

Abstract
We have adopted a special experimental strategy to identify early responsive genes during 12-O-tetradecanoylph-orbol-13-acetate (TPA)-induced macrophage-like differentiation of human myeloid leukemia cells (HL-60). This was performed in cells that were synchronized by nocodazole and treated with TPA in the presence of a protein synthesis inhibitor, cycloheximide, to prevent activation of secondary targets and therefore increase the probability of early transcripts in total RNA pool. The expression alteration was analyzed by microarray and the selection criteria of candidate genes were adjusted by real-time PCR validation to increase its reliability. Finally, 56 genes were identified as early TPA-responsive genes in this multiscreening step approach. Furthermore, upregulation of three candidate genes (NFIL3, SKIL, and JMJD3) was shown to be dosage and time dependent with TPA treatment and was found to be directly regulated by TPA through PKC-dependent signaling. These results revealed that our screenings provide a useful and efficient approach to identify early TPA-responsive genes and these genes might involve the regulation of TPA-induced differentiation program of HL-60 cells as primary targets.
Key words: Early responsive gene, TPA, Cycloheximide, Differentiation, Microarray
INTRODUCTION
The human myeloid leukemia cell line, HL-60, has been widely investigated as a model for studying the genetic and biochemical factors regulating both cell growth and differentiation. HL-60 cells undergo macrophage-like differentiation in response to 12-O-tetra-decanoylphorbol-13-acetate (TPA or PMA) treatment (32,33), and this effect is mainly initiated through PKC signals (24,45). This differentiation program transcriptionally regulates specific target genes and induces phenotype changes in morphology, immunophenotype, and cellular maturation.
Previous studies using molecular biology approaches have identified a number of genes showing altered expression during TPA-triggered macrophage-like differentiation of HL-60 cells, such as EGR1 (18) and the JUN/FOS superfamily (1), some of them might be essential for the onset of differentiation in HL-60 cells (48). With the advance of microarray that can monitor the expression of a large number of genes and the complex gene expression patterns, recent studies have been able to systematically survey cellular transcriptional regulation in TPA-induced differentiating HL-60 cells (36,44,49).
However, many microarray experiments have not clearly distinguished between the primary (direct) and secondary (indirect) targets of TPA. In addition, expression of mRNAs for primary target genes is often expressed at low levels or shows only slight alterations in their expression levels. It is difficult to detect these critical genes because of the limitations of current fluorescence detection system in microarray, and a large number of highly expressed secondary transcripts might mask the low-level signals of primary targets in microarray analysis (9,15,47). An alternative approach is urgently required to study such important primary genes in the transcriptome. By identifying the primary targets of TPA and using these to examine their prospective downstream target genes, it is possible to better construct transcriptional regulatory networks that link key factors to the biological function that they involve.
To overcome these problems of previous microarray study in TPA-induced cell differentiation as mentioned above, we have adopted a new strategy to discover primary TPA target genes using the HL-60 cell differentiation system. Here we have identified 56 early responsive genes as candidate genes directly upregulated by TPA with this new screening strategy. In addition to previously recognized TPA or myeloid differentiation-related genes with high expression levels, we additionally identified 32 more genes with minor changes in their expression levels and have further validated some of these genes that were trans-activated by TPA directly through PKC signaling. Taking all data together, our multiscreening strategy identified the additional primary TPA target genes, which can serve as another base for better understanding the role of TPA during the differentiation of HL-60 cells.
MATERIALS AND METHODS
Cell Culture and Induction of Differentiation
HL-60 cells were cultured at a density of 2 × 105 cells/ml with RPMI-1640 (GIBCO-BRL, Gaithersburg, MD, USA) supplemented with 10% heat-inactivated fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/ml) (GIBCO-BRL) in a humidified atmosphere of 5% CO2 at 37°C. To induce macrophage-like differentiation, HL-60 cells were seeded at a density of 2 × 105 cells/ml and treated with 32 nM TPA (Sigma, St. Louis, MO, USA) for the indicated period; the control group was treated with vehicle (only 0.05% DMSO). Where indicated, 1 μM Go6976 (classic PKC inhibitor) was added to culture media at 1 h before TPA treatment. These specific inhibitors used for the experiments were obtained from Calbiochem Company (La Jolla, CA, USA). Induction of differentiation by TPA was confirmed by transcription of the CD11b marker gene or by increased cell adherence (12,30). The amount of adherent cells was scored in randomly selected microscopic fields under a phase-contrast microscope observation.
RNA Extraction and RT-PCR
Total RNA was isolated using the TRIzol reagent (Invitrogen, USA) followed by DNase I treatment (Promega, Madison, WI, USA) as described by the manufacturer. For RT-PCR, 5 μg of total RNA was reverse transcribed into single-strand cDNA using the oligo-(dT)15 primer and PowerScript reverse transcriptase (Clontech, Palo Alto, CA, USA). GAPDH was used as the internal standard because microarray profiling showed that the signals for GAPDH probe sets were equivalent between our TPA-treated or control cells. The amplified PCR products were separated on a 1.5% agarose gel and stained with ethidium bromide for visualization. The primer sets used for amplification were: sense 5′-CCCTAAGCTGGA GGAGATGATG and antisense 5′-TCATGCTCACT AGGCCACTGAC for EGR1; sense 5′-ATGGCTCT CAGAGTCCTTCTGTTAA and antisense 5′-CAT CAAAGAGAACAAGGTTTTGGA for CD11b; and sense 5′-TGGTATCGTGGAAGGACTCA and antisense 5′-AGTGGGTGTCGCTGTTGAAG for GAPDH.
Schema for Identifying Early TPA-Responsive Genes
We designed a modified screening strategy aimed at enriching early TPA-responsive genes, as outlined in Figure 1. To achieve this aim, a homogeneous cell population was first generated by synchronizing the cells with nocodazole (0.05 μg/ml) for 24 h and then cells were washed twice with culture medium and treated for 30 min with cycloheximide (CHX, 10 μg/ ml). Differentiation was induced by incubation with TPA (32 nM) in the continued presence of cycloheximide, thus excluding the indirect transcriptional effects from the gene product of the primary target gene. Control cells were also synchronized and treated with cycloheximide, but they were treated with 0.05% DMSO instead of TPA. Cells were collected at 1 h post-TPA treatment and subjected for total RNA isolation. In order to minimize the interexperimental variations, we collected and pooled total RNA from three independent experiments for subsequent RT-PCR and microarray analysis. In the present study, microarray analysis was first used as a screening tool to get the preliminary candidate TPA target genes. A subset of these genes was further screened by quantitative real-time PCR (Q-PCR). According to the result of Q-PCR analysis, we adjusted our selection criteria for previous microarray analysis. Thus, only the early TPA-responsive genes are present in this screening, and these genes are more likely to be the primary (direct) targets of TPA in HL-60 cells.
