

Abstract
The aggregation cascade of disease‐related amyloidogenic proteins, terminating in insoluble amyloid fibrils, involves intermediate oligomeric states. The structural and biochemical details of these oligomers have been largely unknown. Here we report crystal structures of variants of the cytotoxic oligomer‐forming segment residues 28–38 of the ALS‐linked protein, SOD1. The crystal structures reveal three different architectures: corkscrew oligomeric structure, nontwisting curved sheet structure and a steric zipper proto‐filament structure. Our work highlights the polymorphism of the segment 28–38 of SOD1 and identifies the molecular features of amyloidogenic entities.
Keywords: amyloid fibril, oligomer, SOD1, ALS, X‐ray crystallography
Short abstract
PDB Code(s): http://firstglance.jmol.org/fg.htm?mol=5WOR, http://firstglance.jmol.org/fg.htm?mol=6B79, http://firstglance.jmol.org/fg.htm?mol=5WMJ;
Abbreviations
- ALS
amyotrophic lateral sclerosis
- REU
Rosetta energy units
- Sc
shape complementarity
- SOD1
superoxide dismutase 1
- TFA
trifluoroacetic acid
Introduction
Amyloid aggregation is implicated in numerous neurodegenerative and systemic diseases1 and many proteins assemble into amyloids to perform essential biological functions.2, 3 Among the various amyloid aggregates believed to form, fibrillar species are the most readily studied, being both stable and having characteristic properties including the cross‐β X‐ray diffraction pattern, binding to specific dyes and birefringence upon binding to Congo Red. The cross‐β diffraction pattern suggests that amyloid fibrils are composed of packed β‐sheets. Indeed, the crystal structures of amyloid‐forming short peptides have revealed a β‐sheet architecture termed steric zipper.4, 5 Their structural and thermodynamic stability suggests that amyloid fibrils represent the energetic end state of the aggregation pathway.
Oligomeric species that evolve prior to fiber deposition are not easily characterized. In animals, the amyloid aggregation cascade of proteins is thought to evolve slowly on a time scale of decades and potentially has multiple intermediate states. Detailed investigation of these intermediates has been challenging, because these states are transient and often heterogeneous. Further, multiple segments in any protein may act synergistically or independently to form different intermediate structures. Some may be precursors of fibrils; others may be off the direct pathway to fibrils, leading to less stable structures that reverse to monomers.6 Thus, the identification of these intermediate states and characterizations of their cytotoxic roles have been limited.
Studies over the last two decades have presented evidence that small oligomers that form transiently in the aggregation pathway exacerbate disease progression by exerting toxicity in neuronal cells.7, 8 This toxic oligomer hypothesis suggests that small oligomers that form either on or off pathway to mature fibrils are the cytotoxic agents.7, 9 The failure of fiber‐inhibiting therapeutics in clinical trials and the low cytotoxicity of amyloid fibers in various model systems suggest that small oligomers may be the toxic intermediate state in at least some amyloid diseases. It is generally accepted that amyloid oligomers are β‐sheet rich, cytotoxic, and bind conformational antibodies such as A11. Structural studies have revealed antiparallel, β‐sheet rich oligomeric structures of proteins such as amyloid‐β, α‐B crystallin, and SOD1.6, 10, 11, 12
ALS is a fatal neurodegenerative disease caused by degeneration of motor neurons that results in paralysis. SOD1, a ubiquitous cytosolic protein is found in insoluble aggregates in a subset of familial ALS patients.13 SOD1 is an exceptionally stable protein, the aggregation of which has been attributed to structural perturbations induced by the familial mutations or oxidative damage accumulated over time.14, 15, 16, 17 Recently, we discovered that SOD1 forms cytotoxic soluble oligomers with the segment 28–38 of SOD1 as the oligomeric core.12 The crystal structure of the segment revealed a twisted β‐sheet termed corkscrew. The segment is cytotoxic to cultured motor neurons and substitutions at Gly33 with bulky residues alleviate toxicity by disrupting oligomer formation. Here we report three new crystal structures of variants of this segment that reveal possible aggregation states. Biochemical characterization of these aggregation states helps differentiate the molecular features of oligomeric and fibrillar structures.
Results
Segment 28–38 of SOD1 has a high aggregation propensity
An important question raised by the discovery of corkscrew is whether it is the only toxic conformation or if the segment forms more than one toxic conformation. Additionally, the presence of familial mutations in the segment led us to investigate the effect of these mutations. Notably, the segment has a lower frequency of familial mutations than other segments of SOD1 and among these mutations only three amino acid substitutions (Ala, Arg, and Val) have been found to date (http://alsod.iop.kcl.ac.uk). Among the various familial mutations found in this segment: Ala substitutes for V29 and V31 and Arg and Val substitute for G37 and L38, we hypothesized that G37R would have a potentially large effect on the aggregation kinetics of this segment due to the change from a small neutral side chain to a large, charged side chain [Fig. 1(A), left]. The pathological mutation, G37R has been previously characterized extensively and shown to increase the aggregation propensity of the full‐length protein leading to rapid disease onset and progression in transgenic mice models.16 We also analyzed the fiber‐forming propensity of the segment using ZipperDB,18 an algorithm that predicts the propensity of six residue segments to form amyloid fibrils [Fig. 1(A), right]. ZipperDB showed two aggregation‐prone sub‐segments—31‐KVWGSI‐36 and 33‐GSIKGL‐38. Thus, we set out to characterize the G37R mutant of the 11‐residue corkscrew as well as the shorter 6‐residue sub‐segments.
Figure 1.
