

Abstract
Transthyretin (TTR) is a homotetrameric protein that is found in the plasma and cerebrospinal fluid. Dissociation of TTR tetramers sets off a downhill cascade of amyloid formation through polymerization of monomeric TTR. Interestingly, TTR has an additional, biologically relevant activity, which pertains to its ability to slow the progression of amyloid beta (Aβ) associated pathology in transgenic mice. In vitro, both TTR and a kinetically stable variant of monomeric TTR (M‐TTR) inhibit the fibril formation of Aβ1‐40/42 molecules. Published evidence suggests that tetrameric TTR binds preferentially to Aβ monomers, thus destabilizing fibril formation by depleting the pool of Aβ monomers from aggregating mixtures. Here, we investigate the effects of M‐TTR on the in vitro aggregation of Aβ1‐42. Our data confirm previous observations that fibril formation of Aβ is suppressed in the presence of sub‐stoichiometric amounts of M‐TTR. Despite this, we find that sub‐stoichiometric levels of M‐TTR are not bona fide inhibitors of aggregation. Instead, they co‐aggregate with Aβ to promote the formation of large, micron‐scale insoluble, non‐fibrillar amorphous deposits. Based on fluorescence correlation spectroscopy measurements, we find that M‐TTR does not interact with monomeric Aβ. Two‐color coincidence analysis of the fluorescence bursts of Aβ and M‐TTR labeled with different fluorophores shows that M‐TTR co‐assembles with soluble Aβ aggregates and this appears to drive the co‐aggregation into amorphous precipitates. Our results suggest that mimicking the co‐aggregation activity with protein‐based therapeutics might be a worthwhile strategy for rerouting amyloid beta peptides into inert, insoluble, and amorphous deposits.
Keywords: amyloid beta, transthyretin, fluorescence correlation spectroscopy, fibrils, co‐aggregation
Introduction
The misfolding of soluble monomeric proteins into stable amyloid fibers is linked to a wide number of neurodegenerative diseases.1 The most well‐known of these is Alzheimer's disease (AD), which affects over five million individuals in the US alone.2 A primary hallmark of AD is the presence of amyloid deposits consisting of proteolytic fragments of the amyloid precursor protein (APP), with the major species being the 40 or 42 residues variants of this peptide designated as amyloid beta (Aβ1–40/42) peptide.3 Since their initial identification, the Aβ1–40/42 systems have been the focus of in vitro and in cell investigations with particular focus on the driving forces and mechanisms of Aβ1–40/42 aggregation.1
Aβ1–40/42 is one of many proteins that undergo misfolding and aggregation to form amyloid fibrils that are also associated with neurodegenerative or systematic disorders. Among the earliest examples of a protein whose misfolding and aggregation were directly linked to disease is the homotetrameric protein transthyretin (TTR), a protein carrier of the thyroid hormone that is abundant in human serum and cerebrospinal fluid (CSF).4, 5 Interestingly, TTR is also relevant to the pathophysiology of Aβ1–40/42 in AD as it is one of three CSF proteins originally identified by Goldgaber et al. to interact with Aβ peptides.6 The other two proteins are apolipoproteins (Apo‐) E and J.6 Of the three, TTR is the most abundant, and is found at concentrations 3‐ to 10‐times higher than Apo‐E and Apo‐J, respectively.7 Early studies suggested that TTR appears to be the principal Aβ sequestering protein.8 In later work, when TTR was co‐expressed as a transgene in muscles of C. elegans expressing Aβ regulated by the same promoter, it suppressed the paralysis phenotype seen when the worms were transgenic only for Aβ.9, 10 It was subsequently shown that genetically programmed tissue‐specific over‐expression of wild‐type human TTR suppressed the behavioral and neuropathologic abnormalities seen in the APP23 transgenic mouse model of human Aβ deposition.11 Additionally, when APP23 and APPswe/PS1ΔE9 transgenic AD model mice were crossed with TTR knockouts, the pace of development of Aβ deposition was accelerated.11, 12
In vitro studies have shown that the wild‐type TTR protein delays or reduces Aβ fibril formation.8, 13, 14, 15, 16, 17, 18, 19, 20, 21 As noted above, TTR can itself drive amyloid formation. The obligatory step driving TTR amyloid fibril formation is the dissociation of tetrameric TTR into monomers.22, 23 Kelly and coworkers have established that dissociation of TTR tetramers is the rate‐limiting step because amyloid formation proceeds as a downhill process upon tetrameter dissociation.4, 5, 24, 25, 26, 27, 28, 29, 30, 31 Recent work showed a genetically engineered, kinetically stable monomeric variant of TTR, designated as M‐TTR,32 also suppresses Aβ fibril formation.18 M‐TTR is different from the monomeric form of TTR that is derived by dissociation of the wild‐type tetramer. The mutant form is a kinetically stable monomer that does not readily aggregate as compared to the monomeric variant of the wild‐type TTR. Interestingly, published studies suggest that M‐TTR is more effective as an inhibitor of Aβ fibril formation than the wild‐type tetramer.17, 18 Additionally, M‐TTR was observed to reduce oligomeric induced toxicity of Aβ to a greater extent than tetrameric TTR in human neuroblastoma cells and rat primary neurons.33
In the context of the CSF, both the tetrameric and monomeric forms of wild‐type TTR have significance, albeit for different reasons. The tetramer is the functional form, whereas the monomer is the pathological form. However, the ability of M‐TTR to inhibit the formation of Aβ fibrils raises the intriguing possibility that tetramer dissociation and the consequent accrual of monomers might have a protective role from the standpoint of Aβ toxicity. Suppression of Aβ fibril formation could also be an alternative way to protect against the toxicity of monomeric TTR while also slowing the progression of Aβ pathology. In light of these intriguing possibilities, which remain conjectures, we sought to obtain a more complete mechanistic understanding of the differences or similarities between how TTR and M‐TTR impact Aβ aggregation. This work builds on previous studies, which suggest that TTR and perhaps M‐TTR act as sensors and scavengers of Aβ oligomers.19
Our focus here is on the Aβ1–42 (Aβ42) alloform since it is the more toxic of the two prominent Aβ species (Aβ1–40 vs. Aβ1–42). Recent studies showed that while the TTR homotetramer binds Aβ1–40/42 monomers, as detected by isothermal titration calorimetry (ITC), M‐TTR does not bind monomeric forms of Aβ1–40/42. Nuclear magnetic resonance (NMR) analysis of mixtures of Aβ monomers and TTR tetramers showed shifted resonances in the TTR molecules, while similar experiments with M‐TTR revealed no detectable interactions.18 These results suggest that the inhibition of Aβ fibril formation might be different for the two TTR species, although the nature of these differences remains unclear.18
Here, we examine how the genetically engineered M‐TTR alters the complex aggregation behavior of Aβ42 in vitro. Our results show that while sub‐stoichiometric levels of M‐TTR suppress Aβ42 fibril formation, these molecules promote the formation of large, micron‐scale amorphous aggregates through a co‐aggregation mechanism. We uncover the mechanistic underpinnings that lead to the co‐aggregation of M‐TTR with Aβ42 and the resulting amorphous deposits. The potent inhibition of fibril formation exhibited by M‐TTR may serve as a model for understanding how molecules can be diverted and re‐routed from fibril formation pathways. The formation of inert amorphous co‐aggregates may be a desirable outcome when compared to the formation of protofibrils and fibrils that can act as seeds and recruit soluble molecules and/or serve as infectious, prion‐like agents.34, 35, 36 Our mechanistic insights regarding M‐TTR may help with the design of protein‐based therapeutics that divert Aβ42 molecules from fibril forming pathways.
