

Abstract
The neurogenic protein Drosophila big brain (BIB), which is involved in the process of neuroblast determination, and the water channel aquaporin-1 (AQP1) are among a subset of the major intrinsic protein (MIP) channels that have been found to show gated monovalent cation channel activity. A glutamate residue in the first trans-membrane (M1) domain is conserved throughout the MIP family. Mutation of this residue to asparagine in BIB (E71N) knocks out ion channel activity, and when coexpressed with BIB wild-type as shown here generates a dominant-negative effect on ion channel function, measured in the Xenopus oocyte expression system using two-electrode voltage clamp. cRNAs for wild-type and mutant BIB or AQP1 channels were injected individually or as mixtures. The magnitude of the BIB ionic conductance response was greatly reduced by coexpression of the mutant E71N subunit, suggesting a dominant-negative mechanism of action. The analogous mutation in AQP1 (E17N) did not impair ion channel activation by cGMP, but did knock out water channel function, although not via a dominant-negative effect. This contrast in sensitivity between BIB and AQP1 to mutation of the M1 glutamate suggests the possibility of interesting structural differences in the molecular basis of the ion permeation between these two classes of channels. The dominant-negative construct of BIB could be a tool for testing a role for BIB ion channels during nervous system development in Drosophila.
Key words: Big brain (BIB), Aquaporin-1 (AQP1), Major intrinsic protein (MIP), Drosophila, Neuroblast, Cation channel
INTRODUCTION
The mammalian aquaporin-1 (AQP1) and the Drosophila big brain (BIB) channels are members of a broad aquaporin-related family known as the major intrinsic protein (MIP) family represented throughout the kingdoms of life (22). BIB serves a novel role, not as a water channel, but in contributing to developmental cell interactions in Drosophila that govern the choice between neuroblast and epidermoblast fate by a process of lateral inhibition (6,20). BIB expressed in oocytes mediates a nonselective monovalent cation channel conductance that is activated in response to endogenous signaling pathways in the oocytes, shows tyrosine phosphorylation, and is modulated by pharmacological agents that alter tyrosine kinase signaling, without any appreciable water channel activity (31). These data suggested a role for membrane depolarization in the neurogenic function of BIB in early development, but genetic or pharmacological tools for testing this hypothesis are lacking.
While most aquaporins are known as pores for water, AQP1 and BIB are among a subset of aquaporin-related channels that have been shown to mediate a gated ion channel function (36). Each subunit of the AQP1 tetramer contains a pore allowing passage of water (1). However, tetrameric assembly is needed to express water channel function (10). A gated monovalent cation channel activity for AQP1 (2) has been proposed to reside in the central pore of the tetramer, lined by residues contributed by the second and fifth transmembrane domains (35,37) and is suggested to serve a regulatory role in transmembrane fluid secretion in choroid plexus (4). The location of the ion channel pore in BIB is not known; however, results of site-directed mutagenesis reported here suggest interesting differences in the sensitivity of the ionic conductance of AQP1 compared with BIB for mutations at a conserved position in the first trans-membrane domain, and could indicate structural differences in the channel design of possible ion conduction pathways.
The MIP channel family carries a characteristic pair of signature motifs (asparagine-proline-alanine; NPA) located in the first cytoplasmic loop (loop B) and the third extracellular loop (loop E) that form the intrasubunit pores (10). In addition, a glutamate residue in the first transmembrane (M1) domain is conserved throughout the MIP family (22). The first test of a functional role for the M1 glutamate was carried out for BIB; mutation to aspartate (E71D) created a gain-of-function block by magnesium (32). Mutation of this residue to asparagine (E71N) abolished the BIB ionic conductance without preventing protein targeting to the plasma membrane (31). These results prompted the idea that the E71N mutation could be tested for a dominant-negative effect on BIB ion channel function. The analogous mutation in the M1 glutamate of AQP1 (E17N) was tested in parallel; the differences in outcome are intriguing, and suggest structural differences in the ion permeation pathways in these two classes of channels.
Crystal structural analyses of several aquaporins have shown they assemble as tetramers of homomeric subunits each with six transmembrane domains and intracellular amino and carboxyl terminal domains (8,28) in a pattern broadly reminiscent of other channels (9). Inherited mutations in ion channels give rise to diverse array of channelopathy diseases, some of which are attributed to a dominant-negative effect; that is, incorporation of a mutant subunit into a complex of otherwise wild type subunits knocks down the expression, targeting, or function of the channel complex (13). Such mutant constructs have been used as tools for the targeted molecular knockdown of specific subtypes of proteins in cells and systems, for example, as was done with the Shaker K+ channel in Drosophila to study the synaptic morphological consequences of enhanced motoneuron excitability (17) and in other studies (14,23,30). In the absence of selective high-affinity pharmacological agents for the aquaporin ion channels, the identification of dominant-negative constructs will be valuable for extending work from the expression system studies to investigating the physiological roles of aquaporin functions in vivo.