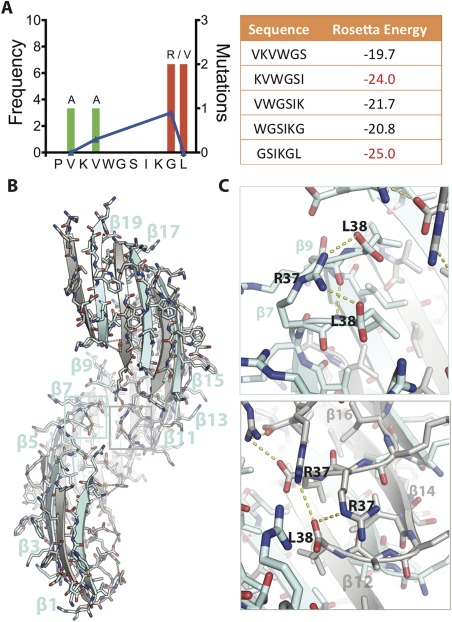
Crystal structure of SOD1 segment harboring a familial mutation, G37R. (A) (Left) Frequency of ALS‐associated mutations in SOD‐1 residues 28–38. Data from http://alsod.iop.kcl.ac.uk. (Right) Computed energies of steric zippers formed by six‐residue segments within segment 28–38 by ZipperDB.18 Segments KVWGSI and GSIKGL have energies of −23 kcal/mol or lower and are predicted to form fibrils. (B) 2.9 Å crystal structure of the toxic oligomeric core of SOD1, residues 28–38 with the familial mutation G37R, reveals a corkscrew assembly of antiparallel β‐strands with a hydrophobic cleft composed of valine and isoleucine residues. Twenty strands form one complete turn with a pitch length of 59 Å and diameter of 38 Å. (C) Arg37 forms multiple inter‐ and intra‐chain interactions that increase the stability of the assembly. (Top) Shown here Arg37 of β7 forms an intra‐chain hydrogen bond with Leu38 and an inter‐chain hydrogen bond with Leu38 of β9. (Bottom) Arg37 also forms inter‐chain hydrogen bonds. Shown here Arg37 of β14 is engaged in hydrogen bonds with Leu38 of β12 and Arg37 of β16. Together these interactions lead to 18 additional hydrogen bonds per turn.
Crystal structure of segment 28–38 with familial mutation G37R reveals a stable corkscrew geometry
We obtained crystals of the segment 28–38 harboring the familial mutation G37R (i.e., the tenth residue in the sequence KVKVWGSIKRL). The peptide subjected to crystallization trials has an additional residue change of P28K needed to increase solubility of the segment. This substitution has been shown previously to contribute minimally to the structure and cytotoxic properties.12 The structure was determined by molecular replacement using the native corkscrew as a search model (sequence KVKVWGSIKGL and PDB ID 5DLI) (Table 1). The structure reveals a twisted β‐sheet with the same antiparallel, out‐of‐register β‐strand construction as the wild‐type corkscrew [Fig. 1(B)]; however, G37R has more strands per full turn of the corkscrew (20 vs. 16), a shorter pitch (59 vs. 71 Å) and larger diameter (38 vs. 25 Å). The shorter pitch is primarily due to R37. The charged side chain of R37 forms hydrogen bonds with the C‐terminal carboxylate (L38) of alternate strands or of the same strand; adding 18 new hydrogen bonds per turn [Fig. 1(C)]. These interactions are not possible with the native Gly residue and support the shorter pitch. An overlay of a single β‐strand of the mutant structure determined here and the corkscrew confirms that the peptide backbone is similar except for deviations near the C‐terminus (Fig. S1, Supporting Information). The overall similarity of the mutant structure to the corkscrew suggests that the segment has a high propensity to form antiparallel, out‐of‐register assemblies although single point substitutions can induce subtle changes in the overall architecture.
Untwisting of the corkscrew segment 28–38 with familial mutation G37R in the presence of orange G
In our efforts to obtain phases for the mutant segment; we crystallized a derivative with a bromo‐allyl valine (B) at position 2 (sequence KBKVWGSIKRL). We obtained crystals in the presence of orange G, a small aromatic dye previously shown to modulate amyloid formation of Alzheimer's disease‐associated proteins like amyloid‐β.19, 20 The crystals displayed a needle‐like morphology distinctly different from the crystals of the mutant peptide KVKVWGSIKRL that had thin plate‐like morphology. The crystals diffracted to 1.8 Å and phases were obtained by molecular replacement using the corkscrew structure as a search model. The structure revealed a curved β‐sheet with the same antiparallel, out‐of‐register β‐sheet construction as the wild‐type and G37R corkscrews, but surprisingly no twist [Fig. 2(A)].
Figure 2.
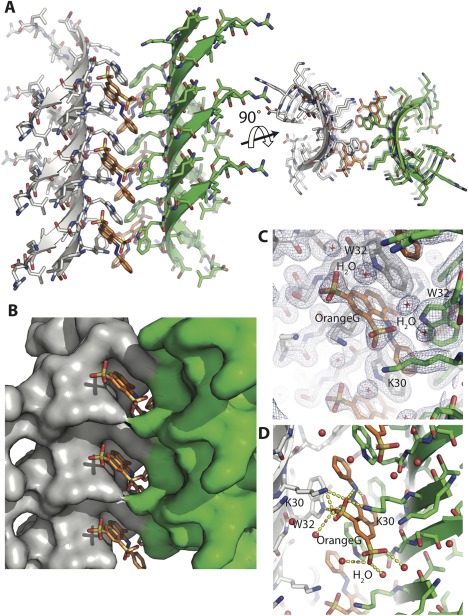
Out‐of‐register mating sheet architecture formed by the segment SOD1 28–38 with the familial mutation G37R. (A) Crystal structure of the segment 28–38 with familial mutation G37R at 1.8 Å resolution reveals a novel architecture of highly curved sheets mating via stacking of aromatic side chains of W32 residue. The curved architecture enables both faces of the sheets to exclude water molecules as shown here in views perpendicular (left) and parallel (right) to the fibril axis. (B) The weak interface composed of W32 stacking forms a pocket in which small molecules can bind. Orange G (shown in orange sticks) was cocrystallized and found to bind in the pocket. (C) Electron density map contoured at 1.0 σ showing clear density for orange G. (D) Orange G stabilizes the assembly by forming a network of hydrogen bonds with K30 and W32 of one sheet and K30 of the opposing sheet. Water molecules are colored red.
