+ imaging studies were performed on tissues from five normoxic and five LTH sheep. Western blot analysis was performed on tissues from six normoxic and six LTH sheep. P < 0.05 was accepted as statistically significant with the actual P value provided, unless otherwise noted.
RESULTS
LTH impairs bradykinin-mediated relaxation.
The first series of studies was designed to determine the extent of the relaxant effects of bradykinin on ovine pulmonary arteries and examine the effects of LTH on this relaxation, based on previous work (1, 6, 38, 70). These relationships were investigated by examining dose-dependent bradykinin-induced relaxation of 10 µM PE-precontracted normoxic and LTH vessels. Dose-response curves and summary results of the data from these studies are shown in Fig. 1. Arteries from LTH sheep had impaired bradykinin-induced relaxation. This was primarily mediated by a subgroup of arteries that contracted in response to bradykinin. Figure 1A shows that bradykinin-induced relaxation was attenuated at intermediate doses in LTH arteries, but the maximum level of relaxation remained unchanged. Figure 1B compares the percentage of normoxic arteries that contracted in response to low concentrations of bradykinin (0%, 0 of 40 arteries) with the percentage of hypoxic arteries that contracted in response to bradykinin (54%, 15 of 28 arteries). Figure 1, C and D, compares the control dose-response curve of normoxic arteries with that of LTH arteries that contracted in response to bradykinin (C) compared with those that did not have any contractile response (D). We also evaluated the proportion of pulmonary arterial rings from normoxic and LTH sheep that contracted in response to bradykinin following treatment with antagonists of NO and prostaglandin synthesis or K+ channel function. None of the arteries from normoxic animals contracted, whereas contractions were observed in arteries from LTH sheep treated with NG-nitro-l-arginine methyl ester (l-NAME, 11 of 15), indomethacin (Indo, 7 of 10), and TEA (2 of 7). The l-NAME, Indo, and TEA treatments failed to alter the proportion of arteries that contracted in response to bradykinin as determined by Chi-square analysis. Figure 1A and Table 1 summarize the data and illustrate that the potency of bradykinin-induced relaxation decreases with LTH. Figure 1A and Table 2 further demonstrate that the maximum level of relaxation is not affected by LTH.
Fig. 1.

Long-term high-altitude hypoxia (LTH) results in a unique contractile response to bradykinin. Dose-response curves of pulmonary arterial rings exposed to 10 pM to1 μM bradykinin in an additive manner normalized to maximum 10 µM phenylephrine (PE)-precontracted tension (%T10 μM PE) for normoxic (filled circles) and LTH (open circles) sheep, comparing all normoxic and hypoxic control data (A), the normoxic dose-response curve with the hypoxic curve representative of arteries that contracted in response to bradykinin (C), and the normoxic curve with the hypoxic curve representative of arteries that did not contract in response to bradykinin (D). B: comparison of the percentage of total normoxic and LTH control pulmonary arterial rings that contracted in response to treatment with bradykinin. Lines show resultant fits with a Hill equation to the dose-response relationships, and markers show means ± SE. The dose-response data were analyzed by 2-way analysis of variance with a Bonferroni posttest analysis for each dose, and the difference in percentages was analyzed using a Chi-squared contingency test. **P < 0.01 and ***P < 0.001, statistical significance between normoxic and hypoxic groups. Normoxic, n = 11/40; LTH animals, n = 16/28.
Table 1.
Long-term hypoxia and potency of bradykinin-induced relaxation
| Normoxic |
Hypoxic |
|||
|---|---|---|---|---|
| Condition | Mean ± SE | n | Mean ± SE | n |
| Control | −8.72 ± 0.11 | 11/40 | −7.34 ± 0.25b | 16/28 |
| l-NAME | −8.70 ± 0.37 | 4/9 | −7.40 ± 0.47 | 7/15 |
| Indo | −7.95 ± 0.13 | 3/11 | −7.21 ± 0.36 | 4/10 |
| TEA | −7.66 ± 0.39a | 3/6 | −7.47 ± 0.13 | 5/7 |
Values are log concentration for the IC50 for bradykinin-induced relaxation for data presented graphically in Figs. 1, 4, 6, and 7; n, no. of arteries studied for each condition in each group. l-NAME, NG-nitro-l-arginine methyl ester; Indo, indomethacin; TEA, tetraethylammonium. aSignificant leftward shift in the IC50 compared with control and bsignificant difference in IC50 compared with normoxic counterpart [with the use of a 95% confidence interval based on the log(inhibitor) vs. response curve fit to the data].
Table 2.
Long-term hypoxia and efficacy of bradykinin-induced relaxation
| Normoxic |
Hypoxic |
|||
|---|---|---|---|---|
| Condition | Mean ± SE | n | Mean ± SE | n |
| Control | 33 ± 2 | 11/40 | 40 ± 6 | 16/28 |
| l-NAME | 57 ± 4a | 4/9 | 56 ± 14 | 7/15 |
| Indo | 36 ± 3 | 3/11 | 47 ± 16 | 4/10 |
| TEA | 52 ± 6a | 3/6 | 24 ± 4 | 5/7 |
Values are percentage of TKmax for bradykinin induced relaxation for data presented graphically in Figs. 1, 4, 6, and 7; n, no. of arteries studied for each condition in each group. l-NAME, NG-nitro-l-arginine methyl ester; Indo, indomethacin; TEA, tetraethylammonium. aSignificant depression in efficacy compared with control [using a 95% confidence interval based on the log(inhibitor) vs. response curve fit to the data].
LTH and endothelial Ca2+ signals.
Because of the reduction of bradykinin relaxation following LTH as presented in Fig. 1 and other work (6, 62), we investigated the possibility that LTH decreased bradykinin-induced Ca2+ signals in endothelial cells. Figure 2 shows Ca2+ activity within the endothelium before and during administration of 1 µM bradykinin in the presence of extracellular Ca2+ but absence of PE preconstriction. Figure 2A shows in situ fluorescence images at various times of endothelial cells in the arterial wall isolated from a normoxic sheep. These images not only show the intracellular Ca2+ but also illustrate that our arterial isolation techniques did not damage the endothelial layer. These images show that the endothelial cells have an ellipsoid shape, which is presumed to be along the direction of blood flow, and that the cells are closely associated with one another, indicating a normal and healthy endothelium in the isolated pulmonary arteries. As shown for the fluorescence intensity trace in Fig. 2B, bradykinin rapidly increased the cytosolic Ca2+ concentration in pulmonary arterial endothelial cells, and these signals decayed slowly back to the baseline. Because the Ca2+ increase was qualitatively similar between normoxic and LTH animals, other representative images are not shown.
Fig. 2.

Long-term hypoxia increases bradykinin-mediated cytosolic Ca2+ responses. Representative images (A) corresponding in time to the trace of fractional fluo 4 fluorescence (B) for four pulmonary arterial myocytes from a normoxic sheep in the presence of extracellular Ca2+. Lowercase letters denote the times at which the images occurred. The colored circles in A correspond to the trace of the same color in B. The brightness of the images in A was adjusted for display purposes. Images were made with a ×40 water immersion Apochromat objective at 1.28 frames/s. Scale bar is 20 μm.
Figure 3 provides summaries of the quantitative analysis for the magnitude and kinetics of the fluorescence Ca2+ signals. These data demonstrate that, following LTH, there was a significant increase in the amplitude (Fig. 3A), area under the curve (Fig. 3B), duration (Fig. 3C), decay (Fig. 3D), and rate of Ca2+ rise (attack, Fig. 3E) in the endothelium.
Fig. 3.

