

Abstract
Misfolding and amyloid aggregation of intrinsically disordered proteins (IDPs) are implicated in a variety of diseases. Studies have shown that membrane plays important roles on the formation of intermediate structures of IDPs that can initiate (and/or speed‐up) amyloid aggregation to form fibers. The process of amyloid aggregation also disrupts membrane to cause cell death in amyloid diseases like Alzheimer's disease and type‐2 diabetes. On the other hand, recent studies reported the membrane fusion properties of amyloid fibers. Remarkably, amyloid‐fibril formation by short peptide fragments of highly abundant prostatic acidic‐phosphatase (PAP) in human semen and are capable of boosting the rate of HIV infection up to 400,000‐fold during sexual contact. Unlike the least toxic fully matured fibers of most amyloid proteins, the semen‐derived enhancer of virus infection (SEVI) amyloid‐fibrils of PAP peptide fragments are highly potent in rendering the maximum rate of HIV infection. This unusual property of amyloid fibers has witnessed increasing number of studies on the biophysical aspects of fiber formation and fiber‐membrane interactions. NMR studies have reported a highly disordered partial helical structure in a membrane environment for the intrinsically disordered PAP peptide that promotes the fusion of the viral membrane with that of host cells. The purpose of this review article is to unify and integrate biophysical and immunological research reported in the previous studies on SEVI. Specifically, amyloid aggregation, dramatic HIV infection enhancing properties, membrane fusion properties, high resolution NMR structure, and approaches to eliminate the enhancement of HIV infection of SEVI peptides are discussed.
Keywords: AIDS, SEVI, amyloid, IDP, inhibition, NMR, membrane fusion, precursor structure
Abbreviations
- AIDS
acquired immunodeficiency syndrome
- Aβ
amyloid‐β
- CD
circular dichroism
- CsgA
bacterial curli protein
- EGCG
epigallocatechin‐3‐gallate
- HIV
human immunodeficiency virus
- IAPP
islet amyloid polypeptide
- IDP
intrinsically disordered protein
- NMR
nuclear magnetic resonance
- PAP
prostatic acidic‐phosphatase
- SEVI
semen‐derived enhancer of viral infection
- TFE
2,2,2‐trifluoroethanol
- TEM
transmission electron microscopy
- ThT
thioflavin
- 2D
two‐dimensional
- 3D
three‐dimensional
Introduction
HIV infections continue to be a major health issue
The human immunodeficiency virus (HIV), a causative agent in acquired immunodeficiency syndrome (AIDS), was first detected in the 1980s.1, 2, 3 AIDS is amongst the top 10 fatal diseases worldwide, and seriously, has killed more than 25 million people.3 Thirty‐three million people still suffer from HIV‐induced AIDS, in particular, in the developing countries: ∼90% of children and ∼68% of all adults are infected with HIV in sub‐Saharan Africa. One out of 200–2000 coital acts results in infection, indicating markedly low sexual infection of HIV virions.4 However, high viral titers that occur during acute transmission accelerate the infection rate by ∼10‐fold.5 Although the concentration of virus particles in seminal fluid is important, semen itself was a key modulator of HIV transmissibility, and enhanced actually the infection of physiologically‐relevant cell types.6, 7 Thus, certain natural cofactors in the ejaculate have been implicated to serve as HIV transmission by enhancing the efficiency of viral infectivity.6
SEVI amyloid dramatically increases the rate of HIV infection up to 4,00,000 times
Innovative studies based on screening, mass spectrometry, and sequencing of a library of all seminal (poly)peptides clarified that one fraction enhanced remarkably HIV infectivity.6 It was eventually elucidated that a proteolytic fragment of prostatic acid phosphatase (PAP),8 which exists in seminal fluid at ∼1–2 mg mL−1, corresponded to residues 248–286 of PAP (PAP248–286). Intriguingly, soluble synthetic PAP248–286 peptide was incapable of potentiating HIV infection; however, aggregated PAP248–286 did augment viral infectivity by up to five orders of magnitude.6 These PAP248–286 aggregates were found to be typical amyloid fibrils, one type of insoluble protein aggregates, on the basis of tinctorial properties of thioflavin T (ThT) and congo red dyes as well as cross‐β structures of X‐ray fiber diffraction (strong reflections at 4.7 and 10.6 Å), and thus are termed semen‐derived enhancer of viral infection (SEVI).
A 39‐amino acid fragment of PAP248–286, SEVI precursor, contains ∼20% of positively‐charged residues with a theoretical pI of 10.2.3 Dual binding capability of SEVI amyloid fibrils to acidic (negatively‐charged) surfaces of the HIV virion and host cell9 was attributable to cationic surfaces of SEVI amyloids. Thus, SEVI amyloids were implicated to function as a connecter between virions and host cells using attractive electrostatic interactions by minimizing repulsive forces between virions and host cells [Fig. 1(A)],9, 10 and thereby promoting fusion of virion. In line with this hypothesis, SEVI amyloid fibrils, formed from a PAP248–286 mutant with positive charges neutralized, largely reduced HIV transmission.9 All these results suggested SEVI amyloid‐mediated enhancement of HIV infection.
Figure 1.
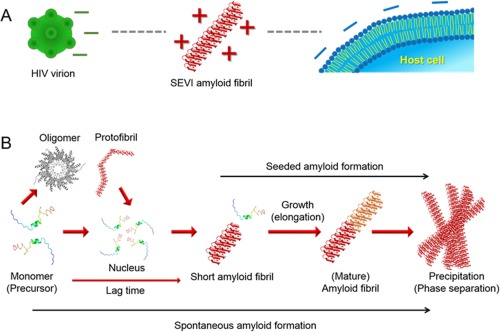
SEVI amyloid‐mediated interaction of HIV with cell and general mechanism of amyloidogenesis. (A) Electrostatic interactions among HIV virion, SEVI amyloid fibril, and host cell. “–” and “+” indicate negative and positive charges, respectively. (B) Nucleation‐dependent two‐step amyloid formation is depicted. Each molecular species and steps during amyloid generation are represented.
Protein misfolding and amyloid aggregation implicated in amyloid diseases
Amyloid fibrils are best studied proteinous fibrillar aggregates as their formation is deeply responsible for more than 40 diseases such as Alzheimer's and Parkinson's diseases and type‐2 diabetes.11, 12, 13 Mature amyloid fibrils consist of several protofillaments containing β‐sheets, which are predominantly stabilized by a number of intermolecular hydrogen bonds among constituent monomers as revealed by several biophysical experiments.11, 14, 15, 16, 17 Spontaneous amyloid fibrillation shows generally the two‐step reaction [Fig. 1(B)]. Amyloid fibrils grow rapidly following nucleation which produces the lag time. Thus, the addition of preformed short amyloid fibrils to monomer solutions as seeds skips the lag phase and solely shows the elongation phase, in seeded amyloid formation. Other types of aggregates such as amorphous aggregates, oligomers, and protofibrils are also detected as on‐ or off‐pathway intermediates.