Results
Effect of M‐TTR on Aβ42 fibril formation
We examined the effect of M‐TTR on the kinetics of fibril formation of Aβ42 by monitoring the rate of increase in thioflavin T (ThT) fluorescence. Figure 1 shows a representative trace for the rate of increase in ThT fluorescence as measured in a solution comprising of 8 µM Aβ42. This kinetic trace shows the characteristic lag and growth phase that is suggestive of a complex nucleated polymerization mechanism for fibril formation. In the presence of 4 µM M‐TTR, we observed a 13‐fold decrease in the ThT fluorescence at the endpoint of the reaction when compared to the absence of M‐TTR. These data are reproducible in varying, sub‐stoichiometric amounts of M‐TTR incubated with Aβ42 at the start of the reaction, thus demonstrating that M‐TTR does indeed suppress fibril formation of Aβ42, in accord with previous findings.18
Figure 1.
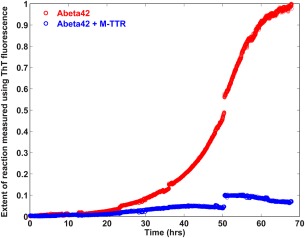
Effect of M‐TTR on Aβ42 fibril formation. Time course of ThT fluorescence of 8 µM unlabeled Aβ42 in absence of M‐TTR and in the presence of 4 µM M‐TTR in PBS, pH 7.4 at room temperature. The mixtures were removed from the fluorimeter at regular intervals and vortexed thoroughly to resuspend the precipitates. The break points in the figure reflect partial precipitation of the solutions in the growth phase and subsequent recovery upon vortexing. The solutions were stirred continuously. The data show that M‐TTR dramatically reduces fibril formation of Aβ42.
Effect of M‐TTR on fibrillar versus non‐fibrillar aggregation of Aβ42
ThT fluorescence is insensitive to the formation of non‐fibrillar aggregates. To measure the possibility of other modes of aggregation, we monitored the rate of loss of tetramethylrhodamine (TMR) fluorescence using dye‐labeled Aβ42 molecules.37 We used labeled Aβ42 molecules where the TMR is covalently attached to a lysine residue on the N‐terminus of Aβ42. TMR molecules self‐quench when they are brought into close proximity, in this case by the oligomerization and/or aggregation of the Aβ42 peptides to which the TMR molecules are attached.37 Unlike the ThT assay, the rate of loss of TMR fluorescence is not limited to the detection of amyloid fibril formation. Instead, any assembly or aggregation process that brings the TMR molecules into close proximity will lead to a quenching of its fluorescence.37, 38 This assay is therefore sensitive to the presence of amyloid‐like and non‐amyloid‐like aggregates and the sensitivity range will span the gamut from oligomers to large aggregates comprising of thousands of molecules. Figure 2 shows a comparison of the rates of loss of TMR fluorescence of TMR‐Aβ42 (2 µM) in the absence versus presence of M‐TTR (1 µM). The kinetics of aggregation of Aβ42 in the absence and presence of M‐TTR are quite similar in the early phases of the reaction, as monitored by the rates of loss of TMR fluorescence. However, we observe significant deviations between the two traces during the growth phase—a feature that is reproducible as shown in Supporting Information, Figure S1. The “growth phase” in a TMR fluorescence assay quenching experiment corresponds to the formation of large aggregates, and this refers to species that are on the order of 1 µm or larger. The apparent aggregation rate in the growth phase is slowed in the presence of M‐TTR.
Figure 2.
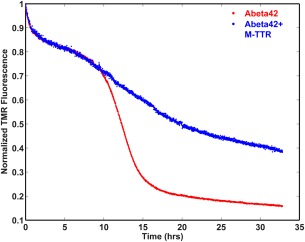
Effect of M‐TTR on overall aggregation kinetics of Aβ42 as probed using the rate of loss of TMR fluorescence. Kinetics of fluorescence of 2 µM TMR‐Aβ42 in the presence or absence of 1 µM M‐TTR in PBS, pH 7.4 buffer at room temperature with continuous stirring. The oligomerization and the lag phases are similar even in presence of M‐TTR. However, the growth phase is considerably slower in presence of M‐TTR. Figure S1 shows the reproducibility of these traces from independent measurements, each with different, albeit sub‐stoichiometric amounts of M‐TTR.