MATERIALS AND METHODS
Oocyte Preparation and Injection
Stage V–VI oocytes were removed from anesthetized adult female Xenopus laevis by partial ovariectomy and defolliculated as described previously (2). On the following day, prepared oocytes were injected with 50 nl of sterile water (control oocytes) or with 50 nl of sterile water containing cRNAs for wild-type or mutant Drosophila BIB, or wild-type or mutant human AQP1, at 1 to 20 ng each or as a mixture (as specified in text) and were incubated for 2 or more days at 18°C in ND96 culture medium (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, 2.5 mM pyruvic acid, 100 U/ml penicillin, and 100 μg/ml streptomycin, pH 7.6) to allow protein expression before recording. Cloned Drosophila melanogaster BIB cDNA was provided by L. and Y. N. Jan (University of California, San Francisco) (20); cloned human AQP1 cDNA was provided by P. Agre (Duke University) (18).
Molecular Methods
Wild-type BIB cDNA previously was subcloned into the Xenopus β-globin expression vector (pXβ-Gev) and modified by addition of an amino-terminal hemagglutanin (HA) epitope tag that did not interfere with channel expression or function (31). The HA-tagged BIB construct is referred to here as BIB wild-type, and was used as the template for site-directed mutagenesis of glutamate (E) to asparagine (N) at position 71 (referenced to the original nontagged Drosophila melanogaster BIB sequence, accession number X53275) using the Stratagene QuikChange kit (Stratagene, La Jolla, CA). The BIB E71N construct was sequenced in full to verify absence of background mutations. BIB cDNA was linearized with SpeI and transcribed in vitro with T3 RNA polymerase. AQP1 in the Xenopus β-globin expression vector was modified by site-directed mutagenesis to create AQP1 E17N (accession number NM_198098) at the equivalent glutamate residue, and sequenced for confirmation. AQP1 cDNA was linearized with BamHI and transcribed with T3 RNA polymerase.
Electrophysiological Recordings
Two-electrode voltage clamp recordings were performed at room temperature with electrodes (0.6–3 MΩ) filled with 3 M KCl. Data were recorded with a GeneClamp 500 (Axon Instruments, Foster City, CA), filtered at 2 kHz and analyzed with pClamp software (Axon Instruments). Secondary data analyses and statistical tests were done with Kaleidagraph 4.0 (Synergy Software, Reading, PA). Standard bath saline contained (in mM): 100 NaCl, 5.0 MgCl2, and 5 HEPES, pH 7.3. For BIB, channel activation was monitored as a function of time after prick initiation of oocyte signaling by brief sweeps of voltage steps repeated every 6 s from a holding potential of −40 mV. The endogenous signaling pathway in oocytes is triggered by electrode insertion (pricking) and involves tyrosine kinase signaling cascades as described previously (31), an effect that is consistent with other studies of prick activation of signaling pathways in oocytes. The ionic current activated in BIB-expressing oocytes is a nonselective monovalent cation conductance that is not seen in control oocytes (31), and was confirmed to be due to BIB and not another endogenous channel pulled along in the protein synthetic process by identification of a single site mutation that created sensitivity to block by extracellular Mg2+ (32). For AQP1, channel activation was monitored with the same voltage protocol, after application of the nitric oxide donor sodium nitroprusside (SNP), which stimulates endogenous soluble guanylate cyclase. The SNP dose (final 0.5–4 mM) applied as a bolus in bath saline was adjusted empirically for each batch of oocytes to provide activation of AQP1-expressing oocytes without appreciable effects on control oocytes. Batches with high levels of endogenous channel activation were not used.
Swelling Assays
Oocytes were placed in 50% hypotonic saline (50 mM NaCl, 5 mM MgCl2, 5 mM HEPES, pH 7.3) at time zero, and images were captured every 2 s for 60 s with a Cohu video camera and frame capture system (Scion Image). Cross-sectional area was used to calculate the increase in size as a function of time, standardized to area of the oocyte in the initial frame at time zero. Swelling rates were determined from the slope of the linear fit of the swelling response, and standardized to the mean swelling rate of the wild-type in the same batch of oocytes. Control oocytes show no appreciable swelling over 5 min or more.
Immunofluorescence Labeling of Intact Oocytes
Two to 3 days after cRNA injections, intact oocytes not subjected to ion channel stimulation were flash frozen in O.C.T. Compound (Electron Microscopy Sciences, Hatfield, PA), fixed in 4% paraformaldehyde for 2–3 h at 4°C, rinsed several times in saline sodium citrate (SSC: 300 mM sodium chloride; 20 mM sodium citrate), and then rinsed in 100 mM glycine. Permeabilization using 0.1% Triton X-100 (in SSC, for 1 h) was followed by overnight incubation at 4°C with monoclonal rat anti-HA antibody (0.5 μg/ ml; clone 3F10, Roche) that detects HA-tagged wild-type and mutant BIB, or with polyclonal rabbit anti-AQP1 antibody at 1:500 dilution (27) that detects the carboxyl terminal domain of wild-type and mutant AQP1. Antibodies were diluted in SSC buffer with 2% goat serum, 1% bovine serum albumin, 0.1% Triton X-100, and 0.02% sodium azide. Oocytes were washed at room temperature in SSC with 0.1% Triton X-100 for 1 h, followed by a 1-h incubation with FITC-conjugated goat anti-rat antibody (1 μg/ml; Zymed) or FITC-conjugated goat anti-rabbit antibody (30 μg/ml; Jackson) in buffer. Oocytes were rinsed for 1 h and imaged with a 10× objective on a Nikon (Tokyo, Japan) PCM 2000 laser scanning confocal microscope. Control and experimental oocytes were prepared in parallel and imaged with identical exposure parameters after confirming exposure settings that produced images that were in a subsaturating range for the strongest signal seen across all samples in the set. Images were grouped into an ensemble montage and minimally processed as a group to convert to grayscale and invert to a light field background before analyzing. Semiquantitative analyses of signal intensities from confocal digital images were done using ImageJ software for Mac OSX (Scion Image) to estimate signal intensity at the plasma membrane, compared with equivalent areas of adjacent regions outside the membrane used to determine background, for estimation of the general relative ratios of protein expression of mutant and wild-type BIB. These calculations were not used to suggest any specific subunit stoichiometry, but to allow an approximation that the level of expression of the BIB mutant subunit was less than that of the wild-type subunit in conditions achieving knockdown of the ion channel function.
