

Abstract
Podocin (NPHS2) is a component of the glomerular slit-diaphragm, with major regulatory functions in renal permeability of proteins. Loss of podocin and decrease in resynthesis may influence the outcome of proteinuric renal disease such as segmental glomerulosclerosis (FSGS), and promoter functionality plays a key role in this process. NPHS2 promoter variants with functional activity may be a part of the problem of podocin resynthesis. We sequenced NPHS2 promoter region from −628 to ATG in a large cohort of 260 nephrotic patients (161 with FSGS) who were presenting proteinuria from moderate to severe and were receiving or had received modular therapies according to their sensitivity to steroids and other immune modulators. Three sequence variants (−236C>T, −52C>G, −26C>G) were identified in our study population that gave an allele frequency below 1% (5 patients out of 520 alleles). Functional implications were shown for each variants that were most evident for −52C>G and −26C>G (−50% of luciferase expression compared to the wild-type sequence, p < 0.01). Consensus analysis for homology of the −52 region with regulatory factors revealed homology for USF1 and the sum of experiments with gel retardation and with cells silenced for USF1 confirmed that this factor regulates NPHS2 expression at this site. In conclusion, three functional variants in NPHS2 promoter have been identified in a large cohort of patients with nephrotic syndrome and FSGS that have a frequency <1%. One of these (i.e., −52C>G) is associated with a poor clinical outcome and evolution to end-stage renal failure. USF1 was identified as the transcriptional factor regulating NPHS2 at this site. Even if not sufficient to cause FSGS per se, these variants could represent modifiers for severity and/or progression of the disease.
Key words: NPHS2 gene, NPHS2 promoter, Nephrotic syndrome, Focal segmental glomerulosclerosis, Proteinuria, Podocin, USF1
NEPHROTIC syndrome (NS) associated with focal segmental glomerulosclerosis (FSGS) is one of the most frequent glomerular diseases in children and adults (2) and is emerging as a leading cause of end-stage renal failure (ESRF). The annual incidence in children has been estimated to be 2.0 to 2.7 per 100,000 in the US with a cumulative prevalence of 16 per 100,000 (31). The basic clinical hallmark of the disease is proteinuria, whose pathogenesis is still debated (10,11). The most recent progress on mechanisms of proteinuria is the characterization of the slit-diaphragm, which is emerging as the prevailing glomerular site for repulsion of proteins. The slit-diaphragm is reminiscent of a tight junction with a differentiate structure that is characterized by an electron dense zipper-like structure where the extracellular proteins nephrin, NEPH-1, P-Cadherin, and FAT (15,23,28) are connected by other specialized proteins (i.e., podocin, CD2AP) to the main cell body. Podocin appears to have a central role in maintaining a functioning glomerular filtration barrier (1,13,14,25,30). It is a raft integral protein of 383 amino acids that forms a membrane invagination and recruits nephrin and CD2AP in these microdomains. Its genetic ablation determines massive proteinuria in mice that die within a few days of life (26). The discovery of mutations of the podocin encoding gene (i.e., NPHS2) in familial nephrotic syndrome represented a breakthrough in the research of mechanisms of nephrotic syndrome (1). Screening studies on NPHS2 mutations involving familial cases and sporadic nephrotic syndrome have now been published (4,5,8,27,32) reporting more than 50 different NPHS2 mutations.
Data also indicate that the expression of slit-diaphragm proteins is regulated, in the presence of proteinuria or pathologic injury. For example, pathologic studies demonstrated that the molecular scaffold of the slit-diaphragm dissembles in animals and humans with proteinuria (8,9,16,20) and in these cases, the protein expression of podocin is diminished in glomeruli (19,29). In addition to the presence of structurally functioning proteins in the slit-diaphragm, a proper quantitative equilibrium among different components is required for the maintenance of the barrier functional integrity. This appears to be a dynamic process in which regulatory mechanisms of every different protein play a role. Data on podocin glomerular mRNA in FSGS and minimal change diseases are controversial. Koop et al. (19) reported on a significant increase in podocin mRNA in both conditions and proposed that this might result from a compensatory reaction of the damaged podocytes. On the contrary, results by Schmid et al. (29) pointed to a net differentiation between FSGS and minimal change disease, the former being characterized by downregulation of podocin mRNA glomerular expression. While it is still unknown what determines podocin loss in such conditions [for a review see (10)] it seems plausible that reparative capacities of the renal barrier strictly depend on their resynthesis potential and the reparative process may eventually influence proteinuria. Promoter functionality plays a key role in this process. NPHS2 promoter has been studied in mice (22,24), while studies on proteinuric diseases in humans have not been published yet, representing a relevant gap in our current knowledge on the topics. Podocin promoter has been located in the 2.5 kb of the 5′ terminus of NPHS2 gene (GenBank accession number AF487463). This human 2.5-kb fragment has been demonstrated to direct podocite-specific expression in transgenic mice (21).