Long-term hypoxia (LTH) augments bradykinin-induced Ca2+ signals. Amplitude of the fractional fluorescence (A), area under the curve (B), duration (C), decay (D), and attack of the Ca2+ event (E) for regions of interest that were automatically detected in normoxic (n = 5/406) and LTH (n = 5/272) sheep arterial myocytes. Bars indicate means ± SE. ***P < 0.001, statistically significant effect of LTH based on Kruskal-Wallis nonparametric 1-way analysis of variance with Dunn's multiple-comparison test.
LTH- and eNOS-dependent relaxation.
The next series of studies investigated the hypothesis that altered bradykinin-induced eNOS activation mediates the uncoupling of endothelial Ca2+ signaling and vasorelaxation. eNOS was inhibited with 100 µM l-NAME (54), and vessels were treated with increasing doses of bradykinin. Figure 4 shows that eNOS inhibition with l-NAME significantly attenuated maximal bradykinin-induced relaxation in normoxic arteries (Fig. 4A). However, Fig. 4B shows that, following LTH, l-NAME only reduced bradykinin relaxation at intermediate doses. The potency of bradykinin-induced relaxation, summarized in Table 1, was unaffected by eNOS inhibition under either normoxic or LTH conditions. Table 2 shows that l-NAME significantly attenuated the efficacy of the bradykinin-mediated relaxation response under normoxic conditions but had no effect on efficacy following LTH.
Fig. 4.

Endothelial nitric oxide synthase (eNOS) or cyclooxygenase (COX) inhibition decreases bradykinin-induced pulmonary arterial relaxation. Dose-response curves of pulmonary arterial rings exposed to 10 pM to 1 μM bradykinin in an additive manner normalized to 10 µM phenylephrine (PE)-precontracted tension (%T10 μM PE) for normoxic (A) and long-term hypoxia (LTH, B) sheep in the presence of DMSO (filled circles) for vehicle control and 100 μM NG-nitro-l-arginine methyl ester (l-NAME, A and B, open circles, dotted line) for eNOS inhibition or 10 μM indomethacin (C and D, open circles, dotted line) to inhibit prostacyclin (PGI2) production. Lines show resultant fits to the dose-response relationships with a Hill equation, and markers show means ± SE. The data were analyzed by 2-way analysis of variance with a Bonferroni posttest analysis for each dose. Statistical significance is noted relative to DMSO control, *P < 0.05, **P < 0.01, and ***P < 0.001. A: normoxic, n = 4/9; B: LTH, n = 7/15; C: normoxic, n = 3/11; D: LTH, n = 4/10.
LTH- and PGI2-dependent relaxation.
To examine the role of PGI2 in bradykinin-induced vasodilation (7), cyclooxygenase (COX)-dependent PGI2 production was inhibited with 10 µM Indo, as we have done previously (6). At this concentration, Indo inhibits both COX-1 and -2 (39, 47). As shown in Fig. 4, bradykinin-induced relaxation at intermediate doses was significantly reduced by COX inhibition under normoxic (Fig. 4C) or LTH (Fig. 4D) conditions. Interestingly, Indo inhibited vasorelaxation to lower concentrations of bradykinin following LTH, an effect that was not observed in arteries from normoxic animals. However, Fig. 4 and Tables 1 and 2 also show that COX inhibition did not affect either the potency or efficacy of bradykinin-mediated vasodilation in either normoxic or LTH vessels.
To further address the impact of LTH on endothelial-dependent vasodilation, arterial segments were treated with the Ca2+ ionophore A-23187, which is often used to directly examine endothelial-dependent relaxation (72). In these studies endothelial Ca2+ signals and arterial reactivity were examined in response to 10 μM A-23187. The summarized data in Fig. 5A show that the area under the Ca2+ release curve during treatment with 10 μM A-23187 was reduced by LTH. Correspondingly, the duration of the fluorescence increase was longer in normoxia compared with LTH (Fig. 5B). LTH also caused a small reduction in the fluorescence amplitude because of A-23187 (Fig. 5C). Even though LTH decreased the Ca2+ responses resulting from A-23187, these responses were still either equivalent to, or substantially greater than, those resulting from bradykinin (Fig. 3). With regard to vascular reactivity, Fig. 5D shows that 10 μM A-23187 relaxed 1 μM serotonin-contracted vessels by roughly 30%. Notably, this relaxation response was unaltered by LTH, although 100 μM l-NAME attenuated the relaxation response in LTH but not normoxic tissues.
Fig. 5.

Long-term hypoxia (LTH) modifies A-23187-mediated relaxation and Ca2+ signals. Area under the curve (A), duration (B), and amplitude of the Ca2+ events (C) for regions of interest that were automatically detected in normoxic (n = 5/553) and LTH (n = 7/563) sheep arterial myocytes. D: relaxation relative to the contraction induced by 1 μM serotonin (5-HT) in normoxic (n = 5/71) and LTH (n = 8/100) sheep pulmonary arteries. Individual data points are shown for normoxic (closed) and LTH (open) recordings, and bars indicate means ± SE †††P < 0.001, statistically significant effect based on a Mann Whitney U-test. **P < 0.01 and ***P < 0.001, statistically significant effect based on a Kruskal-Wallis 1-way ANOVA with a Dunns multiple-comparison test.
NO-induced relaxation and the role of sGC.
Because of the findings of our l-NAME studies and other work (3, 6), we designed a series of experiments to examine the direct impact of NO on pulmonary arterial reactivity. More specifically, we examined the ability of NO to directly relax arteries via the NO donor ProliNO (60). Figure 6 shows 10 µM 5-HT-preconstricted vessels and ProliNO dose-dependent vasodilation in the presence and absence of the sGC inhibitor 10 µM 1H-[1,2,4]oxadia-zolo[4,3-a]quinoxalin-1-one (ODQ) (18). Overall, ProliNO effectively dilated 10 µM 5-HT-preconstricted normoxic and LTH vessels (Fig. 6A). sGC inhibition decreased the potency of NO-induced relaxation by causing a rightward shift of dose-response curves for both normoxic (Fig. 6B) (P < 0.01) and LTH (Fig. 6C) (P < 0.05) groups.
Fig. 6.
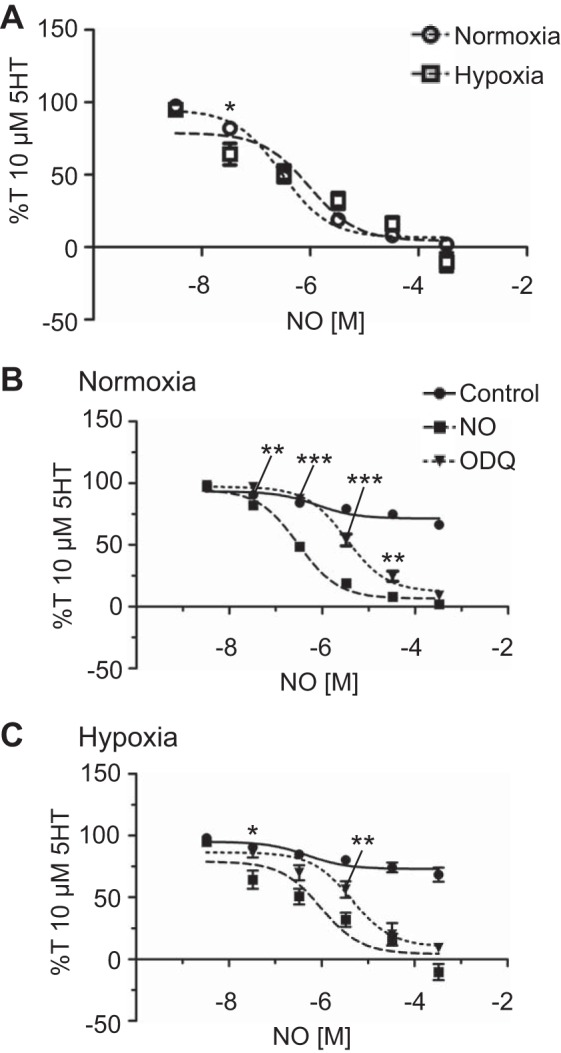
Nitric oxide (NO)-dependent vasorelaxation is preserved following long-term hypoxia (LTH). N-nitrosoproline (ProliNO)-induced isometric tension values normalized to 10 µM serotonin (5-HT)-precontracted tension (%T10 μM 5-HT) for normoxic (open circles, dotted line, n = 5/19) and LTH (open square, n = 7/18) sheep (A), normoxic (n = 5/19, B) and LTH (n = 7/18, C) sheep in the absence (squares) of 1H-[1,2,4]oxadia-zolo[4,3-a]quinoxalin-1-one (ODQ), in the presence of 10 μM ODQ (inverted triangles, dotted line; n = 7/26 normoxic and 4/12 LTH animals), and in the presence of NaOH (filled circles) as a vehicle- and time-matched control (n = 7/32 normoxic and 4/13 LTH animals). Lines show resultant fits with a Hill equation to the dose-response relationships, and markers show means ± SE. The data were analyzed by 2-way analysis of variance with a Bonferroni posttest analysis for each dose. Statistical significance is noted between groups (A) or relative to the presence of ProliNO (B and C). *P < 0.05, **P < 0.01, and ***P < 0.001.
