

Abstract
The 22q11 Deletion Syndrome (22q11DS, also known as DiGeorge or Velo-Cardio-Facial Syndrome) has a variable constellation of phenotypes including life-threatening cardiac malformations, craniofacial, limb, and digit anomalies, a high incidence of learning, language, and behavioral disorders, and increased vulnerability for psychiatric diseases, including schizophrenia. There is still little clear understanding of how heterozygous microdeletion of approximately 30–50 genes on chromosome 22 leads to this diverse spectrum of phenotypes, especially in the brain. Three possibilities exist: 1) 22q11DS may reflect haploinsufficiency, homozygous loss of function, or heterozygous gain of function of a single gene within the deleted region; 2) 22q11DS may result from haploinsufficiency, homozygous loss of function, or heterozygous gain of function of a few genes in the deleted region acting at distinct phenotypically compromised sites; 3) 22q11DS may reflect combinatorial effects of reduced dosage of multiple genes acting in concert at all phenotypically compromised sites. Here, we consider evidence for each of these possibilities. Our review of the literature, as well as interpretation of work from our laboratory, favors the third possibility: 22q11DS reflects diminished expression of multiple 22q11 genes acting on common cellular processes during brain as well as heart, face, and limb development, and subsequently in the adolescent and adult brain.
Key words: 22q11 Deletion Syndrome (22q11DS), Phenotypes, Dosage, Gene expression
22q11DS: ANEUPLOIDY AND DISEASE
Deletion or duplication of large chromosomal domains, which occurs sporadically during oogenesis or spermatogenesis, most frequently results in a nonviable offspring. In human populations, however, there are two notable exceptions to this general rule: 22q11 Deletion Syndrome has an estimated frequency of 1 in 3,000–4,000 live births, and Trisomy 21, even more frequent, occurs in approximately 1 in 1,000 live births (17,53). Both of these fairly common aneuploid disorders result in a broad spectrum of phenotypes that include morphogenetic as well as behavioral and cognitive anomalies (2,14,75). Despite the relatively high incidence of these syndromes and the challenges they present for affected individuals, there is little clear understanding of how loss or gain of multiple genes at the chromosomal level—and resulting expression changes—results in disrupted development or function for limbs, faces, hearts, or brains, the primary sites of change in both Down Syndrome and 22q11DS. For 22q11DS, some insight may be gained by examining the expression patterns and levels of the relatively limited set of deleted 22q11 genes (30–50, as opposed to approximately 500 in Down Syndrome) and their relation to 22q11DS phenotypes.
There has been extensive genetic and genomic characterization of 22q11DS since the deleted genotype and syndrome phenotypes were securely correlated in the early 1990s (10,21,79). 22q11DS results from a minimum deletion of approximately 30 contiguous genes in a 1.5-MB region of chromosome 22; however, 22q11DS is more often accompanied by a larger deletion of 3 MB that includes approximately 50 genes (81). Surprisingly, no robust or consistent phenotypes distinguish the 1.5-MB from the 3-MB deletion (11,43). In fact, the hallmark of 22q11DS genotype/phenotype association studies is the lack of consistent phenotypes: some individuals can have a full set of cardiovascular, morphogenetic, behavioral, and psychiatric anomalies, while others have few, if any, obvious dysmorphologies or behavioral difficulties (16,55,73). This frustrating lack of clear genotype/phenotype correlation in 22q11DS patients suggests several hypotheses: additional genomic or environmental lesions—including mutations in remaining 22q11 alleles, modifier genes, or some sort of gestational stress—may be necessary to reach diverse phenotypic thresholds in 22q11DS. Alternatively, the core 22q11DS phenotypes may reflect consequences of diminished expression of several genes in the deleted region. While these hypotheses are not mutually exclusive, they indicate distinct ways of thinking about 22q11DS: in one case, gene dosage is not the essential variable, while in the other, dosage is key to understanding pathogenesis.