We carried out a sequencing study of the NPHS2 promoter region from −628 to ATG, looking for variants that could affect podocin gene expression, in a large cohort of FSGS patients. In this study we report for the first time the presence of rare variants that reduce podocin gene expression and are associated to a poor outcome.
MATERIALS AND METHODS
Patients
We screened the NPHS2 coding and promoter region in a cohort of 274 patients (154 males and 120 females; mean age at onset of proteinuria 17.1 years; range 0.1–77.4) with sporadic nephrotic syndrome who were presenting or had presented proteinuria from moderate to severe at variable ages and were receiving or had received modular therapies according to their sensitivity to different drugs following consolidated schemes (16,17). Renal histology was available in 210 cases. Overall, 161 had a diagnosis of FSGS based on histological routine criteria, 25 presented mesangial IgM deposition, and 24 minimal change nephropathy. Genomic DNA was extracted according to standard procedures. DNA from 200 normal adults (mean age 38, range 23–46 years) was obtained and sequenced as below. In all cases we obtained the informed consent from patients and in case of children from their parents. Patients with homozygous or compound heterozygous mutations in NPHS2 coding region did not undergo further analysis on promoter.
NPHS2 Coding Region Sequence
Molecular analysis of podocin coding region was performed by direct sequencing as already described. Briefly, exons were amplified by PCR using flanking intronic primers and subjected to automatic sequencing by dye-terminator reaction (Automated sequencer ABI 3100, Applera, Milan, Italy). The primer sequences were selected on the basis of literature reports by different groups (1,3). In particular, for exons 2 and 6 primer design we followed Karle et al. (18), to avoid the presence of a recognized SNP.
NPHS2 Promoter Sequence
Sequencing of NPHS2 promoter from −628 to ATG was done as above; for PCR we utilized the following two primers: forward 5′-GAAAGTTGGGGATGAGGCGA-3′; reverse 5′-CAATCAAAGCTTCCTCAGAGCTGCCGGGCGGCT-3′. PCR conditions were: denaturation at 94°C for 1 min, annealing at 62°C for 1 min, and extension at 72°C for 1 min for 30 cycles.
NPHS2 Promoter Constructs-Luciferase Assay
NPHS2 promoter PCR products were subcloned into the luciferase pGL3 reporter vector (Promega Inc., Madison, WI). All clones obtained bearing a specific mutation (pPL1, pPL2, pPL3, pPL4) and the wild-type clone (pPL5) were sequenced prior to transfection. Luciferase activity was determined in a TD-20/20 luminometer (Turner Designs, Sunnyvale, CA) according to the Promega Luciferase Assay (Promega Inc.) and results were normalized to the cotransfected Renilla luminescence. Results are given as arbitrary units (A.U.) (12). The paired t-test was utilized to compare luciferase expression by different constructs. For each experiment were conducted at least five replicates. Values of p < 0.05 were regarded as significant.
Cell Culture
COS7 (monkey renal fibroblasts) were obtained from LGC Promochem (Milan, Italy). Primary cultures of glomerular epithelial cells (GECs) were established and characterized as previously described (6). Established lines of differentiated GECs were obtained by infection of primary cultures with a hybrid Adeno5/SV40 virus as previously described and characterized as above (6,7). An immortalized GEC line was cultured in DMEM containing 25 mM glucose, 10% inactivated FCS, 100 U/ml penicillin, and 100 mg/ml streptomycin at 37°C and used for these experiments.
