

Abstract
Death effector domain (DED) containing molecules are usually involved in the intracellular apoptosis cascade as executioners or regulators. One of these molecules, DEDD, was identified as a final target of the CD95 signaling pathway by which it would be transferred into the nucleolus to inhibit RNA polymerase I-dependent transcription. Here we describe a longer isoform of DEDD, DEDDl, produced by alternatively splicing, as an immune cell-specific DED-containing molecule. It is only expressed in human T lymphocytes and dendritic cells (DCs), and the mRNA expression in DCs was elevated upon inductive maturation. In cell lines MCF-7 and Jurkat, the overexpression of DEDDl could induce apoptosis more potently than that of DEDD. That DEDDl could bind FADD and cFLIP more potently than DEDD in vivo was revealed by cotransfection and immunoprecipitation. This may explain why DEDDl is a more potent apoptosis inducer, because DED-containing proteins usually induce apoptosis through DED binding. Finally, why DEDD and DEDDl are unstable in the overexpression and other studies may be explained by the finding that they are potential substrates of active caspases.
Key words: Alternative splice variant, Apoptosis, Death effector domain (DED), Dendritic cells
INTRODUCTION
Apoptosis, or programmed cell death, is the most common physiological form of cell death and plays a vital role in embryonic development, tissue remodeling, and homeostasis maintenance within all multicellular organisms (15,27). Genetic and molecular analysis from nematodes to humans has indicated the existence of highly conserved cellular suicide pathways; many of the protein domains that perform critical roles in apoptosis signaling are already present in the much simpler organism. However, higher organisms (i.e., vertebrates) have been revealed to have a major increase in the complexity of the apoptotic molecular machinery, both in terms of the membrane receptors and intracellular molecules. Upon the interaction of surface receptors with their cognate ligands, such proteins would participate in a fine-tuned mechanism to determine whether a cell is to live or die (2,8).
To achieve the delicate balance of the immune system, higher organisms have acquired “death receptors” to respond to environmental “death ligands” so that organisms can actively direct individual cells to self-destruct. CD95 (Fas/Apo 1) is the most important death receptor in physiologic apoptosis in the immune system, evident from the symptoms of certain mouse strains and human patients who have a defective gene for CD95 or CD95L (4,14,20,24,28,33). In the CD95 signaling pathway, death effector domain (DED)-containing proteins have been found to play an obligate role in the initiation and execution of apoptosis (5,32). Ligation of CD95 results in the recruitment of the adaptor molecule FADD by homotypic interaction through its C-terminal death domain (DD) with the DD on the cytoplasmic tail of CD95. Another death receptor, TNF-R I, also transmits a death signal through FADD, but differently from the CD95, TNF-R I recruits FADD through another adaptor TRADD (9,38). FADD, which also contains an N-terminal DED, will interact with DEDs on procaspase-8, thus transducing the activation signal from death receptor to the initiator (9). Thus, the intracellular apoptotic cascade initiator caspase-8 has been activated and will in turn cleave various death substrates and other caspases leading to the execution of apoptosis. Other mammalian DED-containing proteins, such as procaspase-10, c-FLIP, PEA-15, DEDD, and DEDD2, have been shown to be involved in signaling of death receptors (13,16,19,29,30,35,39). DEDD is distinct from other DED-containing molecules in its abundance, high conservation between species, and its function mode, which induces apoptosis from within the nucleus. Overexpression of DEDD acts as a weaker apoptosis inducer, localizes to nucleoli-like structures, activates caspase-6, and specifically inhibits RNA polymerase I-dependent transcription in vivo (31). Further studies suggested that DEDD might represent a novel scaffold protein that directs caspase-3, an apoptotic effector, to certain substrates facilitating ordered degradation (21). Although so many details have been revealed, the distinct role of DEDD still may not be completely elucidated.
In the present report, an alternatively spliced form of DEDD has been found and named DEDDl. Although the difference between these two proteins is only 31 amino acids, DEDDl is the only DC/T-cell-specific DED-containing protein to have ever been found and participates in apoptotic machinery more actively than DEDD. The study of such a molecule will not only establish the physiological role of DEDDl, but also presents a natural model for the functions of DED-containing molecules.
MATERIALS AND METHODS
Identification of DEDDl From Human Dendritic Cells by Subtractive Cloning
Human monocyte-derived DCs were generated and identified as follows. Fresh peripheral blood from normal adults was separated with lymphocyte separation media (ρ = 1.077, Sigma). The cells in the boundary layer were collected and cultured with RPMI-1640 complete medium (Invitrogen) in 35-mm dishes at 5 × 106 cells/ml for 2 h. The cells in suspension were then discarded by swirling the dish softly and the adherent cells were cultured in RPMI-1640 complete medium containing rhGM-CSF (800 U/ml, Sigma) and rhIL-4 (500 U/ml, Sigma) for 5 days. The suspending cells were collected and then cultured for another 1 or 2 days. The cells were human DCs, which were confirmed by cytometry that more than 90% of cells were CD1a+, CD83+, and HLA-DR+>. KLH, a potent antigen, was used to stimulate human DCs. The antigen-pulsed DCs were obtained by incubating normal DC in KLH (10 μg/ml, Sigma) for another 24 h. The strategy used for subtractive cloning has been described elsewhere (42). One of those specifically expressed genes, KE05, was found as an alternatively spliced form of DEDD by using the BLAST algorithm and Ensembl web server (www.ensembl.org).