Figure 1.
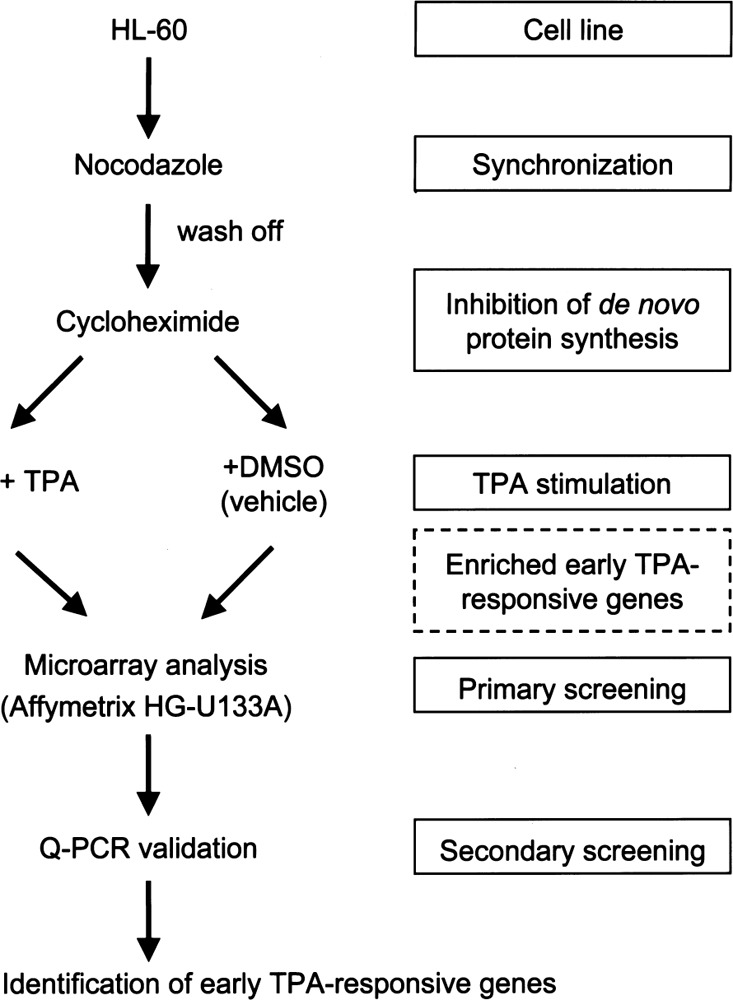
Schema of the experimental design for identifying early TPA-responsive genes. HL-60 cells were synchronized with nocodazole (0.05 μg/ml for 24 h) and then cells were washed with culture medium and followed by treatment with cycloheximide (CHX, 10 μg/ml) for 30 min prior to subsequent stimulation with TPA (32 nM, 1 h) or DMSO (0.05%, 1 h) as the vehicle control. The cells were then collected and used for microarray and realtime PCR analysis.
Microarray Analysis
RNA isolated from cells treated with cycloheximide plus TPA or cycloheximide plus vehicle control as described before was used for global differential gene expression analysis using HG-U133A GeneChip (Affymetrix, Santa Clara, CA, USA). The hybridization and data analysis were performed at UC Davis School of Medicine Microarray Core Facility according to the Affymetrix technical manual. Arrays were analyzed with Affymetrix’s Microarray Suite (MAS 5.0) for background subtraction, normalization, and comparison. To identify differentially expressed transcripts, comparison analysis was performed, and all data were calculated and expressed fold change as a log2 ratio and subjected to a statistical analysis of significance (p < 0.05). The probe set information was obtained using the NetAffx online analysis tool supplied by Affymetrix (http://www.affymetrix.com).
Quantitative Real-Time PCR Assay
Real-time PCR was performed on a Roche Ligh Cycler system using gene-specific primers and DNA Fast-Start Master SYBR Green I mix (Roche, Basel, Switzerland) according to the manufacturer’s protocol. For each sample, the threshold cycle Ct (or Cp) value was used as a measure of the amount of template present in the starting reaction, and the ΔCt value was determined by subtracting the Ct value of reference gene (GAPDH) from the Ct value of target gene. The relative expression of each gene was expressed as −ΔΔCt (ΔCt value of the control sample subtract ΔCt value of the TPA-treated sample) (29). Differences in the mean ± SD (n ≥ 3) between TPA-treated cells and controls were analyzed statistically using Student’s t-test. The real-time PCR was performed using the gene-specific primers (Supplementary Table 1).
SUPPLEMENTARY TABLE 1.