RESULTS
Alignment of the complete N-terminus of AQP1 and the proximal segment of the BIB N-terminus (Fig. 1) illustrates a general homology in amino acid sequence. At the N-terminal side of the first trans-membrane domain (M1), the glutamate residue (17 in AQP1, and 71 in BIB) is conserved across the broad aquaporin-related family of MIP (22). Mutation of the BIB M1 glutamate residue to asparagine (E71N) was shown to disrupt ion channel activation compared with BIB wild-type, when expressed at equivalent membrane protein levels in oocytes (31). The M1 glutamate residue is visualized in the crystal structure of AQP1 [bovine AQP1; MMDB #18789 (28)] as projecting inward towards the individual subunit pore pathway, while residues lining the putative central pore of the tetramer are thought to be located in trans-membrane domains 2 and 5 (37).
Figure 1.
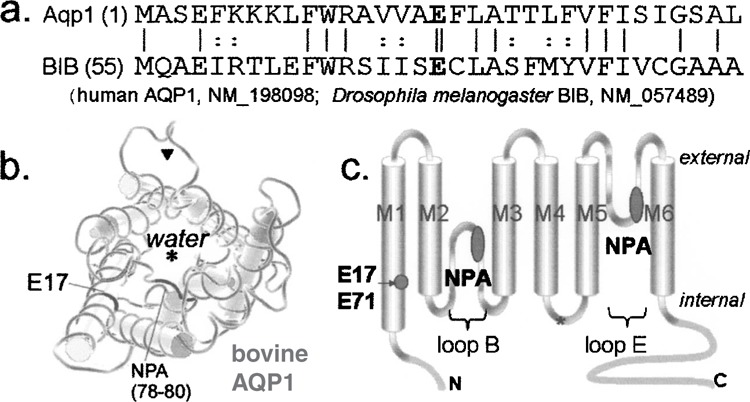
Amino acid sequence homology and location of the conserved glutamate residue of the first transmembrane domain in AQP1 and BIB channels. (a) Alignment of the amino acid sequences of the N terminal and initial first transmembrane domains. The initial 54 amino acids of BIB are not shown. The conserved glutamate (E) highlighted in bold type is at residue 17 in AQP1 and 71 in BIB (double vertical line). Single vertical lines show amino acid identity; dotted lines show amino acid homology. (b) Diagram of a subunit highlighting the positions of the E17 residue and the first signature NPA motif in loop B, generated from public domain structural information (NCBI Molecular Modeling DataBase; MMDB #18789) contributed for bovine AQP1 (28). Asterisk: water pore; arrowhead: proposed ion pore. (c) Cartoon of the general transmembrane topology of an aquaporin subunit, showing the six transmembrane domains (M1–M6) and the conserved glutamate in M1 and asparagine-proline-alanine (NPA) motifs in loops B and E.
Coexpression of BIB E71N with wild-type BIB resulted in impairment of ion channel activity (Fig. 2a), with the magnitude of the knockdown effect correlated to the proportion of mutant cRNA in the injection mixture (Fig. 3). BIB channels are activated in Xenopus oocytes by endogenous signaling pathways involving tyrosine kinase activity, as characterized previously (31). cGMP is an intracellular trigger for AQP1 ion channel activation (2,3). Injection of cRNA for AQP1 E17N at levels giving expression comparable to that of wild-type AQP1 showed no impairment of ion channel activation in response to increased cGMP, induced by application of the nitric oxide donor sodium nitroprusside (Fig. 2b). The approximately linear or weakly voltage-sensitive current–voltage relationships, and the observed reversal potentials at a value expected for a nonselective cat-ionic conductance (Fig. 2c), are properties consistent with the characteristics of BIB and AQP1 channels described previously (31,34).
Figure 2.
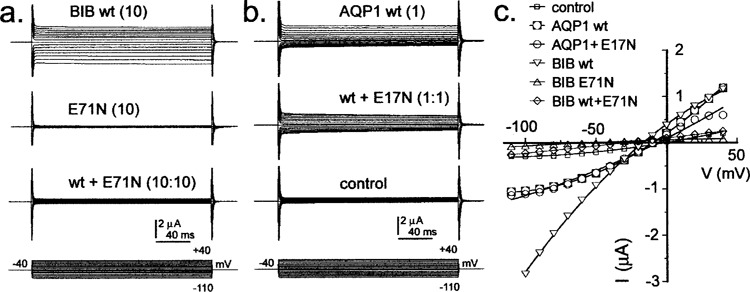
Ionic conductances of wild-type and mutant BIB and AQP1 expressed in Xenopus oocytes and recorded by voltage clamp. (a) Oocytes injected with BIB wild-type (10 ng cRNA) show activation of an ionic current over 10 min following prick stimulation of the oocyte. The mutant E71N alone (10 ng cRNA) does not show an ionic conductance response; coexpression with wild-type effectively suppresses the wild-type function. (b) Oocytes injected with AQP1 wild-type (1 ng cRNA) show an ionic current activated by 7–10 min after application of sodium nitroprusside (1 mM). The response is not impaired by coexpression with the AQP1 E17N mutant (1 ng cRNA). Control oocytes from the same batch show no conductance response to SNP in the same treatment condition. (c) Plots of current–voltage relationships for the same data shown in (a) and (b) illustrate the relatively linear current–voltage relationships and reversal potential consistent with a nonselective cation conductance.