M‐TTR does not significantly alter the overall solubility of Aβ42
The impact of a ligand on aggregation and phase separation can be quantified by measuring the effect of the ligand on the saturation concentration for the large‐scale assembly.39 The saturation concentration c s is the concentration of soluble species including monomers and other soluble aggregates that are in equilibrium with the insoluble aggregates.38, 40 We used the intrinsic TMR fluorescence at the end of the aggregation reaction, measured after incubation for four days to estimate the values of c s for Aβ42 in the absence versus presence of M‐TTR. The results are shown in Figure 3(A). We find that the value of c s for Aβ42 increases from ∼335 nM in the absence of M‐TTR to ∼485 nM in the presence of M‐TTR. Thus, M‐TTR increases the solubility of monomeric Aβ42 molecules by a factor of approximately 1.5, which is a modest effect when compared to the >13‐fold reduction in the extent of Aβ42 fibril formation (Fig. 1). This suggests that M‐TTR does not suppress aggregation per se, although it does suppress fibril formation.
Figure 3.
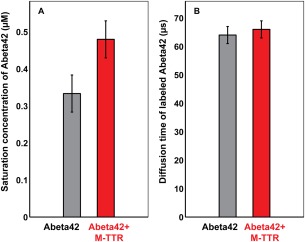
Impact of M‐TTR on Aβ42 solubility and the properties of monomeric Aβ42. Panel A shows the measured saturation concentrations of Aβ42 in the absence and presence of M‐TTR. Panel B shows the measured diffusion times of Aβ42 in the absence and presence of M‐TTR. The error bars in all measurements are standard errors in our estimate of the mean derived from three independent measurements.
Effect of M‐TTR on monomeric Aβ42
To assess the interaction between M‐TTR and Aβ42 we performed fluorescence correlation spectroscopy (FCS) measurements of Alexa488‐labeled Aβ42. Measurements were made in the absence and presence of M‐TTR with freshly purified monomeric Aβ42 isolated by size exclusion chromatography in 3 mM NaOH (see Methods). Experiments were carried out rapidly following NaOH dilution, before the Aβ42 monomer pool could be significantly depleted. If M‐TTR were to bind monomeric Aβ42, we would expect to see a measurable increase in the diffusion time of the fluorescently labeled Aβ42. Figure 3(B) shows that the diffusion of Alexa488‐Aβ42 at time t = 0 is ∼65 µs. This diffusion time, which is in accord with previous measurements,41 does not change in the presence of M‐TTR. Thus, the binding of M‐TTR to predominantly monomeric Aβ42 that would lead to a 1:1 complex must be weak or negligible. These results are consistent with results showing that the early stages of Aβ42 aggregation are not impacted by the presence of M‐TTR (Fig. 2).
Effect of M‐TTR on the morphology of the Aβ42 aggregates
The data presented above indicate that M‐TTR destabilizes Aβ42 fibrillar species while allowing for or even promoting other forms of aggregation. To test this hypothesis, we first used transmission electron microscopy (TEM) to examine the morphology of aggregates collected from the end points of the two experiments summarized in Figure 1. In the absence of M‐TTR, Aβ42 forms fibrils that are several microns long and 6–10 nm wide [Fig. 4(A)]. However, when Aβ42 is incubated with M‐TTR, the aggregates formed are non‐fibrillar [Fig. 4(B)]. The TEM images do not reveal whether these are homotypic aggregates of Aβ42 and/or M‐TTR or co‐aggregates of the two molecules. To answer this question, we used confocal fluorescence microscopy to test for colocalization of M‐TTR and Aβ42 in the precipitates of the M‐TTR‐Aβ42 mixed sample described above. We used samples with mixtures of TMR‐labeled Aβ42, unlabeled Aβ42, Alexa488‐labeled M‐TTR, and unlabeled M‐TTR (see Methods). As shown in the representative images of Figure 5, M‐TTR and Aβ42 are co‐localized in the amorphous aggregates.
Figure 4.

Morphology of the Aβ42 aggregates. (A) Morphology of Aβ42 in the absence and (B) presence of M‐TTR. In the presence of M‐TTR we observe large, micron‐scale amorphous aggregates.
Figure 5.
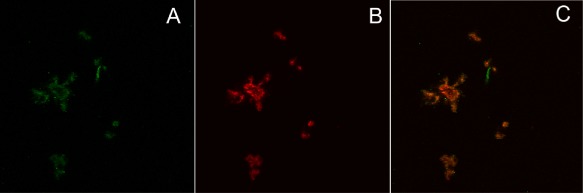
Coprecipitation of Aβ42 and M‐TTR. Confocal microscopy images of precipitates from a solution of 8 µM unlabeled Aβ42 + 0.4 µM TMR‐Aβ42 + 4 µM unlabeled M‐TTR + 0.2 µM Alexa488‐M‐TTR. A and B are images in the M‐TTR (green) and TMR (red) channels whereas C shows the overlap of the two images. The data in C show that Aβ42 and M‐TTR are largely co‐localized in the precipitates. There are regions within the M‐TTR channel that do not overlap with Aβ42. This is attributable to aggregation of M‐TTR that appears to occur independently of its interactions with Aβ42.