GENE-SPECIFIC PRIMERS USED FOR REAL-TIME PCR
| Gene Name | Forward Primer | Reverse Primer |
|---|---|---|
| IL8 | s: TTGGCAGCCTTCCTGATT | as: AACTTCTCCACAACCCTCTG |
| JUN | s: TTGTTTGTTTGGGTATCCTG | as: ATGCAGAAAAGAGGTTAGGG |
| NFIL3 | s: TGGAGAAGACGAGCAACA | as: CTTTGATCTGCATGGCTT |
| SKIL | s: GAGGCTGAATATGCAGGACA | as: GCCTAGTTATCGTCATGC |
| JMJD3 | s: ATCGTGCCCATGATTCAC | as: TCCACATCGCACTCGTTG |
| C140RF92 | s: CGATGTGTGAGGTCTGGT | as: GACACTTTCCCAGCAGAT |
| TNF | s: ACAAGCCTGTAGCCCATGTT | as: AGACGGCGATGCGGCTGATG |
| FOS | s: GGATAGCCTCTCTTACTACCAC | as: TCCTGTCATGGTCTTCACAACG |
| NFKBIA | s: CCAAGCACCCGGATACAG | as: GCTGGCCTCCAAACACAC |
| BCLAF1 | s: GGATGGGATTGTTGAAGATG | as: GTGGGTGCAAGTTCTGCTCT |
| PDE4BC | s: TTGTATCGGCAATGGACAGA | as: GTCTCCCACAATGGATGGA |
| KIAA0286 | s: TTGCCCTTGCCATTATCATC | as: GCCTTTCGGGTTTCTACCTC |
| HSPCA | s: ATGAAACTGCGCTCCTGTCT | as: CTTCTTCCATGCGTGATGTG |
| DDX3X | s: GGTAGCAGCAGAGGATTTGG | as: GCAAAGCAGGCTCAGTTACC |
RESULTS
Confirmation of TPA-Induced Macrophage-Like Differentiation of HL-60 Cells
TPA-induced differentiation of HL-60 cells into macrophage-like cells has been reported (32,33). We first confirmed the effects of TPA on cell adhesion and the expression of CD11b mRNA, which are the hallmarks of macrophage differentiation (12,30). Following 2 days of TPA (32 nM) treatment, most cells (>90%) adhered to the culture vessel and aggregated (Fig. 2A, lower panel). In contrast, control cells treated with vehicle (0.05% DMSO) morphologically resembled untreated cells and were nonadherent (Fig. 2A, upper panel). In concurrence with the cell adherent results, the maturation marker CD11b mRNA was increased after 8 h of TPA treatment and showed a further elevation at 24 h, but it remained low in control cells as evaluated by RT-PCR (Fig. 2B, left panel). In addition, after synchronization by nocodazole (0.05 μg/ml) for 24 h, the expressional kinetics of CD11b mRNA was similar in synchronized and asynchronous cells (Fig. 2B). These results indicated that nocodazole itself did not induce HL-60 cell differentiation. It showed a potential to obtain a homogenous cell population for the initiation of cell differentiation. These characteristics of macrophage-like differentiation of HL-60 cells induced by TPA were therefore consistent with those in previous reports (32,33).
Figure 2.
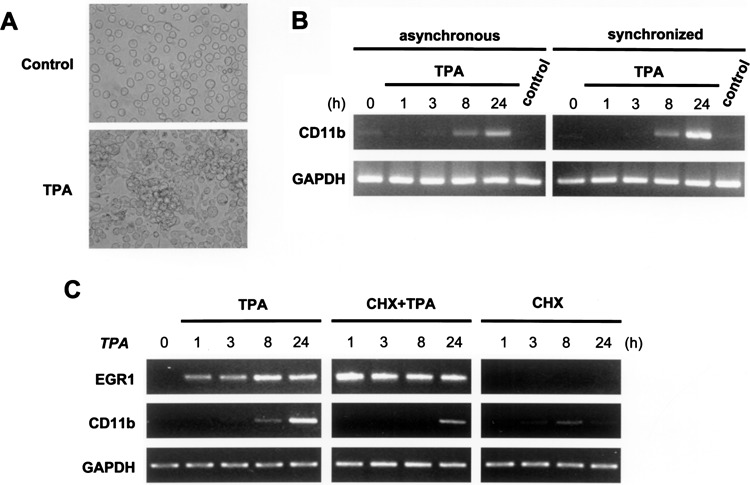
TPA induction of cellular differentiation and the expression of marker genes in HL-60 cells. (A) TPA induced macrophage-like differentiation in HL-60 cells. Morphological changes (aggregation and adherence) were observed in cells treated with TPA (32 nM) for 2 days. (B) The expression of the differentiation marker, CD11b, was increased by TPA treatment and was seen after 8 h of TPA treatment (left panel). Upregulation of CD11b transcription was not affected by nocodazole synchronization (right panel). The kinetic of CD11b transcription was analyzed by RT-PCR after the indicated time (0–24 h) of TPA treatment; control cells were treated with DMSO alone. In the right panel, the cells were synchronized with nocodazole (0.05 μg/ml) for 24 h and then washed off before TPA stimulation. (C) The early and late TPA-responsive genes can be distinguished using the protein synthesis inhibitor cycloheximide. TPA-induced upregulation of the late responsive gene, CD11b, is suppressed by cycloheximide, whereas that of the early responsive gene, EGR1, is not. In this experiment, cells were treated with cycloheximide (CHX, 10 μg/ml) for 30 min, then for 1 h with cycloheximide and TPA.
In addition, with only a short 20-min incubation period with TPA, it was sufficient to commit macrophage-like differentiation in HL-60 cells (33). Thus, the short period of TPA stimulation was sufficient to activate primary target genes, which are important in the initiation of cell differentiation. In order to identify such primary target genes of TPA in HL-60 cells, cells were treated with the protein synthesis inhibitor cycloheximide (CHX, 10 μg/ml) for 30 min prior to, and during, TPA stimulation to prevent the generation of the primary genes’ protein product needed for induction of mRNA expression of secondary targets of TPA. To confirm the ability of cycloheximide to distinguish between primary and secondary target genes, we used the known early TPA-responsive gene, EGR1 (26), and the late responsive gene, CD11b, as indicators. As shown in Figure 2C, upregulation of EGR1 by TPA (32 nM, 0–24 h) was not inhibited by cycloheximide pretreatment, whereas transcription of CD11b was shut down. These results showed TPA treatment in the presence of cycloheximide did not alter the expression of the early responsive genes, but attenuated the expression of the late responsive gene, demonstrating that the use of cycloheximide can efficiently discriminate between the primary and secondary targets of TPA.