22q11DS IN MEN AND MICE I: INDIVIDUAL CANDIDATE GENES AND 22q11 PHENOTYPES
Analysis of 22q11DS was greatly facilitated by sequencing of human chromosome 22—the first human chromosome to be completely sequenced (18)—plus parallel genomic sequencing in the mouse (23,47,70), where a region of mmChr.16 is nearly completely homologous to the 1.5-MB minimally critical deleted region (Fig. 1). Based upon the identification of the full set of transcribed genes in the human 1.5-MB minimally critical deleted region (as well as the larger 3-MB deleted region), several candidate genes were proposed to explain the 22q11DS phenotypic spectrum (12,22,95). The developmental dimensions of the syndrome—particularly the morphogenetic abnormalities—led many investigators to focus on several putative DNA binding or transcription factors including the histone chaperone protein HIRA and the T-box family transcriptional regulator TBX1 [reviewed by (64)]. Initial expression studies in animals confirmed both Hira and Tbx1 as viable candidate genes (12,20,74,92). Both are expressed around mid-gestation (embryonic day 10 in a mouse) in apparent neural crest-derived tissues in the developing aortic arches, as well as in branchial arches and limbs—embryonic structures where disrupted development can generate cardiovascular, craniofacial, and digit anomalies seen in 22q11DS. In contrast, other initial candidate genes, including Ufd1L, have proven more difficult to validate, despite suggestions of a functional role in cardiovascular malformations similar to those in 22q11DS (77,90,94,95). In addition, two genes in the 3-MB typically deleted region—Pcqap, a putative transcriptional regulator, and Crkl, a modulator of Ras signaling—have been suggested as contributors to the 22q11 phenotypic spectrum, perhaps as modulators of genes in the minimal critical deleted region. These genes may indeed be 22q11DS modifiers; nevertheless, neither their deletion nor diminished expression is essential for the core 22q11DS phenotypes. Thus, we focus here on genes in the minimal critical deleted region because most evidence indicates that they play a primary role in 22q11DS.
Figure 1.
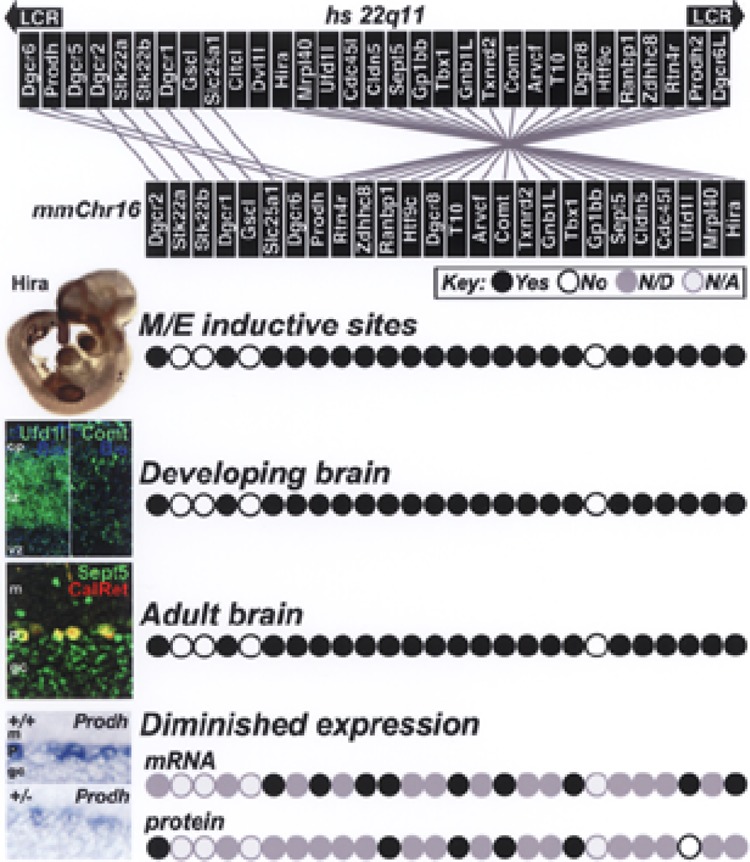
Human 22q11 deletion, and expression profiles of 22q11 genes in mice. First panel: The 1.5-MB minimal critical deleted region associated with 22q11DS. The top diagram shows human 22q11 genes in centromere (left) to telomere (right) order. Below, the homologous region of mmChr.16 shows orthologues in the mouse genome, as well as partial inversions that distinguish the murine region. The murine genome lacks three transcripts (Clctl, Dvl1l, and the noncoding Dgcr5), and also lacks the low copy repeat cassettes (LCRs) that include duplicate copies of Prodh and Dgcr6 found in the human genome. In subsequent panels, circles indicate each mmChr.16 gene, in register with the diagram in the lower part of the first panel. Second to fifth panels: Representative images of 22q11 gene expression at each stage, and summary diagrams of expression profiles of mouse 22q11 orthologues. Second panel: The subset of contiguous orthologues (22/26) expressed selectively or specifically at midgestation at all sites of nonaxial mesenchymal/epithelial (M/E) induction compromised in 22q11DS (aortic arches and heart, branchial arches, limb buds, and forebrain). In the image at left, an E10.5 embryo is shown in which an antibody against Comt has labeled limb buds, aortic arches and heart, branchial arches, and the frontonasal mass/forebrain. Third panel: The same genes expressed in the fetal, neonatal, and adolescent brain. In the image at left, expression of two 22q11 genes, Ufd1l and Comt, is detected immunohistochemically in the embryonic cerebral cortex (E16: cp, cortical plate; iz, intermediate zone; vz, ventricular zone). Fourth panel: The same genes remain expressed in the adult brain. In the image at left, Septin5 (also known as Cdcrel) is localized immunohisto-chemically in Purkinje cells of the adult cerebellum, which have been double-labeled for calretinin (CalRet: m,molecular layer; P, Purkinje cell layer; gc, granule cell layer of the cerebellum). Fifth panel: mRNA expression of at least 11 22q11 orthologues, assessed by quantitative PCR, is diminished by 50% at M/E sites as well as in the fetal, neonatal, and adolescent brain. Protein expression for a subset of five of these 11 genes is also diminished, with the exception of that for Ufd1l (see text). Neither we nor others have evaluated the remaining 15 orthologues (gray circles). The image at left shows cerebellar expression of Prodh2 in the adult wild-type and large deletion cerebellum. First to fourth panels based upon Maynard et al. (51); fifth panel after Meechan et al. (56).