Effect of M‐TTR on the soluble species of Aβ42
Next, we investigated the interactions of M‐TTR with soluble species of Aβ42. Supernatants from samples containing Alexa488‐labeled Aβ42 molecules (0.4 µM) and unlabeled Aβ42 molecules (8 µM) were studied using fluorescence burst analysis and FCS in the absence and presence of M‐TTR (4 µM). Fluorescence burst analysis employs an FCS setup to observe the intensity and frequency (photon counts as a function of time) of fluorophores passing through the confocal volume (bursts). The photon count is proportional to the number of fluorescent molecules passing through the confocal volume, thus providing an assessment of aggregate size. The frequency of bursts of a given magnitude provides a measure of the concentration of aggregates of that size. Figure 6(A) shows the photon count trace obtained from the supernatant of Aβ42 alone. The mean photon count is 0.1 MHz but a large number of fluorescence bursts with counts up to 5 MHz can be observed in this trace. The bursts are produced by soluble aggregates of Aβ42. Figure 6(B) shows the trace of the photon counts from the supernatant of the Aβ42 + M‐TTR mixture. In the presence of M‐TTR there are fewer fluorescent bursts, and those that occur have a lower photon count. Taken together, these results suggest that the concentration of soluble aggregates of Aβ42 is significantly reduced in the presence of M‐TTR.
Figure 6.
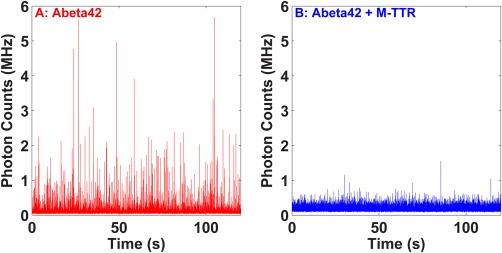
Effect of M‐TTR on the soluble aggregates of Aβ42. Photon count traces from the supernatant of a 0.4 µM Alexa488‐Aβ42 + 8 µM unlabeled Aβ42 solution following aggregation (A) in absence and B) in presence of 4 µM M‐TTR. The solutions were prepared in PBS, pH 7.4 buffer. Incubation was at room temperature with continuous stirring. The supernatants were collected following centrifugation at 2000g. The fluorescence bursts indicate the presence of soluble aggregates of Aβ42. M‐TTR reduces the population of soluble aggregates of Aβ42 in the supernatants.
M‐TTR binds preferentially to soluble Aβ42 aggregates
We investigated whether M‐TTR and Aβ42 interact at the oligomeric level. To examine this possibility, we used two‐color fluorescence burst and coincidence analysis on a sample consisting of Alexa488‐labeled M‐TTR and TMR‐labeled Aβ42. This assay is similar to the fluorescence burst analysis previously described, except that the sample components are labeled with two different fluorophores and the sample is assayed for coincident bursts in the two fluorescent channels. Coincident bursts indicate co‐localization of the constituent biomolecules. We mixed 400 nM TMR‐Aβ42 and 200 nM Alexa488‐M‐TTR with 8 µM unlabeled‐Aβ42 and 4 µM unlabeled M‐TTR. Figure 7 shows the photon count traces in TMR and the Alexa488 channels, respectively. The fluorescence bursts observed in the Alexa488 channel (which are of M‐TTR) correlate with those observed in the TMR channel (which are of Aβ42). Co‐localization of M‐TTR and Aβ42 points to interactions, albeit of unresolved stoichoimetries, between M‐TTR and soluble aggregates of Aβ42 that would appear to be a heterogenous mixture of species.
Figure 7.
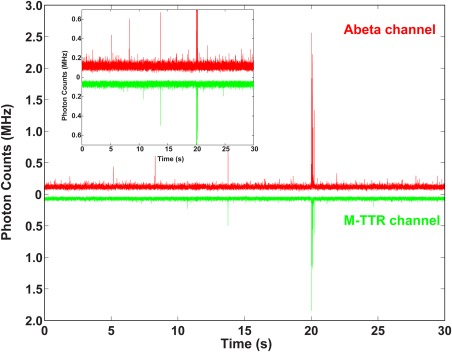
Coincidence of fluorescence bursts of Aβ42 and M‐TTR. Photon counts trace of a Aβ42‐M‐TTR solution in Aβ42 channel (red) and in M‐TTR channel (green) from a solution with 8 µM unlabeled Aβ42 + 0.4 µM TMR‐Aβ42 + 4 µM unlabeled M‐TTR + 0.2 µM Alexa488‐M‐TTR. Fluorescence bursts indicate the presence of the soluble aggregates in the solution. The data show that a fraction of the population of the soluble aggregates contains both Aβ42 and M‐TTR.
Discussion
The overall picture that emerges from our study is as follows: M‐TTR suppresses fibril formation of Aβ42 but it does not bind to monomeric Aβ42, nor does it impact the earliest stages of aggregation. Additionally, M‐TTR has a minimal effect on the solubility limit of Aβ42. Instead, it appears that the ability of M‐TTR to suppress fibril formation of Aβ42 derives from its ability to bind preferentially to soluble aggregates of Aβ42. These soluble aggregates may well be precursors of fibril formation or provide sites for secondary nucleation.42, 43, 44, 45 Our data are consistent with the soluble aggregates being diverted from the fibril‐forming pathway as evidenced by the formation of large, amorphous co‐aggregates with M‐TTR. Therefore, we conclude that instead of being an inhibitor of Aβ42 aggregation, M‐TTR suppresses fibril formation and re‐routes soluble aggregates of Aβ42 molecules into co‐aggregates that are non‐fibrillar and amorphous.