Transient siRNA-Mediated Silencing of USF1
Transient transfection of siRNA for targeting endogenous gene silencing was performed using PEI 25 KDa (Aldrich) and 200 nM siRNA duplex per well. The following Qiagen siRNA oligonucleotides (Hs_USF_1_HP siRNA, catalog number SI00094815) were utilized in the assay: sense r(GAU UAG AGG UCG UCA UCA A) dTdT; antisense r(UUG AUG ACG ACC UCU AAU C) dCdG, which corresponds to base pair between 1092 and 1112 downstream the start codon sequence GAU UAG AGG UCG UCA UCA A. Cells were assayed for 48 h after transfection.
Transfection Experiments
Transfection was performed with 4 μg plasmids following the polyethilenimine (PEI, Sigma-Aldrich, Steinheim, Germany) procedure. Thirty microliters of a 10 μM PEI solution was added to 10 μg DNA and incubated at 37°C for 3 h in 5% CO2. After discarding, cells were incubated with normal medium for 48 h before being processed with the Luciferase assay.
Nuclear Extracts and Cellular Fractionation
Podocytes were scraped with a rubber policeman, after previous wash with PBS, pelleted at 600 × g for 10 min, and resuspended in 200 μl/100 mm plate of 20 mM HEPES, pH 7.9, 1 mM EDTA, 1 mM dithiotreitol, 0.5 mM PMSF, 1 μg/ml of leupeptin, pepstatin, and aprotinin, 1 mM sodium vanadate, 10 mM sodium fluoride, and kept at 4°C for 15 min. Cell suspension was then added with 0.25 vol of 1% NP40 to obtain a final concentration of 0.2% NP40 and incubated at 4°C for 15 min. The cell lysate was then centrifuged at 600 × g for 15 min. The supernatant (S1) was saved for further processing, while the pellet (P1), mostly containing the unbroken nuclei, was resuspended in 1 vol of BLS (20 mM HEPES, pH 7.9, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM dithiotreitol, 0.5 mM PMSF, 80 mM NaCl, 25% glycerol). After extensive mixing, 1 vol of BHS (as in BLS, but containing instead 0.9 M NaCl) was slowly added and incubated at 4°C for 30 min under moderate shaking. P1 was centrifuged at 16,000 × g for 30 min. The supernatant fraction (S2) was collected and used as nuclear extract for gel retardation assay. S1 was then centrifuged at 16,000 × g for 30 min, to obtain a partially purified cytosolic (S3) fraction. The protein concentration was determined using a Blue-Coomassie-based assay.
Electrophoresis Mobility Shift Assay (EMSA)
This assay utilizes using 2–5 μg of nuclear extract proteins in a total volume of 20 μl containing 20 mM Tris-HCl, pH 7.5, 0.1 M NaCl, 0.35 mM dithiotreitol, 0.5 mM EDTA, 0.5 mM PMSF, 10% glycerol, and 0.5 μl of 2 mg/ml of double-stranded poly(dI-dC) (1 g/reaction). The following oligonucleotide sequences of the probes were used: 1) WT, 5′-TGC GCT CCC GTG CCC CTA GC-3′; 2) 52m, 5′-TGC GCT CCG GTG CCC CTA GC-3′; 3) USF E-box, 5′-TGC GCT CAC GTG CCC CTA GC-3′, where the underlined base indicates the change between wild and 52m in 1) and 2) while the effective E-box for USF in 3). The oligonucleotides were labeled at their 5′ ends with [γ32P] ATP (NEN Life Science) using T4 polynucleotide kinase. The binding to the double-stranded oligonucleotides was performed by incubating at 4°C for 30 min 10 fmol of 32P-labeled probes with the previous nuclear extract mix. The DNA–protein complexes were separated by electrophoresis on a 5% polyacrylamide gel and detected by autoradiography of the dried gel. Competitions were performed by addition of 100-fold molar excess of unlabeled double-stranded oligonucleotide competitor to the incubation mixture. Where indicated, the nuclear extracts were preincubated with the specified antibodies (anti-USF1) for 30 min at room temperature and then used for the gel retardation assay. Antibodies against USF1 were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA, USA).
RESULTS AND DISCUSSION
Overall we sequenced NPHS2 promoter region in 260 patients because carriers of homozygous or double heterozygous mutations in coding region were excluded. Three sequence variants of NPHS2 promoter were identified in our study population that gave an allele frequency below 1% (5 patients out of 260); two of them were not found in 400 control chromosomes (Table 1). Variants 1 (−236C>T) and 3 (−52C>G) were found in 1 and 2 unrelated nephrotic patients, respectively, and in none of the cohort of 200 normal subjects; variant 3 (−26C>G) was observed in 2 unrelated patients and in 1 control.