A great deal of genetic analysis in mice has focused on Tbx1 as the primary causal gene for the full spectrum of 22q11DS phenotypes. There is little doubt that homozygous or heterozygous loss of Tbx1 function can lead to cardiovascular anomalies (36,44,57); indeed, mutations in several members of the Tbx gene family are known to disrupt cardiovascular, craniofacial, and limb development (68). Moreover, heterozygous Tbx1 mutants may have mild behavioral deficits, perhaps related to human behavioral pathologies that accompany 22q11DS (66). Nevertheless, it remains unclear whether diminished Tbx1 dosage without broader 22q11 deletion generates the full range of 22q11DS phenotypes, including craniofacial as well as behavioral phenotypes. Human genetic analysis of TBX1, as well as several other candidate genes, has been equivocal at best. In samples of 22q11DS patients with cardiovascular malformations, no clear examples of specific homozygous loss of TBX1 function have been identified (29). Individuals with neither the 1.5- or 3-MB deletion have been diagnosed clinically with VCFS/DiGeorge Syndrome (93)—potentially ideal to evaluate the singular role of TBX1. Five of these individuals had TBX1 point mutations as well as significant DiGeorge clinical phenotypes; however, eight additional phenotypic nondeleted individuals had no TBX1 mutations. A similar analysis of a single family in which VCFS was diagnosed in a mother and her offspring identified a TBX1 loss of function mutation; however, there is significant phenotypic variability in these three family members. Finally, in patients with cardiovascular malformations, but without 22q11 deletion, TBX1 has not emerged as a causal mutation based on analysis of polymorphic alleles (13,72,88).
Beyond these caveats for TBX1 alone as the “heart gene” in 22q11DS, several neurobiological observations suggest that 22q11 genes beyond TBX1 contribute to additional 22q11DS phenotypes. TBX1 has not been identified as a risk gene, independent of 22q11 deletion, for affective disorders (schizophrenia, bipolar disorder, major depression), unlike at least three other 22q11 genes: PRODH, proline dehydrogenase, a mitochondrial enzyme (41,45); COMT, catechol-o-methyl-transferase, a catabolic enzyme for catecholamines (19,82); and ZDHHC8, a protein of unknown function, related to yeast palmitoyl transferase (60). Finally, a recent behavioral study in mice suggests that behavioral anomalies consistent with altered cognitive function (and thus as close to modeling “schizophrenic” symptoms as may be achieved in mice) are seen only in mice carrying the full heterozygous deletion in the homologous region of mm-Chr.16; neither heterozygous nor homozygous Tbx1 mutants had these deficits (46). Thus, while TBX1 remains a viable contributor, it is unlikely that this gene alone, or any other single gene in the 1.5-MB deleted region, can explain the 22q11DS phenotypic spectrum.