Recent studies have proposed that solubility and supersaturation are fundamental factors in understanding protein aggregation in solution.18, 19, 20, 21, 22, 23, 24, 25 Proteins over the solubility limit must form insoluble aggregates such as amyloid fibrils from the thermodynamic point of view. However, apparently soluble proteins prior to the phase transition to insoluble solid states are often detected due to kinetic trapping by the metastability of supersaturation, which is certainly linked to the cause and effect relationship with the nucleation and lag time. Other factors such as biological membranes, hydrophobic surfaces, or agitation/cavitation have shown to promote amyloid formation by disrupting the metastability of supersaturation. Among these, the cell membrane components play crucial roles in the formation of intermediate nucleating structures, kinetics of aggregation, and the generation of polymorphic fibers. Remarkably, the process of amyloid aggregation itself disrupts the cell membrane to execute cell toxicity in various amyloid diseases. This process of amyloid‐induced cell death is implicated in the loss of neuronal cells by the aggregating amyloid‐β (Aβ) and other amyloid proteins in Alzheimer's disease and in the killing of insulin‐producing islet cells by islet amyloid polypeptide (IAPP, or also known as amylin).13, 26, 27
SEVI precursors also showed nucleation‐dependent two‐step amyloid formation under several different conditions6, 28, 29, 30, 31, 32, 33; however, much still remains to be investigated, for instance, detailed aggregation kinetic analysis and cross‐seeding effects on SEVI amyloid formation. Atomistic and secondary structure and morphology of SEVI amyloid fibrils were characterized using X‐ray crystallography, circular dichroism (CD) spectroscopy, Fourier transform infrared spectroscopy, NMR spectroscopy, and transmission electron microscopy (TEM).6, 15, 34, 35 On the one hand, some specific precursor structures of proteins such as α‐synuclein, β2‐microglobulin, SH3 domain, lysozyme, and insulin have been suggested to be amyloidogenic,20, 21, 36, 37, 38, 39 which convinces the consequence of the structural study in understanding amyloidogenesis from the microscopic point of view. Although the residue‐based amyloidogenic propensity of SEVI precursors was examined,32 high‐resolution structural studies of SEVI precursor at various conditions are essential for deeper understanding of the molecular mechanism of SEVI amyloid formation, effects of SEVI on membranes of the HIV virion and host cell, and structure‐based inhibitor development.
We herein describe our recent studies on NMR structures of intrinsically disordered and partially helical SEVI precursors in solution as well as on SEVI‐induced membrane fusion for the remarkable enhancement of HIV infection based on high‐resolution structures. Effects of cross‐seeding of amyloid fibrils of bacterial curli proteins on SEVI amyloid generation will also be addressed. Finally, a series of strategies for suppression, prevention, elimination of SEVI amyloid fibril‐enhanced HIV viral activity are comprehensively surveyed and suggested.
SEVI Induced Membrane Fusion is Key for Dramatic Enhancement of HIV Infection
Since in vivo studies have demonstrated a dramatic enhancement of HIV infection in the presence of SEVI amyloid aggregates, it is very important to understand the mechanism by which SEVI executes its role. Although there are numerous factors in vivo that can contribute to the effect of SEVI on HIV infection, the exact mechanism of SEVI‐enhanced viral infection was unclear from in vivo studies. In vitro experimental evidences have suggested that a synthetic SEVI peptide freshly prepared is ineffective in promoting viral infection whereas the aggregated forms of SEVI were found to exhibit the maximum enhancement of viral infection. Studies have focused on understanding the factors that influence the aggregation kinetics and fibril formation of intrinsically disordered SEVI peptides. For example, in vitro studies focused on evaluating the roles of pH, temperature, peptide concentration, mechanical force, metal ions, membrane, lipid charge, salt, and also the presence of bacterial curli fibers.
By using synthetic lipid bilayers as model cellular membrane, a biophysical study investigated the membrane fusion by SEVI as a mechanism for the observed viral binding and entry into host cells. An interesting observation was that freshly dissolved monomers of SEVI induced membrane disruption with a maximum effect for lipid membranes containing maximum anionic lipids and aggregation of liposomes at acidic pH but not at neutral pH (Fig. 2). This in vitro experimental finding correlated well with SEVI's ability to enhance viral infection under the in vivo acidic vaginal environment (pH is ∼4). Based on mutational studies and binding affinity measurements to liposomes, it has been suggested that the cationic SEVI peptide binds with lipid membrane to cause pH dependent membrane fusion with a maximum effect for pH ∼4. This membrane fusion ability of SEVI was further confirmed by differential scanning calorimetry (DSC) experiments that showed the negative curvature induced by SEVI on DiPoPE lipid bilayers to form inverted hexagonal lipid structures (HII). CD and NMR experiments showed a partial helical structure for the mostly disordered PAP248–286 peptide in the presence of lipid membrane. Based on these experimental evidences, a model was proposed to explain the membrane fusion process, bridging the membranes of HIV virion and target cells, that enhances viral infection by the disordered partial helical structural form of PAP248–286 as illustrated in Figure 3.
Figure 2.
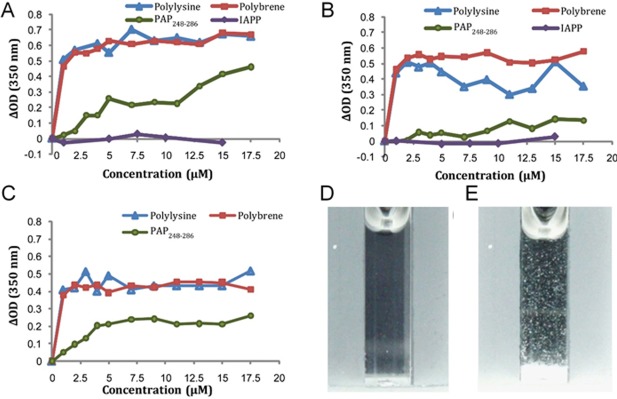
SEVI‐induced aggregation of liposomes by turbidity measurements at pH 4. (A) 7:3 POPC/POPG, (B) viral membrane composition, and (C) host cell membrane composition. (D and E) Photographs of 100 nm 7:3 POPC/POPG liposomes at 500 μM concentration before (D) and after (E) the addition of 17.5 μM PAP248–286. These figures were adopted from a previously published article.28
Figure 3.
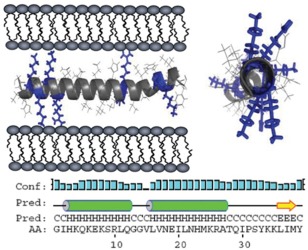
Illustration of SEVI‐induced membrane fusion. Two views of a model α‐helical structure of PAP248–286 showing the distribution of positively charged residues, and the predicted secondary structure of PAP248–286 within full‐length PAP. The letter codes “C, H, and E” denote a residue in random coil, helical, or β‐sheet conformation, respectively. Figure adopted from a previously published article.28
It is remarkable that the membrane fusion process can explain the effect of SEVI in the enhanced HIV infection to target cells. However, additional studies probing various membrane centric physicochemical properties would be useful to fully elucidate the roles of pH, metal ions such as zinc that is highly abundant in semen, anionic lipids, polysaccharides, membrane curvature, and lipid domains. In fact, real time monitoring of the membrane fusion process in liposomes that vary in lipid composition would provide a wealth of information.
Intrinsically Disordered PAP248–286 Adapts a Partial Helical Structure to Promote Membrane Fusion
SEVI‐dependent HIV infection raised a number of very important questions that need to be answered to fully understand the biological process of viral infection and for the development of therapeutic treatments. If SEVI is effective in enhancing HIV infection via membrane fusion only in an aggregated form, what is the role of the secondary and tertiary structures of PAP248–286? What are the different types of intermediate structures formed in the aggregation pathway? What type of interactions initiate the formation of aggregation? How do these intermediate species interact with lipid membrane? Are these species located near the membrane surface or aggregated to form pores? Which intermediate species is effective in causing membrane fusion?
To address these questions and to obtain the complete picture of SEVI's functional role in the viral infection, high‐resolution structure of monomeric PAP248–286 and SEVI aggregates are essential. Monomeric PAP248–286 has been shown to be unstructured in solution conditions, but CD experiments indicated the helix formation upon interaction with lipid membrane. However, CD experimental results are of low resolution and cannot be used to understand the residue specific helicity and the role of the helical structure in the membrane fusion process. To overcome this limitation, NMR experiments were used to investigate the structure of SEVI. Since SEVI induces fusion of liposomes that would pose challenges to perform NMR experiments to determine the high‐resolution structure in a membrane environment, SDS micelles were used to trap the intermediated membrane bound species of SEVI. Two‐dimensional (2D) TOCSY and 2D NOESY experiments were performed to accomplish the resonance assignment of the 39‐residue synthetic peptide and to measure 1H‐1H NOE restraints. Assignment of resonances in the fingerprint region of the 2D NOESY spectrum [Fig. 4(A)], chemical shift index of alpha protons [Fig. 4(B)] and NOE intensity plot [Fig. 4(C)] are shown in Figure 4. The NOE distance constraints were used to determine the three‐dimensional (3D) structure of SEVI (Fig. 5). The membrane‐bound SEVI structure constitutes a short helix between residues V262 and H270, a highly flexible region G248–G261 in the N‐terminus, and a flexible region M271–Y286 with a short helix S279–L283 in the C‐terminus.