Transient Transfection and Immunofluorescence Microscopy
Transient transfection was performed with Lipofectamine™ 2000 (Invitrogen) according to the manufacturer’s instructions. 293T cells and MCF-7 cells were plated (six-well plate) in 2 ml of complete growth medium without antibiotics at 90–95% confluence for transfection. Plasmid DNA (2 μg) carrying the Myc-tagged DEDDl (5 μl of Lipofectamine™ 2000 reagent) was diluted in 250 μl of DMEM. The mixture was first incubated for 20 min at room temperature and then was added to each well. Cells were incubated at 37°C in a CO2 incubator for at least 24 h but no more than 48 h. The cells were then treated with TNF-α (R&D) at 50 ng/μl for 6 h prior to staining. Then the cells were fixed and stained for immunofluorescence assay. Fixation was performed by adding 4% paraformaldehyde for a 10-min incubation followed by a single PBS wash and incubated with −20°C methanol for 5-min permeabilization. Next the cells received three 5-min washes with PBS and then were blocked with PBS containing 10% goat serum and 1% BSA for 60 min. Myc-tag 9B11 monoclonal antibody (Cell Signaling Technology, CST) was diluted in 1% BSA in PBS at 1:2000. The diluted antibody were added to the cells and incubated overnight at 4°C. The primary antibody was removed by three more 5-min washes with PBS. FITC-conjugated anti-mouse IgG antibody was diluted in 1% BSA in PBS at 1:500 and added to cells for an incubation of 45 min in the dark. After another three washes with PBS the cells were ready for visualization by immunofluorescence microscope. Except where indicated, all steps were performed at room temperature.
Northern Blotting and RT-PCR Analysis
Northern blot membranes MTN containing poly(A)+ RNA from multiple tissues (Clontech, BD) were hybridized with a 32P-labeled DNA probe spanning the DEDDl-specific sequence that differs from that of DEDD. Hybridization was performed following the manuals.
The following cultured cells or cell lines were used for mRNA expression analysis: monocyte-derived DC (5-day culture) and its KLH (Sigma) stimulated counterparts (10 mg/ml KLH, stimulated for 24 h); monocyte-derived DC (7-day culture) and stimulated with CD40, IFN-γ, KLH, LPS, OVA, TNF-α; NB4 (promyelocytic leukemia), HL-60 (acute promyelocytic leukemia), U-937 (histiocytic lymphoma), K-562 (chronic myelogenous leukemia), Raji (Burkitt’s lymphoma; B lymphocyte), Daudi (Burkitt’s lymphoma; B lymphoblast), HuT 78 (lymphoma; cutaneous T lymphocyte), Molt-4 (acute lymphoblastic leukemia; T lymphoblast). Total RNA was extracted with TRIZOL reagent (Invitrogen) and then was reverse transcribed by oligo-dT priming according to a standard protocol. RT-PCR with the primer 5′-ATG GGC TGT ACA GCCTG (sense primer) and 5′-GAA CCC GCA GTC TGA TG (antisense primer) for DEDDl, 5′-TCC CAA GGA GAA GCA GAC (sense primer) and 5′-GAG CCA TTG ATG TAG TCA CG (antisense primer) for DEDD, 5′-TCG CTT CAC CGT ATG TTC (sense primer) and 5′-TGG AGG TGC CAT AGC TAT AG (antisense primer) for DEDD2 was performed using Exo-Taq polymerase (Takara). Cycle conditions were 95°C for 10 s, 55°C for 30 s, and 72°C for 30 s for appropriate cycles. Synthesis of cDNA was controlled by amplifying β-actin (sense primer, 5′-GCA TCC TCA CCC TGA AGT AC; antisense primer, 5′-TTC TCC TTA ATG TCA CGC AC). Cycle conditions for β-actin were 95°C for 10 s, 57.5°C for 30 s, and 72°C for 30 s for 25 cycles.
Plasmid Construction
Epitope-tagged expression plasmids were constructed in pcDNA3.1 (Invitrogen) by PCR using primers flanking with FLAG or Myc epitope tag sequences and appropriate restriction sites.
Apoptosis Assays
MCF-7 cells (mammary adenocarcinoma) were maintained in RPMI-1640 (Invitrogen) supplemented with 10% fetal bovine serum. For transient transfection apoptosis assays, 2 × 105 cells in six-well plates (Falcon) were transfected using Lipofectamine™ 2000 with 2–5 μg of plasmid containing cDNA encoding full-length DEDDl or DEDD as described above. In each experiment, the total amount of DNA was normalized using a vector plasmid without cDNA. Twenty-four hours later, both adherent and floating cells were collected and stained with Rhodamine 123 (Rh123) and propidium iodide (PI) (22). The percentages of apoptotic cells were determined by counting the Rh123 low staining and PI-positive cells.
Jurkat T cells (3 × 106/chamber) were electroporated with 30 μg of plasmid DNA for three pulses of 10 ms each at 300 V (ECM 830, BTX technology). The electroporated cells stayed at room temperature for at least 30 min and then were incubated as routine. Twenty-four hours later, the cells were stained with Annexin V and PI to assay for apoptosis (36). If necessary, PHA was added at the final concentration of 1 μg/ml 6 h before apoptotic assays.
Coimmunoprecipitation and Immunoblot Assays
293T cells were seeded in a six-well plate (2 × 105 cells per well) and cotransfected with 2 μg of pcDNA-FLAG-DEDDl, pcDNA-Myc-DEDDl, pcDNA-FLAG-DEDD, or pcDNA-Myc-DEDD, and pcDNA-FLAG-pro-caspase-8, pcDNA-Myc-FADD, pcDNA-Myc-FLIP. The total amount of DNA was normalized using a control vector in each sample. Twenty-four hours after transfection, cells of each well were collected and resuspended in 200 μl of RIPA buffer (50 mM Tris-HCl, pH 7.4; 1% NP-40; 0.5% Na-deoxycholate; 150 mM NaCl; 1 mM EDTA) containing 1 mM PMSF, 1 μg/ml Aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM Na3VO4, and 1 mM NaF. Soluble lysates were incubated with Myc-tag 9B11 monoclonal antibody (1:2000) at 4°C overnight with vigorous agitation and 30 μl of Protein A-Agarose (Santa Cruz) was added to each well for further binding of 3 h. Precipitates were washed two times with RIPA buffer, fractionated by SDS-PAGE, and transferred onto nitrocellulose membranes for immunoblotting using anti-FLAG monoclonal antibody M2 (Sigma) followed by horseradish peroxidase-conjugated goat anti-mouse IgG (Pierce) as secondary antibody. SuperSignal West Products (Pierce) was used for detection.