Identification of Early TPA-Responsive Genes by Microarray Analysis and Validation by Quantitative Real-Time PCR Analysis
We focused on selectively identifying early TPA-responsive genes in the present study, because we hypothesized that some of the early responsive genes might be important in initiation of differentiation of HL-60 cells, and these genes might express in low level or only slightly change in their expression patterns. Thus, we used our modified screening strategy to enrich and collect the early TPA-responsive transcripts for microarray analysis as described in Figure 1. The expression patterns of the TPA-induced genes were assessed by comparing expression in TPA plus cycloheximide-treated cells to that in vehicle plus cycloheximide-treated control using Affymetrix HG-U133A arrays. Based on our primary selection criteria for microarray analysis giving signal log2 ratio ≥ 0.5 and difference ≥ 30, 83 genes were preliminary established to be upregulated by TPA. We selected 18 of the 83 genes from three categories in our primary gene list to validate our microarray screening criteria by quantitative real-time PCR analysis (Q-PCR) (Supplementary Table 2). According to the concurrence between Q-PCR and microarray result, we adjusted our microarray selection criteria and then identified 17 strongly increased genes (difference >200 and log2 ratio ≥ 1.0) and 39 weakly increased genes (difference >50 and log2 ratio ≥ 0.5) as listed in Table 1. These 56 early TPA-responsive genes were candidates of TPA primary target genes. Many of these genes were classified as transcription regulators, cell cycle regulators, apoptosis regulators, cytokines, signaling-related genes, and the others are metabolic, unspecified genes, or ESTs. Based on literature review, 24 of the 56 TPA upregulated genes identified in our study had been recognized as TPA-responsive genes or expressed during myeloid differentiation in previous molecular or microarray studies, such as members of the EGR family (EGR1, EGR2, and EGR3) (18,49), AP1 superfamily [JUN (1,8,23,46), JUNB (5,23,46), FOS (25,46), FOSB (49)], IL8 (42,49), CCL2 (49), CCL3 (37,49), CCL4 (49), TNF (14), MCL1 (20), NFKBIA (49), SAT (7,49), HADHA (49), LDLR (19), PMAIP1 (NOXA) (49), DUSP2 (35,49), CD69 (35), BTG2 (34), SPRY2 (49), CYP1B1 (10), and IER2 (ETR101) (39). Among these 24 genes, 14 genes were classified as strongly increased genes (14 of 17) and 10 were weakly increased genes (10 of 39) in our screening, as shown in Table 1. We identified an additional 32 genes previously not known to be TPA-responsive genes, which exhibited lower expression. Among these, DDX3X, SKIL, SLC7A1, NFIL3, BCLAF1, and JMJD3 were validated by Q-PCR (Supplementary Table 2). The result showed that the early TPA-responsive genes identified by other general approaches expressed at a higher level, and of course, were also efficiently identified by our new screening strategy. Our approach using the combination of the synchronization reagent (nocodazole) and the protein synthesis inhibitor (cycloheximide) can therefore more effectively identify TPA primary regulated genes, which expressed at lower level and/or minor alterations in this HL-60 cell differentiation model system.
SUPPLEMENTARY TABLE 2.
VALIDATION OF MICROARRAY DATA BY Q-PCR
| Gene Symbol | Microarray | Q-PCR −ΔΔCt | ||
|---|---|---|---|---|
| Difference (TPA − Control) | Log2 Ratio (TPA/Control) | 1 h | 3 h | |
| Group I (difference >200) | ||||
| FOS | 595.75 | 3.00 | 3.03* | 4.36* |
| TNF | 1399.75 | 2.80 | 1.79* | 4.32* |
| IL8 | 1536.05 | 2.70 | 3.22* | 6.61* |
| JUN | 343.35 | 1.30 | 1.10* | 3.04* |
| NFKBIA | 1320.85 | 1.20 | 0.79* | 1.13* |
| HSPCA (Hsp90A) | 939.80 | 0.50 | −0.05 | −0.03 |
| Group II (difference 50–200) | ||||
| DDX3X | 56.80 | 0.80 | 0.05* | 1.22* |
| SKIL (SnoN) | 55.35 | 0.70 | 0.03 | 1.73* |
| CCL2 | 61.00 | 0.60 | 0.35* | 1.23* |
| SLC7A1 | 60.65 | 0.60 | −0.16* | 0.85* |
| NFIL3 | 59.65 | 0.60 | 0.64* | 2.56* |
| BCLAF1 (BTF) | 55.90 | 0.50 | −0.12* | 0.80* |
| Group III (difference 30–50) | ||||
| PDE4B | 31.70 | 0.90 | 0.08 | 0.74* |
| C14orf92 | 44.70 | 0.70 | −0.29 | 0.13 |
| LIMD1 | 33.45 | 0.70 | −0.33* | −0.91* |
| TAF1 | 33.15 | 0.70 | −0.43* | 1.09* |
| KIAA0286 | 38.95 | 0.50 | −0.16* | 0.43* |
| JMJD3 | 34.75 | 0.50 | 0.32 | 2.00* |
Eighteen genes showing upregulation in primary microarray analysis of CHX + TPA-treated and CHX + vehicle control were selected for quantitative real-time PCR analysis to validate the primary selection criteria. The Q-PCR is displayed as the differential cycle threshold (indicated as −ΔΔCt) for each gene for triplicate independent experiments.
Significant change in expression (p < 0.05).
TABLE 1.
EARLY TPA-RESPONSIVE GENES UPREGULATED IN HL-60 CELL DIFFERENTIATION
| Gene Symbol | Accession No. | Microarray | Q-PCR −ΔΔCt | Reference(s) | ||
|---|---|---|---|---|---|---|
| Difference | Log2 Ratio | |||||
| (TPA − Control) | TPA/Control) | 1 h | 3 h | |||
| Strongly increased (TPA − control >200, Log2 ≥1.0) | ||||||
| EGR4 | NM_001965 | 227.3 | 4.9 | |||
| DUSP2 | NM_004418 | 799.1 | 3.3 | 35, 49 | ||
| EGR3 | NM_004430 | 421.0 | 3.3 | 18, 49 | ||
| EGR1 | NM_001964 | 1151.7 | 3.1 | 18, 49 | ||