Figure 4.
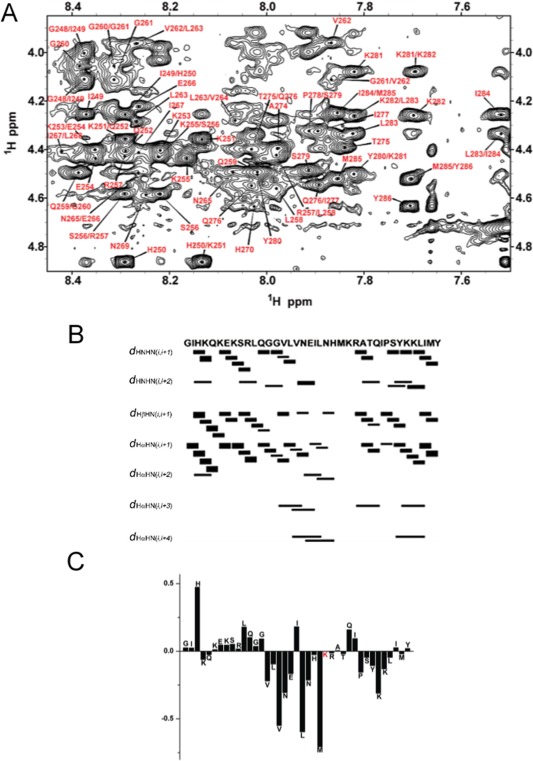
(A) The fingerprint region of 2D 1H‐1H NOESY spectrum of PAP248–286 peptide embedded in SDS micelles. (B and C) NOE connectivity (B) and chemical shift index of alpha protons (C) of PAP248–286. Figures were taken from a previously published article.40
Figure 5.
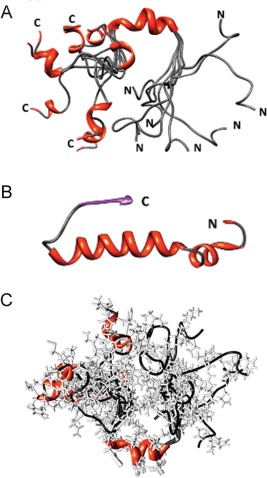
(A) NMR determined α‐helical structures of PAP248–286 in SDS micelles. (B) Structure of PAP248–286 obtained from the crystal structure of human PAP.41 (C) A representation of an ensemble of PAP248–286 conformations. Figures were adopted from a previously published article.40
The helix forming tendency of SEVI was further investigated in 2,2,2‐trifluoroethanol (TFE):water system as reported elsewhere (Fig. 6).40 In TFE solutions, PAP248–286 adopts a largely α‐helical structure that differs substantially from the largely disordered structure in SDS micelles or in aqueous solution but is similar to the conformation PAP248–286 adopts in the crystal structure of the full‐length protein (Fig. 5). The membrane topology determined using paramagnetic quenching of solvent exposed resonances by MnCl2 revealed that most of the residues of SEVI are solvent exposed and weakly bound to the surface of the membrane. This disordered and weakly membrane‐bound structure of SEVI has been proposed to be responsible for the membrane fusion observed in in vitro biophysical studies that enhances the reported dramatic enhancement of HIV viral infection.
Figure 6.
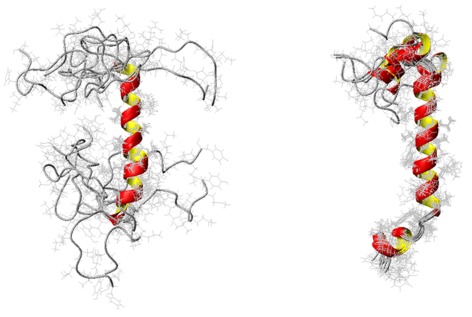
High‐resolution NMR structures of PAP248–286 in 30% TFE (left) and 50% TFE (right). Figures were adopted from a previously published article.40
While the high‐resolution structure is useful to explain the membrane fusion process, more studies are required to understand the roles of membrane components in the formation and stabilization of the disordered helical structure and to obtain a high‐resolution view of the membrane fusion process. Such information would be useful to develop compounds that are capable of suppressing the formation of SEVI and to avoid the membrane fusion process to completely eliminate the SEVI dependent viral infection process. In addition, determining the high‐resolution structure of SEVI in lipid bilayers composed of varying membrane composition, and in viral or host cell membrane composition, could provide more physiologically‐relevant structure that would valuable for the development of potential drugs. Although this is a difficult task due to the challenges posed by the lipid membrane for high‐resolution structural studies, the recently developed nanodiscs can be used to accomplish this task.42, 43 In particular, the recently developed polymers that vary in charge and can form lipid nanodiscs would be valuable to trap the intermediate structures of SEVI in different membrane compositions.44 In addition, the use of “styrene‐free” methacrylate polymer nanodiscs would enable high‐throughput screening of conditions by ThT fluorescence and CD experiments as demonstrated recently.45
Bacterial Curli Protein is a Catalytic Agent for SEVI Formation
The in vivo observation of dramatic enhancement of HIV infection is linked to the amyloid aggregation of SEVI peptides. The rate of amyloid formation is typically nucleation‐dependent and it has been shown to be very slow for SEVI amyloid formation under in vitro conditions. Although SEVI formation can be speeded up by shaking under in vitro conditions, the factors that influence the aggregation in in vivo conditions are unclear. Hartman et al. investigated the role of cross‐seeding on the kinetics of amyloid aggregation of SEVI under in vitro conditions.31 Since bacterial infection is likely in vagina with HIV infection, they specifically examined the cross‐seeding effect of preformed amyloid fibers of Escherichia coli bacterial curli protein, CsgA. They reported that CsgA fibers cross‐seeded by greatly enhancing the rate of aggregation to form SEVI fibrils. The cross‐seeding was found to occur via an unusual mechanism. The preformed CsgA amyloid fiber seeds dramatically enhanced the elongation rate of SEVI. One mole percentage of the seed was sufficient to increase the rate by sixfold. On the other hand, cross‐seeding with CsgA did not change the lag time unlike the significant reduction or complete elimination reported for other amyloid proteins. Interestingly, the SEVI fibers obtained by cross‐seeding with curli proteins were found to be longer than the regular SEVI fibers (Fig. 7). It is also remarkable to note the curli nucleated SEVI amyloid fibers exhibited enhancement of HIV infection.
Figure 7.

Curli nucleation produces longer SEVI fibers. (A and B) TEM images of SEVI fibers formed in the absence of curli (A) and in the presence of 5 mol% CsgA (B). (C) TEM images of CsgA. Amyloid fibrils were grown at a concentration of 440 µM PAP248–286 at 37°C under 1400 rpm orbital shaking for 7 days. Figures were adopted from a previously published article.31
Inhibition of SEVI‐Enhanced HIV Viral Infection
Since the HIV infection continues to be a devastating health issue and as shown by recent studies SEVI's roles are crucial, there is significant interest in the development of approaches to suppress the effect of SEVI. The dramatic enhancement of HIV transmission by SEVI amyloid fibrils can be inhibited at various molecular levels. Some of the important points to be considered in the development of biochemical and biomedical approaches to eliminate the roles of SEVI on HIV infection are: (a) devise strategies to eliminate SEVI or suppress its effects; (b) eliminate HIV virions; (c) suppress membrane fusion. In this section, we comprehensively review the recent studies and possible methods to reduce and prevent HIV infectivity by semen‐derived amyloid fibrils by classifying groups according to their actions (Fig. 8).
Figure 8.
