

Abstract
Prion diseases are infectious neurodegenerative diseases that are capable of cross‐species transmission, thus arousing public health concerns. Seed‐templating propagation of prion protein is believed to underlie prion cross‐species transmission pathology. Understanding the molecular fundamentals of prion propagation is key to unravelling the pathology of prion diseases. In this study, we use coarse‐grained molecular dynamics to investigate the seeding and cross‐seeding aggregation of three prion protein fragments PrP(120–144) originating from human (Hu), bank vole (BV), and Syrian hamster (SHa). We find that the seed accelerates the aggregation of the monomer peptides by eliminating the lag phase. The monomer aggregation kinetics are mainly determined by the structure of the seed. The stronger the hydrophobic residues on the seed associate with each other, the higher the probability that the seed recruits monomer peptides to its surface/interface. For cross‐seeding aggregation, we show that Hu has a strong tendency to adopt the conformation of the BV seed and vice versa; the Hu and BV monomers have a weak tendency to adopt the conformation of the SHa seed. These two findings are consistent with Apostol et al.'s experimental findings on PrP(138–143) and partially consistent with Jones et al.'s finding on PrP(23–144). We also identify several conformational mismatches when SHa cross‐seeds BV and Hu peptides, indicating the existence of a cross‐seeding barrier between SHa and the other two sequences. This study sheds light on the molecular mechanism of seed‐templating aggregation of prion protein fragments underlying the sequence‐dependent transmission barrier in prion diseases.
Keywords: coarse‐grained molecular dynamics, cross‐seeding aggregation, prion protein fragments, amyloid formation, fibril elongation
Introduction
Prion diseases are a family of infectious neurodegenerative diseases that include kuru and Creutzfeldt–Jakob disease (CJD) in humans, scrapie in goats, bovine spongiform encephalopathy (BSE) in cattle, and chronic wasting disease (CWD) in deer.1, 2, 3 The infectious agent in prion disease is the prion protein scrapie (PrPSc) which has diverse β‐sheet‐rich conformations.4 PrPSc replicates without the help of nucleic acids by templating the misfolding and accumulation of normal prion protein into misfolded PrPSc, a mechanism that is similar to seeded polymerization of amyloids.5, 6, 7, 8
The significant threat of prion disease to human health is its potential cross‐species transmission. For example, BSE (also known as Mad Cow Disease) is thought to originate from transmission of a scrapie prion from sheep to cattle.9 Humans can acquire a CJD variant by consuming bovine products contaminated by BSE.9, 10 Experimental mice can acquire variant CJD by intracerebral inoculation of brain homogenates from cattle affected by BSE.11 Cross‐species transmission of prion disease may be related to the direct interaction between the misfolded and cellular forms of prion proteins from different species, leading to cross‐seeding of prion protein aggregation.12, 13 Understanding the molecular mechanism underlying the cross‐seeding aggregation of prion protein could pave the way to elucidating the pathology of cross‐species transmission.
The cross‐seeding efficiency and specificity of full‐length prion protein and its N‐terminal fragments have been investigated in vitro. Vanik et al.14 found that Syrian hamster PrP(23–144) can seed the fibrillation of mouse PrP(23–144) while mouse PrP(23–144) cannot seed the fibrillation of Syrian hamster PrP(23–144), indicating that mouse and Syrian hamster PrP have an asymmetric cross‐seeding barrier.14 They further found14 that two species‐specific mutations (I138M and I139M) are critical in determining the cross‐seeding specificity of PrP(23–144). Experiments by Lee et al.15 showed that residues 109, 112, and 139 determine the seeding and cross‐seeding efficiency of mouse and hamster PrP(108–144). Chuang et al.16 found that the mutation L138M promotes the amyloidogenesis of bovine PrP(108–144) peptide and increases its seeding efficiency while the mutations I139M and N143S do the opposite. Apostol et al.17 found that human and mouse PrP(138–143) fibrils both form parallel steric zippers while hamster PrP(138–143) fibril forms anti‐parallel steric zippers. When cross‐seeded with hamster PrP(138–143) fibril, human and mouse PrP(138–143) fibrils never form anti‐parallel steric zippers.
Molecular simulations have also been applied to investigate the seeding and cross‐seeding mechanism underlying protein aggregation. A number of amyloid β fragments, for example, Aβ(16–22),18 Aβ(35–40),19 and Aβ(17–42),20 have been shown in silico to follow a two‐step “dock‐lock” mechanism when monomer peptides of the same sequence attach to the pre‐formed fibril ends. The cross‐seeding interaction between two different peptides to form hetero‐assemblies have also been investigated. Qi et al. found using molecular dynamics (MD) simulation that monomeric tau protein is stabilized by stretching its conformation and exposing its amyloidogenic motifs in the presence of the Aβ(1–42) protofilament.21 Zhang et al. showed using MD simulation that the rat islet amyloid polypeptide (rIAPP) docks to the end of the human islet amyloid polypeptide (hIAPP) protofilament to form stable rIAPP‐hIAPP assemblies.22 Hu et al. also showed that the U‐shaped Aβ and hIAPP protofilaments can stack on top of each other to form double‐layer assemblies.23
PRIME20 is a knowledge‐based four‐bead‐per‐residue coarse‐grained protein model developed in the Hall group that was specifically designed for discontinuous molecular dynamics (DMD) simulation of protein aggregation. DMD/PRIME20 is able to simulate the dynamic process of large‐scale peptide aggregation from an initial random‐coil configuration to disordered oligomers and then fibrillar structures.24, 25, 26 In recent years, DMD/PRIME20 have been applied to investigate aggregation of prion protein fragments,27 tau protein fragments,28 Aβ(16–22),25 and Aβ(17–42)29 peptides. In addition, it has been extended to investigate the effect of macromolecular crowding,26, 30 and polyphenol inhibition31 on peptide aggregation. In a recent study, we performed coarse‐grained simulations of the spontaneous aggregation of the three prion protein fragments, Hu, BV and SHaPrP(120–144), and found that they form similar polymorphic β‐sheet‐rich fibrillar structures due to the similarity in their amino acid sequences.32 HuPrP(120–144) aggregates to form the S‐, Ω‐ and U‐shaped protofilaments, while BV and SHaPrP(120–144) form the S‐ and Ω‐shaped protofilaments, as shown in Figures 1 and S1. In this study, we investigate the seeding and cross‐seeding aggregation of these three peptides in the presence of the pre‐formed S‐, Ω‐, and U‐shaped protofilaments.