As with both twisted corkscrews, the curvature of the sheet is stabilized by short hydrophobic side chains (valine and isoleucine) pointing into the concave interior and charged and bulky residues pointing outward from the convex exterior; however, all twist is eliminated apparently due to orange G‐mediated contacts between neighboring sheets. Stacked aromatic side chains of W32 intercalate with the aromatic rings of orange G [Fig. 2(B)]. The aromatic rings of orange G stack against the W32 aromatic side chains and the sulfate moieties of orange G form salt bridges with K30 of both sheets [Fig. 2(C, D)]. Together, these interactions result in high values for both computed orange G binding energy (–12.4 kcal/mol) and shape complementarity (0.82). Previously, orange G had been observed bound to a steric zipper composed of a 6‐residue segment of amyloid‐β.21 In that structure aromatic rings of orange G stacked with similar aromatic side chains but the molecule wedged in between the mating β‐sheets. In the structure determined here, orange G fills the cavities in the sides providing additional support to the mating sheets.
Crystal structure of the segment 30‐KVWGSI‐35 reveals a steric zipper
We also obtained diffraction quality crystals of the sub‐segment, 30‐KVWGSI‐35, predicted by ZipperDB to form a steric zipper [Fig 1(A), right]. The 6‐residue segment forms a steric zipper composed of in‐register, antiparallel β‐sheets that are arranged face‐to‐back. It is a Class 7 zipper.22 Notably, the side chains of neighboring sheets do not pack as tightly as most pathogenic steric zippers, due to the conflicting sizes and properties of the side chains in the interface (shape complementarity = 0.64) (Table 2); a void between sheets is filled adventitiously by a trifluoroacetic acid (TFA) molecule. The side chain of W32 extends along the zipper axis and stacks against the backbone of G33 of the strand above it in the sheet [Fig. 3(B)] and results in staggering of the sheets relative to each other [Fig. 3(C)]. Each β‐strand forms six main chain hydrogen bonds with the β‐strands above and below resulting in a uniform pattern of hydrogen bonds.
Figure 3.
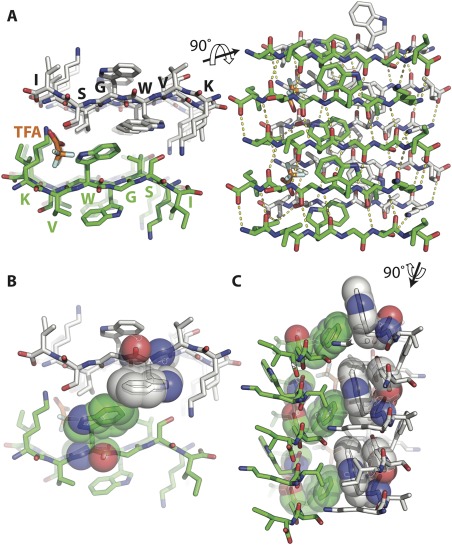
Segment 30‐KVWGSI‐35 of SOD1 forms a steric zipper assembly. (A) The 1.45 Å resolution structure of segment KVWGSI shows two β‐sheets composed of antiparallel β‐strands forming a class 7 steric zipper via face to back stacking. Shown here in views parallel (left) and perpendicular (right) to the fibril axis. Trifluoroacetic acid (TFA) is colored orange. (B) The two sheets do not pack tightly due to the bulky tryptophan side chain in the inner interface. (C) In most steric zippers, side chains are perpendicular to the protofilament axis but here, the W32 side chain protrudes along the fiber axis and aligns with the G33 of the β strand above it as shown in sphere representation. This results in staggering of the sheets relative to each other.
Figure 4.
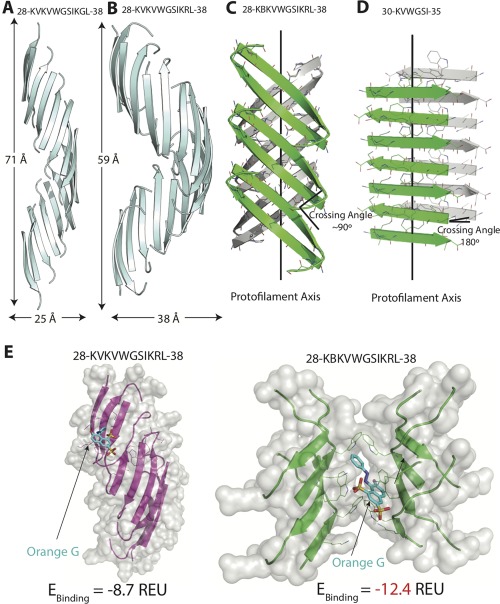
Structural comparison of SOD1 segment 28–38 and its variants. (A) The native 28–38 segment (PDB ID: 5DLI) forms a left‐handed corkscrew‐like assembly of antiparallel, out‐of‐register β‐strands. Sixteen strands form one complete turn with 71 Å pitch and 25 Å diameter. (B) The mutant segment with a familial mutation, G37R forms a corkscrew‐like assembly but with a smaller pitch and larger diameter. Twenty strands form one complete turn with 59 Å pitch and 38 Å diameter. (C) A second form of the variant segment reveals a novel architecture of curved out‐of‐register sheets. The β‐strands are not perpendicular to fibril axis and instead are tilted with a crossing angle of ∼90°. (D) A shorter (6‐residue) segment within 28–38 forms a steric zipper composed of pairs of β‐sheets. The β‐strands are perpendicular to the fibril axis with a 180° crossing angle. (E) Docking simulations of the different structures determined here and the small molecule dye orange G. Computed binding energy is reported in Rosetta Energy Units (REU). Notice that orange G has lower binding energy to curved β‐sheet than to corkscrew single sheet.