22q11DS IN MEN AND MICE II: CONTIGUOUS GENES AND 22q11DS PHENOTYPES
It is also possible that 22q11DS is a contiguous gene syndrome—in which individual deleted genes each yield one aspect of the full set of phenotypes, thus reflecting their action at specific sites [e.g., Williams Syndrome, reviewed by (59)]. It seems possible that either spatially or temporally discrete expression of one 22q11 gene might be correlated with each phenotypic change seen at the apparently disparate sites affected in 22q11DS: the heart, the face, the limbs, and the brain. Expression analysis indicates that the contiguous gene hypothesis is unlikely to be a viable explanation of 22q11DS. Initial expression studies (22,48,49,84,92,95) suggested a general tendency for 22q11 genes to be expressed at multiple sites, rather than one site of phenotypic change (Fig. 1). These sites include the aortic aches, branchial arches, limb buds, frontonasal mass and embryonic brain. All these sites of 22q11DS phenotypes are also sites of neural crest mediated mesenchymal/epithelial (M/E) induction during midgestation [reviewed by (40,50)]. Our systematic evaluation of 32 mouse 22q11 orthologues confirmed a selective pattern of expression at M/E inductive sites for most 22q11 genes: 22 of 26 contiguous mouse orthologues on mmChr.16 are expressed at these sites (51) (Fig. 1) as are five other 22q11 orthologues found at disparate locations in the mouse genome, and at least two orthologues from the 3-MB region: Pcqap and Crkl. Moreover, few 22q11 genes, if any, are uniquely expressed during a particular stage of development or in a particular tissue or cell class—beyond a tendency for 22q11 genes to be expressed in neurons rather than glia in the CNS (Fig. 1). The two exceptions to this rule are T10 (a gene of unknown function) restricted to a subset of Layer V cells in the adult cerebral cortex (see below, Fig. 2), and Slc25a1 (a mitochondrial citrate transporter also known as Ctp) whose expression is limited to a subset of cells in the adult globus pallidus (see below, Fig. 2). Finally, regionally restricted phenotypes (e.g., specific anomalies in the face, heart, limb, or brain alone) have not been correlated with mutation or loss of function of specific 22q11 genes—with the possible exception of the 4th aortic arch phenotype for Tbx1 mutant mice (36,44,57). Thus, regionally restricted action of individual 22q11 genes at discrete phenotypically compromised sites is unlikely to explain the 22q11DS phenotypic spectrum.
Figure 2.
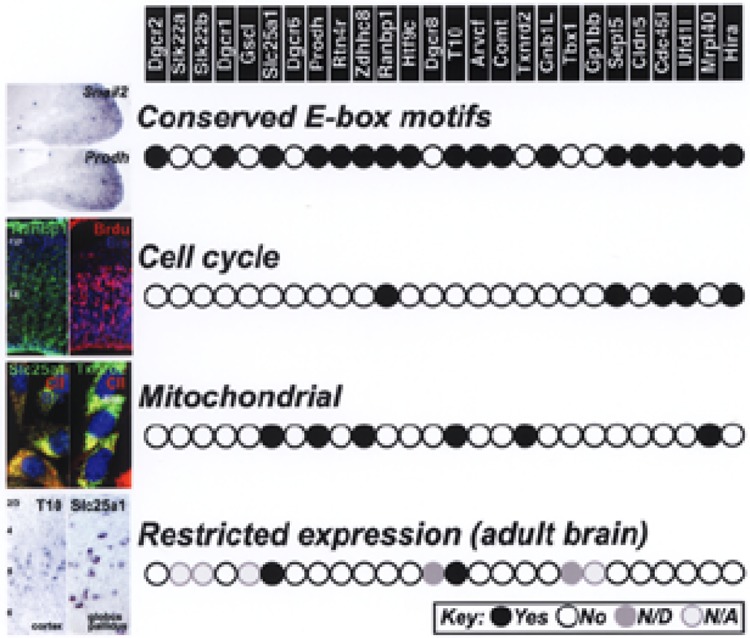
Functional subsets of 22q11 genes. First panel: Eighteen 22q11 genes and murine orthologues have highly conserved E-box binding motifs in their putative upstream regulatory regions. These E-boxes are thought to bind Snail transcription factors, whose expression patterns are coincident with those of 22q11 genes in the neural crest-associated mesenchyme found at sites of M/E induction compromised in 22q11DS. In the image at left, expression of Snail2 and Prodh2 is apparently coincident in the mesenchyme of the maxillary process of E10.5 embryos. Second panel: Five 22q11 genes are thought to be associated with cell cycle control. At least one of these genes, Ranbp1, is robustly expressed in the ventricular (v) and intermediate zone (iz) of the developing forebrain (E16)—the site of mitotically active neural precursors shown labeled after a brief pulse of the thymidine analog bromodeoxyuridine (BrdU; 2 h before sacrifice on E14.5). Ranbp1 is not seen in the cortical plate (cp), the site of postmitotic cortical neurons. Third panel: Six 22q11 genes have apparent mitochondrial localization sequences, suggesting that their expression may be limited to this organelle. The micrographs show coincident localization of two established mitochondrial genes, Slc25a1 and Txnrd2, visualized using GFP fusion proteins, with an antibody against mitochondrial core complex II protein UQCRC2 (CII) in transfected NIH 3T3 cells. Fourth panel: Two of the putative mitochondrial genes, T10 and Slc25a1 have highly specific, limited expression patterns, in layer V of the adult neocortex (far left) and the globus pallidus, respectively. First panel from Meechan et al. (56) and Ghopalikrishna et al., unpublished observations; second panel from Meechan et al., unpublished observations and Maynard et al. (51); third panel from Maynard et al. unpublished observations; fourth panel from Wu, unpublished observations.