Figure 3.
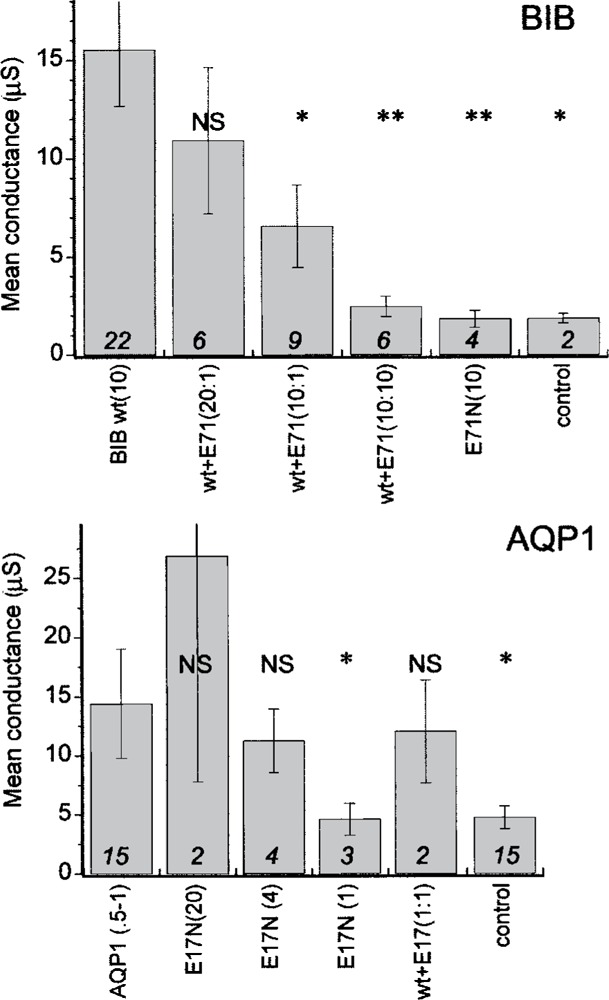
Summary histogram of mean conductance values for wild-type and mutant BIB and AQP1, and the results of coexpression of wild-type and mutant. Amounts of cRNA injected are indicated in parentheses (ng). Final conductances were measured at 8–10 min after stimulation by the channel-appropriate mechanism, and calculated from the linear slope of the current-voltage relationship from −40 to +40 mV. Raw data are compiled as mean ± SE; n values in italics above the j-axis. Significant differences from the respective wild-type groups were assessed by ANOVA and post-hoc Student’s -test (unpaired data with unequal variance) for *p < 0.05 and **p < 0.005. NS, not significant.
Summary histograms (Fig. 3) compiled for data from multiple batches of oocytes show that the coexpression of BIB E71N with wild-type causes a significant dose-dependent block of ionic conductance, and that the expression of the mutant BIB channel alone does not confer any ion channel function, remaining indistinguishable from controls. In contrast, AQP1 E17N expressed as a homomeric channel shows a SNP-induced ionic conductance that correlates in magnitude with the amount of cRNA injected, and does not interfere with the conductance of coexpressed wild-type AQP1 channels.
Confocal immunocytochemistry confirmed that the BIB E71N subunit (carrying an epitope tag identical to that of wild-type) was expressed in the oocyte plasma membrane (Fig. 4), although an approximately 10-fold greater amount of cRNA injection was needed to achieve a level of protein signal intensity that was similar to that of wild-type BIB. Thus, an injection ratio of 10 ng wild-type with 10 ng E71N BIB cRNA would be expected to yield strong expression of wild-type BIB subunits with a minority of BIB mutant subunits, yet the combination produced an effective knockdown of the magnitude of the ionic conductance, compared with the amplitude of the response seen in oocytes injected with the same concentration of cRNA for the BIB wild-type alone (Fig. 3, top panel). These results suggest incorporation of mutant subunits into the tetramer exerts a dominant-negative effect in suppressing ion channel function.
Figure 4.