Bradykinin relaxation and BKCa channels.
K+ channels are important targets of endogenous and therapeutic vasodilators because of their critical role in establishing the myocyte resting membrane potential (35). One of the targets of NO-dependent signaling is the BKCa channel (2), which is important to the regulation of myogenic tone and blood pressure (2, 8, 52). The BKCa channel is activated by ryanodine receptor-dependent Ca2+ sparks and independently regulated by protein kinase A and protein kinase G downstream of sGC and cGMP (2, 59, 61). To gain a better understanding of the role these BKCa channels play in BK-mediated vasodilation, 10 µM PE-precontracted vessels were treated with 1 mM TEA, which blocks roughly one-half of BKCa channel currents without affecting other K+ channels also expressed in the pulmonary vasculature (30, 33). Furthermore, our previous studies in sheep uterine arteries demonstrate that 1 mM TEA has equivalent efficacy of BKCa channel inhibition compared with 100 nM iberiotoxin (30). Figure 7 shows the dose-dependent relaxation of 10 µM PE-preconstricted vessels to bradykinin in the presence or absence of TEA. The basal arterial tension did not increase when arteries were treated with 1 mM TEA in either normoxic or hypoxic arteries (data not shown), indicating that TEA does not inhibit other K+ channels that are important to basal tone. As expected, TEA significantly reduced bradykinin relaxation under normoxic conditions. However, BKCa channel inhibition with TEA had no significant effect on bradykinin-induced vasodilation following LTH. Tables 1 and 2 show that TEA reduced both the potency and efficacy of bradykinin-mediated relaxation under normoxic conditions, but exposure to LTH eliminated these responses.
Fig. 7.

Long-term hypoxia (LTH) decreases the importance of large-conductance voltage- and Ca2+-sensitive K+ (BKCa) channels to bradykinin-mediated arterial relaxation. Dose-response curves of pulmonary arterial rings exposed to 10 pM to 1 μM of bradykinin in an additive manner normalized to 10 µM phenylephrine (PE)-precontracted tension (%T10 μM PE) for normoxic (A) and LTH (B) sheep in the presence of DMSO (filled circles) for vehicle control and in the presence of 1 mM tetraethylammonium (TEA, open circles, dotted line) to inhibit BKCa channels. Lines show resultant fits to the dose-response relationships with a Hill equation, and markers show means ± SE. The data were analyzed by 2-way analysis of variance with a Bonferroni posttest analysis for each dose. Statistical significance is noted relative to DMSO control. **P < 0.01 and ***P < 0.001. Normoxic, n = 3/6; LTH, n = 5/7.
BKCa channel protein expression.
The BKCa channel is formed by four α-subunits and accessory β-subunits (42). Four classes of these β-subunits have been identified, although there may be more (67). β-Subunits can specialize BKCa channel gating by regulating voltage and Ca2+ sensitivity, and the β1-subunit has been identified as the most common β-subunit in vascular smooth muscle (9, 42). Mice with β1-subunit knockdown have been shown to experience increases in arterial tone and blood pressure because of decreased BKCa channel sensitivity and coupling to Ca2+ sparks (51). Furthermore, other studies have shown the BKCa channel activity to be developmentally regulated and differentially affected by Po2 (57). To determine whether the lack of BKCa channel importance following LTH was because of changes in BKCa channel subunit expression, Western immunoblot analysis was conducted for the α- and β1-subunits. This analysis revealed that LTH tissues exhibited a 2.7-fold increase in α-subunit expression without any significant change in β1 expression (Fig. 8).
Fig. 8.
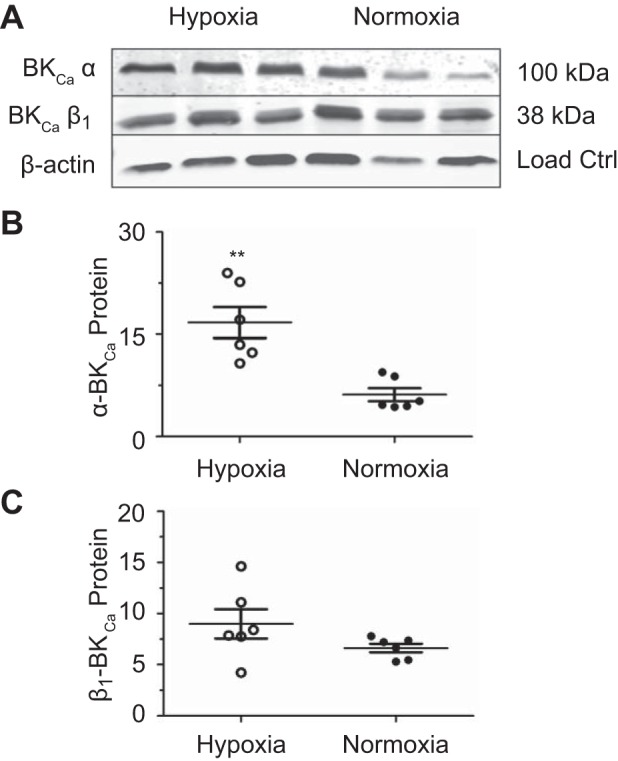
Long-term hypoxia (LTH) increases large-conductance voltage- and Ca2+-sensitive K+ (BKCa) channel α- but not β1-subunit expression. Western blot analysis of pulmonary arteries from normoxic and LTH sheep. A: representative blot of BKCa α- and β1-subunit expression and β-actin to which channel subunit expression was normalized from normoxic and LTH sheep. B: α-subunit expression. C: β1-subunit expression. Protein abundance was normalized to β-actin and expressed as a relative abundance compared with the reference middle cerebral arteries standard, and bars show means ± SE. The data were analyzed by an unpaired t-test with a Welch’s correction for unequal variances for β1. **P < 0.01, statistically significant difference in subunit expression resulting from LTH. Western blots were performed on arteries isolated from 6 normoxic and 6 LTH sheep, with each animal being run in an individual lane.
DISCUSSION
These studies are the first to examine the influence of LTH on bradykinin-mediated pulmonary arterial relaxation of the adult and to investigate the roles of important second-messenger pathways. A pictorial summary of these findings and their relationship to our previous work is displayed in Fig. 9 (6, 26). In the endothelium, we examined the roles of intracellular Ca2+ elevations, eNOS, and COX signaling. In smooth muscle, we evaluated direct stimulation with ProliNO, the role of sGC, and downstream effects on BKCa channels. Our studies suggest LTH damages endothelial mechanisms of arterial relaxation, shifts activation of the bradykinin-signaling axis to smooth muscle, and impairs downstream vasodilatory mechanisms.