Our findings are concordant with so‐called polysteric linkage that has been enunciated in the classical literature on binding and linkage phenomena,39, 46 and in the context of nucleic acid binding proteins.47 This refers to the preferential binding of a ligand to heterogeneous mixtures of soluble aggregates of specific or undefined sizes. Polysteric linkage is a specialized circumstance of polyphasic linkage, where the former refers to preferential binding to specific species without necessarily altering phase boundaries, whereas polyphasic linkage refers to preferential binding that leads to changes in the positions of phase boundaries.39 The concept of polyphasic linkage was recently demonstrated in the context of profilin binding to exon1 spanning constructs of the protein hutingtin.48
The FCS analysis presented here using labeled M‐TTR and Aβ42 provides insights regarding the interactions that cause Aβ42 to supersaturate the soluble phase. Since M‐TTR is larger than Aβ42 monomer (13.8 vs. 4.5 kDa), the stronger Aβ42 signal in the complexes indicates that high Aβ42/M‐TTR molar ratios are most consistent with larger soluble aggregates of Aβ42 being bound by small oligomers (or monomers) of M‐TTR. A precise analysis of the size distribution is confounded by the bleed through from the TMR channel into the Alexa488 channel, which is the result of spectral overlap. Finding dyes that are bright and have minimal overlap is challenging, but new analysis methods and time‐dependent coincidence measurements should allow us to uncover the precise size distributions of Aβ42 aggregates that are targeted by M‐TTR. We observed several fluorescence bursts in the Aβ42 channel that were not associated with corresponding bursts in the M‐TTR channel. This is possibly indicative of soluble aggregates of Aβ42 that were not bound by M‐TTR. Alternatively, it is possible that since the fluorescence bursts are only visible when there are sufficient numbers of labeled molecules in an aggregates, the co‐localization of smaller oligomers may not be detectable. In other words, a soluble aggregate of Aβ42 with a single unlabeled M‐TTR bound would not register coincident bursts and would therefore appear (incorrectly) to be an Aβ42 species that is not bound to M‐TTR.
Our data can be used for crude quantitative estimates of the sizes of the species that are being recognized by M‐TTR. The average brightness quantified in terms of the photon counts per molecule would be 10 kHz for a typical Aβ42 monomer. Figure 6 shows that the photon bursts correspond to counts of ∼1 MHz, which is two orders of magnitude larger than the counts for monomeric Aβ42. Accounting for the fact that only 5% of the Aβ42 molecules are fluorescently labeled, we estimate that the species being recognized by M‐TTR include, on average, approximately 103 Aβ42 molecules. Such species would be analogous to the micellar species observed for Aβ49 and/or precursors of fibrillar species that are about 100 nm in diameter,50 or secondary nuclei that have been proposed to be the determinants of the lag‐phase in Aβ aggregation.42, 43, 44, 45, 51 In this regard, it is worth noting that species in the 25–50 nm range have been observed for huntingtin exon 1 (Httex1) spanning fragments and have been designated as S‐phase species. It is possible that the species that are preferentially recognized by M‐TTR are analogous to the recently observed S‐phase species of Httex1.48
The fact that monomeric TTR (as opposed to the genetically engineered constitutive monomer M‐TTR used in this study) can inhibit fibril formation by Aβ and possibly other amyloid precursors suggests that there may be a general ability for aggregation‐prone proteins to interact in a heterotypic manner, whereas conformational specificity is required for nucleation and templating that leads to fibril formation via homotypic associations.52 We conjecture that oligomers of M‐TTR might display peptide structures that resemble Aβ sequences to interfere with the specific interactions required for fibrils, generating aggregates that are less likely to form fibrils.
Our results are analogous to observations in the field of inorganic crystal formation.53 Mixtures of calcium and orthophosphate will rapidly undergo crystallization to form predominantly needle‐like crystals, with a small number of amorphous aggregates. However, in the presence of several different phosphate and phosphonate salts, the same reaction forms amorphous structures devoid of crystallinity, as indicated by electron microscopy and electron diffraction intensities. These amorphous structures are similar to the early stage assemblies observed during “rapid mixing” experiments of calcium and orthophosphate.54 Since amyloids are semi‐crystalline, we suggest that the introduction of co‐precipitating species is likely a general mechanism to inhibit crystallization, in which these co‐precipitants act to either kinetically or thermodynamically inhibit the conformational re‐arrangements needed to form ordered solids.
The potent inhibition of fibril formation by M‐TTR can serve as a model for understanding how amyloid pathways can be reshaped. Indeed, TTR and M‐TTR have been shown to prevent amyloid formation in other amyloid‐prone proteins, including HypF‐N55 and the curli protein CsgA, a bacterial amyloid forming protein that is responsible for biofilm formation.56 Human wild‐type tetrameric TTR and the engineered M‐TTR inhibit CsgA amyloid formation in vitro. They also inhibit amyloid‐dependent biofilm formation in two different bacterial species. As in other studies, M‐TTR was found to be more potent as an inhibitor of amyloid formation. The fact that TTR is able to prevent amyloid fibril formation in multiple, apparently unrelated amyloidogenic proteins suggests that there are general principles to be gleaned from its mechanism of action. Importantly, these principles are likely to be broadly applicable.
Materials and Methods
Preparation of Aβ42 and TTR
Unlabeled and TMR‐labeled Aβ1–42 peptides, which were chemically synthesized, were purchased from Keck Foundation (Yale University). The peptides were purified by reverse‐phase liquid chromatography using a C18 column in water/acetonitrile media. The purified peptides were lyophilized and resuspended in 6 M GdmHCl. The Aβ42 peptides were further purified by size exclusion chromatography using a Superdex peptide column (GE Healthcare) in 3 mM NaOH. The predominantly monomeric fraction was selected for subsequent experiments. Engineered recombinant monomeric human TTR (F87M/L110M TTR) was produced in an E. coli expression system.32 The protein was purified by gel filtration on a Superdex‐75 column (Amersham‐Pharmacia, Sweden) in 10 mM phosphate buffer (sodium) pH 7.6, 100 mM KCl, 1 mM EDTA and stored at 4°C at a concentration of 0.1–0.5 mg/mL in the dark. The protein was analyzed on a LC‐ESI mass spectrometer to confirm its nature by molecular weight and re‐purified by gel filtration 1–3 days before the assays to ensure that no aggregates were present in the starting material.22 The final M‐TTR solution (42 µM) was prepared in phosphate buffered saline (PBS) at pH 7.4.