Figure 1.
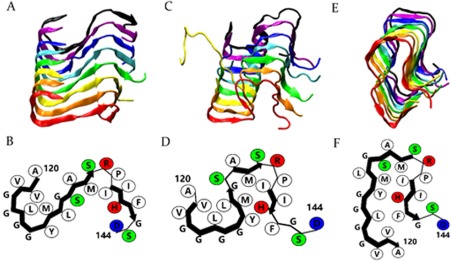
S‐shaped (A), Ω‐shaped (C), and U‐shaped (E) protofilaments formed by HuPrP(120–144) from the 5th, 12th, and 11th runs are shown. B, D, and F, schematic representations of peptide conformations in the S‐, Ω‐, and U‐shaped protofilaments. The sizes of the side chain beads do not reflect the actual radii. Hydrophobic residues (white), positively charged residues (red), negatively charged residues (blue), and polar residues (green) are shown (this figure is reproduced from ref 32).
Highlights of our results are as follows. In comparing seeding versus non‐seeding aggregation, we find that the seed accelerates the aggregation kinetics by eliminating the lag phase for forming a stable nucleus. Analysis of the homogeneous seeding aggregation of HuPrP(120–144) shows that the structure of the seed has a dominant effect on peptide aggregation kinetics. The U‐shaped Hu seed has a higher seeding efficiency than the S‐ and Ω‐shaped Hu seeds because the peptides in the U‐shaped seed have stronger sidechain–sidechain hydrophobic interactions, which help stabilize and template the incoming monomer peptides. From the cross‐seeding simulation of Hu, BV, and SHaPrP(120–144), we find that HuPrP(120–144) has a strong tendency to adopt the conformation of the BV seed and vice versa, indicating that Hu and BVPrP(120–144) may have a low cross‐seeding barrier. Also, the BV seed has high efficiency in templating the Hu and SHa monomer while the SHa seed has low efficiency in templating the Hu, BV monomer. This suggests that SHaPrP(120–144) may have a high cross‐seeding barrier with Hu and BVPrP(120–144). The cross‐seeding efficiency of the Hu, BV, and SHa seeds in simulation is consistent with experimental findings on the species‐dependent seeding specificity of PrP(138–143).17
Results
Seeding versus non‐seeding aggregation of HuPrP(120–144) peptides
Here we apply DMD simulation to investigate the effect of seeding on prion protein peptide aggregation. We first simulate the aggregation of eight initially disordered HuPrP(120–144) peptides in the presence of a pre‐formed S‐shaped HuPrP(120–144) protofilament. The pre‐formed protofilament is regarded as the seed in our simulation. Figure 2 describes the percentage of β‐sheet versus time in seeding and non‐seeding simulation of HuPrP(120–144) peptides. From Figure 2(A), in the absence of the seed, the aggregation of initially disordered HuPrP(120–144) peptides [Fig. 2(B)] has a lag phase before forming a parallel in‐register S‐shaped protofilament [Fig. 2(C)].32 In the presence of the seed, the aggregation lag phase is eliminated and the aggregation of HuPrP(120–144) is significantly accelerated since monomer peptides can directly attach to the preformed seed without overcoming the nucleation barrier. From Figures 2(D) and 2(E), during the seeding aggregation, the pre‐formed seed elongates by recruiting seven out of eight monomeric peptides and templating them into parallel in‐register β‐sheets, essentially adopting the same S‐shaped conformation as the seed. Similarly, the aggregation of BV‐ and SHaPrP(120–144) peptides are also accelerated in the presence of the BV‐ and SHaPrP(120–144) seeds.
Figure 2.
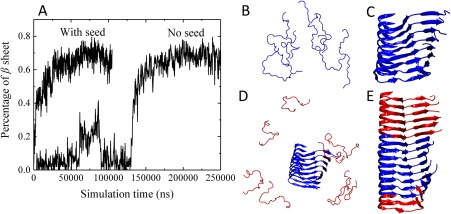
(A) Plot of β‐sheet percentage in HuPrP(120–144) aggregation simulations in the presence and absence of a pre‐formed HuPrP(120–144) seed as a function of time. The percentage of β‐sheet content is calculated by the STRIDE algorithm in the visual molecular dynamics (VMD) software. (B) and (C) are simulation snapshots of the non‐seeding aggregation taken at t = 1 and 198 μs, respectively. (D) and (E) are simulation snapshots of the seeding aggregation taken at t = 1 and 58 μs, respectively.