RESULTS
Identification of DEDDl, a Human-Specific Alternatively Spliced Form of DEDD
An alternatively spliced isoform of DEDD was identified from human dendritic cells by subtractive cloning. The full length sequence is 1256 bp in length and formerly named KE05 (Accession: AF064605) for sequencing (42). It carries an open reading frame from nt 88 to 1134, which results in a protein with predicted size of 348 amino acids whereas DEDD is 317 amino acids. Thus, it is designated DEDDl as the long isoform of DEDD. Blast, the sequence by Ensembl web server (www.ensembl.org), revealed that DEDDl was coded by 4 exons, the same number of exons as DEDD. The difference between the two sequences comes from the change of the 5′ splice site after exon 3. In DEDDl, the 5′ splice site of the intron between exon 3 and exon 4 moves 90 nucleotides downstream and the 3′ splice site of the intron remains the same as DEDD (Fig. 1A). Such a change replaces an Asp (GAT) in DEDD (35) with a peptide consisting of 31 amino acid residues, bringing an immunoreceptor tyrosine-based inhibitory motif (ITIM) IQYIRL to DEDDl.
Figure 1.

Identification of DEDDl, an alternatively spliced form of DEDD. (A) Partial sequence of DEDDl protein deduced form exon 3 and exon 4. The prolonged part of DEDDl is underlined and the putative ITIM is shaded. (B) Alignment of the genome sequence from exon 3 to exon 4 from human (NCBI, AL591806.16, 177237–176402) and mouse (NCBI, AC087229.26, 85551–86323). The exons are underlined and the prolonged part of DEDDl is double underlined.
The exon/intron prediction analysis of full genomic sequences by others indicated the existence of the mouse DEDDl, because the mouse exon 3 was found to be 102 bp longer than human exon 3 of DEDD and still linked to the same exon 4 (31). But according to the chromosomal sequence released at Ensembl website, the longer 102 bp of the mouse exon 3 would present a stop condon TAA in the reading frame (Fig. 1B, arrow). Therefore, the murine counterpart of DEDDl does not exist according to the known genome.
DEDDl Is a Human-Specific Isoform of DEDD That Is Expressed in DC/T Cells
No signals of DEDDl could be detected by Northern blot analysis in human tissues, but adequate β-actin signals suggest DEDDl might be in low abundance in normal tissues (data not shown).
DEDDl was not expressed in any one of the fetal tissues and tumor cell lines detected, including fetal heart, fetal lung, fetal liver, fetal kidney, fetal skeleton muscles, fetal intestine, HeLa, LoVo, HT29, and A172 cells (data not shown). The mRNA expression analysis of DEDDl in hematopoietic cell lines and monocyte-derived DCs revealed that DEDDl mRNA expression pattern is distinct from the DEDDs (Fig. 2A). DEDDl is only expressed in T-cell lines and DCs, whereas DEDD is ubiquitously expressed. These couple of DC templates were used for detecting mRNA expression of DEDD, DEDDl, and DEDD2 simultaneously by PCR. The results showed the mRNA expression of DEDD was not changed by KLH stimulation while the DEDDl was upregulated significantly. The upregulation of DEDD2’s expression was detected but not as significant as that of DEDDl (data not shown).
Figure 2.
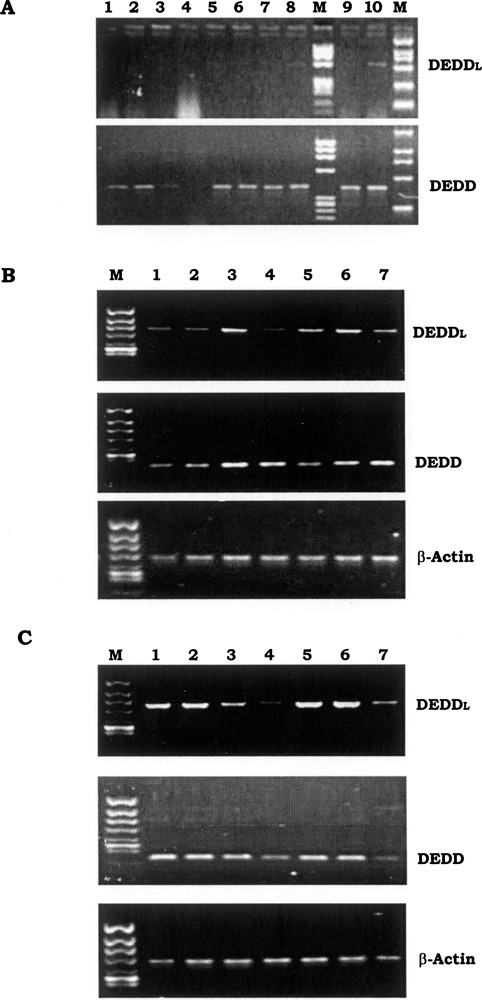
mRNA expression of DEDDl in immune cells. (A) Hematopoietic cell lines and immune cells were arranged in the following order: lane 1, NB4; lane 2, HL-60; land 3, U937; lane 4, K562; lane 5, Raji; lane 6, Daudi; lane 7, HuT78; lane 8, Molt-4; lane 9, DC; lane 10, DC-KLH. (B) Lane 1, DC; lanes 2–4, DC treated with LPS for 1, 8, and 48 h; lanes 5–7, DC treated with TNF-α for 1, 8, and 48 h. (C) Lane 1, T lymphocytes; lanes 2–4, T lymphocytes treated with PHA for 1 and 8 h and 4 days; lanes 5–7, T lymphocytes treated with rIL-2 for 1 and 8 h and 4 days.
We next examined the pattern of DEDDl expression in human DC or T cells and compared it with that of DEDD expression. Human peripheral blood-derived DCs were treated with LPS or TNF-α and human peripheral blood T cells were treated with rIL-2 or PHA. Both DEDDl and DEDD mRNA were induced with a significant increase in DCs by LPS or TNF-α at 8 h. At 48 h the increased DEDDl expression declined to unstimulated levels and the induced DEDD expression was sustained (Fig. 2B). The mRNA of DEDDl and DEDD in human peripheral blood T cells was more abundant than that in DCs and their expression was downregulated by PHA or rIL-2 treatment (the decrease of DEDDl expression happened earlier than that of DEDD) (Fig. 2C).