TABLE 1.
VARIANTS CHARACTERIZED BY CHANGES IN NPHS2 PROMOTER NUCLEOTIDE SEQUENCE
| Variant | No. Patients | Sex | Age at Onset (Years) | Clinical Category | Pathology | Follow-up (Years) | ESRF |
|---|---|---|---|---|---|---|---|
| −236C>T | 1 | M | 6.7 | SRNS | FSGS | 14.8 | no |
| −52C>G | 2 | F | 37.4 | SRNS | MCN | 11.6 | yes |
| M | 9.4 | SRNS | FSGS | 14.7 | yes | ||
| −26C>G | 2 | F | 42.3 | SRNS | FSGS | 10.5 | no |
| F | 47.2 | SRNS | MCN | 8.0 | no |
Three variants characterized by changes in NPHS2 promoter nucleotide sequence were found in five patients with nephrotic syndrome whose clinical features are given in the table. Two variants (−236C>T and −52C>G) were not found in 400 normal chromosomes and the second one was associated with steroid resistance and progression to uremia. The remaining (−26C>G) was also found in one normal chromosome. SRNS, steroid resistant nephrotic syndrome; SSNS, steroid sensitive nephrotic syndrome; FSGS, focal segmental glomerulosclerosis; MCN, minimal change nephropathy.
As reported in Table 1, three out of five patients presented a pathologic picture of FSGS with lack of podocin expression in their glomeruli. As indicated in the introduction, this appears to be an unspecific pattern because most FSGS patients present a reduced expression of podocin in glomeruli, probably for shedding of the protein. It is for this reason that reexpression of podocin in the recovery of the disease probably brings about a key role in evolution.
Functional implications of each sequence variants were studied following consolidated recipes based on subcloning in pGL3 reporter vector and transient transfection experiments in glomerular podocytes and COS7 cells. A significant reduction (−50%; p < 0.01) of luciferase expression compared to the wild-type sequence was determined by the two −52C>G (pPL3) and −26C>G (pPL4) variants, while −236C>T (pPL1) produced few evident changes. The same results were reproduced both in podocytes (Fig. 1a) and in COS 7 cells (not shown). Consensus analysis for homology of −52 variant region with regulatory sequences revealed homology for USF 1 and HIFA. Gel retardation experiments utilizing podocyte extracts and oligonucleotides for degenerate and typical consensus for USF and HIFA were done to characterize the trans-acting elements active at this site. HIFA did not form oligo–protein complexes (not shown) that were instead observed in the case of USF1. The sum of experiment done with various USF1 specific or degenerate sequences, with specific antibodies and with podocytes in which USF had been silenced, is shown in Figures 1b, 2a, and 2b. Figure 2 shows a typical experiment in which degenerate consensus oligos for wild (lane 2) and mutated 52 (lane 1) were incubated with cell extracts and formed a protein–oligo complex only in the former condition, suggesting that the presence of a G in place of a C at this site abolished the interaction. On the other hand, competition with cold wild oligo abolished the binding (lane 3), suggesting a good degree of specificity. In Figure 2a it is shown that also anti-USF antibodies abolished the complex formation (lane 4). Experiments reported in Figure 2b confirmed the finding above and further supported the identity of USF1. In fact, gel retardation with oligo designed to cover the typical consensus sequence of USF was run in parallel to compare the electrophoretic behavior of a typical complex USF1–DNA. It was evident the formation of a marked protein–DNA adduct (lane 3) overwhelms by several factors the DNA–protein complex formed by 52 wild (compare lanes 1 and 3 in Fig. 2b). The presence of anti-USF1 antibodies abolished the formation of the complex and produced a supershift that is represented by a minor band with higher molecular weight and an intensity that is less than 10% of the original adduct. This suggests that only a minor part of the complex forms polymers and justifies the observed lack of supershift formation with wild 52 oligo. Overall our data support the concept that USF1 forms an adduct with a degenerate consensus sequence in NPHS2 promoter and regulates the expression of podocin. To confirm this point, podocytes were silenced for USF1 RNA expression and transfected with wild constructs containing the 52 sequence. As shown in Figure 1b, luciferase expression was downregulated by 50% in this case, strongly supporting USF1 functional implication.