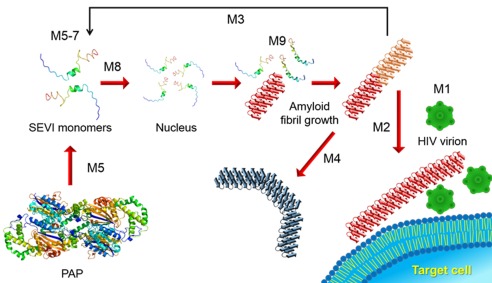
Proposed strategies of SEVI amyloid‐mediated enhancement of HIV infection. Each strategy is indicated by M1–9 (Method 1–9 in the text). Cartoons of all molecular species are represented. SEVI amyloid fibrils, which are capable (red) and incapable (navy blue) of delivering HIV virions to target cells, are distinguished by different colors and shapes.
Removal of HIV virions (Method 1)
One simple way is to get rid of a pathogen of HIV itself using microbicidal compounds. Similar to the development of antimicrobial peptides that exhibit broad‐spectrum antibacterial activities, there is considerable interest in the development of microbicidal compounds that show antiviral activities. Thus, the development of topical microbicides46 is promising to inhibit the sexual transmission of HIV. However, microbicidal compounds contributing to the fast generation of drug‐resistant HIV variants should be taken into consideration. Therefore, it is highly important to investigate the molecular mechanisms of action of such microbicidal compounds against HIV infection and the development of resistance in order to further develop more potent compounds to potentially treat the disease. Some of the questions that can be easily addressed include: (a) Do these microbicidal compounds suppress the formation of SEVI amyloid aggregates? (b) Do these compounds directly interact with SEVI aggregates to suppress their effect HIV infection? (c) Are these compounds capable of interacting directly with the HIV virus to avoid infection? (d) Can these compounds suppress the fusion of viral membrane with host cell membrane?
Development of agents to suppress HIV interaction with SEVI amyloid fibrils (Method 2)
As mentioned earlier, SEVI amyloid aggregates interact with lipid membrane and promote membrane fusion, strategies to inhibit these capabilities of SEVI would be an effective way to reduce the SEVI's enhancement of HIV infection. Since the aggregated forms of SEVI, and not the monomeric species, are potent in the enhancement of HIV infection, altering the chemistry of the exposed regions of SEVI can alter the efficiency of SEVI. For example, coating of SEVI amyloid fibrils by other molecules is expected to act directly on the prevention of SEVI amyloid‐induced augmentation of HIV transmission. This idea is very simple but most likely to show relatively high efficacy against viral infection by blocking accessibility of HIV virions to target cells. Surfaces of SEVI amyloid fibrils are positively charged.32 Therefore, negatively‐charged substances must be effective by weakening attractive electrostatic interactions between HIV virions and SEVI amyloids. Indeed, anionic polymers such as glycosaminoglycans and heparin diminish SEVI‐mediated enhancement of infection.9 Aminoquinoline surfen also antagonized electrostatically the capability of HIV‐SEVI intermolecular interactions.10 Thus, salts, inexpensive and easily applied to the seminal fluid of the male, may also serve as inhibitors as they are counter ions for positively‐charged SEVI fibrils and for negatively‐charged virions and cell surfaces.
Nonpolyanionic small molecules which bind to SEVI amyloid fibrils reduced SEVI activity. BTA‐EG67 and its oligovalent derivatives47 prevented SEVI‐mediated enhancement of HIV infection. Gallic acid showed binding ability to SEVI amyloid fibrils and decreased semen‐mediated enhancement of HIV infection.48 Perturbation of surface electrostatics between cationic SEVI amyloids and HIV virions resulted in a decrease of HIV infectivity although the details on the physicochemical intermolecular interactions are unknown. Meanwhile, a d‐enantiomeric peptide (RPRTRLHTHRNR), an inhibitor of Aβ(1–42) amyloid formation, also attenuated SEVI amyloid‐boosting infection of HIV.49 Importantly, these studies open further opportunities that various small molecules, peptides, and proteins which interact with amyloid fibrils are worth testing as therapeutic agents for HIV acquisition. In this context, substances interacting with surfaces of either virions or cells may show a decrease in HIV infection even if those substance do not bind to SEVI amyloid fibrils. For instance, a curcumin derivative, curcumin diacetate (CurDAc), was implicated to bind to membrane surfaces which, thus, may prevent binding of amyloid fibrils.50
Nanoparticles, which were generated from a polymer comprising moieties of amyloid‐binding 6‐methylbenzothiazole aniline with high affinity for SEVI amyloid fibrils and similar in size and shape to HIV virions, disabled SEVI‐mediated HIV transmission in cells.51 These nanoparticles and polymers were far more efficient for inhibiting SEVI‐enhanced HIV infection than monomeric and pentameric 6‐methylbenzothiazole aniline, proposing a critical role of steric hindrance in the design of effective SEVI neutralizing agents. A very recent study suggested that a nanoparticle formulation of hydrophobic polymers diminished SEVI amyloid‐mediated HIV infection, and that the use of hydrophobic interactions to alter structures of amyloid fibrils at the level of the secondary structures may be an useful approach to neutralize the SEVI function.52
Reverse and dissociate SEVI amyloid fibrils (Method 3)
The presence of SEVI amyloid fibrils is a risky situation in terms of sexual transmission of HIV. Thus, clearance of preformed SEVI amyloids will decrease chances for HIV virions to contact target cells. Atomic structures of amyloid fibrils15, 17 look very strong against exogenous stresses. However, stability of amyloid fibrils is not that high, and rather, is similar to that of globular proteins,53, 54 and is indeed susceptible to surrounding conditions such as pH, ionic strength, metal ions, solvent polarity, pressure, temperature, chemical denaturants, and other peptides/proteins. Amyloid fibrils also showed proteolytic degradation (see below). Amyloid fibrils dissociated (i.e., denatured or depolymerized) to monomers by not only altering the temperature (cold and heat denaturation of amyloid fibrils) but also applying pressure or chemical denaturants (guanidine hydrochloride or urea). In most cases, decreases in solvent polarity by the addition of organic cosolvents such as dimethylsulfoxide and 1,1,1,3,3,3‐hexafluoroisopropanol cause denaturation of amyloid fibrils to monomers.14 As SEVI precursors showed pH‐dependent formation of amyloid fibrils, stable and unstable at neutral and acidic conditions, respectively,29 modulation of pH may be effective dissociate preformed SEVI amyloid fibrils.
However, in terms of clinical application, changing to harsh environmental conditions in order to destruct amyloid fibrils may not be practical. Thus, treatment of seminal fluid containing SEVI amyloid fibrils with additives such as small molecules and proteins/peptides will be a better choice. Epigallocatechin‐3‐gallate (EGCG), the major active constituent of green tea, degraded SEVI amyloid fibrils, inhibited SEVI activity, and abrogated semen‐mediated enhancement of HIV‐1 infectivity.55 NMR studies revealed the site‐specific interactions between SEVI and EGCG and suppression of SEVI amyloid formation by EGCG (Fig. 9)56; specifically, EGCG binds with charged residues, primarily lysine, in the K251–R257 and N269–I277 regions of SEVI to form a pH dependent tight complex. Hsp104 has been implicated as an amyloid‐remodeling nanomachine,57 and showed amazing capability to renatures amorphous protein aggregates destruct various amyloid fibrils.58 Castellano et al. modified Hsp104 to interact with the chambered protease ClpP, which enables coupled remodeling and degradation to irreversibly clear SEVI amyloids.59 Similarly, prostate‐specific antigen and matriptase, which are proteases in seminal plasma degraded SEVI amyloid fibrils.60 A recent study showed disaggregation of Aβ(1–40) amyloid fibrils by nonhemolytic 11‐residue peptide (NAVRWSLMRPF),61 which suggested the possible application of this short peptide for eliminating SEVI amyloid fibrils and peptidomimetic therapeutic agents against amyloid disease.
Figure 9.