Measurement of aggregation kinetics
A 40 µM stock solution of unlabeled Aβ42 or 20 µM stock solution of TMR‐labeled Aβ42 (TMR‐Aβ42) prepared in 3 mM NaOH was diluted to the desired final concentration into PBS pH 7.4 containing 1 mM EDTA, and 5 mM β‐mercaptoethanol (βME). We measured the kinetics of fibril formation by quantifying the gain in ThT fluorescence. The concentration of ThT used was 2 µM. We measured the kinetics of overall aggregation using the TMR fluorescence of TMR‐Aβ42. The samples were incubated inside the fluorimeter in a clean glass test tube with continuous stirring in a temperature‐controlled cuvette holder. Aggregation of unlabeled and TMR‐labeled Aβ42 samples were monitored continuously using fluorescence of ThT (excitation wavelength of 438 nm, emission maximum at 480 nm) or TMR (excitation wavelength of 520 nm, emission maximum of 600 nm). All samples were stirred continuously using a micro stir bar.
Measurement of saturation concentration of Aβ42
Aβ42 peptides, 2 µM of TMR‐Aβ42, in the absence or presence of 1 µM M‐TTR prepared in PBS buffer at pH 7.4 containing 1 mM EDTA, 5 mM βME were incubated for four days at room temperature with continuous stirring. The solutions were centrifuged at 2000g for 20 min. The TMR fluorescence of the supernatants was then recorded. Concentrations of the TMR‐Aβ42 were determined by comparing their fluorescence with that of known concentrations of free TMR. Note that measurements of aggregation kinetics using TMR quenching indicate that aggregate formation plateaus by 24 h, thus four days is sufficient time to reach equilibrium.
Measurement of diffusion time using FCS
Fresh solutions of 400 nM Alexa488‐Aβ42, 4 µM unlabeled Aβ42 were mixed with or without 4 µM unlabeled TTR for FCS measurements. The diffusion times were obtained from the fit of the autocorrelation data using a single diffusion model.
Transmission electron microscopy of aggregates
Aβ42 (8 µM) was incubated in absence or in presence of 4 µM M‐TTR in PBS buffer at pH 7.4 containing 1 mM EDTA, 5 mM βME, and 2 µM ThT for two days at RT with continuous stirring. The aggregation of this solution was monitored using ThT fluorescence. The aggregates were then resuspended in the solution by a brief vortexing. A 10 µL droplet was placed on a Formvar carbon‐coated 200 mesh copper grid (Electron Microscopy Sciences) and allowed to adsorb for 1 min. The grid was washed twice in distilled water. Finally, the grid was negatively stained in 0.5% uranyl acetate for 1 min and then dried in a desiccator overnight at room temperature. The images were collected using a JEOL 100CX transmission electron microscope equipped with an AMT digital camera.
Confocal microscopy of the M‐TTR‐aβ precipitates
Co‐aggregation of soluble Aβ42 and M‐TTR was measured using confocal microscopy in a Zeiss Confocor 2. For these measurements 400 nM TMR‐Aβ42 + 200 nM Alexa488‐M‐TTR + 8 µM unlabeled Aβ42 + 4 µM unlabeled M‐TTR were incubated in PBS buffer at pH 7.4 containing 1 mM EDTA and 5 mM βME were incubated at RT with continuous stirring for two days. A 40 µL aliquot of this sample was placed on a glass cover slip for 1 h prior to imaging. The fluorescence imaging of Alexa488 and TMR were performed in two separate detection channels.
Fluorescence burst analysis
Alexa488‐labeled Aβ42 (400 nM) mixed with unlabeled Aβ42 (8 µM) in PBS buffer at pH 7.4 containing 1 mM EDTA, 5 mM βME, and 2 µM ThT for four days at room temperature with continuous stirring. The aggregation was monitored continuously in the fluorimeter. The solution was centrifuged at 2000g for 20 min. The supernatant was collected for the fluorescence burst measurement by FCS. FCS measurements were performed in a Zeiss confocor 2 microscope equipped with the FCS capability. The photon count traces in the FCS measurement mode was monitored continuously with 100 µs binning time.
Fluorescence burst coincidence analysis
Coincidence of Aβ42 and TTR in the soluble aggregates was measured using coincidence of the fluorescence bursts. In these measurements, 400 nM TMR‐Aβ42 and 200 nM Alexa488‐M‐TTR were mixed with 8 µM unlabeled Aβ42 and 4 µM unlabeled M‐TTR. The solutions prepared in PBS buffer at pH 7.4 containing 1 mM EDTA and 5 mM βME were incubated at room temperature with continuous stirring for two days prior to measurements. The fluorescence of Alexa488 and TMR were monitored in two separate detection channels using FCS.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
We thank our colleagues at Washington University and TSRI including Megan Cohan, Scott Crick, Tyler Harmon, Jeff Kelly, Alex Holehouse, Evan Powers, and Kiersten Ruff for helpful discussions.