22q11DS IN MEN AND MICE III: COINCIDENT EXPRESSION AND COOPERATIVE ACTION
Apparently, neither haploinsufficiency of a single gene nor the independent action of contiguous genes at distinct sites can explain the complex, variable phenotypic spectrum associated with 22q11DS. Two key issues need to be considered to develop a viable alternative to these attractive, but apparently untenable, hypotheses. First, 22q11DS is most likely to be a disorder of gene dosage: some subset of 22q11 genes must be unable to sustain normal development or function when they are expressed at 50% or less of their normal level at phenotypically compromised sites. Second, 22q11DS phenotypes reflect normal synergy between subsets of 22q11 genes, perhaps influenced by modifiers that are disrupted when normal expression levels are not maintained.
Several observations support the relevance of these two concepts to understanding 22q11DS. In mice, there is apparent synergy between at least two 22q11 genes: Prodh2 and Comt for behavioral phenotypes and pharmacological sensitivity to psychoactive drugs (65). In addition, Prodh mutant mice display transcriptional upregulation of Comt in the frontal cortex. Similarly, reduced expression of Dgcr6 can modulate Hira and Tbx1 expression in the aortic arches as well as lead to cardiovascular phenotypes that resemble those in 22q11DS (32), thus suggesting further interactions between 22q11 genes. Finally, our recent study of expression levels of a large subset of 22q11 genes in mice heterozygously deleted for the full set of contiguous 22q11 orthologues [the so-called “large-deletion” mouse (57)] indicates that expression levels of multiple genes are reduced by approximately 50%, both at the message and protein level, with little apparent dosage compensation at phenotypically compromised sites in the embryo as well as the maturing and adult brain (56) (Fig. 1). The only exception, thus far, is apparent translational dosage compensation for Ufd1L. This accords well with other data indicating that organismal viability requires diploid Ufd1L expression levels (38), as well as lack of evidence for phenotypes associated with Ufd1L polymorphisms independent of 22q11 deletion. These observations, as well as the continued absence of strong evidence for consistent 22q11-related phenotypes in patients with single 22q11 gene polymorphisms (25,26,76,91) rather than deletions, accords well with the apparent cooperative nature of dosage-diminished gene action in 22q11DS.
COOPERATIVE GENE FUNCTION, COMMON CELLULAR TARGETS, AND 22q11 PHENOTYPES
Determining the combinatorial contribution of at least 30 (1.5-MB deletion) and potentially 50 (3-MB deletion) genes to the broad range of 22q11DS phenotypes seems a daunting, or even impossible, challenge. Some traction may be gained by evaluating functionally related subsets of 22q11 genes and thus discerning where these genes might normally act in concert to orchestrate specific cellular, physiological, or morphogenetic processes. Such functional groups of 22q11 genes might further illuminate when and where combined diminished dosage leads to emergent phenotypes. We have used bioinformatics as well as cell biological methods to assess whether the transcription of subsets or 22q11 genes is coordinately regulated, and whether groups of genes participate in common cellular processes. Our analysis suggests at least three functional sets of 22q11 genes may have specific phenotypic consequences at distinct stages of development.
The first group of genes is unified by potential parallel transcriptional regulation (Fig. 2). There are 18 22q11 genes, as well as their murine orthologues, with E-box motifs in highly conserved upstream regulatory regions. The degree of conservation of these putative E-boxes and flanking sequences varies between 100% and 65% between mouse and human (56), a degree of conservation consistent with their identity as regulatory sites. We found that at least one of these apparent E-boxes binds Snail, consistent with potential regulation by Snail transcription factors [(56) and unpublished observations]. Moreover, each E-box-containing gene is coincidently expressed in the embryo at sites of 22q11DS phenotypic changes—specifically nonaxial neural crest-derived mesenchymal cells that participate in M/E induction and morphogenesis in the forebrain, branchial arches, heart, and limbs (40,51,56). Snail1 and Snail2 are also expressed selectively and coincidently at these sites (37,56,80). These E-box motifs may also provide regulatory binding sites for other bHLH transcription factors associated with the neural crest, including Twist and Hand1/2, which have more limited expression patterns at some M/E inductive sites. It remains to be determined whether these or other neural crest-associated transcriptional regulators are either upstream modulators or downstream targets of 22q11 genes. The significance of shared regulatory motifs and coincident expression patterns depends upon the role of Snail transcription factors, or other neural crest regulators, in controlling gene expression, epithelial-to-mesenchymal transitions, and neural crest differentiation (61,62,78). It is possible that selective expression of 22q11 genes in neural crest-derived mesenchyme reflects in part concerted regulation by Snail transcription factors. If this is the case, Snail polymorphisms (or polymorphisms in genes that regulate Snail action) might modulate 22q11DS phenotypes, providing a potential basis for some of the phenotypic variability seen in 22q11DS (83,85).