Variant peptides show distinct fiber diffraction patterns
We performed diffraction experiments on aligned, insoluble species of native corkscrew‐forming peptide (KVKVWGSIKGL and PDB ID 5DLI) as well as the familial mutant crystallized here (KVKVWGSIKRL and KBKVWGSIKRL). Residues 28–38 and G37R mutant peptides produce a distinct diffraction ring at 4.6 Å (Fig. 5). As shown in the radial profiles, no strong reflections are observed at 10 Å indicating a β‐sheet rich structure that is devoid of mated pairs of sheets. Interestingly, the segment forming untwisted structure (KBKVWGSIKRL) gives a distinct different diffraction pattern with multiple rings notably at 4.75 and 9.5 Å. We confirmed that the 9.5 Å reflection is not due to the tris salt by collecting diffraction patterns of the salt (Fig. S2, Supporting Information). However, it is possible that the 9.5 Å reflection is due to an unidentified contaminant. We also collected the diffraction patterns after shaking (which presumably creates fibrils) and found that the native 28–38 segment shows rings at 4.6 and 11.9 Å. The segment KBKVWGSIKRL shows diffraction at 4.75, 9.5, and 13.1 Å indicative of a mixture of different β‐sheet structures. The absence of reflections near at 10 Å in the diffraction pattern of corkscrew suggests that it is the predominant oligomeric intermediate state.
Figure 5.
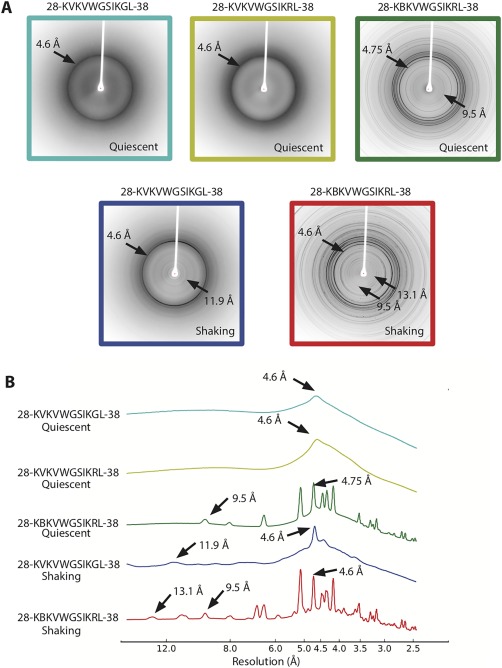
Comparison of diffraction patterns of the different variant segments. (A) Top: 28–38 and G37R mutant segments show diffuse diffraction rings at 4.6 Å. The G37R segment forming out‐of‐register pairs of sheets reveals sharp diffraction rings at 4.75 and 9.5 Å. Bottom: The segment 28–38 under shaking conditions reveals diffraction rings consistent with cross‐β structure, indicative of pairs of sheets. (B) Radial profiles of the diffraction patterns with peaks at or around 4 and 11 Å labeled.
Discussion
Building on our discovery of segment 28–38 as the toxic oligomeric core of SOD1, here we further characterize the segment by determining three atomic structures of its variants and characterizing their biochemical properties. The new structures we determined reveal three additional aggregation states: in addition to the original corkscrew, we find (1) a soluble corkscrew oligomer stabilized by additional hydrogen bonds and altered pitch, constructed from an antiparallel, out‐of‐register β‐sheet; (2) an untwisted antiparallel, out‐of‐register mating β‐sheet; and (3) a steric zipper representing the fibrillar β‐sheet state (Figure 4).
The crystal structure of the G37R mutant segment provides a molecular explanation for the enhanced toxicity associated with this pathological mutant. G37R was one of the first mutations found in familial ALS patients.13 In transgenic mouse models, overexpression of the G37R mutant protein leads to rapid disease onset and progression16 and in cell culture models it has been shown to increase oligomer formation and toxicity.23 The atomic structure of the 28–38 segment with the mutation G37R forms a corkscrew assembly composed of antiparallel out‐of‐register β‐strands. The change from a small glycine residue to a large arginine residue causes 18 new hydrogen bonds per turn stabilizing the corkscrew assembly. This stabilization of the corkscrew oligomers could explain the increased toxicity of the G37R mutant protein in animal and cell culture model systems.
The formation of corkscrew by the wild‐type and G37R mutant SOD1 segment 28–38 suggests that the corkscrew architecture is robust and may accommodate a range of other sequences with the same pattern. That is, structural homologs might alternate small hydrophobic residues with bulky or like‐charged residues, forming the concave and convex surfaces of the corkscrew. There is evidence for antiparallel β‐sheet rich oligomers by other amyloid proteins although the structures of these toxic oligomers are not known.24, 25 It is possible that the corkscrew architecture is a common oligomeric state of amyloid proteins and may be involved in pathology of other neurological amyloid diseases. The so‐far undefined stoichiometry of corkscrew oligomers may be consistent with a variety of corkscrew‐like oligomers.