DEDDl Induced Apoptosis More Potently Than DEDD in MCF-7 and Jurkat T Cells
Mitochondrial transmembrane potential (ΔΨm) decreases early during apoptosis, thus enabling cells entering apoptosis to be detected by using ΔΨm probes such as Rhodamine 123 (Rh123) and 3,3′-dihexiloxa dicarbocyanine [DiOC6(3)]. The ΔΨm probes are membrane-permeable lipophilic cationic fluorochromes that will accumulate in mitochondria of live cells so that normal cells are highly stained with Rh123 in contrast with the lower staining of apoptotic cells because of the decreased retention of Rh123 in apoptotic mitochondria. Propidium iodide (PI) was used to detect late apoptotic cells in combination with the earlier Rh123 apoptotic detection of MCF-7 cells, which was used to study the potential role of DEDDl in apoptosis, shown to be sensitive to Rh123/PI double staining. In Figure 3A, area I represents PI-positive cells and area II represents Rh123 low staining cells. The percentages of apoptotic cells in MCF-7 cells transfected with DEDDl and DEDD gene were increased from 15% with the control vector to 31.5% and 16.4%, respectively (Fig. 3a, b, c). This is consistent with other reports that DEDD is a weak apoptosis inducer and indicates that DEDDl could be more potent in induction of apoptosis than DEDD. Because C-DEDD showed antiapoptotic function, that DEDDl is more potent than DEDD in apoptosis induction may be explained by the stretch part of DEDDl that affects C-DEDD function by changing its folding mode or by recruiting other unknown molecules to interfere with it. Following with this hypothesis, upon TNF-α treatment, the apoptosis cells were increased significantly (Fig. 3d, e). The decrease in retention of Rh123 was retarded in DEDD compared with DEDDl in the groups of 1-h TNF-α treatment (Fig. 3f, g; arrow).
Figure 3.
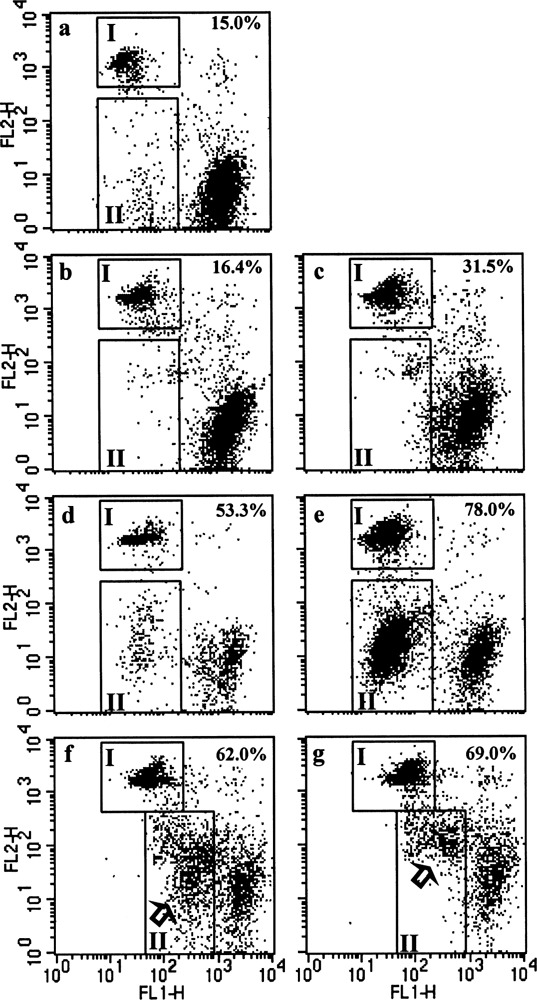
DEDDl is a more potent apoptosis inducer than DEDD. MCF-7 cells were transfected with DEDDl or DEDD genes and treated with TNF-α for indicated times. The percentage of apoptosis cells indicated were counted by PI-positive (I) plus Rh123 low staining (II) cells.
Jurkat T cells were transfected with DEDD or DEDDl genes and double stained with Annexin-V and PI after a 6-h treatment of PHA. As shown in Figure 4, the percentage of Annexin-V/PI double positive cells in DEDDl transfected cells (23.1%) was much higher than that in DEDD transfected cells (8.11%). The percentage of Annexin-V-positive cells in two groups showed no significant difference (DEDDl 30.67% vs. DEDD 31.13%).
Figure 4.
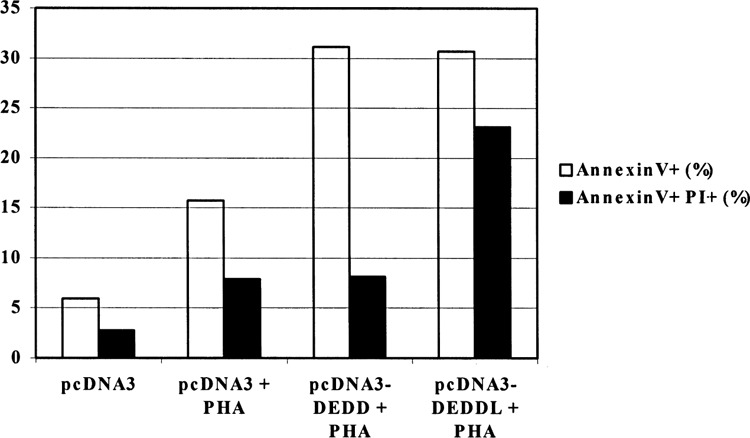
Comparison of the apoptosis induction between Jurkat T cells transfected with DEDDl or DEDD.
DEDDl Translocated Into Nuclei Upon Activation
In DEDDl, the three nuclear localization signals (NLS) identified in DEDD are intact. When a Myc-tagged DEDDl was transfected into 293T cells and MCF-7 cells, immunofluorescence assays showed DEDDl distributed both in the cytoplasm and nuclei (Fig. 5A, C). It seems that overexpression induced DEDDl accumulation into nucleus spontaneously. After TNF-α treatment, DEDDl were no longer uniformly distributed within the nucleoplasm, but accumulated in distinct globular structures or segregated into sharply circumscribed masses that abut the nuclear membrane (Fig. 5B, D). The results indicate that DEDDl may play a role in the nucleus during apoptosis or transcriptional regulation.