The second subset of 22q11 genes comprises five that may modulate the cell cycle (Fig. 2). These genes are the small GTPase binding protein Ranbp1 (15); the mini-chromosome maintenance complex associating Cdc45l (33); the septin Sept5/Cdcrel (54), which is homologous to a protein involved in yeast cytokinesis; Hira, a DNA/histone chaperone protein (31); and Ufd1l, which regulates ubiquitination, particularly during mitosis (3). Ufd1l in large deletion mice, however, is apparently translationally dosage compensated [see above and (56)]. Some of these genes are expressed in ventricular or subventricular regions of the developing forebrain neuroepithelium (50,51) (Fig. 1), and thus are at least in the right place to contribute to modulation of the cell cycle in mitotically active neural precursors. Moreover, expression of at least two of these genes—Ranbp1 and Hira—is significantly increased during peak neurogenesis in the embryonic brain (50). Finally, these genes may interact quantitatively with additional cell cycle regulators, thus leading to aberrant regulation that can be further modulated by mutations in cell cycle genes beyond the 22q11 minimal critical deleted region. Accordingly, it is possible that combined diminished dosage might impact cell numbers via differences in proliferation or cell death, or alter the influence of the cell cycle on differentiation and cellular diversity (8,9,52), perhaps without a significant change in cell numbers (56).
The third subset comprises six genes, known or believed to be limited to mitochondria based upon inferred amino acid mitochondrial localization motifs (Fig. 2). These include Slc25a1 (Ctp), Thior2, and Prodh2, established mitochondrial proteins (4,6,63), plus Mrpl40/Nlvcf, T10, and Zdhhc8. The presumably selective expression and activity of each in mitochondria, as well as reduced expression that occurs following 22q11 deletion (56), could lead to a combined deleterious effect on bioenergetic functions or cellular metabolism. In the embryo, such effects could alter inductive signaling, cell migration, apoptosis, or metabolism, thus compromising morphogenesis and differentiation. In the developing and mature brain, modest changes in mitochondrial function could impact synapse formation and maintenance. Indeed, expression levels of two of these genes—Prodh2 and Zdhhc8—reach a peak during the most robust phase of CNS synaptogenesis and remain highly expressed in the adult brain (56). Diminished expression in the adult brain following 22q11 deletion may enhance vulnerability to stress-related and other neurodegenerative insults, thus further increasing susceptibility for behavioral and psychiatric disease in 22q11DS (5,34). Intriguingly, this set of putative mitochondrial genes includes the only two that we have found to have highly restricted expression patterns in the adult brain: T10 in large apparent pyramidal cells in Layer V of the cerebral cortex, and CTP in the globus pallidus, the site of some of the most tonically and presumably metabolically active neurons in the brain (Fig. 2). Moreover, this subset includes two of three genes—Prodh2 and Zdhhc8 (but not Comt)—for which polymorphisms have been independently associated with schizophrenia and other affective disorders in some samples of non-22q11-deleted patient populations (35,41,45,60).
WHEN IS HALF NOT ENOUGH? DOSAGE CHANGES, CONCERTED ACTION, AND 22q11DS PHENOTYPES
The genomic lesion in 22q11DS as well as details of 22q11 gene expression and function indicate that changes in gene dosage for multiple genes (i.e., most or not all genes in the minimal critical deleted region) are the essential contributor to 22q11 phenotypes. It remains difficult, however, to assess dosage-sensitive functions of 30 or more genes. It is unlikely that 22q11DS phenotypes reflect altered expression of genes flanking the deleted region due to local chromosomal instability. Expression levels of adjacent genes neither diminish nor increase as a result of deletion of the entire region (either the “large deletion mouse,” the Df1 mouse, or the “small deletion mouse”) (39,42,56,57,69). Nevertheless, loci beyond 22q11 may modify 22q11DS phenotypes, and thus explain the high degree of phenotypic variability. This sort of modulation, however, further complicates the association of 22q11 genes with specific aspects of the phenotypic spectrum. A few candidate modifiers, many related to major signaling pathways at sites of M/E induction, have been suggested (1,7,24,86,87). At least one of these modifiers, sonic hedgehog, can modulate expression of a 22q11 gene: Tbx1; another, Fgf8, is apparently regulated by Tbx1. Additional candidates, however, must be interpreted cautiously because coincident phenotypes do not necessarily suggest genetic interaction. The essential requirement, not yet fully satisfied in many published studies, is that putative modifiers either disrupt 22q11 gene dosage or have regulatory influences on shared downstream targets.