References
- 1. Knowles TP, Vendruscolo M, Dobson CM (2014) The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 15:384–396. [DOI] [PubMed] [Google Scholar]
- 2. Knowles TP, Vendruscolo M, Dobson CM (2015) 2015 Alzheimer's disease facts and figures. Alzheimer's Dement 11:332–384. [DOI] [PubMed] [Google Scholar]
- 3. Glenner GG, Wong CW (1984) Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120:885–890. [DOI] [PubMed] [Google Scholar]
- 4. McCutchen SL, Lai Z, Miroy GJ, Kelly JW, Colon W (1995) Comparison of lethal and nonlethal transthyretin variants and their relationship to amyloid disease. Biochemistry 34:13527–13536. [DOI] [PubMed] [Google Scholar]
- 5. Peterson SA, Klabunde T, Lashuel HA, Purkey H, Sacchettini JC, Kelly JW (1998) Inhibiting transthyretin conformational changes that lead to amyloid fibril formation. Proc Natl Acad Sci USA 95:12956–12960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goldgaber D, Schwarzman AI, Bhasin R, Gregori L, Schmechel D, Saunders AM, Roses AD, Strittmatter WJ (1993) Sequestration of amyloid beta‐peptide. Ann NY Acad Sci 695:139–143. [DOI] [PubMed] [Google Scholar]
- 7. Merched A, Serot JM, Visvikis S, Aguillon D, Faure G, Siest G (1998) Apolipoprotein E, transthyretin and actin in the CSF of Alzheimer's patients: relation with the senile plaques and cytoskeleton biochemistry. FEBS Lett 425:225–228. [DOI] [PubMed] [Google Scholar]
- 8. Schwarzman AL, Goldgaber D (1996) Interaction of transthyretin with amyloid beta‐protein: binding and inhibition of amyloid formation. Ciba Found Symp 199:146–160. [DOI] [PubMed] [Google Scholar]
- 9. Dostal V, Link CD (2010) Assaying beta‐amyloid toxicity using a transgenic C. elegans model. J Visual Exper 44:e2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Link CD (1995) Expression of human beta‐amyloid peptide in transgenic Caenorhabditis elegans . Proc Natl Acad Sci USA 92:9368–9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Buxbaum JN, Ye Z, Reixach N, Friske L, Levy C, Das P, Golde T, Masliah E, Roberts AR, Bartfai T (2008) Transthyretin protects Alzheimer's mice from the behavioral and biochemical effects of Abeta toxicity. Proc Natl Acad Sci USA 105:2681–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Choi SH, Leight SN, Lee VM, Li T, Wong PC, Johnson JA, Saraiva MJ, Sisodia SS (2007) Accelerated Abeta deposition in APPswe/PS1deltaE9 mice with hemizygous deletions of TTR (transthyretin). J Neurosci 27:7006–7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schwarzman AL, Tsiper M, Wente H, Wang A, Vitek MP, Vasiliev V, Goldgaber D (2004) Amyloidogenic and anti‐amyloidogenic properties of recombinant transthyretin variants. Amyloid 11:1–9. [DOI] [PubMed] [Google Scholar]
- 14. Schwarzman AL, Tsiper M, Gregori L, Goldgaber D, Frakowiak J, Mazur‐Kolecka B, Taraskina A, Pchelina S, Vitek MP (2005) Selection of peptides binding to the amyloid b‐protein reveals potential inhibitors of amyloid formation. Amyloid 12:199–209. [DOI] [PubMed] [Google Scholar]
- 15. Costa R, Ferreira‐da‐Silva F, Saraiva MJ, Cardoso I (2008) Transthyretin protects against A‐beta peptide toxicity by proteolytic cleavage of the peptide: a mechanism sensitive to the Kunitz protease inhibitor. PloS One 3:e2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Costa R, Goncalves A, Saraiva MJ, Cardoso I (2008) Transthyretin binding to A‐Beta peptide—impact on A‐Beta fibrillogenesis and toxicity. FEBS Lett 582:936–942. [DOI] [PubMed] [Google Scholar]
- 17. Du J, Murphy RM (2010) Characterization of the interaction of beta‐amyloid with transthyretin monomers and tetramers. Biochemistry 49:8276–8289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li X, Zhang X, Ladiwala AR, Du D, Yadav JK, Tessier PM, Wright PE, Kelly JW, Buxbaum JN (2013) Mechanisms of transthyretin inhibition of beta‐amyloid aggregation in vitro. J Neurosci 33:19423–19433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang DT, Joshi G, Cho PY, Johnson JA, Murphy RM (2013) Transthyretin as both a sensor and a scavenger of beta‐amyloid oligomers. Biochemistry 52:2849–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mangrolia P, Yang DT, Murphy RM (2016) Transthyretin variants with improved inhibition of beta‐amyloid aggregation. Prot Eng Des Select 29:209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lu X, Brickson CR, Murphy RM (2016) TANGO‐inspired design of anti‐amyloid cyclic peptides. ACS Chem Neurosci 7:1264–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reixach N, Deechongkit S, Jiang X, Kelly JW, Buxbaum JN (2004) Tissue damage in the amyloidoses: transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc Natl Acad Sci USA 101:2817–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Foss TR, Kelker MS, Wiseman RL, Wilson IA, Kelly JW (2005) Kinetic stabilization of the native state by protein engineering: implications for inhibition of transthyretin amyloidogenesis. J Mol Biol 347:841–854. [DOI] [PubMed] [Google Scholar]
- 24. Hurshman AR, White JT, Powers ET, Kelly JW (2004) Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry 43:7365–7381. [DOI] [PubMed] [Google Scholar]
- 25. Foss TR, Wiseman RL, Kelly JW (2005) The pathway by which the tetrameric protein transthyretin dissociates. Biochemistry 44:15525–15533. [DOI] [PubMed] [Google Scholar]
- 26. Johnson SM, Wiseman RL, Sekijima Y, Green NS, Adamski‐Werner SL, Kelly JW (2005) Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: a focus on the transthyretin amyloidoses. Acc Chem Res 38:911–921. [DOI] [PubMed] [Google Scholar]
- 27. Wiseman RL, Green NS, Kelly JW (2005) Kinetic stabilization of an oligomeric protein under physiological conditions demonstrated by a lack of subunit exchange: implications for transthyretin amyloidosis. Biochemistry 44:9265–9274. [DOI] [PubMed] [Google Scholar]
- 28. Wiseman RL, Johnson SM, Kelker MS, Foss T, Wilson IA, Kelly JW (2005) Kinetic stabilization of an oligomeric protein by a single ligand binding event. J Am Chem Soc 127:5540–5551. [DOI] [PubMed] [Google Scholar]