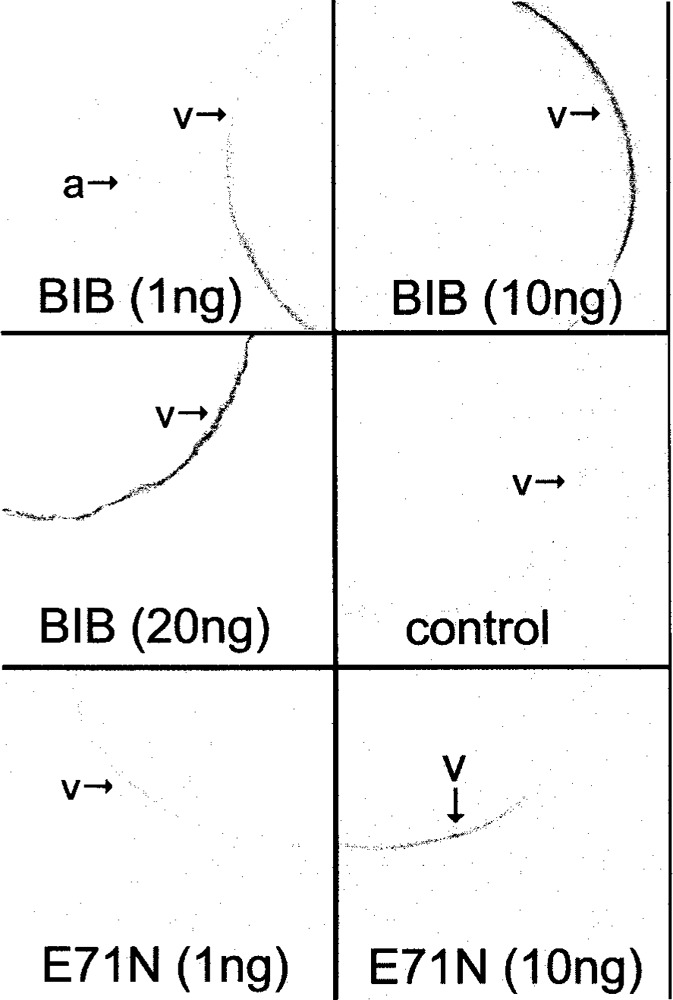
Confocal images of oocytes expressing BIB wild-type and mutant channels. Oocytes with BIB wild-type and mutant at 1 and 10 ng and control were all handled in parallel and imaged under identical conditions, using antibody against the HA epitope tag. The image for the oocyte with BIB at 20 ng was from a separate preparation, and not used in the semiquantitative analysis of signal intensity (see Materials and Methods for details). BIB is expressed predominantly in the vegetal hemisphere of the oocyte (v) and is not apparent in the animal hemisphere (a) as illustrated in the top left panel capturing two oocytes in the same field of view.
Because the individual subunit pathways subserve water channel activity in AQP1 (19,29), we assessed whether the E17N mutation in AQP1 influenced osmotic water permeability (Fig. 5). As in BIB, the mutation of the M1 glutamate in AQP1 decreased the efficiency of protein expression in the membrane. Using immunocytochemistry and confocal imaging, we determined that injection of 20 ng of cRNA for AQP1 E17N yielded a signal intensity for membrane protein similar to that seen for wild-type AQP1 injected at 1 ng per oocyte (Fig. 5). It is interesting that the oocytes expressing AQP1 E17N (20 ng) failed to show osmotic water flux, remaining at a level not significantly different from control. Coexpression of AQP1 wild-type and E17N (co-injected at 1 and 20 ng, respectively) significantly decreased osmotic water permeability compared with AQP1 wild-type alone (1 ng), but did not show a dominant-negative suppression of the wild-type water channel function. While the E17N mutation does not appear to grossly compromise ion channel function in AQP1, the water channel activity of the individual subunit pore is disabled by the mutation.
Figure 5.
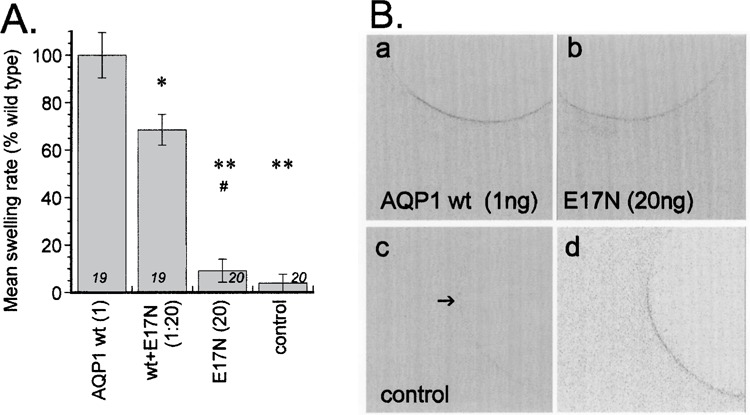
Osmotic water permeability of the AQP1 E17N mutant channel, and the effects of coexpression with wild-type AQP1. (A) Summary histogram of the osmotic water permeability of oocytes expressing wild-type AQP1 (1 ng cRNA), the lack of water channel activity in the mutant E17N (20 ng cRNA), and the moderately inhibitory effect of mutant when coexpressed with wild-type. Data are mean ± SE; n values in italics above the x-axis. Significant differences from the AQP1 wild-type were assessed by ANOVA and post-hoc Student’s t-tests (unpaired data with unequal variance) for *p < 0.01 and **p < 0.001. #, not significantly different from control. (B) Confocal images of oocytes expressing wild-type and mutant AQP1 channels, illustrating that comparable levels of protein were expressed in the oocyte membrane using 1 ng of wild-type or 20 ng of the E17N mutant. Panels (a)–(c) show images from oocytes prepared and imaged under identical conditions; panel (d) is from a longer exposure image to show the low level of background.