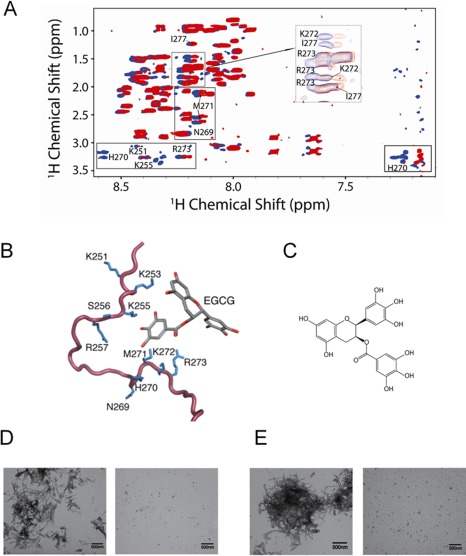
EGCG:PAP248–286 interaction and inhibition of SEVI amyloid formation by EGCG. (A) Overlaid 2D 1H‐1H TOCSY spectra of PAP248–286 alone (blue) and EGCG bound to peptide (red, 1:1 molar ratio). (B) Specific interactions between EGCG and the side‐chains of monomeric PAP248–286 in two regions (K251–R257 and N269–I277) of primarily charged residues, particularly lysine. (C) Chemical structure of EGCG. (D and E) TEM image of PAP248–286 incubated alone for 4 days (left) and coincubated for 4 days with EGCG at a 1:5 molar ratio (right) at pH 7.3 (D) and 6 (E). Figures were adopted from a previously published article.56
Remodeling/deformation or changes of SEVI aggregates to form HIV infection‐incompetent strains (Method 4)
Changes in structures and morphologies of amyloid fibrils (remodeling and/or deformation of amyloids) as well as transitions among different types of proteins aggregates such as amyloid fibrils, protofibrils, oligomers, and amorphous aggregates are often observed when environmental conditions change or exogenous additives are supplemented. Remodeling and transition of preformed amyloid fibrils of Aβ(1–40) peptides to protofibrillar aggregates on model membranes were observed.62 Thus, if clearance of SEVI amyloid fibrils is imperfect, that is, complete denaturation to precursor proteins in soluble monomeric states, remodeling and deformation of SEVI amyloid fibrils to other types of amyloid fibrils which are incapable of elevating HIV infection by taking advantages of nature of polymorphs of amyloid fibrils will be alternative. In addition, conversion to nonamyloid aggregates without HIV virion binding capability will be also effective.
Castellano et al. showed remodeling of several seminal amyloid fibrils including SEVI amyloids by EGCG with diminished viral infectivity in cell culture.63 Hsp104 and an enhanced engineered variant, Hsp104A503V, directly remodel SEVI and PAP85–120 fibrils into nonamyloid forms.59 Investigating proper conditions which cause remodeling and/or deformation of HIV infection‐enhancing SEVI amyloids with cytotoxicity to nontoxic strains without HIV infection‐enhancing activity.
Development of proteases of SEVI precursors and inhibitors of PAP proteolysis (Method 5)
Amyloidogenesis is remarkably context‐dependent. Changes in environmental conditions, sole single mutation, or chemical modification alters greatly amyloidogenicity of precursors,64, 65, 66 which makes precursors amyloid‐incompetent. SEVI precursors show high amyloidogenicity as a series of experimental results and prediction demonstrated.67 In addition, they do not take solid 3D structures and thus share similar properties of intrinsically disordered proteins.68 In these contexts, cleavage and degradation of SEVI precursors by proteases may eliminate or decelerate their amyloidogenicity and thereby inhibiting proviral activity. Martellini et al. showed a possibility of this concept by using subphysiological concentrations of two prominent proteases of seminal plasma, prostate‐specific antigen, and matriptase.60 These two proteases degraded physiological and supraphysiological concentrations of naturally occurring PAP peptides including SEVI precursors.
The cleavage of amyloid precursor protein by beta secretase and gamma secretase produces Aβ peptides, which are thought to play important roles in Alzheimer's disease by forming amyloid fibrils.69, 70, 71 Thus, tremendous studies and endeavors have focused on establishing strategies as possible interventions against amyloid‐induced neurodegenerative disease. Inhibitors of beta secretase and gamma secretase have been intensively investigated as one of promising ways to suppress the onset of disease.72 Likewise, inhibition of degradation of PAP by proteolysis which produces seminal amyloid‐competent fragments will be fundamental blocking of the enhancement of HIV viral activity. Thus, discovery and development of proper protease inhibitors are required. Alternatively, stabilization and protection of PAP by adding effective molecules may play an important role in decreasing activity of proteases.
Increasing solubility of SEVI precursors (Method 6)
Protein aggregation must occur over solubility limit which changes with environmental conditions.20, 21, 23 Thus, SEVI precursors under solubility conditions keep soluble and do not form amyloid fibrils. In these conditions, no enhancement of HIV viral activity is expected. Several algorithms are available to predict solubility of proteins.73 Various factors in seminal fluid influence the solubility of SEVI precursors. For instance, a decrease in pH, if any, will increase solubility due to the increase in the number of positive charges. Indeed, SEVI precursors did not form amyloid fibrils at acidic conditions in spite of amyloid generation at neutral pH.29 Structural changes are also ascribed to the change in solubility.23 Exposures of hydrophobic regions of proteins in aqueous solution decrease the solubility and thereby promoting aggregation. Well‐tailored additives which bind to hydrophobic regions and simultaneously can form hydrogen bonds with water molecules recover solubility. Single amino acid residues of arginine and lysine are interesting solubilizers for protein solutes and can inhibit the formation of several amyloid fibrils.74 These residues will help increase the solubility of SEVI precursors and appear to be a promising supplement to antiretroviral microbicides to reduce sexual transmission of HIV‐1.
Stabilization of SEVI precursors (Method 7)
Maintaining of stable structures of amyloidogenic precursors which prohibits triggering of aggregation as the equilibrium shifts to monomers under the two‐state transition between monomers and amyloid fibrils. Stabilization of transthyretin tetramers and insulin dimers in neutral pH inhibit their amyloid formation.75, 76 Binding of small molecules to amyloidogenic SEVI precursors stabilized and inhibited amyloid fibrillation. EGCG was found to interact with SEVI precursors by forming a covalent bond between lysine side chains and EGCG (Fig. 9).56 Binding of ADS‐J1 to SEVI precursors obviously inhibited amyloidogenesis in a dose dependent manner.77 A recent study demonstrated that brazilin, a natural compound that is isolated from Caesalpinia sappan, bound to SEVI precursors and inhibited amyloid formation at lower concentrations than EGCG.78 In addition, nonhemolytic 11‐residue peptides inhibited the formation of Aβ(1–40) amyloid fibrils by interacting with the N‐terminal part and the central hydrophobic cluster of Aβ(1–40).61
Kinetic trapping of SEVI precursors by metastability of supersaturation (Method 8)
Protein aggregation is thermodynamically and kinetically controlled. As described above, aggregation initiates when proteins are over solubility. However, in reality, this supersaturated state (i.e., metastability of supersatuaration) kinetically traps proteins from nucleation to form amyloids and still keeps proteins apparently soluble.20, 21, 23 Thus, the lag time is often observed prior to burst generation of amyloid formation. Agitation and hydrophobic surfaces disrupt the metastability of supersatuaration and promote amyloid formation through nucleation.
Thus, increases in the metastability of SEVI precursors by the addition of other molecules will be a useful strategy for inhibiting SEVI amyloidogenesis even over solubility limit by taking advantage of the kinetic nature of nucleation‐dependent amyloid formation. Slowing down the amyloid formation without changing the concentration of residual monomers in equilibrium evidences the metastability of supersaturation‐limited amyloid formation in terms of kinetics. Thus, a number of effectors which extend the lag time of amyloid formation would be related to the metastability of supersaturation.