Figure 5.
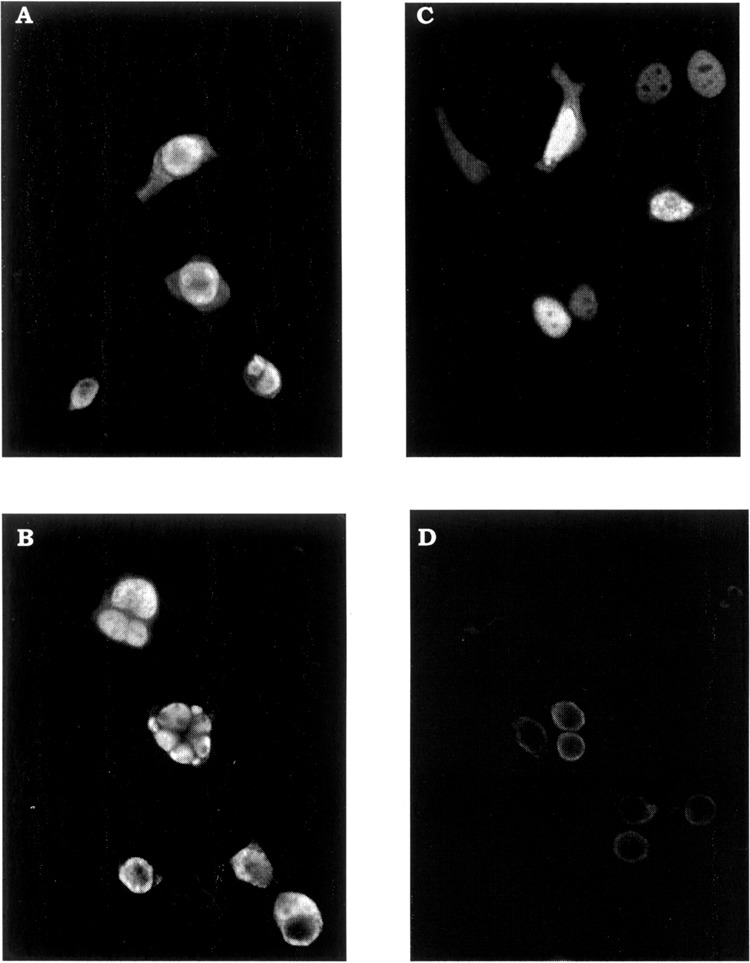
Cellular localization of DEDDl. Cells were transfected with Myc-tagged DEDDl and subjected to immunofluorescence staining with 9B11 monoclonal anti-Myc antibody followed with FITC-labeled secondary antibody. (A) Transfected 293T cells. (B) TNF-α-treated transfected 293T cells. (C) Tranfected MCF-7 cells. (D) TNF-α-treated transfected MCF-7 cells.
DEDDl Can Bind Typical DED-Containing Molecules In Vivo
The DEDDl protein was tested for interaction with other DED-containing proteins in vivo by coimmunoprecipitations when DEDDl, FADD, procaspase-8, and cFLIP were expressed in 293T cells with appropriate epitope tags. The same test was performed on DEDD as a control. DEDDl was capable of binding FADD and cFLIP, noticeably much stronger than was DEDD (Fig. 6A), whereas neither isoform could bind procaspase-8 under this condition (Fig. 6B). It was also revealed that DEDDl and DEDD could bind each other (Fig. 6C).
Figure 6.
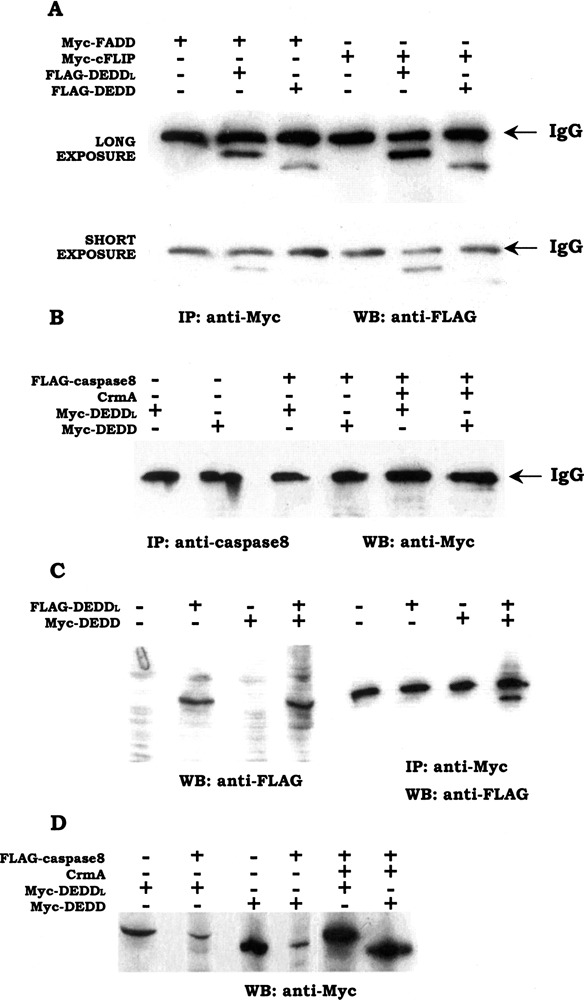
Interaction of DEDDl with other DED-containing proteins. (A) DEDDl binds with FADD and cFLIP much stronger than does DEDD, especially with cFLIP. 293T cells were transiently transfected with various combinations of plasmids encoding FLAG-DEDDl, FLAG-DEDD, Myc-FADD, and Myc-cFLIP. Cell lysates were prepared and immunoprecipitations were performed using anti-Myc monoclonal antibodies (9B11), followed by Western blot analysis using anti-FLAG M2 monoclonal antibody. To show the difference clearly, the same blot was exposed to film for a short period and long period as indicated. (B) DEDDl and DEDD cannot bind caspase-8 in vivo. 293T cells were transfected with DEDDl or DEDD combined with caspase-8 with or without the presence of crmA. Cell lysates were immunprecipitated by using the anti-caspase-8 monoclonal antibody and the following immunoblotting was perfomed with anti-Myc antibody. (C) DEDDl and DEDD bind each other. (D) Caspase-8 can degrade DEDDl and DEDD.