Homogeneous seeding aggregation of PrP(120–144)
We investigate the effect of polymorphic Hu seeds (U‐, S‐, and Ω‐shaped) on HuPrP(120–144) aggregation kinetics and structures. The average potential energy versus time of HuPrP(120–144) aggregation in the presence of the S, Ω and U‐shaped seeds from five independent runs are shown in Figure 3(A). From Figure 3(A), the potential energy of the system using the U‐shaped seed decreases faster than those of the S‐ and Ω‐shaped seed, indicating that the U‐shaped seed has the highest seeding efficiency among the three Hu seeds. In Table 1, we list the average number of HuPrP(120–144) peptides that adopt the seed conformation, partially adopt the seed conformation or adopt other conformations by the end of the aggregation. The large error bars in the average number of peptides adopting the conformation of the seed indicates large deviations in the number of peptides adopting the conformation of the seed among the five independent runs for each type of seeding simulation. We find that the average numbers of HuPrP(120–144) peptides that adopt the conformation of the Ω‐ and U‐shaped seeds are slightly higher than the number of peptides to adopt that conformation of the S‐shaped seed. As shown in Figure 3(B), peptides in the original U and Ω‐shaped seeds have stronger sidechain–sidechain hydrophobic interactions than in the S‐shaped seed. The explanation is that the U‐shaped seed has the most stable hydrophobic core among the three seeds, leading to high efficiency in stabilizing and templating the monomer peptide when attaching to it.
Figure 3.
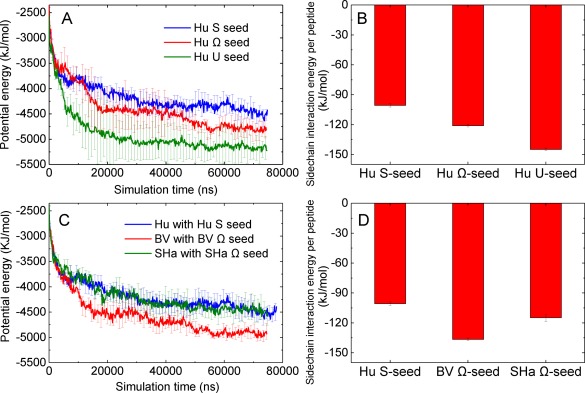
(A) Potential energy versus time of HuPrP(120–144) peptides aggregating in the presence of U‐, S‐, Ω‐shaped Hu seeds. (B) Average sidechain interaction energy per peptide of U‐, S‐, Ω‐shaped HuPrP(120–144) protofilaments (seeds). (C) Potential energy versus time of Hu, BV, and ShaPrP(120–144) peptides aggregating in the presence of S‐shaped Hu, Ω‐shaped BV, and Ω‐shaped Sha seeds. (D) Average sidechain interaction energy per peptide of S‐shaped Hu, Ω‐shaped BV, and Ω‐shaped Sha seeds.
Table 1.
Number of HuPrP(120–144) Peptides that Adopt the Peptide Conformation in the Seed or Other Conformations During the Aggregation in the Presence of the S‐, Ω‐, and U‐Shaped Hu Seeds
| Seed | # Hu monomers that adopt seed conformation | # Hu monomers that partially adopt seed conformation | # Hu monomers that adopt other conformations |
|---|---|---|---|
| Hu S‐shaped | 4.0 ± 2.7 | 1.2 ± 1.6 | 2.8 ± 1.9 |
| Hu Ω‐shaped | 5.6 ± 1.8 | 1.4 ± 1.1 | 1.2 ± 1.8 |
| Hu U‐shaped | 6.0 ± 2.1 | 0.0 ± 0.0 | 2.0 ± 2.1 |
In addition, we compare the aggregation rate of Hu, BV, and SHaPrP(120–144) in the presence of the S‐shaped Hu, Ω‐shaped BV, and Ω‐shaped SHaPrP(120–144) seeds, respectively. As shown in Figure 3(C), the potential energy of the BV monomer with the BV seed decreases fastest among the three systems. From Table 2, we find that the number of BVPrP(120–144) peptides adopting the conformation of the Ω‐shaped BV seed is higher than those of Hu and ShaPrP(120–144) adopting the conformation of their corresponding seeds. This indicates that the Ω‐shaped BVPrP(120–144) seed has higher homogeneous seeding efficiency than the S‐shaped Hu and Ω‐shaped SHa seeds. The reason is that, from Figure 3(D), BVPrP(120–144) peptide in the original Ω‐shaped BV seed has stronger sidechain interaction energy than those of the S‐shaped Hu and Ω‐shaped SHa seeds.
Table 2.