Amyloid capping (Method 9)
Another intriguing method to target deleterious effects of SEVI is the structure/computation‐based peptide inhibitor design which caps the growing end of amyloid fibrils with high specificity.15, 16, 67 This approach calculates the binding energy based on the steric‐zipper structure of amyloid(‐like) fibrils formed using hexapeptide segments of amyloidogenic proteins and sequences of amino acid residues,67 and was applied to the inhibition of SEVI amyloid formation. Amyloidogenic hexapeptide segment of PAP248–286 (G260‐N265, GGVLVN) was predicted by Zipper DB67 and inhibited SEVI (full‐length PAP248–286) amyloid formation.15 Interestingly, a nontoxic and serum stable‐designed heptapeptide, KR7 (KPWWPRR‐NH2), blocked primarily the elongation step of insulin amyloid fibrils,79 suggesting the potential availability of peptide‐based inhibitors for SEVI amyloid formation.
Summary and Future Directions
HIV infection continues to be a major health issue, and the determination of the remarkable roles of SEVI amyloid is a major break‐through for the development of compounds to eliminate HIV infection. In this review article, we have summarized the previous studies that reported the kinetics of amyloid aggregation of PAP peptide fragments and their membrane fusion properties, the effect of cross‐seeding with curli bacterial amyloid protein, in vivo studies on the role of SEVI amyloid aggregates, and approaches to eliminate the role of SEVI. Although bacterial curli protein cross‐seeds the aggregation to form potent SEVI amyloid aggregates, more in vitro and in vivo studies are essential to fully evaluate the factors that influence the aggregation of PAP peptide fragments and the formation of SEVI amyloid aggregates. For example, a systematic investigation of the effects of molecular crowding on the formation SEVI would provide valuable insights into the mechanism underlying the rate of increase of HIV infection by SEVI. Since zinc is highly abundant in semen, and zinc is known to play very important roles in amyloid aggregation, it would be useful to evaluate the role of zinc and other metals on the formation of SEVI and its effect on HIV infection.
While the development of small molecule compounds to suppress SEVI's ability to boost HIV infection is exciting, it is important to fully investigate the role of molecular and structural interactions among SEVI, membrane, and HIV virion. It is worthwhile to examine the small molecule compounds that have already been tested for the suppression of amyloid aggregation and cellular toxicity of Aβ, IAPP, or other amyloid proteins.80, 81, 82 Unlike the amyloid fibers of most other amyloid proteins, the very unique functional properties of SEVI fibers are exciting and related studies could provide enormous molecular insights into the function of a variety of other pathogens. It is also worthwhile to examine the effect of HIV virions on the membrane fusion effects of other amyloid proteins and the implications in other amyloid diseases.
Acknowledgments
The authors thank the contributions of Dr. Jeffrey Brender and Dr. Kevin Hartman for the original studies on SEVI in the Ramamoorthy (RAMYLOID) group.
References
- 1. Weiss RA, Jaffe HW (1990) Duesberg, HIV and AIDS. Nature 345:659–660. [DOI] [PubMed] [Google Scholar]
- 2. Schechter MT, Craib KJ, Montaner JS, Lee TN, O'Shaughnessy MV, Gelmon KA (1993) Aetiology of AIDS. Lancet 341:1222–1223. [DOI] [PubMed] [Google Scholar]
- 3. Castellano LM, Shorter J (2012) The surprising role of amyloid fibrils in HIV infection. Biology 1:58–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gray RH, Wawer MJ, Brookmeyer R, Sewankambo NK, Serwadda D, Wabwire‐Mangen F, Lutalo T, Li X, vanCott T, Quinn TC (2001) Probability of HIV‐1 transmission per coital act in monogamous, heterosexual, HIV‐1‐discordant couples in Rakai, Uganda. Lancet 357:1149–1153. [DOI] [PubMed] [Google Scholar]
- 5. Pilcher CD, Tien HC, Eron JJ, Jr , Vernazza PL, Leu SY, Stewart PW, Goh LE, Cohen MS (2004) Brief but efficient: acute HIV infection and the sexual transmission of HIV. J Infect Dis 189:1785–1792. [DOI] [PubMed] [Google Scholar]
- 6. Roan NR, Greene WC (2007) A seminal finding for understanding HIV transmission. Cell 131:1044–1046. [DOI] [PubMed] [Google Scholar]
- 7. Olsen JS, Brown C, Capule CC, Rubinshtein M, Doran TM, Srivastava RK, Feng C, Nilsson BL, Yang J, Dewhurst S (2010) Amyloid‐binding small molecules efficiently block SEVI (semen‐derived enhancer of virus infection)‐ and semen‐mediated enhancement of HIV‐1 infection. J Biol Chem 285:35488–35496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Muniyan S, Chaturvedi NK, Dwyer JG, Lagrange CA, Chaney WG, Lin MF (2013) Human prostatic acid phosphatase: structure, function and regulation. Int J Mol Sci 14:10438–10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Roan NR, Munch J, Arhel N, Mothes W, Neidleman J, Kobayashi A, Smith‐McCune K, Kirchhoff F, Greene WC (2009) The cationic properties of SEVI underlie its ability to enhance human immunodeficiency virus infection. J Virol 83:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roan NR, Sowinski S, Munch J, Kirchhoff F, Greene WC (2010) Aminoquinoline surfen inhibits the action of SEVI (semen‐derived enhancer of viral infection). J Biol Chem 285:1861–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee YH, Goto Y (2012) Kinetic intermediates of amyloid fibrillation studied by hydrogen exchange methods with nuclear magnetic resonance. Biochim Biophys Acta 1824:1307–1323. [DOI] [PubMed] [Google Scholar]
- 12. Knowles TP, Vendruscolo M, Dobson CM (2014) The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 15:384–396. [DOI] [PubMed] [Google Scholar]
- 13. Kotler SA, Walsh P, Brender JR, Ramamoorthy A (2014) Differences between amyloid‐beta aggregation in solution and on the membrane: insights into elucidation of the mechanistic details of Alzheimer's disease. Chem Soc Rev 43:6692–6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoshino M, Katou H, Hagihara Y, Hasegawa K, Naiki H, Goto Y (2002) Mapping the core of the beta(2)‐microglobulin amyloid fibril by H/D exchange. Nat Struct Biol 9:332–336. [DOI] [PubMed] [Google Scholar]
- 15. Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJ, McFarlane HT, Madsen AO, Riekel C, Eisenberg D (2007) Atomic structures of amyloid cross‐beta spines reveal varied steric zippers. Nature 447:453–457. [DOI] [PubMed] [Google Scholar]
- 16. Sievers SA, Karanicolas J, Chang HW, Zhao A, Jiang L, Zirafi O, Stevens JT, Munch J, Baker D, Eisenberg D (2011) Structure‐based design of non‐natural amino‐acid inhibitors of amyloid fibril formation. Nature 475:96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, Courtney JM, Kim JK, Barclay AM, Kendall A, Wan W, Stubbs G, Schwieters CD, Lee VM, George JM, Rienstra CM (2016) Solid‐state NMR structure of a pathogenic fibril of full‐length human alpha‐synuclein. Nat Struct Mol Biol 23:409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yoshimura Y, Lin Y, Yagi H, Lee YH, Kitayama H, Sakurai K, So M, Ogi H, Naiki H, Goto Y (2012) Distinguishing crystal‐like amyloid fibrils and glass‐like amorphous aggregates from their kinetics of formation. Proc Natl Acad Sci U S A 109:14446–14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ciryam P, Tartaglia GG, Morimoto RI, Dobson CM, Vendruscolo M (2013) Widespread aggregation and neurodegenerative diseases are associated with supersaturated proteins. Cell Rep 5:781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ling X, Fang W, Lee YH, Araujo PT, Zhang X, Rodriguez‐Nieva JF, Lin Y, Zhang J, Kong J, Dresselhaus MS (2014) Raman enhancement effect on two‐dimensional layered materials: graphene, h‐BN and MoS2. Nano Lett 14:3033–3040. [DOI] [PubMed] [Google Scholar]