| FOS | BC004490 | 595.8 | 3.0 | 3.03* | 4.36* | 25, 46 |
| TNF | NM_000594 | 1399.8 | 2.8 | 1.79* | 4.32* | 14 |
| IL8 | AF043337 | 1536.1 | 2.7 | 3.22* | 6.61* | 42, 49 |
| EGR2 | NM_000399 | 291.8 | 2.6 | 18, 49 | ||
| JUNB | NM_002229 | 848.1 | 1.7 | 5, 23, 46 | ||
| FOSB | NM_006732 | 201.7 | 1.7 | 49 | ||
| IER2 (ETR101) | NM_004907 | 2729.5 | 1.4 | 39 | ||
| JUN | BG491844 | 343.4 | 1.3 | 1.10* | 3.04* | 1, 8, 23, 46 |
| NFKBIA | AI078167 | 1320.9 | 1.2 | 0.79* | 1.13* | 49 |
| TNFAIP3 | NM_006290 | 219.4 | 1.2 | |||
| MCL1 | NM_021960 | 397.7 | 1.0 | 20 | ||
| TIPARP | AL556438 | 344.4 | 1.0 | |||
| CYP1B1 | AU144855 | 260.5 | 1.0 | 10 | ||
| Weakly increased (TPA − control >50, Log2 ≥0.5) | ||||||
| CD69 | L07555 | 53.1 | 4.0 | 35 | ||
| HNRPA1 | AI144007 | 75.5 | 1.7 | |||
| est | BC003629 | 59.0 | 1.7 | |||
| IER3 | NM_003897 | 112.4 | 1.6 | |||
| CCL3 | NM_002983 | 118.5 | 1.3 | 37, 49 | ||
| ZFP36 | NM_003407 | 165.3 | 1.0 | |||
| BTG2 | NM_006763 | 101.7 | 1.0 | 34 | ||
| HIS1 | AW193511 | 67.3 | 1.0 | |||
| EIF4A1 | U79273 | 180.1 | 0.9 | |||
| est | AV764378 | 82.1 | 0.9 | |||
| SRPR | BG474541 | 61.2 | 0.9 | |||
| PPP1R15A | NM_014330 | 128.4 | 0.8 | |||
| NXT2 | AF201942 | 99.3 | 0.8 | |||
| C14orf43 | NM_018678 | 98.7 | 0.8 | |||
| EIF5 | BE552334 | 58.6 | 0.8 | |||
| DDX3X | R60068 | 56.8 | 0.8 | 0.05* | 1.22* | |
| SAT | BE971383 | 103.3 | 0.7 | 7, 49 | ||
| KCNAB2 | AF044253 | 89.6 | 0.7 | |||
| SKIL (SnoN) | BF725121 | 55.4 | 0.7 | 0.03 | 1.73* | |
| SFRS7 | AA524053 | 54.5 | 0.7 | |||
| HADHA | BG472176 | 154.0 | 0.6 | 49 | ||
| LDLR | NM_000527 | 134.0 | 0.6 | 19 | ||
| PMAIP1 (NOXA) | AI857639 | 131.8 | 0.6 | 49 | ||
| WEE1 | AJ277546 | 77.5 | 0.6 | |||
| PPP1R10 | NM_002714 | 75.3 | 0.6 | |||
| CCL2 | S69738 | 61.0 | 0.6 | 0.35* | 1.23* | 49 |
| PRKAR1A | Ml 8468 | 60.7 | 0.6 | |||
| SLC7A1 | AA527433 | 60.7 | 0.6 | −0.16* | 0.85* | |
| NFIL3 | NM_005384 | 59.7 | 0.6 | 0.64* | 2.56* | |
| SPRY2 | NM_005842 | 136.9 | 0.5 | 49 | ||
| IXNDC | NM_030755 | 134.9 | 0.5 | |||
| CCL4 | NM_002984 | 119.6 | 0.5 | 49 | ||
| IDI1 | NM_004508 | 105.8 | 0.5 | |||
| PBEF1 | BF575514 | 76.4 | 0.5 | |||
| RAD21 | BG289967 | 76.3 | 0.5 | |||
| HNRPD | W74620 | 73.0 | 0.5 | |||
| RRM1 | AI692974 | 62.9 | 0.5 | |||
| BCLAF1 | BE963370 | 55.9 | 0.5 | −0.12* | 0.80* | |
| JMJD3 | AA521267 | 34.8 | 0.5 | 0.32 | 2.00* | |
The genes showing upregulation detected in this comparison microarray analysis of TPA-treated and vehicle control in the presence of cycloheximide in synchronized HL-60 cells are listed. The TPA induction schema and detailed microarray analysis are described in Materials and Methods.
Significantly (p < 0.05) upregulated by TPA-treated over vehicle-treated cells.
Upregulation of NFIL3, SKIL, and JMJD3 by TPA Through PKC Signaling Pathway in HL-60 Cells
We selectively focused on three unreported TPA primary regulated genes (NFIL3, SKIL, and JMJD3), which showed significant increases in expression at lower level and/or minor alterations both in the microarray and Q-PCR analysis in this HL-60 cell differentiation system under nocodazole synchronization and cycloheximide treatment conditions. These genes with transcription regulatory activity (SKIL and NFIL3) or with chromatin remodeling activity (JMJD3) might be important in the regulation of TPA-dependent differentiation in HL-60 cells. First, we demonstrated their transcriptional upregulation in response to TPA was also shown in the absence of nocodazole and cycloheximide in HL-60 cells (Fig. 3A, left panel). The changes of these three genes was significantly increased after 3 h post-TPA treatment, in good agreement with the Q-PCR results obtained in nocodazole- and cycloheximide-treated cells. The expression of these three genes showed a greater increase with TPA stimulation in the presence of cycloheximide than in the absence of cycloheximide treatment (Fig. 3A, right panel). They displayed as typically early responsive genes, whose expression was induced without the requirement for protein synthesis. Thus, NFIL3, SKIL, and JMJD3 can be classified as new early TPA-responsive genes in HL-60 cells.
Figure 3.
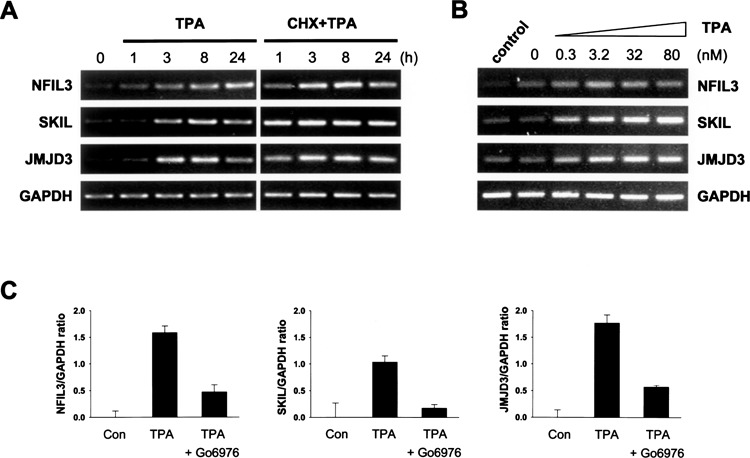
Expression pattern of TPA upregulated genes in HL-60 cells in the absence of cycloheximide. (A) Upregulation of NFIL3, SKIL, and JMJD3 is not suppressed by cycloheximide treatment. Cells were pretreated with cycloheximide (CHX, 10 Jig/ml) for 30 min and then were treated with TPA (32 nM) for 0–24 h, and gene expression was determined by RT-PCR. (B) Upregulation of NFIL3, SKIL, and JMJD3 by TPA is shown to be dose dependent. Cells were treated for 3 h with 0–80 nM of TPA and gene expression was determined by RT-PCR. GAPDH was used as control for normalization. (C) The TPA-induced upregulated expression of NFIL3, SKIL, and JMJD3 is decreased by Go6976 treatment. Cells were pretreated with classic PKC inhibitor, Go6976 (1 μM) for 1 h prior to TPA stimulation to block PKC signaling. Q-PCR assay was used to determine the altered expression of the genes in the TPA-treated cells relative to that in untreated cells, normalized with GAPDH and calculated by ΔΔCt method. The results are the mean ± SD for three independent experiments.