Number of Hu, BV, and SHaPrP(120–144) Peptides that Adopt the Seed Conformation During the Seeding and Cross‐Seeding Aggregation
| Seed/Monomer peptide | # Hu monomers that adopt seed conformation | # BV monomers that adopt seed conformation | # SHa monomers that adopt seed conformation |
|---|---|---|---|
| Hu S‐shaped seed | 4.0 ± 2.7 | 7.0 ± 0.7 | 4.8 ± 2.4 |
| BV Ω‐shaped seed | 6.0 ± 0.8 | 6.2 ± 1.3 | 6.4 ± 1.3 |
| SHa Ω‐shaped seed | 2.8 ± 1.9 | 3.2 ± 2.6 | 3.0 ± 2.5 |
Cross‐seeding aggregation of Hu, BV, and SHaPrP(120–144) peptides
We also performed simulations of six types of cross‐seeding aggregation for the Hu, BV, and SHaPrP(120–144) peptides. Similar to the homogeneous seeding simulation results, our cross‐seeding simulation results also showed that the structure of the seed has a major effect on the monomer aggregation kinetics which is dominant over the monomer‐seed sequence difference. Figure 4 plots the potential energy profiles of three homogeneous seeding and six cross‐seeding aggregation simulations. From Figure 4(A), in the presence of the S‐shaped Hu seed, Hu, BV, and SHaPrP(120–144) peptides all have similar aggregation kinetics regardless of the fact that HuPrP(120–144) has higher aggregation propensity than BV and SHaPrP(120–144),32 indicating that the sequence differences of the monomer play a minor role on the cross‐seeding efficiency. Lee et al.15 had a similar experimental finding that using SHaPrP(108–144) to seed the fibrillation of its M139I mutant is as efficient as homologous seeding. In addition, Figures 4(B–D) and S2 show that the aggregation rates of the Hu, BV and SHaPrP(120–144) peptides in the presence of the Ω‐shaped BV seed are always higher than those in the presence of the S‐ and Ω‐shaped Hu seeds and the Ω‐shaped SHa seed. This indicates that the Ω‐shaped BV seed has the highest cross‐seeding efficiency among the three seeds. We also notice from Figure 3(D) that the peptides in the original Ω‐shaped BV seed have the strongest sidechain–sidechain interaction energy among the three seeds, indicating that the Ω‐shaped BV seed has a strong hydrophobic core. Thus, the stronger the hydrophobic residues associate with each other within the seed, the higher the probability that the seed recruits monomer peptides on its surface/interface and, hence the faster it elongates.
Figure 4.
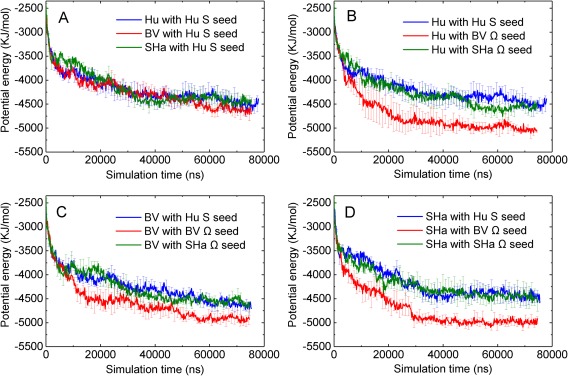
(A) Plot of potential energy versus simulation time of HuPrP(120–144) peptides aggregation in the presence of S‐shaped Hu, Ω‐shaped BV, and Ω‐shaped SHa seed. (B–D) Plots of potential energy versus simulation time of Hu, BV, SHaPrP(120–144) peptides aggregation in the presence of S‐shaped Hu, Ω‐shaped BV, and Ω‐shaped SHa seed.
For the cross‐seeding aggregation, we compare the tendencies of the monomers to adopt the seed conformation. In Table 2, we list the average number of Hu, BV and SHaPrP(120–144) peptides that adopt the seed conformation from five independent runs. We found that Hu and BVPrP(120–144) have a relatively low cross‐seeding barrier. From Table 2, when the HuPrP(120–144) is cross‐seeded with the Ω‐shaped BV seed, most of the HuPrP(120–144) peptides adopt the peptide conformation of the BV seed; when the BVPrP(120–144) is cross‐seeded with the S‐shaped Hu seed, most of the BVPrP(120–144) peptides adopt the peptide conformation of the Hu seed. In addition, we notice that BVPrP(120–144) peptides adopt the conformation (7.0 ± 0.7 out of 8) of Hu S‐shaped seed slightly more than they adopt the conformation of the Ω‐shaped BV seed (6.2 ± 1.3 out of 8). Since these two numbers are within standard deviation of each other, we think that the tendency of BVPrP(120–144) peptide to adopt the conformation of the S‐shaped Hu is similar to its tendency to adopt the conformation of the Ω‐shaped BV seeds. Note that mouse (Mo) prion has the same PrP(120–144) sequence as bank vole (BV). This is consistent with experimental finding by Apostol et al.17 that the crystal structure of fibrils formed by Hu and MoPrP(138–143) are similar, but are different from those formed by SHaPrP(138–143). Both the HuPrP(138–143) monomer with the pre‐formed MoPrP(138–143) fibril crystals and vice versa show seeding. This is also consistent with the experimental finding by Jones et al.33 that the human and mouse PrP(23–144) fibrils both have twisted morphologies and that one can be used to seed the amyloid formation of the other. The major reason for the weak tendency of Hu and BV to adopt the conformation of the Ω‐shaped SHa seed is that conformation of the Ω‐shaped SHa seed is hard for a monomer to adopt. From Table 2, not only Hu and BV but SHa have weak tendencies to adopt the conformation of the Ω‐shaped SHa seed. Also, most of the SHa monomers adopt the BV seed conformation but most of the BV monomers do not adopt the SHa seed, which is inconsistent with the finding on PrP(23–144) by Vanik et al.14
To evaluate the impact of seeding on the three mutated residues of Hu, BV, and SHaPrP(120–144) peptides, we calculated the interaction energies experienced by each of the three mutated residues on the monomer peptides with all other residues in the system averaged over the entire trajectory in Figure 5. Firstly, compared with the non‐seeding simulations [Fig. 8(A) from ref. 32], the interaction energies experienced by residue 138 on monomer Hu, BV and SHa in the seeding simulations are all increased, while those of residues 139 and 143 decreased. Secondly, residue 138 has the strongest interaction energy with the seeds regardless of whether it is on the Hu, BV or SHa peptides. It also seems to be the most affected by the type of seed and has its strongest interaction energy in the presence of the BV seed, regardless of whether it is on Hu, BV and SHa peptides. Residue 139 is modestly affected by the seed and residue 143 seems hardly affected by the seeds.