Fig. 9.

Influence of long-term hypoxia (LTH) on bradykinin-induced vasorelaxation in adult pulmonary vessels. The schematic provides an overview of the findings of the current studies on adult arteries. The data indicate that LTH has a significant influence on bradykinin-induced vasorelaxation involving modification to the function of both the endothelium (above the broken line) and smooth muscle (below the broken line). The influence of LTH is shown on the right side of each pathway step. These observed changes are codified as functional increases (green circles, “+”), decreases (red circles, “−”), or no changes (open circles, “N”). Pathways are depicted as either a coupled sequence (straight lines) or through a multistep process (broken lines), where intermediate pathway steps were not evaluated. BKCa, large-conductance voltage- and Ca2+-sensitive K+ channel; RyR, ryanodine receptor; sGC, soluble guanylate cyclase.
Adult pulmonary arteries experienced decreased potency of, and sensitivity to, bradykinin following LTH as delineated by the data presented in Fig. 1A and Tables 1 and 2 and summarized in the schematic of Fig. 9. What is more, a substantial proportion of the LTH vessels had a unique contractile response to bradykinin (Fig. 1, B–D). Although studies in conduit pulmonary arteries of adult sheep gestated under hypoxic conditions have shown impaired bradykinin-induced relaxation (38), to our knowledge we are the first to report LTH-induced contraction to bradykinin in endothelium-intact pulmonary arteries. There are a number of possible explanations for this unique constrictor response that will be the focus of future investigations. Bradykinin activates two subtypes of G protein-coupled receptors. The B2R is constitutively expressed in the endothelium and smooth muscle while the bradykinin type 1 receptor (B1R) is typically induced in smooth muscle by tissue injury, inflammation, or oxidative stress (41, 44, 55). Although both bradykinin receptor subtypes have been linked to vessel relaxation and constriction in various tissues and vascular beds, B2R is largely associated with endothelium-induced relaxation while B1R is largely associated with smooth muscle contraction (41, 44, 55).
Even though bradykinin- and eNOS-dependent relaxation decreased following LTH, Ca2+ signals in the pulmonary endothelium were elevated. These data suggest that LTH causes bradykinin Ca2+ signaling to uncouple from eNOS-dependent vasorelaxation. The changes in Ca2+ signaling might include dysfunction in spatial-temporal coupling based on the characteristics of the Ca2+ signals (14, 45). The rates of Ca2+ rise and decay and the overall duration of the Ca2+ events increase with LTH. These findings, together with the significant increases in the Ca2+ spike amplitude and area under the curve, suggest that LTH causes complex changes in Ca2+ release, influx, sequestration, and extrusion. This may include alterations in the expression or activation of inositol trisphosphate receptors or Ca2+-permeable ion channels at the plasma membrane, extrusion of Ca2+ from the cytosol by plasma membrane or endoplasmic reticulum Ca2+-ATPases, or Ca2+ storage in the endoplasmic reticulum (23, 24). Even still, these imaging studies are limited, since they were performed during inhibition of rho kinase, myosin light chain kinase, and actin polymerization, which could have a wide variety of effects on endothelial Ca2+ signals through changes in ion channel gating, Ca2+ sequestration, or extrusion (11, 28).
The uncoupling of the augmented bradykinin-induced Ca2+ signals from eNOS-dependent vasorelaxation could be the result of a number of processes (13, 25, 64). eNOS expression could decrease following LTH, or there could be more complex changes in eNOS function, since many factors besides Ca2+ determine eNOS activity. For example, there could be suppression and dephosphorylation of amino acid residues that sensitize eNOS to calmodulin and Ca2+ (19, 25). Concurrently, autoinhibitory domains that prevent calmodulin binding and thus inactivate eNOS could be phosphorylated (19, 25). Additionally, binding partners such as 90-kDa heat shock protein or calmodulin may have less interaction, or there could be increases in the abundance of cofactors such as l-arginine, flavin adenine dinucleotide, flavin mononucleotide, and tetrahydrobiopterin that decrease NO production directly or indirectly (15, 45, 64). Resolving the mechanisms that govern the LTH-induced decrease in bradykinin-mediated relaxation through Ca2+-eNOS uncoupling is important but was outside the scope of the present experiments.
The current studies were focused on assessing bradykinin-mediated relaxation in a stepwise fashion, as delineated in Fig. 9. Associatively, NO is well established as being vital to pulmonary vasorelaxation (13, 27, 45, 48). Consequently, we expected the finding that eNOS is important to bradykinin-mediated relaxation (Fig. 4). However, the shift in eNOS-mediated bradykinin dose dependence from high doses in normoxic animals to low doses following LTH suggests LTH activates other bradykinin-mediated vasodilatory pathways. Furthermore, the previous finding that newborn porcine pulmonary resistance arteries relax in response to bradykinin despite NOS inhibition supports the theory that LTH induces a loss of endothelium-mediated vasodilation, resulting in an unmasking of vasoconstriction caused by bradykinin receptors on the smooth muscle in our sheep (4).
PGI2 signaling pathways are activated by bradykinin in parallel with NO signaling as presented in Fig. 9. Although we did not evaluate PGI2-signaling pathways in their entirety, our Indo studies were informative. COX activity and subsequent PGI2 production are known to be important to endothelium-dependent relaxation in a number of vasculatures (46, 53). Previous studies on goats and newborn sheep demonstrated relaxation of pulmonary arteries and veins in response to direct stimulation with PGI2 agonists and precursors (10, 17). Whereas LTH seemed to increase the importance of PGI2 to bradykinin-mediated relaxation during initial doses, Indo had no effect on maximal relaxation (Fig. 4D), potency (Table 1), or efficacy (Table 2). Even still, the failure of COX inhibition to attenuate bradykinin relaxation in normoxic arteries was somewhat surprising, but it could be an indicator that bradykinin does not fully activate endogenous PGI2 synthesis pathways or that there is a potential change in Indo sensitivity. Nonetheless, our current results are in general agreement with previous studies done on endothelium-intact bovine supernumerary pulmonary arteries where Indo had no effect on bradykinin relaxation (68). On the whole, our Indo studies provide further support for the theory that LTH damages the pulmonary endothelium and promotes bradykinin activation of arterial smooth muscle.
Because bradykinin did not relax arteries back to their resting tension and the data suggest there is enhanced endothelium-independent vasorelaxation following LTH, we examined the effects of direct activation of endothelium with A-23187 (Fig. 5) and smooth muscle with NO (Fig. 6). As expected, treatment of arterial segments with A-23187 caused Ca2+ responses in the endothelium of normoxic and LTH tissues. The prediction was that the resultant vasorelaxation would be sensitive to l-NAME and lessened by LTH. Instead, only the LTH vessels were sensitive to l-NAME, and LTH failed to attenuate the relaxation response to A-23187. This suggests that LTH modifies endothelial relaxation pathways in discrete ways that may include differential activation of Ca2+-dependent pathways within the endothelium. Because l-NAME influenced the relaxation responses to A-23187 preferentially in LTH vessels we presume that these modifications include changes in eNOS function. However, we cannot exclude that there may also be alterations in PGI2-dependent dilation or other Ca2+-regulated vasodilatory signaling pathways. Potentially the differential Ca2+ responses contribute to the differences between normoxia and LTH. This is further supported by our studies where ProliNO both intensified and normalized the relaxation responses of 5-HT-precontracted vessels, where vessels from normoxic and LTH animals relaxed to a similar extent (Fig. 6A). Inhibition of sGC successfully reduced the potency of NO but did not attenuate the overall level of relaxation (Fig. 6, B and C). Previous studies from our laboratory and others have shown that sGC is important to endothelial-activated NO-dependent relaxation, suggesting that sGC and subsequent cGMP generation could be more important to vessel relaxation when coupled with upstream mechanisms in the endothelium (6, 18, 68). 5-HT was selected to be the agonist because we found it to be more stable than phenylephrine in preliminary studies and because it allows us to build from and draw comparisons between the results on adult arteries with previous studies conducted on fetal and newborn endothelium (6). Overall, the ProliNO data further illustrate that bradykinin does not fully activate vasorelaxant pathways and that there is substantial NO-sensitive and sGC-insensitive reserve. This sGC-insensitive reserve likely includes protein nitrosylation, which is a key NO-dependent signaling mechanism (37).