DISCUSSION
Work presented here is the first to characterize a mutation of BIB that exerts a dominant-negative suppression of its ion channel activity, and the first to suggest that a mutation in AQP1 can be used to disable water channel function while leaving the ion channel function intact. The mechanism of BIB signaling in the fly is unknown. Our data suggest the dominant-negative construct as a possible genetic tool for in vivo dissection of the mechanism of action of BIB in nervous system development. In Drosophila, loss-of-function mutations in the big brain gene cause a neurogenic phenotype, similar to mutations in genes such as Notch and Delta that cause effects classified by a loss of lateral inhibition and an overproduction of neuroblasts (12,21). A proposed ion channel function for BIB, prompted by sequence comparisons with other channels (20), was confirmed by demonstration that BIB is a tyrosine-kinase-sensitive monovalent cation channel when expressed in Xenopus oocytes (31) that shows voltage-dependent block by external divalent cations (32). In the developing Drosophila embryo, BIB is expressed in cells within proneural cell clusters, and mediates a lateral inhibition signal that suppresses neuroblast formation and promotes the alternative epidermoblast fate (6). Future work using expression of the dominant-negative construct of BIB in developing Drosophila embryos will be of interest for testing the hypothesis that the ion channel function of BIB is involved in lateral inhibition and cell fate determination during neurogenesis.
Loss-of-function mutations of BIB result in overgrowth of the fly nervous system at the expense of epidermis, approximately doubling the number of neuronal precursors and progeny in the embryonic peripheral nervous system (21). Genetic studies showed that BIB does not fit into an interacting cascade but plays a parallel role in cell–cell communication that is synergistic with the function of other neurogenic genes such as Notch and Delta (6). BIB, expressed in regions that give rise to neural precursors, is concentrated in apical adherens junctions in the plasma membrane and in small cytoplasmic vesicles. The activity of BIB is required in epidermal precursors to prevent neural development, supporting the proposal that BIB is a channel protein that serves a necessary function in the response of these precursor cells to the lateral inhibition signal (6).
Dominant-negative mutations have been demonstrated previously for classes of aquaporin channels including AQP0, AQP1, AQP2, and AQP4. Mutations in AQP0 are associated with dominantly inherited cataracts; the clinical features of the opacities in the crystalline lens of eye vary by family. Characterized in Xenopus oocytes, AQP0 mutations of glutamate to glycine at position 134 (E134G), or threonine to arginine at 138 (T138R), cause a loss of water channel activity when coexpressed with wild-type, thought to be due to impaired trafficking to the plasma membrane (7). Mutation of AQP1 from alanine to methionine at position 73 (A73M) results in a nonfunctional channel when coexpressed with wild-type, suppressing water channel activity by an unknown mechanism (16). AQP2 channels couple vasopressin (antidiuretic hormone) signaling to an increase in water reabsorption in kidney. Inherited frameshift mutations in the carboxyl terminal domain of AQP2 cause a nonsense extension of the amino acid sequence, associated with autosomal dominant nephrogenic diabetes insipidus. In Xenopus oocytes, surface expression of the mutant AQP2 channel is reduced compared to wild-type, suggesting the loss is in part a defect in trafficking; heteromeric channel assembly of wild-type and mutant subunits results in reduced water permeability via a dominant-negative effect (11). AQP2 subunits carrying a partial deletion of the carboxyl terminal show a dominant-negative effect in recombinant knock-in mice, and impairment of normal apical sorting of AQP2 even under conditions of dehydration (25). However, wild-type channels can also show nonapical targeting after chronic treatment with vasopressin for an extended period (5). AQP4 subunits with a double mutation to proline at leucine 246 and tyrosine 247 (L246P + Y247P) cause a dominant-negative effect on AQP4-mediated transcellular water flow in rodent liver cholangiocyte cells (26). AQP4 subunits with mutations to tryptophan at glycine 72 (G72W) or alanine 188 (A188W) are targeted to oocyte plasma membrane as shown by immunofluorescence, but do not mediate appreciable water flux. Coexpression with wild-type causes a dominant-negative effect, suggesting the individual water channels located in each subunit do not function autonomously, but are affected by interactions with other monomers in the tetramer (24).
The difference between BIB and AQP1 with respect to sensitivity to a dominant-negative influence of glutamate in the first transmembrane domain M1 is intriguing. Mutation of E71 in BIB impairs ion channel function, whereas the equivalent mutation in AQP1 at E17 does not prevent ion channel activity. The differential effects of mutation on BIB and AQP1 might offer insights into the structural basis of ion permeation in these classes of channels. The central pore in AQP1, lined by transmembrane domains M2 and M5, has been suggested as the site of gated ion permeation (35,37). In contrast, ion channel activity in another aquaporin, AQP6, has been suggested to involve flux through the individual subunit pores (33), though AQP6 channel function also is influenced by mutation of asparagine 60 predicted to be located at the crossing point of M2 and M5 (15), so a central pore contribution cannot be entirely ruled out.