Figure 5.
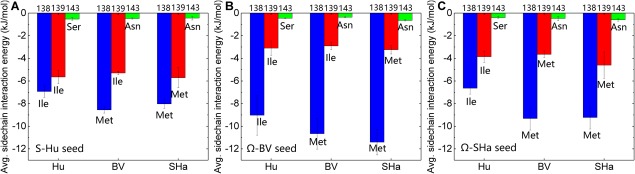
Plots of the average side chain interaction energy (kJ/mol) experienced by residues 138, 139, and 143 of monomer Hu, BV, and SHaPrP(120–144) peptides in the presence of (A) S‐shaped Hu, (B) Ω‐shaped BV, and (C) Ω‐shaped SHa seeds, respectively.
We also noticed from Table 2 that only a small portion of the Hu and BV monomers adopt the conformation of the SHa seed, indicating a strong cross‐seeding barrier between SHaPrP(120–144) and the other two sequences. This is also consistent with experimental findings by Apostol et al.17 that when seeded with SHaPrP(138–143) fibril, Hu and MoPrP(138–143) cannot reproduce the morphology of the SHaPrP(138–143) fibril.
Structural mismatch in homogeneous and heterogeneous PrP(120–144) fibril
We observed a number of different mismatches between the conformations of the monomers after they attach to the seed and the original seed conformation in the seeded and cross‐seeded protofilaments. Figure 6(A) shows that for Hu aggregating in the presence of the SHa seed, four Hu peptides and one SHa peptide form a parallel in‐register β‐sheet but with their C‐terminal (residues 137Pro to 144Asp) detached from the Ω‐shaped SHa seed. This molecular‐level structural mismatch is similar to the hybridized fibril model proposed by Makarava et al.34 who found that when mouse full length prion protein is seeded with 30% hamster fibrils, a hybrid Mo‐SHa fibril is formed having two distinct Mo and SHa domains connected by a shared common local structural motif. We also found some other types of structural mismatch during the seeding and cross‐seeding aggregation simulations. From Figure 6(B), in the case of BV aggregation with the SHa seed, residues 120Ala to 131Gly of BV adopt a “triangle” conformation which is a mirror image of the conformation adopted by residues 120Ala to 131Gly of peptides in the seed. From Figure 6(C), in the case of BV aggregating with the SHa seed, one BV monomer adopts a configuration that is antiparallel to the seed conformation. This conformation mismatch causes the other three BV peptides to adopt the S‐shaped conformation. From Figure 6(D), in the case of SHa aggregation with the BV seed, one SHa monomer adopts the seed conformation but only near the N‐terminal region (residues 120Ala to 131Gly), leaving its tail (132Ser to 144Asp) to float in a disordered conformation. From Figure 6(E) in the case of SHa aggregation with the SHa seed, two SHa monomers adopt Ω‐shaped conformations that are slightly different from the original Ω‐shaped SHa seed. Our finding from simulation indicates that during seeding and cross‐seeding aggregation, the seed may not only template monomer peptides to form the same conformation as the seed but may also induce monomer peptides to form new conformations which may eventually elongate into fibrils with new morphology, or new “strains”.
Figure 6.
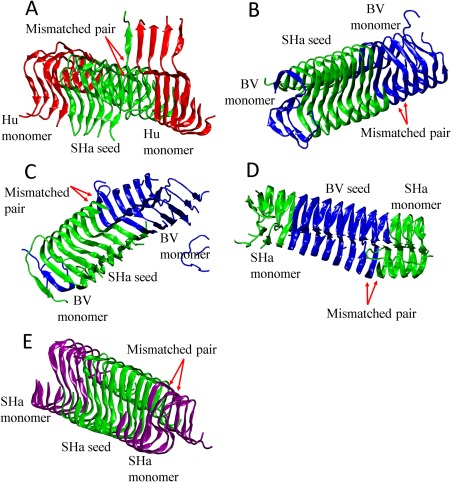
Five types of structural mismatches found in homogeneous and heterogeneous protofilaments formed by Hu, BV, and SHaPrP(120–144) peptides. Simulation snapshot of (A) Hu with the Ω‐shaped SHa protofilament from the 2nd run; (B) BV with the Ω‐shaped SHa protofilament from the 3rd run; (C) BV with the Ω‐shaped SHa protofilament from the 4th run; (D) SHa with the Ω‐shaped BV protofilament from the 2nd run; (E) SHa with the Ω‐shaped SHa protofilament from the 1st run. Hu, BV, and SHaPrP(120–144) peptides in A–E are colored in red, blue and green, respectively, except that the SHa(120–144) monomer peptides in E are colored in purple.