Previous studies in porcine pulmonary resistance arteries have shown that multiple K+ channel subtypes are involved in mediating bradykinin-induced relaxation (4). Our laboratory has shown BKCa channels to be particularly important to bradykinin-induced vasorelaxation in newborn sheep pulmonary arteries, but, to our knowledge, we are the first to examine the importance of BKCa channels to bradykinin relaxation in the adult pulmonary vasculature (6). The finding that BKCa channels play a significant part in mediating bradykinin-induced relaxation but lose this role following LTH is consistent with our previous findings in the newborn.
Hypoxia-induced impairments of BKCa channel activity are frequently mediated by channel dysfunction and changes in subunit expression (6, 30, 40). In this study, we inhibited BKCa channel activity in myography studies and conducted Western immunoblots to investigate the possibility that BKCa channel expression decreases in parallel with the hypoxia-induced changes in BKCa channel-mediated relaxation. Surprisingly, the hypoxia-induced changes in BKCa channel α- and β1-subunit expression did not parallel the hypoxia-induced changes in channel function. Although our myography studies found that the BKCa channel no longer has a functional role in bradykinin-induced relaxation following LTH, our Western blot analysis revealed the expression of the α-subunit of the channel increases following LTH. Our work parallels findings in chronic obstructive pulmonary disease patients, who have upregulated BKCa abundance (50). Similarly, previous work has shown upregulation of α-subunit expression in hypoxic-cultured sheep pulmonary vascular smooth muscle cells and in pulmonary arterial smooth muscle cells in chronic hypoxic rats (56). However, the current studies did not ascertain whether BKCa channels are expressed on vascular smooth muscle or on endothelial cells, which are also important for vasodilatory responses (58, 71) and upregulated by chronic hypoxia in rat pulmonary arterial endothelium (31).
The dichotomy between increased BKCa channel α-subunit expression and diminished channel function is fascinating and can be the result of any one of a number of mechanisms. Potentially, LTH negatively affects channel function by diminishing the ability of protein kinase G to phosphorylate and activate channels (65), or the phosphorylation status of the protein could be modified in other ways (75). Additionally, there could be dysregulation of BKCa α-/BKCa β-colocalization akin to what we found in cerebral vessels (66). Furthermore, the BKCa channel can be expressed not only on the plasma membrane but also on the mitochondrial membrane (73). Because our Western blot analyses measured subunit content in whole cell lysates, we cannot discern whether LTH is upregulating BKCa channel expression on the mitochondrial membrane or preventing proper channel trafficking to the plasma membrane and thereby causing changes in cellular metabolism and excitability.
Another potential explanation for the disconnect between BKCa channel expression and function is that LTH causes structural changes that negatively impact channel sensitivity and the ability to regulate K+ flux. We previously showed LTH increases Ca2+ spark activity, indicating that the loss of BKCa channel function is likely because of changes in the channel itself and not with the occurrence of Ca2+ sparks (26). Potentially, the dramatic increase in α-subunit relative to β1-subunit expression could impair proper channel gating because β1-subunits assist in regulating the Ca2+ and voltage dependence of the channel. Alternatively, there could be changes in the proximity of the Ca2+ spark relative to BKCa channels on the plasma membrane (29). LTH could also induce an increase in the expression of BKCa β4, which is a negative modulator of channel activation, as occurs in rat following 24-h exposures to hypoxia (74). These changes would not necessarily be detected by our Western blot analysis but would affect the attachment and interaction of the subunits, thereby influencing channel activation and K+ flux.
Perspectives and Significance
The current series of studies significantly advance our knowledge of the impact of LTH on bradykinin-induced pulmonary arterial relaxation in sheep, which like many humans have modest responses to high-altitude exposure (48, 69). The data illustrate that both eNOS-sensitive and -insensitive pathways contribute significantly to bradykinin-mediated vasorelaxation. Additionally, the data illuminate the importance of conducting studies in the whole animal in addition to isolated cells. The changes induced by LTH stress are complex and systemic. As such, short-term stress induced during in vitro experimentation does not provide a clear picture of the multifaceted physiological adaptations and malformations that take place. The data suggest that LTH damages the vasodilatory function of the pulmonary endothelium by uncoupling Ca2+ signaling and eNOS activity. Such endothelial damage seemingly shifts activation of bradykinin receptors to the smooth muscle, which is linked to downstream loss of BKCa channel function. On the whole, our results show that LTH blunts normal bradykinin-induced relaxation via complex changes to the eNOS-BKCa channel-signaling pathway in sheep. These substantial changes brought on by LTH provide important insights into the mechanisms that compromise pulmonary arterial function in individuals and species that respond modestly to LTH and thus may be contributing factors in the development of pulmonary hypertension. Further investigation is required to delineate the general applicability of these results to other species, including humans. Improved understanding of these mechanisms may prove important to developing novel and improved therapies for pulmonary hypertension.
GRANTS
This material is based on work supported by the National Science Foundation under Grant MRI 0923559 and National Institutes of Health (NIH) Grants HD-069746 (S. M. Wilson), R01-NS-076945 (W. J. Pearce), HL-095973 (A. B. Blood), P01-HD-031226 and R01-HD-003807 (L. D. Longo), and P01-HD-083132 (L. Zhang). Additional funding for research reported in this publication was supported by NIH under award number 2-R25-GM-060507. C. Blum-Johnston was a summer research fellow in the Apprenticeship Bridge to College and Undergraduate Training Programs through the Initiative for Maximizing Student Development Program in the Center for Health Disparities and Molecular Medicine. R. Opsahl was a summer research fellow with the Walter E. McPherson Program in the Loma Linda University School of Medicine. Additional support for the Advanced Imaging and Microscopy Core was provided by the Loma Linda University School of Medicine.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or National Science Foundation.
AUTHOR CONTRIBUTIONS
C.B.-J., R.B.T., C.W., R.O., M.R., S.M., A.B., Q.B., R.W., A.B.B., L.Z., L.D.L., W.J.P., and S.M.W. analyzed data; C.B.-J., R.B.T., C.W., R.O., M.R., S.M., A.B., Q.B., R.W., A.B.B., L.Z., L.D.L., W.J.P., and S.M.W. interpreted results of experiments; C.B.-J., R.B.T., C.W., R.O., M.R., S.M., A.B., Q.B., R.W., A.B.B., L.Z., L.D.L., W.J.P., and S.M.W. prepared figures; C.B.-J., R.B.T., C.W., R.O., M.R., S.M., A.B., Q.B., R.W., A.B.B., L.Z., L.D.L., W.J.P., and S.M.W. drafted manuscript; C.B.-J., R.B.T., C.W., R.O., M.R., S.M., A.B., Q.B., R.W., A.B.B., L.Z., L.D.L., W.J.P., and S.M.W. edited and revised manuscript; C.B.-J., R.B.T., C.W., R.O., M.R., S.M., A.B., Q.B., R.W., A.B.B., L.Z., W.J.P., and S.M.W. approved final version of manuscript; R.B.T., A.B.B., L.Z., L.D.L., W.J.P., and S.M.W. conceived and designed research; R.B.T., M.R., S.M., A.B., Q.B., R.W., and S.M.W. performed experiments.