The structure of the G37R variant segment, KBKVWGSIKRL suggests out‐of‐register β‐sheets may be a common architecture of amyloid proteins. Instead of the extended β‐sheet conformation seen in steric zippers and corkscrew, here the strands are curved forming a dry interface on both faces of the strands. This structure belongs to the growing class of out‐of‐register steric zippers.6, 10, 26 KDWSFY, a short segment of β2‐microglobulin and NFGAILS, a short segment of islet amyloid polypeptide (IAPP) were previously reported to form out‐of‐register sheet structures.6, 26 An out‐of‐register sheet structure of a 11‐residue segment of IAPP was recently determined.27 Notably, the IAPP structure was composed of a single sheet and is nontoxic. The structure was found to be labile to thermal denaturation suggesting that it is the mating of pairs of sheets that imparts stability to amyloid. The 11‐residue structure reported here is composed of pairs of sheets and is to date the longest segment crystallized in an out‐of‐register mating sheet conformation. Similar to the structure of KDWSFY, the strands of the two mating sheets of the zipper are not parallel to each other but form a crossing angle of ∼90° and each β‐strand forms nine hydrogen bonds with one β‐strand and seven with another. This network of alternating weak and strong interfaces has been seen with all out‐of‐register structures determined thus far. Previously, we suggested that out‐of‐register assemblies are off pathway to in‐register fibrils.6, 10 The out‐of‐register KBKVWGSIKRL fibril structure presumably represents the end of a pathway distinct from that leading to in‐register fibrils.
Steric zipper structure of the segment KVWGSI suggests a weak role of this segment in full‐length SOD1 fibrillation. Structural studies of SOD1 aggregates have been limited to structures of full‐length mutants with or without metals bound,28, 29, 30, 31 which suggest that most mutations destabilize the native structure. However, these studies do not illustrate the structure of pathogenic SOD1 aggregates. Previously, we obtained crystal structures of short segments from the C‐terminus of the protein.32 We also showed that mutations in the segment 28–38 (G33 or I35) do not disrupt fiber formation although G33 substitutions alleviate toxicity by disrupting oligomer formation.12, 32 The structure of KVWGSI determined here shows a steric zipper with poor shape complementarity (Sc = 0.64) compared to segments in the C‐terminus; GVIGIAQ (Sc = 0.84) and DSVISLS (Sc = 0.77). Taken together, our work suggests that the C‐terminus forms the fibril spine of SOD1 aggregates while the segment 28–38 forms the toxic oligomeric core.
Our fiber diffraction studies provide clues of the molecular architecture of the oligomer and fiber conformations in solution. Diffraction of the native corkscrew forming segment KVKVWGSIKGL and the G37R mutant KVKVWGSIKRL showed similar fiber diffraction patterns with a diffuse ring at 4.6 Å. The absence of strong reflections at 10 Å suggests single sheet architecture of the oligomer, different from the classical amyloid cross‐β diffraction, which shows reflections at 4.8 and 10.4 Å. Indeed, under shaking conditions (which would promote robust fibril growth) the diffraction pattern of KVKVWGSIKGL showed diffraction at 4.6 and 11.9 Å indicative of pairs of β‐sheets.
Knowledge of the atomic structures of amyloid oligomers offers the possibility of designed inhibitors of toxicity. To date rational design of small molecule modulators of aggregation have been limited.33, 34, 35 Congo‐red, thioflavin T, curcumin, and EGCG and their derivatives have been shown to modulate both fibril and oligomer formation and can be used as probes for detecting amyloid deposits,36, 37, 38, 39 but the exact mechanism of action of these small molecules has not been discovered. Cocrystal structures of orange G with amyloid‐forming segments of amyloid‐β and tau and of curcumin with a segment from Tau have been obtained.21 In all these structures, the small molecules are arranged in a similar orientation; their long axes parallel to the fiber axis, charged groups engaged in multiple hydrogen bonds with charged side chains such as lysine and aromatic rings forming pi stacking with aromatic side chains.
The structure of out‐of‐register fibril of SOD1 determined here provides the atomic information for developing small molecule modulators of SOD1 aggregation. Docking simulations suggest that orange G has a high propensity to bind the mating sheet interface (binding energy = −12.4 REU) compared to the twisted single sheet corkscrew structure (binding energy = −8.7 REU). Based on the structure and our computational energy predictions, we hypothesize that binding of orange G stabilizes the curved mating sheet architecture and may drive the equilibrium from cytotoxic corkscrew oligomers. Although we cannot rule out if orange G stabilizes other nontoxic intermediates, this structure suggests a potential mechanism of rescue of cytotoxicity by small molecules. We speculate that small molecules designed to stabilize fibrillar mating sheet architectures can alleviate cytotoxicity of aggregated proteins. Our lab has used this strategy to reduce the toxicity of amyloid‐β, a protein associated with Alzheimer's disease.33 Our work here offers the possibility of designing similar molecules against SOD1 cytotoxicity.
In summary, our structural studies of the SOD1 segment of residues 28–38 suggest that oligomers of amyloid proteins are composed of single twisted sheets whereas fibrillar species are composed of mated β‐sheets.
Materials and Methods
Crystallization
All peptides were commercially obtained from Genscript with more than 98% purity. All data were collected at the Advanced Photon Source (Chicago, IL) on beamline 24‐ID‐E and data were processed using DENZO and SCALEPACK or XDS.40 Structures were built using COOT.41 Model refinement was performed using REFMAC42 and BUSTER.43 Figures were generated using PyMOL.44
28‐KVKVWGSIKRL38: Crystals of segment 28–38 with P28K and G37R substitution were grown by hanging drop vapor diffusion using VDX plates (Hampton Research, Aliso, Viejo, CA). Lyophilized peptide was dissolved to 50 mg/mL in 50 mM Tris‐base buffer, 1 mM Orange G. The reservoir solution contained 0.3M sodium malonate and 15% PEG 3350. Crystallization drops were prepared by mixing peptide with reservoir in a 3:1 ratio, in a total volume of 4 μL. The crystals were subsequently mounted with CrystalCap HT Cryoloops (Hampton Research, Aliso Viejo, CA) and flash frozen in liquid nitrogen. Four strands composed of the core of the corkscrew structure PDB ID: 5DLI, residues VKVWGSI were used as the initial model for molecular replacement.