Discussion and Conclusion
Coarse‐grained molecular dynamic simulations were used to investigate the early‐stage aggregation of three prion protein fragments Hu, BV, and SHaPrP(120–144). In a previous article, we had investigated the spontaneous aggregation (nucleation) of PrP(120–144) peptides, similar to the study by Cheon et al.29 for Aβ(17–42) peptides. In that article, we showed that the sequence differences in residues 138, 139, and 143 in Hu, BV and SHaPrP(120–144) cause these peptides to have different aggregation propensities (Hu > BV > SHa). It also causes these peptides to form various fibrillar structures including the S‐, Ω‐ and U‐shaped protofilaments. In this article, we investigated the early‐stage seeding aggregation of these three peptides. We show that the seeding aggregation kinetics depends primarily on the structure of the seed, or the hydrophobicity of the seed core. The hydrophobic sidechain‐sidechain attraction, along with the backbone hydrogen bonding interaction, are the driving forces for seeding and cross‐seeding aggregation. The stronger the hydrophobic residues associate with each other within the seed, the higher the probability that the seed recruits monomer peptides on its surface/interface, and hence the faster it elongates. We also surmise that in experiments, initially PrP(120–144) forms polymorphic fibrillar oligomers. The ones with the lower energies are likely to have faster elongation rates.
Table 3.
Amino Acid Sequence of Three Prion Protein Fragments
| 120 | 130 | 140 | 144 | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Human | A | V | V | G | G | L | G | G | Y | M | L | G | S | A | M | S | R | P | I | I | H | F | G | S | D |
| Bank Vole | A | V | V | G | G | L | G | G | Y | M | L | G | S | A | M | S | R | P | M | I | H | F | G | N | D |
| Syrian Hamster | A | V | V | G | G | L | G | G | Y | M | L | G | S | A | M | S | R | P | M | M | H | F | G | N | D |
Note: mouse prion has the same PrP(120–144) sequence as bank vole. The letters colored in red are residues mutated from human sequence to bank vole and Syrian hamster sequences.
In addition, conformational mismatches between the monomer and the seed are commonly observed during the fibril elongation, explaining the polymorphism in the final morphology of the macroscopic fibril. An implication of these conformational mismatches is that even when the monomer and the seed consist of the same prion protein, the seeded aggregation may still generate new fibril structures, which could give rise to new variants of prion diseases. Within a single population, for example, human, various strains of CJD are believed to be caused by the same prion protein but with diverse PrPSc conformations.35
In the cross‐seeding simulation of PrP(120–144), mutations on residues 138, 139, and 143 between species give rise to differences in the seeding efficiency, specificity and fibril morphology, suggesting that there is a species‐dependent seeding barrier for prion proteins. On the one hand, HuPrP(120–144) has a strong tendency to adopt the conformation of the BV seed and vice versa. On the other hand, the Hu and BV monomers have relatively weak tendencies to adopt the conformation of the SHa seed. The mismatch between the sequences of the SHa seed and the Hu and BV monomer causes structural mismatches and decreases thermodynamic stability of the cross‐seeded fibril, which leads to a decrease in the cross‐seeding aggregation rate. These simulation results are consistent with the experimental findings for PrP(23–144)14, 33 and PrP(138–143).17 In addition, bank vole is a unique species that has been shown in vivo to be a universal acceptor of various prion diseases.36 This indicates that BVPrP has no inter‐species transmission barrier and can likely be subject to various infectious PrPSc from other species.
The small system size in the current simulations (eight peptides plus a pre‐formed octameric seed) allow us to investigate the elongation of the PrP(120–144) protofilament. As revealed from our and other investigators' simulations of fibrillation of short amyloidogenic peptides like Aβ(16–22),25, 26 the complete aggregation pathway includes oligomerization, nucleation and fibril elongation stages. To investigate the complete aggregation pathway of Hu, BV, and SHaPrP(120–144) peptides, we may need to perform larger scale simulations of hundreds of peptides and fit the simulation data to the self‐consistent solution of a master equation of the fibril growth by Cohen et al.37 to predict the primary nucleation rate, the secondary nucleation rate and the fibril elongation rate.
Materials and Methods
DMD and PRIME20 force field
Discontinuous molecular dynamics (DMD), a fast alternative to traditional molecular dynamics, is employed as our main simulation method.38 PRIME20 is a four‐bead‐per‐residue coarse‐grained protein model developed in the Hall group that was specifically designed for DMD simulation of protein aggregation.39 The PRIME20 force field models the 20 different amino acids (each contains NH, Cα, CO, and sidechain sphere R) with 20 different sets of geometric parameters. Specifically, each sidechain bead of the 20 amino acids has a distinct hard sphere diameter (effective van der Waals radius) and has distinct sidechain‐to‐backbone distances (R‐Cα, R‐NH, R‐CO). The potential function between two amino acid sidechain beads is modeled as a square well potential which contains two parameters, square well width and square well depth. We reduce the total number of the 210 independent square well depths between 20 different amino acids to 19 parameters while maintaining 210 independent square well widths to ensure physically meaningful pair interaction energies and reasonably accurate results in discriminating decoys from native structures in the PDB database. The backbone hydrogen bonding interactions are also modeled as a directional square well potential. All the other non‐bonded interactions are modeled as hard sphere interactions. A detailed description of the derivation of the geometric and energetic parameters of the PRIME20 model is given in our earlier study.24, 39, 40 The main difference between our force field and most of the atomistic (Amber, OPLS, CHARMM, etc.) or coarse‐grained protein force fields (MARTINI,41 OPEP,42 and AWSEM43) is that PRIME20 models the non‐bonded interactions (backbone hydrogen bonding and sidechain–sidechain interactions) as discontinuous potentials while other models use continuous potentials, like the Lennard‐Jones or Columbic potentials. The square well potential for modeling sidechain–sidechain interaction in PRIME20 is a sum of the van der Waals, charge‐charge and hydrophobic interactions; these contributions cannot be split separately. In addition, PRIME20 does not take into account the effect of water/counter‐ions as it is an implicit solvent model. Instead the PRIME20 forced field is focused on predicting the structural changes of peptides in their oligomeric and fibrillar state.