DEDDl and DEDD were also shown be unstable when overexpressed in 293T cells with procaspase-8 cotransfection, whereas they could attain a stable overexpression with the presence of crmA (Fig. 6D). The degradation of DEDDl and DEDD occurred whenever the host cells underwent induced or spontaneous apoptosis.
DISCUSSION
Programmed cell death is a fundamental physiological process in the developing embryo during morphogenesis or synaptogenesis and in the adult animal during tissue renewal and immune response (20). Typically, the immune system produces more cells than ultimately needed to satisfy the receptor repertoire selection during lymphocyte maturation and somatic selection during the adaptive immune response against pathogens (4). The elimination of extra cells by apoptosis is part of the normal life of higher organisms to maintain homeostasis. The immune system is a society of interacting cells consisting of T and B lymphocytes, NK cells, macrophages, and professional APCs at different differentiational levels. It is widely recognized that immune cells can choose different pathways to die under different environmental conditions according to their differentiation status.
In this report, a human-specific DED-containing protein was identified and designated as DEDDl because it is an alternatively spliced form of DEDD and is produced by acquiring 31 more amino acids from the third intron than DEDD. That there is no mouse counterpart of DEDDl indicates DEDDl is developed during late evolution. DEDDl is also distinct from DEDD and other DED-containing proteins in tissue distribution. DEDDl is shown only expressed in DC and T cell lines. Supported with the report that a longer splice form was detected in peripheral human T cells, we conclude that DEDDl is the DC/T cell-specific form of DEDD. This confirms the hypothesis presented by Schickling et al., that DEDD has alternatively spliced forms that may be expressed in a tissue-specific fashion, as with many other apoptosis signaling molecules (31). DEDD is a highly conserved protein with 98.7% homology between human and mouse. Usually, only proteins that act at intracellular key control points are so highly conserved between human and mouse, such as cytochrome C (91.3%); ribosomes subunit L7 (95.9%), L12 (98.8%), L19 (99.5%), L28 (97.8%), S2 (98.2%), S16 (97.3%); Histones H3 and H4 (100%). Although many functional details of DEDD have been revealed, they could not piece together a crucial role that required such high conservation. DEDD was suggested to be a regulator of FADD intracellular apoptotic signaling by interacting with caspase-3 or caspase-6, or interfering with DNA transcription (31,35,37). DEDDl is at least an immune cell-specific regulator in the apoptotic cascade because it binds FADD and cFLIP effectively.
Generally, living systems operate under interactive selective pressures (10). The immune system differs from other biological systems in that it is under threat from foreign pathogens or from within by malignant cellular change (i.e., the much accelerated evolutionary encounters) (26). Apart from somatic evolution adopted against these threats during the lifetime of an individual, it is not surprising that higher species have developed their own particular mechanisms to fulfill immune defense. That DEDDl has no murine counterpart suggests that it may have been developed late in species evolution by the immune system under increased threats. Apoptosis is such a central regulatory feature of the immune system that it is not surprising that immune system would develop its own exclusive molecules. DEDDl provides a model that they have come from ubiquitous molecules by alternative splicing. Although the excess part is only 31 amino acids, it brings three tyrosines and a typical ITIM that implies its function in the immune system.
Deletion of excessive expanded lymphocytes by apoptosis occurs at the peak or the down phase of the immune response and is called activation-induced cell death (AICD) (1,7,11,17,34,37). Antigen presenting cells (APCs) initiate and modulate the activity and function of T lymphocytes and in turn activated T cells influence APC function through surface molecules, including death receptors (CD95, CD40, TNF RI) and cognate ligands (6,23,25,41). APCs exhibit resistance to apoptosis during their role as immune response initiators. This resistance is subsequently switched off to downregulate the response (3). The genes upregulated, including DEDDl, upon DC maturation may participate in the postactivation elimination. The DEDDl mRNA expression in DCs was enhanced upon TNF-α and LPS stimulation, reached its highest level 8 h after treatment, and then declined, whereas the expression of DEDDl in peripheral CD4+ T lymphocytes was decreased upon PHA and IL-2 stimulation. This result indicates that DEDDl would participate in the CD95 signaling pathway because DCs would become resistant to CD95-mediated apoptosis upon TNF-α and LPS treatment and T lymphocytes would become sensitive to CD95-mediated apoptosis when activated with PHA and IL-2 (3,18,40).
It was indicated that the C-terminal half of DEDD has antiapoptotic activity because removing 60% of the C-terminus of DEDD resulted in a protein that was more potent in induction of apoptosis than wild-type DEDD. DEDDl is shown to induce apoptosis more potently than DEDD. This potency may be due to the higher efficiency of DEDDl in binding with other DED-containing proteins, especially cFLIP, and the influence of the stretch part of DEDDl on folding the C-terminal half of DEDD or recruiting other molecules to interfere with itself. Furthermore, the expressions of DEDDl and DEDD are shown to be unstable. When they are cotransfected with caspase-8, their expression is decreased, which can be reversed by crmA. The fact that such degradation also occurs in cultured cells under spontaneous apoptosis suggests that DEDDl and DEDD may be potential substrates of active caspases. DEDD has several expected cleavage sites of caspases and also fits the criteria that caspase substrates are abundant and involved in DNA metabolism (12). It also may explain why DEDD is ubiquitinated and cannot be detected in cytoplasm sometimes.
In this report, an alternatively spliced form of DEDD, DEDDl, was identified to be a DC/T-specific DED-containing protein that is expressed exclusively in humans. It can induce apoptosis more potently than DEDD when overexpressed, probably because DEDDl binds other DED-containing proteins more effectively. Higher conservation of DEDD between human and mouse implies its still unknown role in the intracellular control points. Accelerating investigations suggest DED proteins may do more than simply engage the caspase-8-dependent apoptosis machinery; thus, DEDD and DEDDl may be revealed to perform a crucial role in cell biology.