Analysis of most mouse single mutants of 22q11 orthologues, with the exception of Tbx1 heterozygous null mice, indicates that diminished dosage of individual genes has few discernible phenotypic consequences (27,28,60,65,67,74). Thus, understanding the consequences of diminished dosage of functional subsets of 22q11 genes provides a novel opportunity to evaluate how diminished expression of multiple genes might compromise specific cell functions that underlie the 22q11DS phenotypic spectrum. Diminished expression of these genes most likely occurs on a “per cell” basis (although not necessarily by 50% in every cell) (56) across widely distributed cellular populations in affected tissues, especially in the brain. Limited expression patterns of others, like T10 or Slc25a1, indicate that critical populations of fore-brain neurons—in this instance layer 5 cortical cells and cells in the basal ganglia—might be focally compromised by diminished dosage. Thus, expression changes following 22q11 deletion might lead to variable threshold effects for specific cell classes at distinct times. Such changes may yield phenotypic change in subsets of neurons while sparing others. This may reflect differentiation or transcriptional state of some cells, as well as variably diminished expression levels of some 22q11 genes on a cell-by-cell basis.
Our observations define apparent functional subsets of 22q11 genes, and we suggest that these subsets compromise distinct phases of neural development, potentially leading to anomalous circuit differentiation and, consequently, vulnerability for behavioral and psychiatric disorders, including schizophrenia. Our preliminary observations indicate that several 22q11 genes with highly conserved E-box motifs—including Arvcf, Wdvcf, and Cldn5, all of which are expressed at M/E inductive sites during early gestation—are dramatically regulated by inductive signaling molecules including retinoic acid, sonic hedgehog, Fgfs, and Bmps. Dosage changes for this subset of 22q11 genes, perhaps modulated by Snail, Twist, or other neural crest-associated regulatory factors, might compromise signaling capacity of the mesenchyme and subsequent morphogenetic interactions at M/E sites compromised in 22q11DS (Fig. 3). Intriguingly, clinical observation suggests that limb and craniofacial anomalies are significantly more frequent in schizophrenic patients (presumably not all 22q11 deleted), suggesting possible phenotypic convergence (30,89). Subsequently, during neural development, at least two of five putative cell cycle genes are maximally expressed during mid to late gestation, coincident with peak neurogenesis and cell migration, while two of six mitochondrial genes reach maximal expression during early postnatal life, coincident with peak synaptogenesis (56). Dosage changes of these 22q11 genes may compromise critical sequential developmental processes, due to convergent modulation of nodal cellular mechanisms: cell cycle kinetics during neurogenesis and migration; mitochondrial integrity, bioenergetics, and cell survival during synaptogenesis (Fig. 3). Once again, independent evidence suggests that several behavioral and psychiatric diseases may reflect slight anomalies in neurogenesis and migration (71) or synapse addition and elimination (58). Accordingly, dosage-dependent changes at distinct times might lead to cumulative morphogenetic, neurogenic, and connectivity changes that establish forebrain circuits with increased vulnerability for schizophrenia. Thus, it may be possible to recognize when, and where, half is not enough, and begin to connect the consequences of the 22q11DS genomic lesion—diminished gene dosage—with its complex and variable phenotypic expression.
Figure 3.
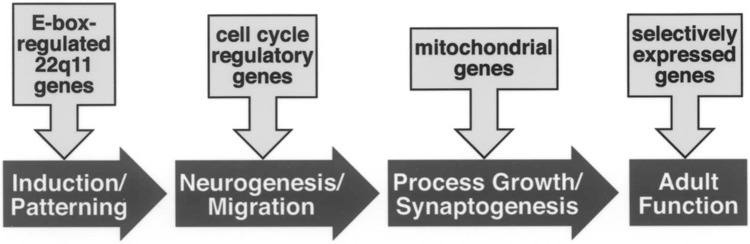
A schematic of major phases of neural development, and the potential impact of dosage changes for functionally related subsets of 22q11 genes. At each stage, diminished dosage of several related 22q11 genes might compromise essential cellular processes: M/E signaling during induction and patterning; cell cycle kinetics during neurogenesis and migration; mitochondrial function, metabolism, and cell survival during synaptogenesis. Moreover, ongoing diminished expression of specific 22q11 genes in subsets of mature neurons might further compromise circuit function in the brains of 22q11DS, enhancing vulnerability for psychotic disorders including schizophrenia.