Figure 1.
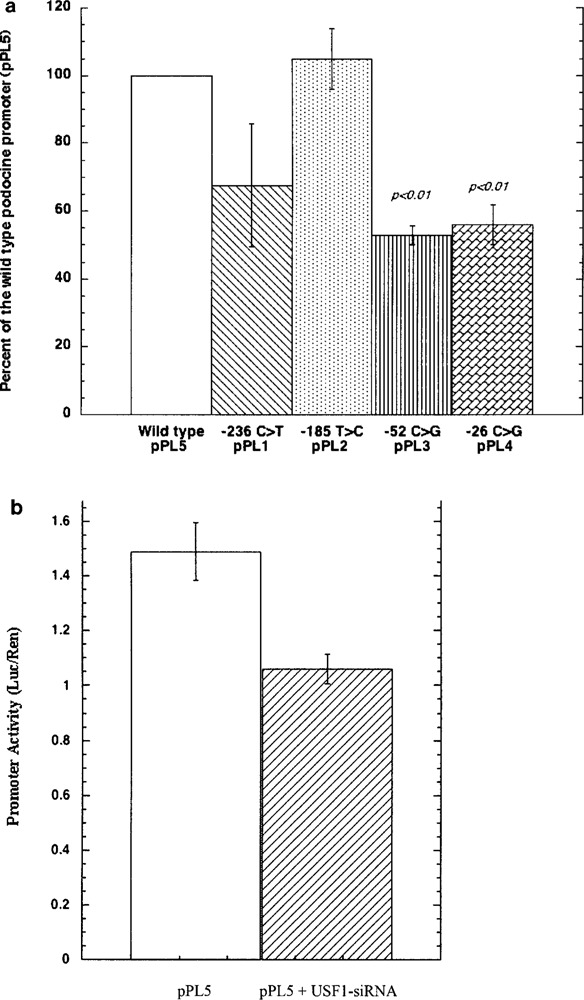
(a) Luciferase activity in podocytes transfected with pPL 1–5 constructs in which the following three variants of NPHS2 promoter were subcloned: pPL1-236C > T; pPL2-185T > L; pPL3-52C > G; pPL4-26C > G; pPL5 wild-type. Results were given in arbitrary units (AU) of Luciferase normalized by Renilla activity. (b) Transfection with pPL5 wild-type was done in normal podocytes and in podocytes silenced for USF1 RNA, showing reduction of luciferase expression by 50% in this case.
Figure 2.
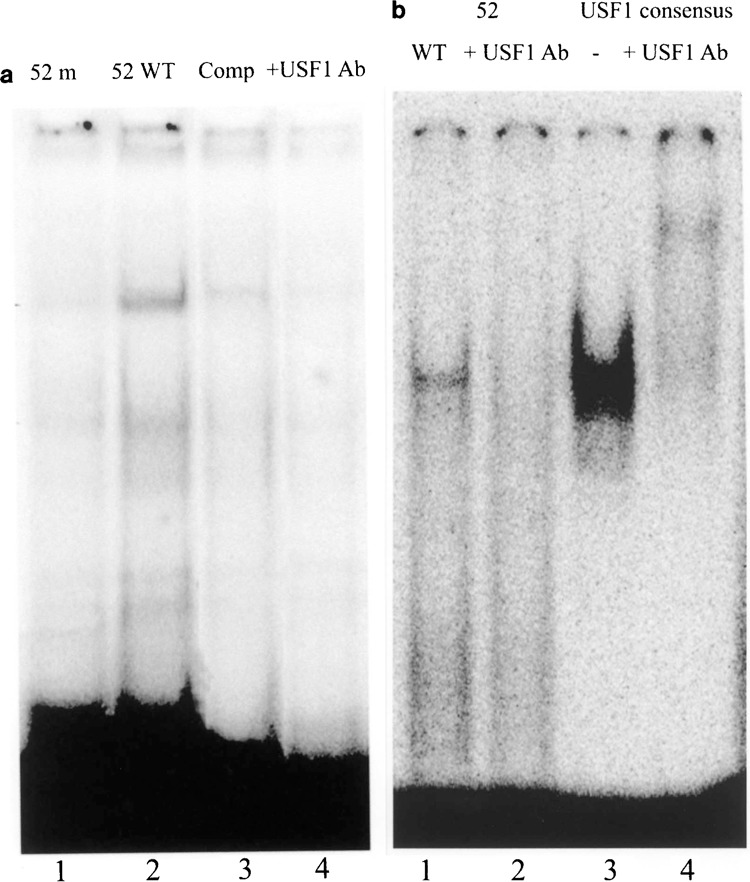
(a) Podocyte extracts were incubated with the wild-type (5′-TGC GCT CCC GTG CCC CTA GC-3′) and 52m (5′-TGC GCT CCG GTG CCC CTA GC-3′) 32P labeled oligonucleotide as indicated in Materials and Methods in the absence and in the presence of cold oligonucleotide 100 molar excess or of anti-USF1 antibodies. Electrophoresis in nondenaturing condition was run as described in Materials and Methods. (b) Experiments with oligonucleotide giving the E-box for USF1 (5′-TGC GCT CAC GTG CCC CTA GC-3′) and specific antibodies showed, as expected, the formation of a marked gel retardation adduct that was abolished by anti-USF antibodies. A clear supershift product was produced in this case even it was less evident in respect to the original adduct (less than 10%).