- 29. Wiseman RL, Powers ET, Kelly JW (2005) Partitioning conformational intermediates between competing refolding and aggregation pathways: insights into transthyretin amyloid disease. Biochemistry 44:16612–16623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hurshman Babbes AR, Powers ET, Kelly JW (2008) Quantification of the thermodynamically linked quaternary and tertiary structural stabilities of transthyretin and its disease‐associated variants: the relationship between stability and amyloidosis. Biochemistry 47:6969–6984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lim KH, Dasari AKR, Ma R, Hung I, Gan Z, Kelly JW, Fitzgerald MC (2017) Pathogenic mutations induce partial structural changes in the native beta‐sheet structure of transthyretin and accelerate aggregation. Biochemistry 56:4808–4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jiang X, Smith CS, Petrassi HM, Hammarstrom P, White JT, Sacchettini JC, Kelly JW (2001) An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry 40:11442–11452. [DOI] [PubMed] [Google Scholar]
- 33. Cascella R, Conti S, Mannini B, Li X, Buxbaum JN, Tiribilli B, Chiti F, Cecchi C (2013) Transthyretin suppresses the toxicity of oligomers formed by misfolded proteins in vitro. Biochim Biophys Acta 1832:2302–2314. [DOI] [PubMed] [Google Scholar]
- 34. Morales R, Bravo‐Alegria J, Duran‐Aniotz C, Soto C (2015) Titration of biologically active amyloid‐beta seeds in a transgenic mouse model of Alzheimer's disease. Sci Rep 5:9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morales R, Callegari K, Soto C (2015) Prion‐like features of misfolded Abeta and tau aggregates. Virus Res 207:106–112. [DOI] [PubMed] [Google Scholar]
- 36. Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC, Thorpe JR, Serpell LC, Miller TM, Grinberg LT, Seeley WW, Diamond MI (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82:1271–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garai K, Frieden C (2013) Quantitative analysis of the time course of A beta oligomerization and subsequent growth steps using tetramethylrhodamine‐labeled A beta. Proc Natl Acad Sci USA 110:3321–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Crick SL, Ruff KM, Garai K, Frieden C, Pappu RV (2013) Unmasking the roles of N‐ and C‐terminal flanking sequences from exon 1 of huntingtin as modulators of polyglutamine aggregation. Proc Natl Acad Sci USA 110:20075–20080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wyman J, Gill SJ (1980) Ligand‐linked phase changes in a biological system: applications to sickle cell hemoglobin. Proc Natl Acad Sci USA 77:5239–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Garai K, Sahoo B, Sengupta P, Maiti S (2008) Quasihomogeneous nucleation of amyloid beta yields numerical bounds for the critical radius, the surface tension, and the free energy barrier for nucleus formation. J Chem Phys 128:045102. [DOI] [PubMed] [Google Scholar]
- 41. Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, Bu G, Frieden C, Holtzman DM (2013) ApoE influences amyloid‐beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc Natl Acad Sci USA 110:E1807–E1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cohen SI, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM, Knowles TP (2013) Proliferation of amyloid‐beta42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci USA 110:9758–9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Arosio P, Cukalevski R, Frohm B, Knowles TP, Linse S (2014) Quantification of the concentration of Abeta42 propagons during the lag phase by an amyloid chain reaction assay. J Am Chem Soc 136:219–225. [DOI] [PubMed] [Google Scholar]
- 44. Meisl G, Yang X, Hellstrand E, Frohm B, Kirkegaard JB, Cohen SI, Dobson CM, Linse S, Knowles TP (2014) Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Abeta40 and Abeta42 peptides. Proc Natl Acad Sci USA 111:9384–9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Arosio P, Knowles TP, Linse S (2015) On the lag phase in amyloid fibril formation. Phys Chem Chem Phys 17:7606–7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Colosimo A, Brunori M, Wyman J (1976) Polysteric linkage. J Mol Biol 100:47–57. [DOI] [PubMed] [Google Scholar]
- 47. Wong I, Lohman TM (1995) Linkage of protein assembly to protein‐DNA binding. Methods Enzymol 259:95–127. [DOI] [PubMed] [Google Scholar]
- 48. Posey AE, Ruff KM, Harmon TS, Crick SL, Li A, Diamond MI, Pappu RV (2018) Profilin reduces aggregation and phase separation of huntingtin N‐terminal fragments by preferentially binding to soluble monomers and oligomers. J Biol Chem DOI: 10.1074/jbc.RA117.000357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yong W, Lomakin A, Kirkitadze MD, Teplow DB, Chen SH, Benedek GB (2002) Structure determination of micelle‐like intermediates in amyloid beta ‐protein fibril assembly by using small angle neutron scattering. Proc Natl Acad Sci USA 99:150–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pallitto MM, Murphy RM (2001) A mathematical model of the kinetics of beta‐amyloid fibril growth from the denatured state. Biophys J 81:1805–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Šarić A, Buell AK, Meisl G, Michaels TCT, Dobson CM, Linse S, Knowles Tuomas PJ, Frenkel D (2016) Physical determinants of the self‐replication of protein fibrils. Nat Phys 12:874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Buxbaum JN, Johansson J (2017) Transthyretin and BRICHOS: the paradox of amyloidogenic proteins with anti‐amyloidogenic activity for Abeta in the central nervous system. Front Neurosci 11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Francis MD (1969) The inhibition of calcium hydroxypatite crystal growth by polyphosphonates and polyphosphates. Calc Tiss Res 3:151–162. [DOI] [PubMed] [Google Scholar]
- 54. Brès EF, Moebus G, Kleebe HJ, Pourroy G, Werkmann J, Ehret G (1993) High resolution electron microscopy study of amorphous calcium phosphate. J Cryst Growth 129:149–162. [Google Scholar]
- 55. Cappelli S, Penco A, Mannini B, Cascella R, Wilson MR, Ecroyd H, Li X, Buxbaum JN, Dobson CM, Cecchi C, Relini A, Chiti F (2016) Effect of molecular chaperones on aberrant protein oligomers in vitro: super‐versus sub‐stoichiometric chaperone concentrations. Biol Chem 397:401–415. [DOI] [PubMed] [Google Scholar]
- 56. Jain N, Aden J, Nagamatsu K, Evans ML, Li X, McMichael B, Ivanova MI, Almqvist F, Buxbaum JN, Chapman MR (2017) Inhibition of curli assembly and Escherichia coli biofilm formation by the human systemic amyloid precursor transthyretin. Proc Natl Acad Sci USA 114:12184–12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information