28‐KBKVWGSIKRL‐38: Lyophilized peptide (KBKVWGSIKRL, B represents the non‐natural amino acid bromo‐allyl valine) was dissolved to 50 mg/mL in 50 mM Tris‐base buffer with 1 mM Orange G. The reservoir solution contained 0.1M HEPES pH 7.5, 0.2M sodium citrate and 15% MPD. Needle‐like crystals appeared overnight and were directly flash frozen from the original screening plate.
30‐KVWGSI‐35: Lyophilized peptide was dissolved to 50 mg/mL in water and filtered by 0.1 μm filter. Crystals were grown by hanging drop vapor diffusion using VDX plates (Hampton Research, Aliso, Viejo, CA). Crystallization drops were prepared by mixing peptide with reservoir in a 2:1 ratio, in a total volume of 3 μL. The reservoir solution contained 4M sodium formate. Crystals were mounted on pulled glass capillaries without any cryoprotectant. A pair of antiparallel β‐strands was used as an initial model for molecular replacement.
Fiber diffraction
Peptide segments were dissolved in 50 mM tris‐base buffer at 25 mg/mL and incubated at 37°C without agitation to form oligomers for four days. For fibril preparations, the peptide segments were incubated at 37°C in Torrey Pine shakers for four days. Samples were mounted by pipetting 2 μL drops between glass rods and dried overnight. Diffraction data was collected on a Rigaku FR‐E generator with Raxis imaging plate. The crystal to specimen distance was 200 mm.
Ligand docking
Three‐dimensional structures of the ligands used for docking simulation were downloaded from the PubChem Compound Database (CID = 24181578).45 We generated the ligand perturbation ensemble using the method described previously.33 A torsion angle deviation of ±5° for each rotatable bond of the ligand was applied to generate 100 conformations for each ligand. Ligand docking was performed using the HighResDocker protocol in Rosetta version 3.5, using the talaris2014 energy function.46 Docking poses were sampled in a 7 Å box centered at the known binding site of Orange G to the variant form 1, or to the corresponding residues in the corkscrew structure. Small ligand perturbations were coupled with cycles of side chain repacking, in which rotamers of side chains around the ligand are optimized in a Monte‐Carlo minimization algorithm. Ligand poses were ranked based on lowest binding energy.
Table 1.
Data Collection and Refinement Statistics
| KVKVWGSIKRL (Form 1) | KBKVWGSIKRL (Form 2) | KVWGSI | |
|---|---|---|---|
| Beam line | APS 24‐ID‐E | APS 24‐ID‐E | APS 24‐ID‐E |
| Resolution (Å) | 2.9 | 1.80 | 1.45 |
| Total unique reflections | 5444 | 2330 | 1738 |
| Total reflections observed | 27,350 | 7194 | 11,632 |
| Space group | P3121 | C2 | P212121 |
| R sym | 18.3% (90.7%) | 21.1% (197.9%) | 16.6% (55.0%) |
| I/σ | 8.9 (1.0) | 4.06 (0.48) | 8.6 (1.6) |
| Completeness | 98.9% | 96.6% | 89.6% |
| Wavelength (Å) | 0.9791 | 0.9791 | 0.9791 |
| Unit cell dimensions | |||
| a b c (Å) | 59.61 59.61 98.92 | 56.94 11.64 44.93 | 9.51 20.28 44.24 |
| α β γ (°) | 90.0 90.0 120.0 | 90.0 127.3 90.0 | 90.0 90.0 90.0 |
| Refinement | |||
| Resolution (Å) | 45.8–2.8 | 35.7–1.8 | 22.1–1.4 |
| Reflections for refinement | 5422 | 2327 | 1564 |
| R free/R work (%) | 25.0/21.3 | 26.7/23.0 | 17.0/17.1 |
| Molecules in the asymmetric unit | 10 | 2 | 2 |
| Solvent content (%) | 71 | 50 | 24 |
| Matthews coefficient | 4.19 | 2.45 | 1.61 |
| Total water molecules | 14 | 16 | 6 |
| Glycerol molecule | 1 | 0 | 0 |
| Malonate molecules | 3 | 0 | 0 |
| TFA molecule | 0 | 0 | 1 |
| Orange G | 0 | 1 | 0 |
| Rmsd bond length (Å) | 0.010 | 0.010 | 0.020 |
| Rmsd angles (°) | 1.18 | 1.25 | 2.00 |
| Ramachandran plot | |||
| Allowed | 96 | 94 | 100 |
| Generous (%) | 4.44 | 5.56 | 0 |
| Disallowed (%) | 0 | 0 | 0 |
Table 2.
Comparison of Shape Complementarity (Sc) and Buried Surface Area (Ab) of Oligomers of SOD1 Segments
| KVKVWGSIKRL Form 1 | KBKVWGSIKRL Form 2 | KVWGSI | |
|---|---|---|---|
| Sc | 0.76 | 0.76 | 0.64 |
| A b (Å2) | 1195 | 1047 | 621 |
| A b/Residue (Å2) | 109 | 95 | 104 |
For the Sc calculation, we examined the interface between one chain and the remaining chains of the assembly.
A b values were calculated using AREAIMOL. The area buried was calculated by subtracting the solvent accessible surface area of one chain of the assembly from the total solvent accessible surface area of an isolated chain.