- 21. Muta H, Lee YH, Kardos J, Lin Y, Yagi H, Goto Y (2014) Supersaturation‐limited amyloid fibrillation of insulin revealed by ultrasonication. J Biol Chem 289:18228–18238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Terakawa MS, Yagi H, Adachi M, Lee YH, Goto Y (2015) Small liposomes accelerate the fibrillation of amyloid beta (1–40). J Biol Chem 290:815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin Y, Kardos J, Imai M, Ikenoue T, Kinoshita M, Sugiki T, Ishimori K, Goto Y, Lee YH (2016) Amorphous aggregation of cytochrome c with inherently low amyloidogenicity is characterized by the metastability of supersaturation and the phase diagram. Langmuir 32:2010–2022. [DOI] [PubMed] [Google Scholar]
- 24. Kinoshita M, Kakimoto E, Terakawa MS, Lin Y, Ikenoue T, So M, Sugiki T, Ramamoorthy A, Goto Y, Lee YH (2017) Model membrane size‐dependent amyloidogenesis of Alzheimer's amyloid‐beta peptides. Phys Chem Chem Phys 19:16257–16266. [DOI] [PubMed] [Google Scholar]
- 25. Kundra R, Ciryam P, Morimoto RI, Dobson CM, Vendruscolo M (2017) Protein homeostasis of a metastable subproteome associated with Alzheimer's disease. Proc Natl Acad Sci U S A 114:E5703–E5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brender JR, Salamekh S, Ramamoorthy A (2012) Membrane disruption and early events in the aggregation of the diabetes related peptide IAPP from a molecular perspective. Acc Chem Res 45:454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Patel HR, Pithadia AS, Brender JR, Fierke CA, Ramamoorthy A (2014) In search of aggregation pathways of IAPP and other amyloidogenic proteins: finding answers through NMR spectroscopy. J Phys Chem Lett 5:1864–1870. [DOI] [PubMed] [Google Scholar]
- 28. Brender JR, Hartman K, Gottler LM, Cavitt ME, Youngstrom DW, Ramamoorthy A (2009) Helical conformation of the SEVI precursor peptide PAP248–286, a dramatic enhancer of HIV infectivity, promotes lipid aggregation and fusion. Biophys J 97:2474–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. French KC, Makhatadze GI (2012) Core sequence of PAPf39 amyloid fibrils and mechanism of pH‐dependent fibril formation: the role of monomer conformation. Biochemistry 51:10127–10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Olsen JS, DiMaio JT, Doran TM, Brown C, Nilsson BL, Dewhurst S (2012) Seminal plasma accelerates semen‐derived enhancer of viral infection (SEVI) fibril formation by the prostatic acid phosphatase (PAP248–286) peptide. J Biol Chem 287:11842–11849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hartman K, Brender JR, Monde K, Ono A, Evans ML, Popovych N, Chapman MR, Ramamoorthy A (2013) Bacterial curli protein promotes the conversion of PAP248–286 into the amyloid SEVI: cross‐seeding of dissimilar amyloid sequences. PeerJ 1:e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Elias AK, Scanlon D, Musgrave IF, Carver JA (2014) SEVI, the semen enhancer of HIV infection along with fragments from its central region, form amyloid fibrils that are toxic to neuronal cells. Biochim Biophys Acta 1844:1591–1598. [DOI] [PubMed] [Google Scholar]
- 33. Selmani V, Robbins KJ, Ivancic VA, Lazo ND (2015) K114 (trans, trans)‐bromo‐2,5‐bis(4‐hydroxystyryl)benzene is an efficient detector of cationic amyloid fibrils. Protein Sci 24:420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Easterhoff D, DiMaio JT, Doran TM, Dewhurst S, Nilsson BL (2011) Enhancement of HIV‐1 infectivity by simple, self‐assembling modular peptides. Biophys J 100:1325–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qiao X, Jeon J, Cole AL, Matos JO, Bautista S, Castillo J, Hung I, Gan Z, Tatulian SA, Cole AM, Chen B (2015) Morphology‐dependent HIV‐enhancing effect of semen‐derived enhancer of viral infection. Biophys J 108:2028–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhu M, Fink AL (2003) Lipid binding inhibits alpha‐synuclein fibril formation. J Biol Chem 278:16873–16877. [DOI] [PubMed] [Google Scholar]
- 37. Anderson VL, Ramlall TF, Rospigliosi CC, Webb WW, Eliezer D (2010) Identification of a helical intermediate in trifluoroethanol‐induced alpha‐synuclein aggregation. Proc Natl Acad Sci U S A 107:18850–18855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Neudecker P, Robustelli P, Cavalli A, Walsh P, Lundstrom P, Zarrine‐Afsar A, Sharpe S, Vendruscolo M, Kay LE (2012) Structure of an intermediate state in protein folding and aggregation. Science 336:362–366. [DOI] [PubMed] [Google Scholar]
- 39. Terakawa MS, Lee YH, Kinoshita M, Lin Y, Sugiki T, Fukui N, Ikenoue T, Kawata Y, Goto Y (2018) Membrane‐induced initial structure of alpha‐synuclein control its amyloidogenesis on model membranes. Biochim Biophys Acta 1860:757–766. [DOI] [PubMed] [Google Scholar]
- 40. Nanga RP, Brender JR, Xu J, Hartman K, Subramanian V, Ramamoorthy A (2009) Three‐dimensional structure and orientation of rat islet amyloid polypeptide protein in a membrane environment by solution NMR spectroscopy. J Am Chem Soc 131:8252–8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jakob CG, Lewinski K, Kuciel R, Ostrowski W, Lebioda L (2000) Crystal structure of human prostatic acid phosphatase. Prostate 42:211–218. [DOI] [PubMed] [Google Scholar]
- 42. Denisov IG, Sligar SG (2017) Nanodiscs in membrane biochemistry and biophysics. Chem Rev 117:4669–4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rodriguez Camargo DC, Korshavn KJ, Jussupow A, Raltchev K, Goricanec D, Fleisch M, Sarkar R, Xue K, Aichler M, Mettenleiter G, Walch AK, Camilloni C, Hagn F, Reif B, Ramamoorthy A (2017) Stabilization and structural analysis of a membrane‐associated hIAPP aggregation intermediate. Elife 6:e31226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ravula T, Hardin NZ, Ramadugu S, Cox SJ, Ramamoorthy A (2018) pH resistant monodispersed polymer‐lipid nanodiscs. Angew Chem Int Ed 57:1342–1345. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 45. Yasuhara K, Arakida J, Ravula T, Ramadugu SK, Sahoo B, Kikuchi JI, Ramamoorthy A (2017) Spontaneous lipid nanodisc fomation by amphiphilic polymethacrylate copolymers. J Am Chem Soc 139:18657–18663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Garg AB, Nuttall J, Romano J (2009) The future of HIV microbicides: challenges and opportunities. Antivir Chem Chemother 19:143–150. [DOI] [PubMed] [Google Scholar]
- 47. Capule CC, Brown C, Olsen JS, Dewhurst S, Yang J (2012) Oligovalent amyloid‐binding agents reduce SEVI‐mediated enhancement of HIV‐1 infection. J Am Chem Soc 134:905–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. LoRicco JG, Xu CS, Neidleman J, Bergkvist M, Greene WC, Roan NR, Makhatadze GI (2016) Gallic acid is an antagonist of semen amyloid fibrils that enhance HIV‐1 infection. J Biol Chem 291:14045–14055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Widera M, Klein AN, Cinar Y, Funke SA, Willbold D, Schaal H (2014) The D‐amino acid peptide D3 reduces amyloid fibril boosted HIV‐1 infectivity. AIDS Res Ther 11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pithadia AS, Bhunia A, Sribalan R, Padmini V, Fierke CA, Ramamoorthy A (2016) Influence of a curcumin derivative on hIAPP aggregation in the absence and presence of lipid membranes. Chem Commun 52:942–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sheik DA, Brooks L, Frantzen K, Dewhurst S, Yang J (2015) Inhibition of the enhancement of infection of human immunodeficiency virus by semen‐derived enhancer of virus infection using amyloid‐targeting polymeric nanoparticles. ACS Nano 9:1829–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sheik DA, Chamberlain JM, Brooks L, Clark M, Kim YH, Leriche G, Kubiak CP, Dewhurst S, Yang J (2017) Hydrophobic nanoparticles reduce the beta‐sheet content of SEVI amyloid fibrils and inhibit SEVI‐enhanced HIV infectivity. Langmuir 33:2596–2602. [DOI] [PubMed] [Google Scholar]