Whether and how downregulation of podocin influences the extent and outcome of nephrotic syndrome is matter of debate. Indeed, podocin expression is very low in patients with FSGS, suggesting that the process of regeneration is crucial for the clinical outcome. While the common finding of low podocin expression in glomeruli prevents us to define a potential correlation between rare mutation in NPHS2 promoter and podocin expression in renal biopsies it also allows to speculate on the pathogenesis and perpetuation of proteinuria because it is fully possible that low regulation lasting over years could represent a reason for progression to renal failure.
In summary, original results of a sequencing screening study on NPHS2 promoter demonstrate for the first time that heterozygous carriers of mutation at this site, although rare, may be found in nephrotic syndrome. Functional studies here described demonstrated that the functional implication of at least two variants, one of which (−52) abolishes the binding with USF1 that forms a DNA–protein adduct in the case of the wild type. This functional variant is associated with a poor clinical outcome with evolution to end-stage renal failure. Thus, even if not sufficient to cause the disease per se, these variants could represent modifier factors for severity and/or progression of the disease in FSGS and/or in other proteinuric diseases as other primary glomerulonephritis and diabetic nephropathy.
ACKNOWLEDGMENTS
This work was done with the financial support of the Italian Ministry of Health and from Renal Child Foundation. Authors also acknowledge Fondazione Mara Wilma e Bianca Querci for the financial support to the project “Nuove evoluzioni sulla multifattorialità della sindrome nefrosica.”
REFERENCES
- 1. Boute N.; Gribouval O.; Roselli S.; Benessy F.; Lee H.; Fuchshuber A.; Dahan K.; Gubler M. C.; Niaudet P.; Antignac C. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat. Genet. 24:349–354; 2000. [DOI] [PubMed] [Google Scholar]
- 2. Braden G. L.; Mulhern J. G.; O’Shea M. H.; Nash S. V.; Ucci A. A. Jr.; Germain M. J. Changing incidence of glomerular diseases in adults. Am. J. Kidney Dis. 35:878–883; 2000. [DOI] [PubMed] [Google Scholar]
- 3. Caridi G.; Bertelli R.; Carrea A.; Di Duca M.; Catarsi P.; Artero M.; Carraro M.; Zennaro C.; Candiano G.; Musante L.; Seri M.; Ginevri F.; Perfumo F.; Ghiggeri G. M. Prevalence, genetics, and clinical features of patients carrying podocin mutations in steroid-resistant nonfamilial focal segmental glomerulo-sclerosis. J. Am. Soc. Nephrol. 12:2742–2746; 2001. [DOI] [PubMed] [Google Scholar]
- 4. Caridi G.; Bertelli R.; Di Duca M.; Dagnino M.; Emma F.; Onetti Muda A.; Scolari F.; Miglietti N.; Mazzucco G.; Murer L.; Carrea A.; Massella L.; Rizzoni G.; Perfumo F.; Ghiggeri G. M. Broadening the spectrum of diseases related to podocin mutations. J. Am. Soc. Nephrol. 14:1278–1286; 2003. [DOI] [PubMed] [Google Scholar]
- 5. Caridi G.; Bertelli R.; Scolari F.; Sanna-Cherchi S.; Di Duca M.; Ghiggeri G. M. Podocin mutations in sporadic focal-segmental glomerulosclerosis occurring in adulthood. Kidney Int. 64:365; 2003. [DOI] [PubMed] [Google Scholar]