Supporting information
Supporting Information
Acknowledgments
We thank Michael Collazo, Duilio Cascio, and staff at Argonne Photon Source (APS), Northeastern Collaborative Access Team beamline 24‐ID‐E. The beamline is funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). The Pilatus 6M detector is funded by a NIH‐ORIP HEI grant (S10 RR029205). APS is a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE‐AC02–06CH11357. SS is supported by the UCLA Graduate Division Dissertation Year Fellowship. We thank HHMI and NIH (AG054022) for support. Atomic coordinates and structure factors have been deposited in the Protein Data Bank as 5WOR for KVKVWGSIKRL form 1, 6B79 for KBKVWGSIKRL form 2 and 5WMJ for KVWGSI.
References
- 1. Eisenberg D, Jucker M (2012) The amyloid state of proteins in human diseases. Cell 148:1188–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fowler DM, Koulov AV, Balch WE, Kelly JW (2007) Functional amyloid‐from bacteria to humans. Trends Biochem Sci 32:217–224. [DOI] [PubMed] [Google Scholar]
- 3. Chiti F, Dobson CM (2006) Protein misfolding, functional amyloid, and human disease. Ann Rev Biochem 75:333–366. [DOI] [PubMed] [Google Scholar]
- 4. Nelson R, Sawaya MR, Balbirnie M, Madsen A, Riekel C, Grothe R, Eisenberg D (2005) Structure of the cross‐β spine of amyloid‐like fibrils. Nature 435:773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJW, McFarlane HT, Madsen AO, Riekel C, Eisenberg D (2007) Atomic structures of amyloid cross‐β spines reveal varied steric zippers. Nature 447:453–457. [DOI] [PubMed] [Google Scholar]
- 6. Liu C, Zhao M, Jiang L, Cheng P‐N, Park J, Sawaya MR, Pensalfini A, Gou D, Berk AJ, Glabe CG, Nowick J, Eisenberg D (2012) Out‐of‐register β‐sheets suggest a pathway to toxic amyloid aggregates. Proc Natl Acad Sci 109:20913–20918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, Condron MM, Bitan G, Teplow DB, Shea JE, Ruotolo BT, Robinson CV, Bowers MT (2009) Amyloid‐β 2 protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer's disease. Nat Chem 1:326–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300:486–489. [DOI] [PubMed] [Google Scholar]
- 9. Riek R, Eisenberg DS (2016) The activities of amyloids from a structural perspective. Nature 539:227–235. [DOI] [PubMed] [Google Scholar]
- 10. Laganowsky A, Liu C, Sawaya MR, Whitelegge JP, Park J, Zhao M, Pensalfini A, Soriaga AB, Landau M, Teng PK, Cascio D, Glabe C, Eisenberg D (2012) Atomic view of a toxic amyloid small oligomer. Science 335:1228–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu C, Sawaya MR, Cheng PN, Zheng J, Nowick JS, Eisenberg D (2011) Characteristics of amyloid‐related oligomers revealed by crystal structures of macrocyclic β‐sheet mimics. J Am Chem Soc 133:6736–6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sangwan S, Zhao A, Adams KL, Jayson CK, Sawaya MR, Guenther EL, Pan AC, Ngo J, Moore DM, Soriaga AB, Do TD, Goldschmidt L, Nelson R, Bowers MT, Koehler CM, Shaw DE, Novitch BG, Eisenberg DS (2017) Atomic structure of a toxic, oligomeric segment of SOD1 linked to amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci U S A 114:8770–8775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng H‐X, Rahmani Z, Krizus A, McKenna‐Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung W‐Y, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak–Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH Jr (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62. [DOI] [PubMed] [Google Scholar]
- 14. Sangwan S, Eisenberg DS (2016) Perspective on SOD1 mediated toxicity in amyotrophic lateral sclerosis*. Postepy Biochemii 1:362–369. [PubMed] [Google Scholar]
- 15. Guareschi S, Cova E, Cereda C, Ceroni M, Donetti E, Bosco DA, Trotti D, Pasinelli P (2012) An over‐oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc Natl Acad Sci U S A 109:5074–5079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL (1995) An adverse property of a familial ALS‐linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14:1105–1116. [DOI] [PubMed] [Google Scholar]
- 17. Valentine JS, Hart PJ (2003) Misfolded CuZnSOD and amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 100:3617–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goldschmidt L, Teng PK, Riek R, Eisenberg D (2010) Identifying the amylome, proteins capable of forming amyloid‐like fibrils. Proc Natl Acad Sci U S A 107:3487–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pollack SJ, Sadler IIJ, Hawtin SR, Tailor VJ, Shearman MS (1995) Sulfonated dyes attenuate the toxic effects of β‐amyloid in a structure‐specific fashion. Neurosci Lett 197:211–214. [DOI] [PubMed] [Google Scholar]
- 20. Necula M, Kayed R, Milton S, Glabe CG (2007) Small molecule inhibitors of aggregation indicate that amyloid‐β oligomerization and fibrillization pathways are independent and distinct. J Biol Chem 282:10311–10324. [DOI] [PubMed] [Google Scholar]
- 21. Landau M, Sawaya MR, Faull KF, Laganowsky A, Jiang L, Sievers SA, Liu J, Barrio JR, Eisenberg D (2011) Towards a pharmacophore for amyloid. PLoS Biol 9:e1001080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eisenberg DS, Sawaya MR (2017) Structural studies of amyloid proteins at the molecular level. Ann Rev Biochem 86:69–95. [DOI] [PubMed] [Google Scholar]