One explanation of the results for the difference between BIB and AQP1 is that the glutamate residue in M1 indirectly stabilizes a putative pore structure at the center of the tetramer in both BIB and AQP1, but that the structural consequence of the mutation in AQP1 is less severe. Alternatively, it is conceivable that BIB might differ from AQP1 by utilizing individual subunit pores for ion permeation. This would be consistent with the observation that the water channel function in AQP1 E17N is impaired, because this residue appears from crystal structure to project inward in the individual subunits towards the water pore domains. If BIB carries ion current through individual subunit pores rather than a central pore, why wouldn’t the subunits function independently of one another, in contrast to the observed dominant negative effect? The idea that individual subunit pores could be sensitive to a dominant negative influence from the tetrameric composition has precedent from studies of AQP1 and AQP4 water channel function (16,24), in which certain mutations can impact the water channel function of wild-type subunits incorporated in the tetrameric complex. Characterization of the molecular nature of the pores and barriers for ion permeation in aquaporin channels and the development of dominant negative constructs will be of interest in expanding our knowledge of this ancient and multifunctional family of membrane proteins.
ACKNOWLEDGMENTS
Thanks to D. Bitner, K. Louie and A. Marble for technical assistance, and to S. Srinivasan for assistance with pilot studies. Supported by NIH R01 GM059986.
REFERENCES
- 1. Agre P.; Preston G. M.; Smith B. L.; Jung J. S.; Raina S.; Moon C.; Guggino W. B.; Nielsen S. Aquaporin CHIP: The archetypal molecular water channel. Am. J. Physiol. 265:F463–476; 1993. [DOI] [PubMed] [Google Scholar]
- 2. Anthony T. L.; Brooks H. L.; Boassa D.; Leonov S.; Yanochko G. M.; Regan J. W.; Yool A. J. Cloned human aquaporin-1 is a cyclic GMP-gated ion channel. Mol. Pharmacol. 57:576–588; 2000. [DOI] [PubMed] [Google Scholar]
- 3. Boassa D.; Yool A. J. A fascinating tail: Cyclic GMP activation of aquaporin-1 ion channels. Trends Pharmacol. Sci. 23:558–562; 2002. [DOI] [PubMed] [Google Scholar]
- 4. Boassa D. L.; Stamer W. D.; Yool A. J. Ion channel function of aquaporin-1 natively expressed in choroid plexus. J. Neurosci. 26:7811–7819; 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brown D. The ins and outs of aquaporin-2 trafficking. Am. J. Physiol. Renal Physiol. 284:F893–901; 2003. [DOI] [PubMed] [Google Scholar]
- 6. Doherty D.; Jan L. Y.; Jan Y. N. The Drosophila neurogenic gene big brain, which encodes a membrane-associated protein, acts cell autonomously and can act synergistically with Notch and Delta. Development 124:3881–3893; 1997. [DOI] [PubMed] [Google Scholar]
- 7. Francis P.; Chung J. J.; Yasui M.; Berry V.; Moore A.; Wyatt M. K.; Wistow G.; Bhattacharya S. S.; Agre P. Functional impairment of lens aquaporin in two families with dominantly inherited cataracts. Hum. Mol. Genet. 9:2329–2334; 2000. [DOI] [PubMed] [Google Scholar]
- 8. Fu D.; Libson A.; Miercke L. J.; Weitzman C.; Nollert P.; Krucinski J.; Stroud R. M. Structure of a glycerol-conducting channel and the basis for its selectivity. Science 290:481–486; 2000. [DOI] [PubMed] [Google Scholar]
- 9. Jan L. Y.; Jan Y. N. Tracing the roots of ion channels. Cell 69:715–718; 1992. [DOI] [PubMed] [Google Scholar]
- 10. Jung J. S.; Preston G. M.; Smith B. L.; Guggino W. B.; Agre P. Molecular structure of the water channel through aquaporin CHIP. The hourglass model. J. Biol. Chem. 269:14648–14654; 1994. [PubMed] [Google Scholar]
- 11. Kuwahara M.; Iwai K.; Ooeda T.; Igarashi T.; Ogawa E.; Katsushima Y.; Shinbo I.; Uchida S.; Terada Y.; Arthus M. F.; Lonergan M.; Fujiwara T. M.; Bichet D. G.; Marumo F.; Sasaki S. Three families with autosomal dominant nephrogenic diabetes insipidus caused by aquaporin-2 mutations in the C-terminus. Am. J. Hum. Genet. 69:738–748; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lehmann R.; Jimenez F.; Dietrich U. On the phenotype and development of mutants of early neurogenesis in Drosophila melanogaster. Rouxs Arch. Dev. Biol. 192:62–74; 1983. [DOI] [PubMed] [Google Scholar]
- 13. Lehmann-Horn F.; Jurkat-Rott K. Voltage-gated ion channels and hereditary disease. Physiol. Rev. 79:1317–1372; 1999. [DOI] [PubMed] [Google Scholar]
- 14. Li M.; Jan Y. N.; Jan L. Y. Specification of subunit assembly by the hydrophilic amino-terminal domain of the Shaker potassium channel. Science 257:1225–1230; 1992. [DOI] [PubMed] [Google Scholar]
- 15. Liu K.; Kozono D.; Kato Y.; Agre P.; Hazama A.; Yasui M. Conversion of aquaporin 6 from an anion channel to a water-selective channel by a single amino acid substitution. Proc. Natl. Acad. Sci. USA 102:2192–2197; 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mathai J. C.; Agre P. Hourglass pore-forming domains restrict aquaporin-1 tetramer assembly. Biochemistry 38:923–928; 1999. [DOI] [PubMed] [Google Scholar]