- 6. Conaldi P. G.; Biancone L.; Bottelli A.; De Martino A.; Camussi G.; Toniolo A. Distinct pathogenic effects of group B coxsackieviruses on human glomerular and tubular kidney cells. J. Virol. 71:9180–9187; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Conaldi P. G.; Biancone L.; Bottelli A.; Wade-Evans A.; Racusen L. C.; Boccellino M.; Orlandi V.; Serra C.; Camussi G.; Toniolo A. HIV-1 kills renal tubular epithelial cells in vitro by triggering an apoptotic pathway involving caspase activation and Fas upregulation. J. Clin. Invest. 102:2041–2049; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Doublier S.; Ruotsalainen V.; Salvidio G.; Lupia E.; Biancone L.; Conaldi P. G.; Reponen P.; Tryggvason K.; Camussi G. Nephrin redistribution on podocytes is a potential mechanism for proteinuria in patients with primary acquired nephrotic syndrome. Am. J. Pathol. 158:1723–1731; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Doublier S.; Salvidio G.; Lupia E.; Ruotsalainen V.; Verzola D.; Deferrari G.; Camussi G. Nephrin expression is reduced in human diabetic nephropathy: Evidence for a distinct role for glycated albumin and angiotensin II. Diabetes. 52:1023–1030; 2003. [DOI] [PubMed] [Google Scholar]
- 10. Ghiggeri G. M.; Artero M.; Carraro M.; Perfumo F. Permeability plasma factors in nephrotic syndrome: More than one factor, more than one inhibitor. Nephrol. Dial. Transplant. 16:882–885; 2001. [DOI] [PubMed] [Google Scholar]
- 11. Ghiggeri G. M.; Carraro M.; Vincenti F. Recurrent focal glomerulosclerosis in the era of genetics of podocyte proteins: Theory and therapy. Nephrol. Dial. Transplant. 19:1036–1040; 2004. [DOI] [PubMed] [Google Scholar]
- 12. Giampuzzi M.; Botti G.; Di Duca M.; Arata L.; Ghiggeri G.; Gusmano R.; Ravazzolo R.; Di Donato A. Lysyl oxidase activates the transcription activity of human collagene III promoter. Possible involvement of Ku antigen. J. Biol. Chem. 275:36341–36349; 2000. [DOI] [PubMed] [Google Scholar]
- 13. Huber T. B.; Kottgen M.; Schilling B.; Walz G.; Benzing T. Interaction with podocin facilitates nephrin signaling. J. Biol. Chem. 276:41543–41546; 2001. [DOI] [PubMed] [Google Scholar]
- 14. Huber T. B.; Simons M.; Hartleben B.; Sernetz L.; Schmidts M.; Gundlach E.; Saleem M. A.; Walz G.; Benzing T. Molecular basis of the functional podocinnephrin complex: Mutations in the NPHS2 gene disrupt nephrin targeting to lipid raft microdomains. Hum. Mol. Genet. 12:3397–3405; 2003. [DOI] [PubMed] [Google Scholar]
- 15. Inoue T.; Yaoita E.; Kurihara H.; Shimizu F.; Sakai T.; Kobayashi T.; Ohshiro K.; Kawachi H.; Okada H.; Suzuki H.; Kihara I.; Yamamoto T. FAT is a component of glomerular slit diaphragms. Kidney Int. 59:1003–1012; 2001. [DOI] [PubMed] [Google Scholar]
- 16. International Study of Kidney Disease in Children. Primary nephrotic syndrome in children: clinical significance of histopathologic variants of minimal change and of diffuse mesangial hypercellularity. Kidney Int. 20:765–771; 1981. [DOI] [PubMed] [Google Scholar]
- 17. International Study of Kidney Disease in Children. Prospective, controlled trial of cyclophosphamide therapy in children with nephrotic syndrome. Lancet 2:423–427; 1974. [PubMed] [Google Scholar]
- 18. Karle S. M.; Uetz B.; Ronner V.; Glaeser L.; Hildebrandt F.; Fuchshuber A. Novel mutations in NPHS2 detected in both familial and sporadic steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol. 13:388–393; 2002. [DOI] [PubMed] [Google Scholar]