- 23. McAlary L, Aquilina JA, Yerbury JJ (2016) Susceptibility of mutant SOD1 to form a destabilized monomer predicts cellular aggregation and toxicity but not in vitro aggregation propensity. Front Neurosci 10:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bucciantini M, Calloni G, Chiti F, Formigli L, Nosi D, Dobson CM, Stefani M (2004) Prefibrillar amyloid protein aggregates share common features of cytotoxicity. J Biol Chem 279:31374–31382. [DOI] [PubMed] [Google Scholar]
- 25. Celej MS, Sarroukh R, Goormaghtigh E, Fidelio GD, Ruysschaert J‐M, Raussens V (2012) Toxic prefibrillar α‐synuclein amyloid oligomers adopt a distinctive antiparallel β‐sheet structure. Biochem J 443:719–726. [DOI] [PubMed] [Google Scholar]
- 26. Soriaga AB, Sangwan S, MacDonald R, Sawaya MR, Eisenberg D (2016) Crystal structures of IAPP amyloidogenic segments reveal a novel packing motif of out‐of‐register beta sheets. J Phys Chem B 120:5810–5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krotee P, Rodriguez JA, Sawaya MR, Cascio D, Reyes FE, Shi D, Hattne J, Nannenga BL, Oskarsson ME, Philipp S, Griner S, Jiang L, Glabe CG, Westermark GT, Gonen T, Eisenberg DS (2017) Atomic structures of fibrillar segments of hIAPP suggest tightly mated β‐sheets are important for cytotoxicity. eLife 6:e19273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Elam JS, Taylor AB, Strange R, Antonyuk S, Doucette PA, Rodriguez JA, Hasnain SS, Hayward LJ, Valentine JS, Yeates TO, Hart PJ (2003) Amyloid‐like filaments and water‐filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat Struct Biol 10:461–467. [DOI] [PubMed] [Google Scholar]
- 29. Hough MA, Grossmann JG, Antonyuk SV, Strange RW, Doucette PA, Rodriguez JA, Whitson LJ, Hart PJ, Hayward LJ, Valentine JS, Hasnain SS (2004) Dimer destabilization in superoxide dismutase may result in disease‐causing properties: structures of motor neuron disease mutants. Proc Natl Acad Sci U S A 101:5976–5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sea K, Sohn SH, Durazo A, Sheng Y, Shaw BF, Cao X, Taylor AB, Whitson LJ, Holloway SP, Hart PJ, Cabelli DE, Gralla EB, Valentine JS (2015) Insights into the role of the unusual disulfide bond in copper‐zinc superoxide dismutase. J Biol Chem 290:2405–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wright GSA, Antonyuk SV, Kershaw NM, Strange RW, Samar Hasnain S (2013) Ligand binding and aggregation of pathogenic SOD1. Nat Commun 4:1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ivanova MI, Sievers SA, Guenther EL, Johnson LM, Winkler DD, Galaleldeen A, Sawaya MR, Hart PJ, Eisenberg DS (2014) Aggregation‐triggering segments of SOD1 fibril formation support a common pathway for familial and sporadic ALS. Proc Natl Acad Sci U S A 111:197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jiang L, Liu C, Leibly D, Landau M, Zhao M, Hughes MP, Eisenberg DS (2013) Structure‐based discovery of fiber‐binding compounds that reduce the cytotoxicity of amyloid beta. eLife 2:e00857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Porat Y, Abramowitz A, Gazit E (2006) Inhibition of amyloid fibril formation by polyphenols: Structural similarity and aromatic interactions as a common inhibition mechanism. Chem Biol Drug Des 67:27–37. [DOI] [PubMed] [Google Scholar]
- 35. Young LM, Ashcroft AE, Radford SE (2017) Small molecule probes of protein aggregation. Curr Opin Chem Biol 39:90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen P, Kayed R, Glabe CG, Frautschy SA, Cole GM (2005) Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem 280:5892–5901. [DOI] [PubMed] [Google Scholar]
- 37. Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, Engemann S, Pastore A, Wanker EE (2008) EGCG redirects amyloidogenic polypeptides into unstructured, off‐pathway oligomers. Nat Struct Mol Biol 15:558–566. [DOI] [PubMed] [Google Scholar]
- 38. Maezawa I, Hong HS, Liu R, Wu CY, Cheng RH, Kung MP, Kung HF, Lam KS, Oddo S, LaFerla FM, Jin L‐W (2008) Congo red and thioflavin‐T analogs detect Aβ oligomers. J Neurochem 104:457–468. [DOI] [PubMed] [Google Scholar]
- 39. Poli G, Ponti W, Carcassola G, Ceciliani F, Colombo L, Dall'Ara P, Gervasoni M, Giannino ML, Martino PA, Pollera C, Villa S, Salmona M (2003) In vitro evaluation of the anti‐prionic activity of newly synthesized congo red derivatives. Arzneimittelforschung 53:875–888. [DOI] [PubMed] [Google Scholar]
- 40. Kabsch W (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry land cell constants. J Appl Cryst 26:795–800. [Google Scholar]
- 41. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Cryst 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 42. Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN (2004) REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Cryst 60:2184–2195. [DOI] [PubMed] [Google Scholar]
- 43. Bricogne G, Blanc E, Brandl M, Flensbueg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C (2011) BUSTER version 1.10.0. Cambridge, United Kingdom: Global Phasing Ltd.
- 44. DeLano WL (2002). The PyMOL Molecular Graphics System. Schrödinger LLC http://wwwpymolorg.; Version 1.:http://www.pymol.org. doi:citeulike‐article‐id:240061
- 45.PubChem. PubChem Compound. National Center for Biotechnology Information, U.S. National Library of Medicine. 2016. doi:CID=445639
- 46. Lemmon G, Meiler J (2012) Rosetta ligand docking with flexible XML protocols. Methods Mol Biol 819:143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information