There is a dose dependency of the cell adherent, the terminal differentiation marker in TPA-induced HL-60 cells with only a short period TPA treatment (33). Figure 3B shows that upregulation of NFIL3, SKIL, and JMJD3 mRNA by TPA (up to 80 nM, for 3 h) was also in a dose-dependent manner. Because TPA is a well-known PKC activator, and acute exposure to TPA can prolong activation of the classic and novel PKC isoforms (2,3,6,24,41,45). In order to determine whether this dose-dependent effect was resulting from the direct activation of their transcription by TPA through PKC, we pretreated HL-60 cells with classic PKC inhibitor Go6976 (1 μM) for 1 h and subsequently treated with TPA. We found that TPA-induced increase in NFIL3, SKIL, and JMJD3 mRNA levels was significantly suppressed by treatment with classic PKC inhibitor during TPA stimulation (Fig. 3C) by Q-PCR assay. These results show that the upregulation of NFIL3, SKIL, and JMJD3 by TPA is dose dependent and that this effect is directly mediated by activation of the PKC signaling pathway in HL-60 cells. We suggest that these TPA primary target genes may play an important role as early regulatory mediators during TPA-induced macrophage-like differentiation in HL-60 cells through the PKC signaling pathway.
DISCUSSION
Most genes regulated during myeloid differentiation are controlled at the transcription level (31), and such transcriptional regulation is the result of the comprehensive interactions of a few key transcription factors but not a single “master” regulator (31,40,48). Therefore, it is beneficial to identify these unrecognized regulators in order to determine the molecular mechanisms of controlled myeloid differentiation. Genes that show immediate expression alterations might be important in the initiation stage of cell differentiation, whereas genes that show delayed expression changes might be involved in the progression of differentiation, maturation, and/or apoptosis (22). Because the early responsive genes might play key regulatory roles, they deserve much more attention. In addition, it is helpful to construct transcriptional regulatory networks by using these primary targets to examine their prospective downstream target genes, which could link these key factors to their biological function. Therefore, we tried to identify additional key regulatory factors that have not been found in previous studies and might be involved in the initiation stage of differentiation.
The early responsive genes are likely to be primarily regulated at the transcription level, as their induction does not require protein synthesis, but only the modification of preexisting transcriptional modulators. In contrast, the delayed responsive genes require protein synthesis for induction and might depend on the prior induction of other genes (11,13). Such concerns are usually addressed by blocking protein synthesis using cycloheximide. There have been reports of the use of cycloheximide in the microarray analysis of primary targets of specific genes, such as p53 (17) and c-myc (27). In this study, we aimed to identify early TPA-responsive genes in treated HL-60 cells in the presence of cycloheximide. Our data showed that a subset of genes showing a minor change in transcription might be more effectively identified in the presence of cycloheximide than in its absence. If we take JUN as an example, it is immediately induced during monocytic differentiation (23), but it has never been detected at an earlier time period of TPA stimulation by previous microarray approaches. In our study, JUN mRNA was induced in TPA-treated HL-60 cells in the presence of cycloheximide. The transcription of JUN was not affected by cycloheximide treatment alone, but the half-life of the JUN transcript was prolonged by TPA treatment, resulting in its mRNA accumulation (38). We therefore suggest that it is beneficial to adopt cycloheximide treatment in our experimental strategy, so cycloheximide can reduce/block signals generated from late responsive genes and therefore enhance the probability of early responsive gene detection, which might be expressed at lower levels and with a shorter half-life. Therefore, this new approach can identify additional factors that have never been reported in early TPA-induced HL60 cells.
This is the first report to show that the expression of three transcription-regulated genes, NFIL3, SKIL, and JMJD3 is immediately upregulated during TPA-induced cellular differentiation in HL-60 cells. We demonstrated that the TPA-induced upregulation of these three genes was slightly increased and the expressional alteration can be further enhanced by cycloheximide cotreatment and that this expression pattern was very similar to that of the prototype early responsive genes, EGR1 (26) and JUN (23), in TPA-induced HL-60 cell differentiation. Except for the novel gene JMJD3, NFIL3, also known as E4BP4, is a member of the bZIP transcription factor superfamily, and it acts as a transcription activator of IL3 and also is upregulated by IL3 and therefore plays an important role in IL3-mediated survival (16,21). The other gene, SKIL, also known as SnoN, has also been found to involve in the regulation of cell growth and differentiation through TGF-β signaling pathway (4,43) and its expression level was also enhanced in the differentiation of murine tri-lineage early myeloid progenitors (28). SKIL might act as a transcription repressor of the CD11b gene, because the differentiation marker CD11b expression was significantly increased in 1,25-dihydroxyvitamin D3-induced monocytic differentiation in HL-60 cells with SKIL shRNA knock down transduction (data not shown). Although these transcription-related genes were recognized in this study as primary TPA-regulated genes, their exact biological roles in regulating HL-60 cell differentiation is not yet clear and further investigations are needed.
ACKNOWLEDGMENTS
This study was supported, in part, by research grants provided by the Academia Sinica. We thank Dr. Chang-Jen Huang from the Institute of Biological Chemistry, Academia Sinica, for his critical review and suggestions.