- 53. Ikenoue T, Lee YH, Kardos J, Saiki M, Yagi H, Kawata Y, Goto Y (2014) Cold denaturation of alpha‐synuclein amyloid fibrils. Angew Chem Int Ed Engl 53:7799–7804. [DOI] [PubMed] [Google Scholar]
- 54. Ikenoue T, Lee YH, Kardos J, Yagi H, Ikegami T, Naiki H, Goto Y (2014) Heat of supersaturation‐limited amyloid burst directly monitored by isothermal titration calorimetry. Proc Natl Acad Sci U S A 111:6654–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hauber I, Hohenberg H, Holstermann B, Hunstein W, Hauber J (2009) The main green tea polyphenol epigallocatechin‐3‐gallate counteracts semen‐mediated enhancement of HIV infection. Proc Natl Acad Sci U S A 106:9033–9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Popovych N, Brender JR, Soong R, Vivekanandan S, Hartman K, Basrur V, Macdonald PM, Ramamoorthy A (2012) Site specific interaction of the polyphenol EGCG with the SEVI amyloid precursor peptide PAP(248–286). J Phys Chem B 116:3650–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shorter J (2008) Hsp104: a weapon to combat diverse neurodegenerative disorders. Neurosignals 16:63–74. [DOI] [PubMed] [Google Scholar]
- 58. DeSantis ME, Leung EH, Sweeny EA, Jackrel ME, Cushman‐Nick M, Neuhaus‐Follini A, Vashist S, Sochor MA, Knight MN, Shorter J (2012) Operational plasticity enables hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell 151:778–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Castellano LM, Bart SM, Holmes VM, Weissman D, Shorter J (2015) Repurposing Hsp104 to antagonize seminal amyloid and counter HIV infection. Chem Biol 22:1074–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Martellini JA, Cole AL, Svoboda P, Stuchlik O, Chen LM, Chai KX, Gangrade BK, Sorensen OE, Pohl J, Cole AM (2011) HIV‐1 enhancing effect of prostatic acid phosphatase peptides is reduced in human seminal plasma. PLoS One 6:e16285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ghosh A, Pradhan N, Bera S, Datta A, Krishnamoorthy J, Jana NR, Bhunia A (2017) Inhibition and degradation of amyloid beta (Abeta40) fibrillation by designed small peptide: a combined spectroscopy, microscopy, and cell toxicity study. ACS Chem Neurosci 8:718–722. [DOI] [PubMed] [Google Scholar]
- 62. Korshavn KJ, Satriano C, Lin Y, Zhang R, Dulchavsky M, Bhunia A, Ivanova MI, Lee YH, La Rosa C, Lim MH, Ramamoorthy A (2017) Reduced lipid bilayer thickness regulates the aggregation and cytotoxicity of amyloid‐beta. J Biol Chem 292:4638–4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Castellano LM, Hammond RM, Holmes VM, Weissman D, Shorter J (2015) Epigallocatechin‐3‐gallate rapidly remodels PAP85–120, SEM1(45–107), and SEM2(49–107) seminal amyloid fibrils. Biol Open 4:1206–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Volpatti LR, Vendruscolo M, Dobson CM, Knowles TP (2013) A clear view of polymorphism, twist, and chirality in amyloid fibril formation. ACS Nano 7:10443–10448. [DOI] [PubMed] [Google Scholar]
- 65. Sidhu A, Segers‐Nolten I, Subramaniam V (2014) Solution conditions define morphological homogeneity of alpha‐synuclein fibrils. Biochim Biophys Acta 1844:2127–2134. [DOI] [PubMed] [Google Scholar]
- 66. Kinoshita M, Lin Y, Nakatsuji M, Inui T, Lee YH (2017) Kinetics and polymorphs of yeast prion Sup35NM amyloidogenesis. Int J Biol Macromol 102:1241–1249. [DOI] [PubMed] [Google Scholar]
- 67. Goldschmidt L, Teng PK, Riek R, Eisenberg D (2010) Identifying the amylome, proteins capable of forming amyloid‐like fibrils. Proc Natl Acad Sci U S A 107:3487–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, Fuxreiter M, Gough J, Gsponer J, Jones DT, Kim PM, Kriwacki RW, Oldfield CJ, Pappu RV, Tompa P, Uversky VN, Wright PE, Babu MM (2014) Classification of intrinsically disordered regions and proteins. Chem Rev 114:6589–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta‐peptide. Nat Rev Mol Cell Biol 8:101–112. [DOI] [PubMed] [Google Scholar]
- 70. Vivekanandan S, Brender JR, Lee SY, Ramamoorthy A (2011) A partially folded structure of amyloid‐beta(1–40) in an aqueous environment. Biochem Biophys Res Commun 411:312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hamley IW (2012) The amyloid beta peptide: a chemist's perspective. Role in Alzheimer's and fibrillization. Chem Rev 112:5147–5192. [DOI] [PubMed] [Google Scholar]
- 72. Citron M (2010) Alzheimer's disease: strategies for disease modification. Nat Rev Drug Discov 9:387–398. [DOI] [PubMed] [Google Scholar]
- 73. Trainor K, Broom A, Meiering EM (2017) Exploring the relationships between protein sequence, structure and solubility. Curr Opin Struct Biol 42:136–146. [DOI] [PubMed] [Google Scholar]
- 74. Schrader T, Bitan G, Klarner FG (2016) Molecular tweezers for lysine and arginine—powerful inhibitors of pathologic protein aggregation. Chem Commun 52:11318–11334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Eisele YS, Monteiro C, Fearns C, Encalada SE, Wiseman RL, Powers ET, Kelly JW (2015) Targeting protein aggregation for the treatment of degenerative diseases. Nat Rev Drug Discov 14:759–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yoshihara H, Saito J, Tanabe A, Amada T, Asakura T, Kitagawa K, Asada S (2016) Characterization of novel insulin fibrils that show strong cytotoxicity under physiological pH. J Pharm Sci 105:1419–1426. [DOI] [PubMed] [Google Scholar]
- 77. Xun T, Li W, Chen J, Yu F, Xu W, Wang Q, Yu R, Li X, Zhou X, Lu L, Jiang S, Li L, Tan S, Liu S (2015) ADS‐J1 inhibits semen‐derived amyloid fibril formation and blocks fibril‐mediated enhancement of HIV‐1 infection. Antimicrob Agents Chemother 59:5123–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Li M, Dong X, Liu Y, Sun Y (2017) Brazilin inhibits prostatic acidic phosphatase fibrillogenesis and decreases its cytotoxicity. Chem Asian J 12:1062–1068. [DOI] [PubMed] [Google Scholar]
- 79. Ratha BN, Ghosh A, Brender JR, Gayen N, Ilyas H, Neeraja C, Das KP, Mandal AK, Bhunia A (2016) Inhibition of insulin amyloid fibrillation by a novel amphipathic heptapeptide: mechanistic details studied by spectroscopy in combination with microscopy. J Biol Chem 291:23545–23556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ramamoorthy A, Lim MH (2013) Structural characterization and inhibition of toxic amyloid‐beta oligomeric intermediates. Biophys J 105:287–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pithadia A, Brender JR, Fierke CA, Ramamoorthy A (2016) Inhibition of IAPP aggregation and toxicity by natural products and derivatives. J Diabetes Res 2016:2046327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Krotee P, Griner SL, Sawaya MR, Cascio D, Rodriguez JA, Shi D, Philipp S, Murray K, Saelices L, Lee J, Seidler P, Glabe CG, Jiang L, Gonen T, Eisenberg DS (2018) Common fibrillar spines of amyloid‐beta and human islet amyloid polypeptide revealed by micro electron diffraction and inhibitors developed using structure‐based design. J Biol Chem 293:2888–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]