ACKNOWLEDGMENTS
We thank Dr. Xiaoqing Sun for providing the 293T cell line and the cDNA of procaspase-8, FADD, cFLIP; and Dr. Jun Wu for Jurkat T cell line. The authors thank Caroline K. Hu for her help preparing the manuscript. This research was supported by funds from the National Science Foundation of China (Grant 30271210) and the Special Funds for Authors of Excellent Doctoral Dissertations from National Department of Education (Grant 200258).
REFERENCES
- 1. Alderson M. R.; Tough T. W.; Davis-Smith T.; Braddy S.; Falk B.; Schooley K. A.; Goodwin R. G.; Smith C. A.; Ramsdell F.; Lynch D. H. Fas ligand mediates activation-induced cell death in human T lymphocytes. J. Exp. Med. 181:71–77; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aravind L.; Dixit V. M.; Koonin E. V. Apoptotic molecular machinery: Vastly increased complexity in vertebrates revealed by genome comparisons. Science 291:1279–1284; 2001. [DOI] [PubMed] [Google Scholar]
- 3. Ashany D.; Savir A.; Bhardwaj N.; Elkon K. B. Dendritic cells are resistant to apoptosis through the Fas (CD95/APO-1) pathway. J. Immunol. 163:5303–5311; 1999. [PubMed] [Google Scholar]
- 4. Ashkenazi A.; Dixit V. M. Death receptors: Signaling and modulation. Science 281:1305–1308; 1998. [DOI] [PubMed] [Google Scholar]
- 5. Barnhart B. C.; Lee J. C.; Alappat E. C.; Peter M. E. The death effector domain protein family. Oncogene 22:8634–8644; 2003. [DOI] [PubMed] [Google Scholar]
- 6. Bjorck P.; Banchereau J.; Flores-Romo L. CD40 ligation counteracts Fas-induced apoptosis of human dendritic cells. Int. Immunol. 9:365–372; 1997. [DOI] [PubMed] [Google Scholar]
- 7. Brunner T.; Mogil R. J.; LaFace D.; Yoo N. J.; Mahboubi A.; Echeverri F.; Martin S. J.; Force W. R.; Lynch D. H.; Ware C. F. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature 373:441–444; 1995. [DOI] [PubMed] [Google Scholar]
- 8. Chao D. T.; Korsmeyer S. J. Bcl-2 family: Regulators of cell death. Annu. Rev. Immunol. 16:395–419; 1998. [DOI] [PubMed] [Google Scholar]
- 9. Chinnaiyan A. M.; O’Rourke K.; Yu G. L.; Lyons R. H.; Garg M.; Duan D. R.; Xing L.; Gentz R.; Ni J.; Dixit V. M. Signal transduction by DR3, a death domain-containing receptor related to TNFR-1 and CD95. Science 274:990–992; 1996. [DOI] [PubMed] [Google Scholar]
- 10. Cohn M. The immune system: A weapon of mass destruction invented by evolution to even the odds during the war of DNAs. Immunol. Rev. 185:24–38; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dhein J.; Walczak H.; Baumler C.; Debatin K. M.; Krammer P. H. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95). Nature 373:438–441; 1995. [DOI] [PubMed] [Google Scholar]
- 12. Earnshaw W. C.; Martins L. M.; Kaufmann S. H. Mammalian caspases: Structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 68:383–424; 1999. [DOI] [PubMed] [Google Scholar]
- 13. Fernandes-Alnemri T.; Armstrong R. C.; Krebs J.; Srinivasula S. M.; Wang L.; Bullrich F.; Fritz L. C.; Trapani J. A.; Tomaselli K. J.; Litwack G.; Alnemri E. S. In vitro activation of CPP32 and Mch3 by Mch4, a novel human apoptotic cysteine protease containing two FADD-like domains. Proc. Natl. Acad. Sci. USA 93:7464–7469; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fisher G. H.; Rosenberg F. J.; Straus S. E.; Dale J. K.; Middleton L. A.; Lin A. Y.; Strober W.; Lenardo M. J.; Puck J. M. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 81:935–946; 1995. [DOI] [PubMed] [Google Scholar]
- 15. Hengartner M. O. The biochemistry of apoptosis. Nature 407:770; 2000. [DOI] [PubMed] [Google Scholar]
- 16. Irmler M.; Thome M.; Hahne M.; Schneider P.; Hofmann K.; Steiner V.; Bodmer J. L.; Schroter M.; Burns K.; Mattmann C.; Rimoldi D.; French L. E.; Tschopp J. Inhibition of death receptor signals by cellular FLIP. Nature 388:190–195; 1997. [DOI] [PubMed] [Google Scholar]
- 17. Ju S. T.; Panka D. J.; Cui H.; Ettinger R.; el-Khatib M.; Sherr D. H.; Stanger B. Z.; Marshak-Rothstein A. Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature 373:444–448; 1995. [DOI] [PubMed] [Google Scholar]
- 18. Kirchhoff S.; Muller W. W.; Krueger A.; Schmitz I.; Krammer P. H. TCR-mediated up-regulation of c-FLIPshort correlates with resistance toward CD95-mediated apoptosis by blocking death-inducing signaling complex activity. J. Immunol. 165:6293–6300; 2000. [DOI] [PubMed] [Google Scholar]
- 19. Kitsberg D.; Formstecher E.; Fauquet M.; Kubes M.; Cordier J.; Canton B.; Pan G.; Rolli M.; Glowinski J.; Chneiweiss H. Knock-out of the neural death effector domain protein PEA-15 demonstrates that its expression protects astrocytes from TNFalpha-induced apoptosis. J. Neurosci. 19:8244–8251; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kramer P. H. CD95’s deadly mission in the immune system. Nature 407:789–795; 2000. [DOI] [PubMed] [Google Scholar]