- 17. Mosca T. J.; Carrillo R. A.; White B. H.; Keshishian H. Dissection of synaptic excitability phenotypes by using a dominant-negative Shaker K+ channel subunit. Proc. Natl. Acad. Sci. USA 102:3477–3482; 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Preston G. M.; Agre P. Isolation of the cDNA for erythrocyte integral membrane protein of 28 kilodaltons: Member of an ancient channel family. Proc. Natl. Acad. Sci. USA 88:11110–11114; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Preston G. M.; Jung J. S.; Guggino W. B.; Agre P. The mercury-sensitive residue at cysteine 189 in the CHIP28 water channel. J. Biol. Chem. 268:17–20; 1993. [PubMed] [Google Scholar]
- 20. Rao Y.; Jan L. Y.; Jan Y. N. Similarity of the product of the Drosophila neurogenic gene big brain to transmembrane channel proteins. Nature 345:163–167; 1990. [DOI] [PubMed] [Google Scholar]
- 21. Rao Y.; Bodmer R.; Jan L. Y.; Jan Y. N. The big brain gene of Drosophila functions to control the number of neuronal precursors in the peripheral nervous system. Development 116:31–40; 1992. [DOI] [PubMed] [Google Scholar]
- 22. Reizer J.; Reizer A.; Saier M. H. Jr. The MIP family of integral membrane channel proteins: Sequence comparisons, evolutionary relationships, reconstructed pathway of evolution, and proposed functional differentiation of the two repeated halves of the proteins. Crit. Rev. Biochem. Mol. Biol. 28:235–257; 1993. [DOI] [PubMed] [Google Scholar]
- 23. Ribera A. B.; Pacioretty L. M.; Taylor R. S. Probing molecular identity of native single potassium channels by overexpression of dominant negative subunits. Neuropharmacology 35:1007–1016; 1996. [DOI] [PubMed] [Google Scholar]
- 24. Shi L. B.; Verkman A. S. Selected cysteine point mutations confer mercurial sensitivity to the mercurial-insensitive water channel MIWC/AQP-4. Biochemistry 35:538–544; 1996. [DOI] [PubMed] [Google Scholar]
- 25. Sohara E.; Rai T.; Yang S. S.; Uchida K.; Nitta K.; Horita S.; Ohno M.; Harada A.; Sasaki S.; Uchida S. Pathogenesis and treatment of autosomal-dominant nephrogenic diabetes insipidus caused by an aquaporin 2 mutation. Proc. Natl. Acad. Sci. USA 103:14217–14222; 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Splinter P. L.; Masyuk A. I.; Marinelli R. A.; LaRusso N. F. AQP4 transfected into mouse cholangiocytes promotes water transport in biliary epithelia. Hepatology 39:109–116; 2004. [DOI] [PubMed] [Google Scholar]
- 27. Stamer W. D.; Seftor R. E.; Snyder R. W.; Regan J. W. Cultured human trabecular meshwork cells express aquaporin-1 water channels. Curr. Eye Res. 14:1095–1100; 1995. [DOI] [PubMed] [Google Scholar]
- 28. Sui H.; Han B. G.; Lee J. K.; Walian P.; Jap B. K. Structural basis of water-specific transport through the AQP1 water channel. Nature 414:872–878; 2001. [DOI] [PubMed] [Google Scholar]
- 29. Tajkhorshid E.; Nollert P.; Jensen M. O.; Miercke L. J.; O’Connell J.; Stroud R. M.; Schulten K. Control of the selectivity of the aquaporin water channel family by global orientational tuning. Science 296:525–530; 2002. [DOI] [PubMed] [Google Scholar]
- 30. Tu L.; Santarelli V.; Deutsch C. Truncated K+ channel DNA sequences specifically suppress lymphocyte K+ channel gene expression. Biophys. J. 68:147–156; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yanochko G. M.; Yool A. J. Regulated cationic channel function in Xenopus oocytes expressing Drosophila big brain. J. Neurosci. 22:2530–2540; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yanochko G. M.; Yool A. J. Block by extracellular divalent cations of Drosophila big brain channels expressed in Xenopus oocytes. Biophys. J. 86:1470–1478; 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yasui M.; Hazama A.; Kwon T. H.; Nielsen S.; Guggino W. B.; Agre P. Rapid gating and anion permeability of an intracellular aquaporin. Nature 402:184–187; 1999. [DOI] [PubMed] [Google Scholar]
- 34. Yool A. J.; Stamer W. D.; Regan J. W. Forskolin stimulation of water and cation permeability in aquaporin 1 water channels. Science 273:1216–1218; 1996. [DOI] [PubMed] [Google Scholar]
- 35. Yool A. J.; Weinstein A. M. New roles for old holes: Ion channel function in aquaporin-1. News Physiol. Sci. 17:68–72; 2002. [DOI] [PubMed] [Google Scholar]
- 36. Yool A. J.; Stamer W. D. Novel roles for aquaporins as gated ion channels. In: Maui R. A., ed. Molecular and cellular insights to ion channel biology. Amsterdam: Elsevier; 2004:351–379. [Google Scholar]
- 37. Yu J.; Yool A. J.; Schulten K.; Tajkhorshid E. Mechanism of gating and ion conductivity of a possible tetrameric pore in aquaporin-1. Structure 14:1411–1423; 2006. [DOI] [PubMed] [Google Scholar]