REFERENCES
- 1. Auwerx J.; Staels B.; Sassone-Corsi P. Coupled and uncoupled induction of fos and jun transcription by different second messengers in cells of hematopoietic origin. Nucleic Acids Res. 18:221–228; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carter C. A. Protein kinase C as a drug target: Implications for drug or diet prevention and treatment of cancer. Curr. Drug Targets 1:163–183; 2000. [DOI] [PubMed] [Google Scholar]
- 3. Castagna M.; Takai Y.; Kaibuchi K.; Sano K.; Kikkawa U.; Nishizuka Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J. Biol. Chem. 257:7847–7851; 1982. [PubMed] [Google Scholar]
- 4. Cohen S. B.; Nicol R.; Stavnezer E. A domain necessary for the transforming activity of SnoN is required for specific DNA binding, transcriptional repression and interaction with TAF(II)110. Oncogene 17:2505–2513; 1998. [DOI] [PubMed] [Google Scholar]
- 5. Datta R.; Sherman M. L.; Stone R. M.; Kufe D. Expression of the jun-B gene during induction of monocytic differentiation. Cell Growth Differ. 2:43–49; 1991. [PubMed] [Google Scholar]
- 6. Edashige K.; Sato E. F.; Akimaru K.; Kasai M.; Utsumi K. Differentiation of HL-60 cells by phorbol ester is correlated with up-regulation of protein kinase C-alpha. Arch. Biochem. Biophys. 299:200–205; 1992. [DOI] [PubMed] [Google Scholar]
- 7. Gavin I. M.; Glesne D.; Zhao Y.; Kubera C.; Huberman E. Spermine acts as a negative regulator of macrophage differentiation in human myeloid leukemia cells. Cancer Res. 64:7432–7438; 2004. [DOI] [PubMed] [Google Scholar]
- 8. Gaynor R.; Simon K.; Koeffler P. Expression of c-jun during macrophage differentiation of HL-60 cells. Blood 77:2618–2623; 1991. [PubMed] [Google Scholar]
- 9. Gunji W.; Kai T.; Sameshima E.; Iizuka N.; Katagi H.; Utsugi T.; Fujimori F.; Murakami Y. Global analysis of the expression patterns of transcriptional regulatory factors in formation of embryoid bodies using sensitive oligonucleotide microarray systems. Biochem. Biophys. Res. Commun. 325:265–275; 2004. [DOI] [PubMed] [Google Scholar]
- 10. Guo M.; Joiakim A.; Dudley D. T.; Reiners J. J. Suppression of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-mediated CYP1A1 and CYP1B1 induction by 12-O-tetradecanoylphorbol-13-acetate: Role of transforming growth factor beta and mitogen-activated protein kinases. Biochem. Pharmacol. 62:1449–1457; 2001. [DOI] [PubMed] [Google Scholar]
- 11. Herschman H. R. Primary response genes induced by growth factors and tumor promoters. Annu. Rev. Biochem. 60:281–319; 1991. [DOI] [PubMed] [Google Scholar]
- 12. Hickstein D. D.; Smith A.; Fisher W.; Beatty P. G.; Schwartz B. R.; Harlan J. M.; Root R. K.; Locksley R. M. Expression of leukocyte adherence-related glycoproteins during differentiation of HL-60 promyelocytic leukemia cells. J. Immunol. 138:513–519; 1987. [PubMed] [Google Scholar]
- 13. Hofbauer R.; Denhardt D. T. Cell cycle-regulated and proliferation stimulus-responsive genes. Crit. Rev. Eukaryot. Gene Expr. 1:247–300; 1991. [PubMed] [Google Scholar]
- 14. Horiguchi J.; Spriggs D.; Imamura K.; Stone R.; Luebbers R.; Kufe D. Role of arachidonic acid metabolism in transcriptional induction of tumor necrosis factor gene expression by phorbol ester. Mol. Cell. Biol. 9:252–258; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ideker T.; Thorsson V.; Ranish J. A.; Christmas R.; Buhler J.; Eng J. K.; Bumgarner R.; Goodlett D. R.; Aebersold R.; Hood L. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science 292:929–934; 2001. [DOI] [PubMed] [Google Scholar]
- 16. Ikushima S.; Inukai T.; Inaba T.; Nimer S. D.; Cleveland J. L.; Look A. T. Pivotal role for the NFIL3/E4BP4 transcription factor in interleukin 3-mediated survival of pro-B lymphocytes. Proc. Natl. Acad. Sci. USA 94:2609–2614; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kannan K.; Amariglio N.; Rechavi G.; Jakob-Hirsch J.; Kela I.; Kaminski N.; Getz G.; Domany E.; Givol D. DNA microarrays identification of primary and secondary target genes regulated by p53. Oncogene 20:2225–2234; 2001. [DOI] [PubMed] [Google Scholar]
- 18. Kharbanda S.; Nakamura T.; Stone R.; Hass R.; Bernstein S.; Datta R.; Sukhatme V. P.; Kufe D. Expression of the early growth response 1 and 2 zinc finger genes during induction of monocytic differentiation. J. Clin. Invest. 88:571–577; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kosaka S.; Takahashi S.; Masamura K.; Kanehara H.; Sakai J.; Tohda G.; Okada E.; Oida K.; Iwasaki T.; Hattori H.; Kodama T.; Yamamoto T.; Miyamori I. Evidence of macrophage foam cell formation by very low-density lipoprotein receptor: interferon-gamma inhibition of very low-density lipoprotein receptor expression and foam cell formation in macrophages. Circulation 103:1142–1147; 2001. [DOI] [PubMed] [Google Scholar]
- 20. Kozopas K. M.; Yang T.; Buchan H. L.; Zhou P.; Craig R. W. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc. Natl. Acad. Sci. USA 90:3516–3520; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kuribara R.; Kinoshita T.; Miyajima A.; Shinjyo T.; Yoshihara T.; Inukai T.; Ozawa K.; Look A. T.; Inaba T. Two distinct interleukin-3-mediated signal pathways, Ras-NFIL3 (E4BP4) and Bcl-xL, regulate the survival of murine pro-B lymphocytes. Mol. Cell. Biol. 19:2754–2762; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee K. H.; Chang M. Y.; Ahn J. I.; Yu D. H.; Jung S. S.; Choi J. H.; Noh Y. H.; Lee Y. S.; Ahn M. J. Differential gene expression in retinoic acid-induced differentiation of acute promyelocytic leukemia cells, NB4 and HL-60 cells. Biochem. Biophys. Res. Commun. 296:1125–1133; 2002. [DOI] [PubMed] [Google Scholar]