Simulation Procedure
We simulate the seeding and cross‐seeding aggregation of the Hu, BV and SHaPrP(120–144) peptides in the presence of a pre‐formed β‐sheet‐rich protofilament. The S‐, Ω‐, and U‐shaped protofilaments formed by spontaneous aggregation of eight Hu, BV, and SHaPrP(120–144) peptides in the previous study32 are used as seeds. The simulations are performed in the canonical ensemble (fixed number of particles, constant volume, and temperature). The Andersen thermostat is implemented to maintain the system at a constant temperature.44 The initial velocity for every bead in the system is generated based on a Maxwell‐Boltzmann distribution centered at the desired reduced temperature. The system is initialized by randomly placing a pre‐formed β‐sheet‐rich octamer surrounded by eight monomeric peptides in a cubic box with box length equal to 110.0 Å corresponding to a total peptide concentration of 20 mM. The reduced temperature is defined to be T *= k B T/ɛHB, where the hydrogen bonding energy, ɛHB = 12.47 kJ/mol. The reduced temperature T * is chosen to be 0.195, which corresponds to 330 K in real temperature.32 We have five independent runs for each of the 14 seeding and cross‐seeding simulation. Each simulation lasts for at least 75 μs.
Supporting information
Supporting Information
Acknowledgments
This study was supported by National Institutes of Health Grant R01 EB006006 and in part by National Science Foundation Research Triangle Materials Research Science and Engineering Centers Grant DMR‐1121107 and in part by National Science Foundation CBET 1743432.
References
- 1. Prusiner SB (1991) Molecular‐biology of prion diseases. Science 252:1515–1522. [DOI] [PubMed] [Google Scholar]
- 2. Miller G (2009) Could they all be prion diseases?. Science 326:1337–1339. [DOI] [PubMed] [Google Scholar]
- 3. Andréoletti O, Lacroux C, Chabert A, Monnereau L, Tabouret G, Lantier F, Berthon P, Eychenne F, Lafond‐Benestad S, Elsen J‐M, Schelcher F (2002) PrPSc accumulation in placentas of ewes exposed to natural scrapie: influence of foetal PrP genotype and effect on ewe‐to‐lamb transmission. J Gen Virol 83:2607–2616. [DOI] [PubMed] [Google Scholar]
- 4. Soto C, Estrada L, Castilla J (2006) Amyloids, prions and the inherent infectious nature of misfolded protein aggregates. Trends Biochem Sci 31:150–155. [DOI] [PubMed] [Google Scholar]
- 5. Walker LC, Jucker M (2015) Neurodegenerative diseases: expanding the prion concept. Annu Rev Neurosci 38:87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang ZW, Fletterick RJ, Cohen FE, Prusiner SB (1993) Conversion of alpha‐helices in to beta‐sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA 90:10962–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alper T, Cramp WA, Haig DA, Clarke MC (1967) Does the agent of scrapie replicate without nucleic acid?. Nature 214:764–766. [DOI] [PubMed] [Google Scholar]
- 8. Jarrett JT, Lansbury PT (1993) Seeding “one‐dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer's disease and scrapie?. Cell 73:1055–1058. [DOI] [PubMed] [Google Scholar]
- 9. Prusiner SB (1997) Prion diseases and the BSE crisis. Science 278:245–251. [DOI] [PubMed] [Google Scholar]
- 10. Parchi P, Giese A, Capellari S, Brown P, Schulz‐Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti B, Gambetti P, Kretzschmar H (1999) Classification of sporadic Creutzfeldt‐Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46:224–233. [PubMed] [Google Scholar]
- 11. Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H, Bostock CJ (1997) Transmissions to mice indicate that/‘new variant/’ CJD is caused by the BSE agent. Nature 389:498–501. [DOI] [PubMed] [Google Scholar]
- 12. Morales R, Moreno‐Gonzalez I, Soto C (2013) Cross‐seeding of misfolded proteins: Implications for etiology and pathogenesis of protein misfolding diseases. PLoS Pathog 9:e1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sharma A, Bruce KL, Chen BX, Gyoneva S, Behrens SH, Bommarius AS, Chernoff YO (2016) Contributions of the prion protein sequence, strain, and environment to the species barrier. J Biol Chem 291:1277–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vanik DL, Surewicz KA, Surewicz WK (2004) Molecular basis of barriers for interspecies transmissibility of mammalian prions. Mol Cell 14:139–145. [DOI] [PubMed] [Google Scholar]
- 15. Lee LYL, Chen RPY (2007) Quantifying the sequence‐dependent species barrier between hamster and mouse prions. J Am Chem Soc 129:1644–1652. [DOI] [PubMed] [Google Scholar]
- 16. Chuang CC, Liao TY, Chen EHL, Chen RPY (2013) How do amino acid substitutions affect the amyloidogenic properties and seeding efficiency of prion peptides. Amino Acids 45:785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Apostol MI, Wiltzius JJW, Sawaya MR, Cascio D, Eisenberg D (2011) Atomic structures suggest determinants of transmission barriers in mammalian prion disease. Biochemistry 50:2456–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nguyen PH, Li MS, Stock G, Straub JE, Thirumalai D (2007) Monomer adds to preformed structured oligomers of Aβ‐peptides by a two‐stage dock–lock mechanism. Proc Natl Acad Sci USA 104:111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. O'Brien EP, Okamoto Y, Straub JE, Brooks BR, Thirumalai D (2009) Thermodynamic perspective on the dock‐lock growth mechanism of amyloid fibrils. J Phys Chem B 113:14421–14430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Han W, Schulten K (2014) Fibril elongation by Aβ17–42: kinetic network analysis of hybrid‐resolution molecular dynamics simulations. J Am Chem Soc 136:12450–12460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qi R, Luo Y, Wei G, Nussinov R, Ma B (2015) Aβ “stretching‐and‐packing” cross‐seeding mechanism can trigger tau protein aggregation. J Phys Chem Lett 6:3276–3282. [Google Scholar]