ACKNOWLEDGMENTS
We thank Kurt Vrancken and Dr. Taiming Liu for technical assistance with the ProliNO studies. We thank Michael Francis and Mark S. Taylor from the Department of Physiology and Cell Biology University of South Alabama College of Medicine for assistance with LCPro.
REFERENCES
- 1.Adnot S, Raffestin B, Eddahibi S, Braquet P, Chabrier PE. Loss of endothelium-dependent relaxant activity in the pulmonary circulation of rats exposed to chronic hypoxia. J Clin Invest 87: 155–162, 1991. doi: 10.1172/JCI114965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Archer SL, Huang JM, Hampl V, Nelson DP, Shultz PJ, Weir EK. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc Natl Acad Sci USA 91: 7583–7587, 1994. doi: 10.1073/pnas.91.16.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci USA 74: 3203–3207, 1977. doi: 10.1073/pnas.74.8.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aschner JL, Smith TK, Kovacs N, Pinheiro JM, Fuloria M. Mechanisms of bradykinin-mediated dilation in newborn piglet pulmonary conducting and resistance vessels. Am J Physiol Lung Cell Mol Physiol 283: L373–L382, 2002. doi: 10.1152/ajplung.00032.2002. [DOI] [PubMed] [Google Scholar]
- 5.Blood AB, Terry MH, Merritt TA, Papamatheakis DG, Blood Q, Ross JM, Power GG, Longo LD, Wilson SM. Effect of chronic perinatal hypoxia on the role of rho-kinase in pulmonary artery contraction in newborn lambs. Am J Physiol Regul Integr Comp Physiol 304: R136–R146, 2013. doi: 10.1152/ajpregu.00126.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blum-Johnston C, Thorpe RB, Wee C, Romero M, Brunelle A, Blood Q, Wilson R, Blood AB, Francis M, Taylor MS, Longo LD, Pearce WJ, Wilson SM. Developmental acceleration of bradykinin-dependent relaxation by prenatal chronic hypoxia impedes normal development after birth. Am J Physiol Lung Cell Mol Physiol 310: L271–L286, 2016. doi: 10.1152/ajplung.00340.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bradbury DA, Newton R, Zhu YM, Stocks J, Corbett L, Holland ED, Pang LH, Knox AJ. Effect of bradykinin, TGF-β1, IL-1β, and hypoxia on COX-2 expression in pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 283: L717–L725, 2002. doi: 10.1152/ajplung.00070.2002. [DOI] [PubMed] [Google Scholar]
- 8.Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science 256: 532–535, 1992. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- 9.Brenner R, Peréz GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature 407: 870–876, 2000. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- 10.Cassin S, Winikor I, Tod M, Philips J, Frisinger J, Jordan J, Gibbs C. Effects of prostacyclin on the fetal pulmonary circulation. Pediatr Pharmacol (New York) 1: 197–207, 1981. [PubMed] [Google Scholar]
- 11.Chakrabarti R, Ji WK, Stan RV, de Juan Sanz J, Ryan TA, Higgs HN. INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J Cell Biol 217: 251–268, 2018. doi: 10.1083/jcb.201709111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chand N, Mahoney TP Jr, Diamantis W, Sofia RD. Pharmacological modulation of bradykinin-, acetylcholine- and calcium ionophore A23187-induced relaxation of rabbit pulmonary arterial segments. Eur J Pharmacol 137: 173–177, 1987. doi: 10.1016/0014-2999(87)90219-6. [DOI] [PubMed] [Google Scholar]
- 13.Dudzinski DM, Michel T. Life history of eNOS: partners and pathways. Cardiovasc Res 75: 247–260, 2007. doi: 10.1016/j.cardiores.2007.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Francis M, Qian X, Charbel C, Ledoux J, Parker JC, Taylor MS. Automated region of interest analysis of dynamic Ca2+ signals in image sequences. Am J Physiol Cell Physiol 303: C236–C243, 2012. doi: 10.1152/ajpcell.00016.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fulton D, Fontana J, Sowa G, Gratton JP, Lin M, Li KX, Michell B, Kemp BE, Rodman D, Sessa WC. Localization of endothelial nitric-oxide synthase phosphorylated on serine 1179 and nitric oxide in Golgi and plasma membrane defines the existence of two pools of active enzyme. J Biol Chem 277: 4277–4284, 2002. doi: 10.1074/jbc.M106302200. [DOI] [PubMed] [Google Scholar]
- 16.Gao Y, Portugal AD, Negash S, Zhou W, Longo LD, Usha Raj J. Role of Rho kinases in PKG-mediated relaxation of pulmonary arteries of fetal lambs exposed to chronic high-altitude hypoxia. Am J Physiol Lung Cell Mol Physiol 292: L678–L684, 2007. doi: 10.1152/ajplung.00178.2006. [DOI] [PubMed] [Google Scholar]
- 17.Gao Y, Zhou H, Ibe BO, Raj JU. Prostaglandins E2 and I2 cause greater relaxations in pulmonary veins than in arteries of newborn lambs. J Appl Physiol (1985) 81: 2534–2539, 1996. doi: 10.1152/jappl.1996.81.6.2534. [DOI] [PubMed] [Google Scholar]
- 18.Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol Pharmacol 48: 184–188, 1995. [PubMed] [Google Scholar]
- 19.Ghosh S, Gupta M, Xu W, Mavrakis DA, Janocha AJ, Comhair SA, Haque MM, Stuehr DJ, Yu J, Polgar P, Naga Prasad SV, Erzurum SC. Phosphorylation inactivation of endothelial nitric oxide synthesis in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 310: L1199–L1205, 2016. doi: 10.1152/ajplung.00092.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med 333: 214–221, 1995. doi: 10.1056/NEJM199507273330403. [DOI] [PubMed] [Google Scholar]
- 21.Giang M, Papamatheakis DG, Nguyen D, Paez R, Blum Johnston C, Kim J, Brunnell A, Blood Q, Goyal R, Longo LD, Wilson SM. muscarinic receptor activation affects pulmonary artery contractility in sheep: the impact of maturation and chronic hypoxia on endothelium-dependent and endothelium-independent function. High Alt Med Biol 17: 122–132, 2016. doi: 10.1089/ham.2015.0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glasgow RE, Buga GM, Ignarro LJ, Chaudhuri G, Heymann MA. Endothelium-derived relaxing factor as a mediator of bradykinin-induced perinatal pulmonary vasodilatation in fetal sheep. Reprod Fertil Dev 9: 213–216, 1997. doi: 10.1071/R96084. [DOI] [PubMed] [Google Scholar]
- 23.Goyal R, Angermann JE, Ostrovskaya O, Buchholz JN, Smith GD, Wilson SM. Enhanced capacitative calcium entry and sarcoplasmic-reticulum calcium storage capacity with advanced age in murine mesenteric arterial smooth muscle cells. Exp Gerontol 44: 201–207, 2009. doi: 10.1016/j.exger.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goyal R, Creel KD, Chavis E, Smith GD, Longo LD, Wilson SM. Maturation of intracellular calcium homeostasis in sheep pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 295: L905–L914, 2008. doi: 10.1152/ajplung.00053.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greif DM, Sacks DB, Michel T. Calmodulin phosphorylation and modulation of endothelial nitric oxide synthase catalysis. Proc Natl Acad Sci USA 101: 1165–1170, 2004. doi: 10.1073/pnas.0306377101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hadley SR, Blood Q, Rubalcava M, Waskel E, Lumbard B, Le P, Longo LD, Buchholz JN, Wilson SM. Maternal high-altitude hypoxia and suppression of ryanodine receptor-mediated Ca2+ sparks in fetal sheep pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 303: L799–L813, 2012. doi: 10.1152/ajplung.00009.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hampl V, Herget J. Role of nitric oxide in the pathogenesis of chronic pulmonary hypertension. Physiol Rev 80: 1337–1372, 2000. doi: 10.1152/physrev.2000.80.4.1337. [DOI] [PubMed] [Google Scholar]