- 23. Lord K. A.; Abdollahi A.; Hoffman-Liebermann B.; Liebermann D. A. Proto-oncogenes of the fos/jun family of transcription factors are positive regulators of myeloid differentiation. Mol. Cell. Biol. 13:841–851; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Macfarlane D. E.; Manzel L. Activation of beta-isozyme of protein kinase C (PKC beta) is necessary and sufficient for phorbol ester-induced differentiation of HL-60 promyelocytes. Studies with PKC beta-defective PET mutant. J. Biol. Chem. 269:4327–4331; 1994. [PubMed] [Google Scholar]
- 25. Muller R.; Curran T.; Muller D.; Guilbert L. Induction of c-fos during myelomonocytic differentiation and macrophage proliferation. Nature 314:546–548; 1985. [DOI] [PubMed] [Google Scholar]
- 26. Nguyen H. Q.; Hoffman-Liebermann B.; Liebermann D. A. The zinc finger transcription factor Egr-1 is essential for and restricts differentiation along the macrophage lineage. Cell 72:197–209; 1993. [DOI] [PubMed] [Google Scholar]
- 27. O’Connell B. C.; Cheung A. F.; Simkevich C. P.; Tam W.; Ren X.; Mateyak M. K.; Sedivy J. M. A large scale genetic analysis of c-Myc-regulated gene expression patterns. J. Biol. Chem. 278:12563–12573; 2003. [DOI] [PubMed] [Google Scholar]
- 28. Pearson-White S.; Deacon D.; Crittenden R.; Brady G.; Iscove N.; Quesenberry P. J. The ski/sno protooncogene family in hematopoietic development. Blood 86:2146–2155; 1995. [PubMed] [Google Scholar]
- 29. Pfaffl M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rosmarin A. G.; Weil S. C.; Rosner G. L.; Griffin J. D.; Arnaout M. A.; Tenen D. G. Differential expression of CD11b/CD18 (Mo1) and myeloperoxidase genes during myeloid differentiation. Blood 73:131–136; 1989. [PubMed] [Google Scholar]
- 31. Rosmarin A. G.; Yang Z.; Resendes K. K. Transcriptional regulation in myelopoiesis: Hematopoietic fate choice, myeloid differentiation, and leukemogenesis. Exp. Hematol. 33:131–143; 2005. [DOI] [PubMed] [Google Scholar]
- 32. Rovera G.; O’Brien T. G.; Diamond L. Induction of differentiation in human promyelocytic leukemia cells by tumor promoters. Science 204:868–870; 1979. [DOI] [PubMed] [Google Scholar]
- 33. Rovera G.; Santoli D.; Damsky C. Human promyelocytic leukemia cells in culture differentiate into macrophage-like cells when treated with a phorbol diester. Proc. Natl. Acad. Sci. USA 76:2779–2783; 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ryu M. S.; Lee M. S.; Hong J. W.; Hahn T. R.; Moon E.; Lim I. K. TIS21/BTG2/PC3 is expressed through PKC-delta pathway and inhibits binding of cyclin B1-Cdc2 and its activity, independent of p53 expression. Exp. Cell Res. 299:159–170; 2004. [DOI] [PubMed] [Google Scholar]
- 35. Segel G. B.; Woodlock T. J.; Xu J.; Li L.; Felgar R. E.; Ryan D. H.; Lichtman M. A.; Wang N. Early gene activation in chronic leukemic B lymphocytes induced toward a plasma cell phenotype. Blood Cells Mol. Dis. 30:277–287; 2003. [DOI] [PubMed] [Google Scholar]
- 36. Seo J.; Kim M.; Kim J. Identification of novel genes differentially expressed in PMA-induced HL-60 cells using cDNA microarrays. Mol. Cells 10:733–739; 2000. [DOI] [PubMed] [Google Scholar]
- 37. Sharma V.; Xu M.; Ritter L. M. Biochemical characterization of MIP-1 alpha nuclear protein. Biochem. Biophys. Res. Commun. 248:716–721; 1998. [DOI] [PubMed] [Google Scholar]
- 38. Sherman M. L.; Stone R. M.; Datta R.; Bernstein S. H.; Kufe D. W. Transcriptional and post-transcriptional regulation of c-jun expression during monocytic differentiation of human myeloid leukemic cells. J. Biol. Chem. 265:3320–3323; 1990. [PubMed] [Google Scholar]
- 39. Shimizu N.; Ohta M.; Fujiwara C.; Sagara J.; Mochizuki N.; Oda T.; Utiyama H. A gene coding for a zinc finger protein is induced during 12-O-tetradeca-noylphorbol-13-acetate-stimulated HL-60 cell differentiation. J. Biochem. (Tokyo) 111:272–277; 1992. [DOI] [PubMed] [Google Scholar]
- 40. Shivdasani R. A.; Orkin S. H. The transcriptional control of hematopoiesis. Blood 87:4025–4039; 1996. [PubMed] [Google Scholar]
- 41. Slapak C. A.; Kharbanda S.; Saleem A.; Kufe D. W. Defective translocation of protein kinase C in multi-drug-resistant HL-60 cells confers a reversible loss of phorbol ester-induced monocytic differentiation. J. Biol. Chem. 268:12267–12273; 1993. [PubMed] [Google Scholar]
- 42. Steube K. G.; Meyer C.; Drexler H. G. Multiple regulation of constitutive and induced interleukin 8 secretion in human myelomonocytic cell lines. Cytokine 12:1236–1239; 2000. [DOI] [PubMed] [Google Scholar]
- 43. Stroschein S. L.; Wang W.; Zhou S.; Zhou Q.; Luo K. Negative feedback regulation of TGF-beta signaling by the SnoN oncoprotein. Science 286:771–774; 1999. [DOI] [PubMed] [Google Scholar]
- 44. Tamayo P.; Slonim D.; Mesirov J.; Zhu Q.; Kitareewan S.; Dmitrovsky E.; Lander E. S.; Golub T. R. Interpreting patterns of gene expression with self-organizing maps: methods and application to hematopoietic differentiation. Proc. Natl. Acad. Sci. USA 96:2907–2912; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tonetti D. A.; Henning-Chubb C.; Yamanishi D. T.; Huberman E. Protein kinase C-beta is required for macrophage differentiation of human HL-60 leukemia cells. J. Biol. Chem. 269:23230–23235; 1994. [PubMed] [Google Scholar]
- 46. Tonetti D. A.; Horio M.; Collart F. R.; Huberman E. Protein kinase C beta gene expression is associated with susceptibility of human promyelocytic leukemia cells to phorbol ester-induced differentiation. Cell Growth Differ. 3:739–745; 1992. [PubMed] [Google Scholar]
- 47. Tucker C. L.; Gera J. F.; Uetz P. Towards an understanding of complex protein networks. Trends Cell Biol. 11:102–106; 2001. [DOI] [PubMed] [Google Scholar]
- 48. Valledor A. F.; Borras F. E.; Cullell-Young M.; Celada A. Transcription factors that regulate monocyte/ macrophage differentiation. J. Leukoc. Biol. 63:405–417; 1998. [DOI] [PubMed] [Google Scholar]
- 49. Zheng X.; Ravatn R.; Lin Y.; Shih W. C.; Rabson A.; Strair R.; Huberman E.; Conney A.; Chin K. V. Gene expression of TPA induced differentiation in HL-60 cells by DNA microarray analysis. Nucleic Acids Res. 30:4489–4499; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]