- 21. Lee J. C.; Schickling O.; Stegh A. H.; Oshima R. G.; Dinsdale D.; Cohen G. M.; Peter M. E. DEDD regulates degradation of intermediate filaments during apoptosis. J. Cell. Biol. 158:1051–1066; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lizard G.; Fournel S.; Genestier L.; Dgedin N.; Chaput C.; Flacher M.; Mutin M.; Panaye G.; Revillard J. P. Kinetics of plasma membrane and mitochondrial alterations in cells undergoing apoptosis. Cytometry 21:275–283; 1995. [DOI] [PubMed] [Google Scholar]
- 23. Matsue H.; Edelbaum D.; Hartmann A. C.; Morita A.; Bergstresser P. R.; Yagita H.; Okumura K.; Takashima A. Dendritic cells undergo rapid apoptosis in vitro during antigen-specific interaction with CD4+ T cells. J. Immunol. 162:5287–5298; 1999. [PubMed] [Google Scholar]
- 24. Nagata S. Human autoimmune lymphoproliferative syndrome, a defect in the apoptosis-inducing Fas receptor: A lesson from the mouse model. J. Hum. Genet. 43:2–8; 1998. [DOI] [PubMed] [Google Scholar]
- 25. Nieda M.; Kikuchi A.; Nicol A.; Koezuka Y.; Ando Y.; Ishihara S.; Lapteva N.; Yabe T.; Tokunaga K.; Tadokoro K.; Juji T. Dendritic cells rapidly undergo apoptosis in vitro following culture with activated CD4+ Valpha 24 natural killer T cells expressing CD40L. Immunology 102:137–145; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Phillips R. E. Immunology taught by Darwin. Nat. Immunol. 3:987–989; 2002. [DOI] [PubMed] [Google Scholar]
- 27. Rich T.; Allen R. L.; Wyllie A. H. Defying death after DNA damage. Nature 407:777; 2000. [DOI] [PubMed] [Google Scholar]
- 28. Rieux-Laucat F.; Le Deist F.; Hivroz C.; Roberts I. A.; Debatin K. M.; Fischer A.; de Villartay J. P. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science 268:1347–1349; 1995. [DOI] [PubMed] [Google Scholar]
- 29. Roth W.; Stenner-Liewen F.; Pawlowski K.; Godzik A.; Reed J. C. Identification and characterization of DEDD2, a death effector domain-containing protein. J. Biol. Chem. 277:7501–7508; 2002. [DOI] [PubMed] [Google Scholar]
- 30. Scaffidi C.; Schmitz I.; Krammer P. H.; Peter M. E. The role of c-FLIP in modulation of CD95-induced apoptosis. J. Biol. Chem. 274:1541–1548; 1999. [DOI] [PubMed] [Google Scholar]
- 31. Schickling O.; Stegh A. H.; Byrd J.; Peter M. E. Nuclear localization of DEDD leads to caspase-6 activation through its death effector domain and inhibition of RNA polymerase I dependent transcription. Cell Death Differ. 8:1157–1168; 2001. [DOI] [PubMed] [Google Scholar]
- 32. Schulze-Osthoff K.; Ferrari D.; Los M.; Wesselborg S.; Peter M. E. Apoptosis signaling by death receptors. Eur. J. Biochem. 254:439–459; 1998. [DOI] [PubMed] [Google Scholar]
- 33. Siegel R. M.; Frederiksen J. K.; Zacharias D. A.; Chan F. K.; Johnson M.; Lynch D.; Tsien R. Y.; Lenardo M. J. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science 288:2354–2357; 2000. [DOI] [PubMed] [Google Scholar]
- 34. Singer G. G.; Abbas A. K. The fas antigen is involved in peripheral but not thymic deletion of T lymphocytes in T cell receptor transgenic mice. Immunity 1:365–371; 1994. [DOI] [PubMed] [Google Scholar]
- 35. Stegh A. H.; Schickling O.; Ehret A.; Scaffidi C.; Peterhansel C.; Hofmann T. G.; Grummt I.; Krammer P. H.; Peter M. E. DEDD, a novel death effector domain-containing protein, targeted to the nucleolus. EMBO J. 17:5974–5986; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Engeland M.; Nieland L. J.; Ramaekers F. C.; Schutte B.; Reutelingsperger C. P. Annexin V-affinity assay: A review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry 31:1–9; 1998. [DOI] [PubMed] [Google Scholar]
- 37. van Parijs L.; Ibraghimov A.; Abbas A. K. The roles of costimulation and Fas in T cell apoptosis and peripheral tolerance. Immunity 4:321–328; 1996. [DOI] [PubMed] [Google Scholar]
- 38. Varfolomeev E. E.; Boldin M. P.; Goncharov T. M.; Wallach D. A potential mechanism of “cross-talk” between the p55 tumor necrosis factor receptor and Fas/APO1: Proteins binding to the death domains of the two receptors also bind to each other. J. Exp. Med. 183:1271–1275; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vincenz C. A.; Dixit V. M. Fas-associated death domain protein interleukin-1β-converting enzyme 2 (FLICE2), an ICE/Ced-3 homologue, is proximally involved in CD95- and p55-meiated death signaling. J. Biol. Chem. 272:6578–6583; 1997. [DOI] [PubMed] [Google Scholar]
- 40. Wesch D.; Marx S.; Kabelitz D. Monocyte-dependent death of freshly isolated T lymphocytes: Induction by phorbolester and mitogens and differential effects of catalase. J. Immunol. 161:1248–1256; 1998. [PubMed] [Google Scholar]
- 41. Yokota A.; Oikawa A.; Matsuda C.; Shinohara N.; Eshima K. Cell-mediated Fas-based lysis of dendritic cells which are apparently resistant to anti-Fas antibody. Microbial. Immunol. 47:285–293; 2003. [DOI] [PubMed] [Google Scholar]
- 42. Zhao Z.; Huang X.; Li N.; Chen S.; Cao X. Direct cloning of cell differential expression genes with full-length by a new strategy based on the multiple rounds of “long distance” PCR and magnetic beads mediated subtraction. J. Biotech. 73:35–41; 1999. [DOI] [PubMed] [Google Scholar]