- 19. Koop K.; Eikmans M.; Baelde H. J.; Kawachi H.; De Heer E.; Paul L. C.; Bruijn J. A. Expression of podocyte-associated molecules in acquired human kidney diseases. J. Am. Soc. Nephrol. 14:2063–2071; 2003. [DOI] [PubMed] [Google Scholar]
- 20. Luimula P.; Sandstrom N.; Novikov D.; Holthofer H. Podocyte-associated molecules in puromycin aminonucleoside nephrosis of the rat. Lab. Invest. 82:713–718; 2002. [DOI] [PubMed] [Google Scholar]
- 21. Moeller M. J.; Sanden S. K.; Soofi A.; Wiggins R. C.; Holzman L. B. Podocyte-specific expression of cre recombinase in transgenic mice. Genesis 35:39–42; 2003. [DOI] [PubMed] [Google Scholar]
- 22. Moeller M. J.; Sanden S. K.; Soofi A.; Wiggins R. C.; Holzman L. B. Two gene fragments that direct podocyte-specific expression in transgenic mice. J. Am. Soc. Nephrol. 13:1561–1567; 2002. [DOI] [PubMed] [Google Scholar]
- 23. Reiser J.; Kriz W.; Kretzler M.; Mundel P. The glomerular slit diaphragm is a modified adherens junction. J. Am. Soc. Nephrol. 11:1–8; 2000. [DOI] [PubMed] [Google Scholar]
- 24. Rohr C.; Prestel J.; Heidet L.; Hosser H.; Kriz W.; Johnson R. L.; Antignac C.; Witzgall R. The LIM-homeodomain transcription factor Lmx1b plays a crucial role in podocytes. J. Clin. Invest. 109:1073–1082; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roselli S.; Gribouval O.; Boute N.; Sich M.; Benessy F.; Attie T.; Gubler M. C.; Antignac C. Podocin localizes in the kidney to the slit diaphragm area. Am. J. Pathol. 160:131–139; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roselli S.; Heidet L.; Sich M.; Henger A.; Kretzler M.; Gubler M. C.; Antignac C. Early glomerular filtration defect and severe renal disease in podocin-deficient mice. Mol. Cell. Biol. 24:550–560; 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ruf R. G.; Lichtenberger A.; Karle S. M.; Haas J. P.; Anacleto F. E.; Schultheiss M.; Zalewski I.; Imm A.; Ruf E. M.; Mucha B.; Bagga A.; Neuhaus T.; Fuchshuber A.; Bakkaloglu A.; Hildebrandt F. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J. Am. Soc. Nephrol. 15:722–732; 2004. [DOI] [PubMed] [Google Scholar]
- 28. Ruotsalainen V.; Ljungberg P.; Wartiovaara J.; Lenkkeri U.; Kestila M.; Jalanko H.; Holmberg C.; Tryggvason K. Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc. Natl. Acad. Sci. USA 96:7962–7967; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schmid H.; Henger A.; Cohen C. D.; Frach K.; Grone H. J.; Schlondorff D.; Kretzler M. Gene expression profiles of podocyte-associated molecules as diagnostic markers in acquired proteinuric diseases. J. Am. Soc. Nephrol. 14:2958–2966; 2003. [DOI] [PubMed] [Google Scholar]
- 30. Schwarz K.; Simons M.; Reiser J.; Saleem M. A.; Faul C.; Kriz W.; Shaw A. S.; Holzman L. B.; Mundel P. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J. Clin. Invest. 108:1621–1629; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sharples P. M.; Poulton J.; White R. H. Steroid responsive nephrotic syndrome is more common in Asians. Arch. Dis. Child. 60:1014–1017; 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weber S.; Gribouval O.; Esquivel E. L.; Moriniere V.; Tete M. J.; Legendre C.; Niaudet P.; Antignac C. NPHS2 mutation analysis shows genetic heterogeneity of steroid-resistant nephrotic syndrome and low post-transplant recurrence. Kidney Int. 66:571–579; 2004. [DOI] [PubMed] [Google Scholar]