- 28.Hartzell CA, Jankowska KI, Burkhardt JK, Lewis RS. Calcium influx through CRAC channels controls actin organization and dynamics at the immune synapse. eLife 5: e14850, 2016. doi: 10.7554/eLife.14850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herrera GM, Nelson MT. Differential regulation of SK and BK channels by Ca(2+) signals from Ca(2+) channels and ryanodine receptors in guinea-pig urinary bladder myocytes. J Physiol 541: 483–492, 2002. doi: 10.1113/jphysiol.2002.017707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu XQ, Xiao D, Zhu R, Huang X, Yang S, Wilson SM, Zhang L. Chronic hypoxia suppresses pregnancy-induced upregulation of large-conductance Ca2+-activated K+ channel activity in uterine arteries. Hypertension 60: 214–222, 2012. doi: 10.1161/HYPERTENSIONAHA.112.196097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hughes JM, Riddle MA, Paffett ML, Gonzalez Bosc LV, Walker BR. Novel role of endothelial BKCa channels in altered vasoreactivity following hypoxia. Am J Physiol Heart Circ Physiol 299: H1439–H1450, 2010. doi: 10.1152/ajpheart.00124.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ignarro LJ, Harbison RG, Wood KS, Kadowitz PJ. Activation of purified soluble guanylate cyclase by endothelium-derived relaxing factor from intrapulmonary artery and vein: stimulation by acetylcholine, bradykinin and arachidonic acid. J Pharmacol Exp Ther 237: 893–900, 1986. [PubMed] [Google Scholar]
- 33.Iwatsuki N, Petersen OH. Action of tetraethylammonium on calcium-activated potassium channels in pig pancreatic acinar cells studied by patch-clamp single-channel and whole-cell current recording. J Membr Biol 86: 139–144, 1985. doi: 10.1007/BF01870780. [DOI] [PubMed] [Google Scholar]
- 34.Kamitomo M, Alonso JG, Okai T, Longo LD, Gilbert RD. Effects of long-term, high-altitude hypoxemia on ovine fetal cardiac output and blood flow distribution. Am J Obstet Gynecol 169: 701–707, 1993. doi: 10.1016/0002-9378(93)90646-Z. [DOI] [PubMed] [Google Scholar]
- 35.Ko EA, Han J, Jung ID, Park WS. Physiological roles of K+ channels in vascular smooth muscle cells. J Smooth Muscle Res 44: 65–81, 2008. [DOI] [PubMed] [Google Scholar]
- 36.León-Velarde F, Maggiorini M, Reeves JT, Aldashev A, Asmus I, Bernardi L, Ge RL, Hackett P, Kobayashi T, Moore LG, Penaloza D, Richalet JP, Roach R, Wu T, Vargas E, Zubieta-Castillo G, Zubieta-Calleja G. Consensus statement on chronic and subacute high altitude diseases. High Alt Med Biol 6: 147–157, 2005. doi: 10.1089/ham.2005.6.147. [DOI] [PubMed] [Google Scholar]
- 37.Lima B, Forrester MT, Hess DT, Stamler JS. S-nitrosylation in cardiovascular signaling. Circ Res 106: 633–646, 2010. doi: 10.1161/CIRCRESAHA.109.207381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu J, Gao Y, Negash S, Longo LD, Raj JU. Long-term effects of prenatal hypoxia on endothelium-dependent relaxation responses in pulmonary arteries of adult sheep. Am J Physiol Lung Cell Mol Physiol 296: L547–L554, 2009. doi: 10.1152/ajplung.90333.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Llorens O, Perez JJ, Palomer A, Mauleon D. Differential binding mode of diverse cyclooxygenase inhibitors. J Mol Graph Model 20: 359–371, 2002. doi: 10.1016/S1093-3263(01)00135-8. [DOI] [PubMed] [Google Scholar]
- 40.Lorca RA, Prabagaran M, England SK. Functional insights into modulation of BKCa channel activity to alter myometrial contractility. Front Physiol 5: 289, 2014. doi: 10.3389/fphys.2014.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marceau F. Kinin B1 receptors: a review. Immunopharmacology 30: 1–26, 1995. doi: 10.1016/0162-3109(95)00011-H. [DOI] [PubMed] [Google Scholar]
- 42.Meera P, Wallner M, Jiang Z, Toro L. A calcium switch for the functional coupling between alpha (hslo) and beta subunits (KV,Ca beta) of maxi K channels. FEBS Lett 382: 84–88, 1996. doi: 10.1016/0014-5793(96)00151-2. [DOI] [PubMed] [Google Scholar]
- 43.Moore LG, Niermeyer S, Zamudio S. Human adaptation to high altitude: regional and life-cycle perspectives. Am J Phys Anthropol 107, Suppl 27: 25–64, 1998. doi: 10.1002/(SICI)1096-8644(1998)107:27+<25::AID-AJPA3>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 44.Morand-Contant M, Anand-Srivastava MB, Couture R. Kinin B1 receptor upregulation by angiotensin II and endothelin-1 in rat vascular smooth muscle cells: receptors and mechanisms. Am J Physiol Heart Circ Physiol 299: H1625–H1632, 2010. doi: 10.1152/ajpheart.00735.2009. [DOI] [PubMed] [Google Scholar]
- 45.Mutchler SM, Straub AC. Compartmentalized nitric oxide signaling in the resistance vasculature. Nitric Oxide 49: 8–15, 2015. doi: 10.1016/j.niox.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohlmann P, Martínez MC, Schneider F, Stoclet JC, Andriantsitohaina R. Characterization of endothelium-derived relaxing factors released by bradykinin in human resistance arteries. Br J Pharmacol 121: 657–664, 1997. doi: 10.1038/sj.bjp.0701169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Palomer A, Cabré F, Pascual J, Campos J, Trujillo MA, Entrena A, Gallo MA, García L, Mauleón D, Espinosa A. Identification of novel cyclooxygenase-2 selective inhibitors using pharmacophore models. J Med Chem 45: 1402–1411, 2002. doi: 10.1021/jm010458r. [DOI] [PubMed] [Google Scholar]
- 48.Papamatheakis DG, Blood AB, Kim JH, Wilson SM. Antenatal hypoxia and pulmonary vascular function and remodeling. Curr Vasc Pharmacol 11: 616–640, 2013. doi: 10.2174/1570161111311050006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Papamatheakis DG, Patel JJ, Blood Q, Merritt TT, Longo LD, Wilson SM. Depolarization-dependent contraction increase after birth and preservation following long-term hypoxia in sheep pulmonary arteries. Pulm Circ 2: 41–53, 2012. doi: 10.4103/2045-8932.94832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peinado VI, París R, Ramírez J, Roca J, Rodriguez-Roisin R, Barberà JA. Expression of BK(Ca) channels in human pulmonary arteries: relationship with remodeling and hypoxic pulmonary vasoconstriction. Vascul Pharmacol 49: 178–184, 2008. doi: 10.1016/j.vph.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 51.Plüger S, Faulhaber J, Fürstenau M, Löhn M, Waldschütz R, Gollasch M, Haller H, Luft FC, Ehmke H, Pongs O. Mice with disrupted BK channel beta1 subunit gene feature abnormal Ca(2+) spark/STOC coupling and elevated blood pressure. Circ Res 87: E53–E60, 2000. doi: 10.1161/01.RES.87.11.e53. [DOI] [PubMed] [Google Scholar]