- 22. Zhang M, Hu R, Liang G, Chang Y, Sun Y, Peng Z, Zheng J (2014) Structural and energetic insight into the cross‐seeding amyloid assemblies of human IAPP and rat IAPP. J Phys Chem B 118:7026–7036. [DOI] [PubMed] [Google Scholar]
- 23. Hu R, Zhang M, Chen H, Jiang B, Zheng J (2015) Cross‐seeding interaction between β‐amyloid and human islet amyloid polypeptide. ACS Chem Neurosci 6:1759–1768. [DOI] [PubMed] [Google Scholar]
- 24. Nguyen HD, Hall CK (2004) Molecular dynamics simulations of spontaneous fibril formation by random‐coil peptides. Proc Natl Acad Sci USA 101:16180–16185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheon M, Chang I, Hall CK (2011) Spontaneous formation of twisted A beta(16–22) fibrils in large‐scale molecular‐dynamics simulations. Biophys J 101:2493–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Latshaw DC, Cheon M, Hall CK (2014) Effects of macromolecular crowding on amyloid beta (16–22) aggregation using coarse‐grained simulations. J Phys Chem B 118:13513–13526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wagoner VA, Cheon M, Chang I, Hall CK (2011) Computer simulation study of amyloid fibril formation by palindromic sequences in prion peptides. Proteins Struct Funct Bioinf 79:2132–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheon M, Chang I, Hall CK (2012) Influence of temperature on formation of perfect tau fragment fibrils using PRIME20/DMD simulations. Protein Sci 21:1514–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cheon M, Hall CK, Chang I (2015) Structural conversion of A beta(17–42) peptides from disordered oligomers to U‐shape protofilaments via multiple kinetic pathways. PLoS Comput Biol 11:e1004258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Latshaw DC, Hall CK (2015) Effects of hydrophobic macromolecular crowders on amyloid beta (16–22) aggregation. Biophys J 109:124–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang Y, Latshaw DC, Hall CK (2017) Aggregation of Aβ(17–36) in the presence of naturally occurring phenolic inhibitors using coarse‐grained simulations. J Mol Biol 429:3893–3908. [DOI] [PubMed] [Google Scholar]
- 32. Wang Y, Shao Q, Hall CK (2016) N‐terminal prion protein peptides (PrP(120–144)) form parallel in‐register β‐sheets via multiple nucleation‐dependent pathways. J Biol Chem 291:22093–22105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jones EM, Surewicz WK (2005) Fibril conformation as the basis of species‐ and strain‐dependent seeding specificity of mammalian prion amyloids. Cell 121:63–72. [DOI] [PubMed] [Google Scholar]
- 34. Makarava N, Ostapchenko VG, Savtchenko R, Baskakov IV (2009) Conformational switching within individual amyloid fibrils. J Biol Chem 284:14386–14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morales R, Abid K, Soto C (2007) The prion strain phenomenon: molecular basis and unprecedented features. Biochim Biophys Acta Mol Basis Dis 1772:681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Watts JC, Giles K, Patel S, Oehler A, DeArmond SJ, Prusiner SB (2014) Evidence that bank vole PrP is a universal acceptor for prions. PLoS Pathog 10:e1003990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cohen SIA, Vendruscolo M, Dobson CM, Knowles TPJ (2011) Nucleated polymerization with secondary pathways. II. Determination of self‐consistent solutions to growth processes described by non‐linear master equations. J Chem Phys 135:065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Alder BJ, Wainwright TE (1959) Studies in molecular dynamics. 1. General method. J Chem Phys 31:459–466. [Google Scholar]
- 39. Smith AV, Hall CK (2001) Alpha‐helix formation: discontinuous molecular dynamics on an intermediate‐resolution protein model. Proteins Struct Funct Genet 44:344–360. [DOI] [PubMed] [Google Scholar]
- 40. Cheon M, Chang I, Hall CK (2010) Extending the PRIME model for protein aggregation to all 20 amino acids. Proteins Struct Funct Bioinf 78:2950–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Monticelli L, Kandasamy SK, Periole X, Larson RG, Tieleman DP, Marrink SJ (2008) The MARTINI coarse‐grained force field: extension to proteins. J Chem Theory Comput 4:819–834. [DOI] [PubMed] [Google Scholar]
- 42. Chebaro Y, Pasquali S, Derreumaux P (2012) The coarse‐grained OPEP force field for non‐amyloid and amyloid proteins. J Phys Chem B 116:8741–8752. [DOI] [PubMed] [Google Scholar]
- 43. Davtyan A, Schafer NP, Zheng WH, Clementi C, Wolynes PG, Papoian GA (2012) AWSEM‐MD: protein structure prediction using coarse‐grained physical potentials and bioinformatically based local structure biasing. J Phys Chem B 116:8494–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Andersen HC (1980) Molecular dynamics simulations at constant pressure and/or temperature. J Chem Phys 72:2384–2393. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information