- 52.Porter VA, Rhodes MT, Reeve HL, Cornfield DN. Oxygen-induced fetal pulmonary vasodilation is mediated by intracellular calcium activation of KCa channels. Am J Physiol Lung Cell Mol Physiol 281: L1379–L1385, 2001. doi: 10.1152/ajplung.2001.281.6.L1379. [DOI] [PubMed] [Google Scholar]
- 53.Quinn S, O’Brien C, McLoughlin P. Role of cyclooxygenase and haemoxygenase products in nitric oxide-independent vasodilatation in the porcine ciliary artery. Eye (Lond) 17: 628–636, 2003. doi: 10.1038/sj.eye.6700437. [DOI] [PubMed] [Google Scholar]
- 54.Rees DD, Palmer RM, Schulz R, Hodson HF, Moncada S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br J Pharmacol 101: 746–752, 1990. doi: 10.1111/j.1476-5381.1990.tb14151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Regoli D. Neurohumoral regulation of precapillary vessels: the kallikrein-kinin system. J Cardiovasc Pharmacol 6, Suppl 2: S401–S412, 1984. doi: 10.1097/00005344-198406002-00015. [DOI] [PubMed] [Google Scholar]
- 56.Resnik E, Herron J, Fu R, Ivy DD, Cornfield DN. Oxygen tension modulates the expression of pulmonary vascular BKCa channel α- and β-subunits. Am J Physiol Lung Cell Mol Physiol 290: L761–L768, 2006. doi: 10.1152/ajplung.00283.2005. [DOI] [PubMed] [Google Scholar]
- 57.Rhodes MT, Porter VA, Saqueton CB, Herron JM, Resnik ER, Cornfield DN. Pulmonary vascular response to normoxia and KCa channel activity is developmentally regulated. Am J Physiol Lung Cell Mol Physiol 280: L1250–L1257, 2001. doi: 10.1152/ajplung.2001.280.6.L1250. [DOI] [PubMed] [Google Scholar]
- 58.Riddle MA, Hughes JM, Walker BR. Role of caveolin-1 in endothelial BKCa channel regulation of vasoreactivity. Am J Physiol Cell Physiol 301: C1404–C1414, 2011. doi: 10.1152/ajpcell.00013.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robertson BE, Schubert R, Hescheler J, Nelson MT. cGMP-dependent protein kinase activates Ca-activated K channels in cerebral artery smooth muscle cells. Am J Physiol Cell Physiol 265: C299–C303, 1993. doi: 10.1152/ajpcell.1993.265.1.C299. [DOI] [PubMed] [Google Scholar]
- 60.Saavedra JE, Southan GJ, Davies KM, Lundell A, Markou C, Hanson SR, Adrie C, Hurford WE, Zapol WM, Keefer LK. Localizing antithrombotic and vasodilatory activity with a novel, ultrafast nitric oxide donor. J Med Chem 39: 4361–4365, 1996. doi: 10.1021/jm960616s. [DOI] [PubMed] [Google Scholar]
- 61.Sadoshima J, Akaike N, Kanaide H, Nakamura M. Cyclic AMP modulates Ca-activated K channel in cultured smooth muscle cells of rat aortas. Am J Physiol Heart Circ Physioll 255: H754–H759, 1988. doi: 10.1152/ajpheart.1988.255.4.H754. [DOI] [PubMed] [Google Scholar]
- 62.Sage SO, Adams DJ, van Breemen C. Synchronized oscillations in cytoplasmic free calcium concentration in confluent bradykinin-stimulated bovine pulmonary artery endothelial cell monolayers. J Biol Chem 264: 6–9, 1989. [PubMed] [Google Scholar]
- 63.Sartori C, Allemann Y, Scherrer U. Pathogenesis of pulmonary edema: learning from high-altitude pulmonary edema. Respir Physiol Neurobiol 159: 338–349, 2007. doi: 10.1016/j.resp.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 64.Takahashi S, Mendelsohn ME. Synergistic activation of endothelial nitric-oxide synthase (eNOS) by HSP90 and Akt: calcium-independent eNOS activation involves formation of an HSP90-Akt-CaM-bound eNOS complex. J Biol Chem 278: 30821–30827, 2003. doi: 10.1074/jbc.M304471200. [DOI] [PubMed] [Google Scholar]
- 65.Thorpe RB, Stockman SL, Williams JM, Lincoln TM, Pearce WJ. Hypoxic depression of PKG-mediated inhibition of serotonergic contraction in ovine carotid arteries. Am J Physiol Regul Integr Comp Physiol 304: R734–R743, 2013. doi: 10.1152/ajpregu.00212.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thorpe RB, Williams JM, Pearce WJ. Chronic hypoxia downregulates colocalization of PKG with its target BK channel proteins in ovine cerebral arteries. FASEB J 30: 985.982, 2016. [Google Scholar]
- 67.Toro L, Li M, Zhang Z, Singh H, Wu Y, Stefani E. MaxiK channel and cell signalling. Pflugers Arch 466: 875–886, 2014. doi: 10.1007/s00424-013-1359-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tracey A, Bunton D, Irvine J, MacDonald A, Shaw AM. Relaxation to bradykinin in bovine pulmonary supernumerary arteries can be mediated by both a nitric oxide-dependent and -independent mechanism. Br J Pharmacol 137: 538–544, 2002. doi: 10.1038/sj.bjp.0704890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tucker A, Rhodes J. Role of vascular smooth muscle in the development of high altitude pulmonary hypertension: an interspecies evaluation. High Alt Med Biol 2: 173–189, 2001. doi: 10.1089/152702901750265288. [DOI] [PubMed] [Google Scholar]
- 70.Tulloh RM, Hislop AA, Boels PJ, Deutsch J, Haworth SG. Chronic hypoxia inhibits postnatal maturation of porcine intrapulmonary artery relaxation. Am J Physiol Heart Circ Physiol 272: H2436–H2445, 1997. doi: 10.1152/ajpheart.1997.272.5.H2436. [DOI] [PubMed] [Google Scholar]
- 71.Vang A, Mazer J, Casserly B, Choudhary G. Activation of endothelial BKCa channels causes pulmonary vasodilation. Vascul Pharmacol 53: 122–129, 2010. doi: 10.1016/j.vph.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 72.Williams JM, Pearce WJ. Age-dependent modulation of endothelium-dependent vasodilatation by chronic hypoxia in ovine cranial arteries. J Appl Physiol (1985) 100: 225–232, 2006. doi: 10.1152/japplphysiol.00221.2005. [DOI] [PubMed] [Google Scholar]
- 73.Xu W, Liu Y, Wang S, McDonald T, Van Eyk JE, Sidor A, O’Rourke B. Cytoprotective role of Ca2+- activated K+ channels in the cardiac inner mitochondrial membrane. Science 298: 1029–1033, 2002. doi: 10.1126/science.1074360. [DOI] [PubMed] [Google Scholar]
- 74.Zhang R, Sun H, Liao C, Yang H, Zhao B, Tian J, Dong S, Zhang Z, Jiao J. Chronic hypoxia in cultured human podocytes inhibits BKCa channels by upregulating its β4-subunit. Biochem Biophys Res Commun 420: 505–510, 2012. doi: 10.1016/j.bbrc.2012.03.021. [DOI] [PubMed] [Google Scholar]
- 75.Zhu S, Browning DD, White RE, Fulton D, Barman SA. Mutation of protein kinase C phosphorylation site S1076 on α-subunits affects BKCa channel activity in HEK-293 cells. Am J Physiol Lung Cell Mol Physiol 297: L758–L766, 2009. doi: 10.1152/ajplung.90518.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]