ACKNOWLEDGMENTS
This work was supported by NICHD (HD42182) and a NARSAD independent investigator award to A.-S.L., a NARSAD young investigator award to T.M., and a Silvio M. Conte Research for Mental Disorders Award (MH64065). D.M. was a fellow of the UNC Developmental Biology Training Program (NIH HD46369).
REFERENCES
- 1. Bachiller D.; Klingensmith J.; Shneyder N.; Tran U.; Anderson R.; Rossant J.; De Robertis E. M. The role of chordin/Bmp signals in mammalian pharyngeal development and DiGeorge syndrome. Development 130(15):3567–3578; 2003. [DOI] [PubMed] [Google Scholar]
- 2. Bassett A. S.; Chow E. W. 22q11 deletion syndrome: A genetic subtype of schizophrenia. Biol. Psychiatry 46(7):882–891; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bays N. W.; Hampton R. Y. Cdc48-Ufd1-Npl4: Stuck in the middle with Ub. Curr. Biol. 12(10):R366–371; 2002. [DOI] [PubMed] [Google Scholar]
- 4. Bender H. U.; Almashanu S.; Steel G.; Hu C. A.; Lin W. W.; Willis A.; Pulver A.; Valle D. Functional consequences of PRODH missense mutations. Am. J. Hum. Genet. 76(3):409–420; 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ben-Shachar D.; Laifenfeld D. Mitochondria, synaptic plasticity, and schizophrenia. Int. Rev. Neurobiol. 59:273–296; 2004. [DOI] [PubMed] [Google Scholar]
- 6. Biterova E. I.; Turanov A. A.; Gladyshev V. N.; Barycki J. J. Crystal structures of oxidized and reduced mitochondrial thioredoxin reductase provide molecular details of the reaction mechanism. Proc. Natl. Acad. Sci. USA 102(42):15018–15023; 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brown C. B.; Wenning J. M.; Lu M. M.; Epstein D. J.; Meyers E. N.; Epstein J. A. Cre-mediated excision of Fgf8 in the Tbx1 expression domain reveals a critical role for Fgf8 in cardiovascular development in the mouse. Dev. Biol. 267(1):190–202; 2004. [DOI] [PubMed] [Google Scholar]
- 8. Calegari F.; Haubensak W.; Haffner C.; Huttner W. B. Selective lengthening of the cell cycle in the neurogenic subpopulation of neural progenitor cells during mouse brain development. J. Neurosci. 25(28):6533–6538; 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Calegari F.; Huttner W. B. An inhibition of cyclin-dependent kinases that lengthens, but does not arrest, neuroepithelial cell cycle induces premature neurogenesis. J. Cell Sci. 116(Pt. 24):4947–4955; 2003. [DOI] [PubMed] [Google Scholar]
- 10. Carey A. H.; Roach S.; Williamson R.; Dumanski J. P.; Nordenskjold M.; Collins V. P.; Rouleau G.; Blin N.; Jalbert P.; Scambler P. J. Localization of 27 DNA markers to the region of human chromosome 22q11-pter deleted in patients with the DiGeorge syndrome and duplicated in the der22 syndrome. Genomics 7(3):299–306; 1990. [DOI] [PubMed] [Google Scholar]
- 11. Carlson C.; Sirotkin H.; Pandita R.; Goldberg R.; McKie J.; Wadey R.; Patanjali S. R.; Weissman S. M.; Anyane-Yeboa K.; Warburton D.; Scambler P.; Shprintzen R.; Kucherlapati R.; Morrow B. E. Molecular definition of 22q11 deletions in 151 velocardio-facial syndrome patients. Am. J. Hum. Genet. 61(3):620–629; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chieffo C.; Garvey N.; Gong W.; Roe B.; Zhang G.; Silver L.; Emanuel B. S.; Budarf M. L. Isolation and characterization of a gene from the DiGeorge chromosomal region homologous to the mouse Tbx1 gene. Genomics 43(3):267–277; 1997. [DOI] [PubMed] [Google Scholar]
- 13. Conti E.; Grifone N.; Sarkozy A.; Tandoi C.; Marino B.; Digilio M. C.; Mingarelli R.; Pizzuti A.; Dallapiccola B. DiGeorge subtypes of nonsyndromic conotruncal defects: Evidence against a major role of TBX1 gene. Eur. J. Hum. Genet. 11(4):349–351; 2003. [DOI] [PubMed] [Google Scholar]
- 14. Demczuk S.; Aurias A. DiGeorge syndrome and related syndromes associated with 22q11.2 deletions. A review. Ann. Genet. 38(2):59–76; 1995. [PubMed] [Google Scholar]
- 15. Di Fiore B.; Ciciarello M.; Mangiacasale R.; Palena A.; Tassin A. M.; Cundari E.; Lavia P. Mammalian RanBP1 regulates centrosome cohesion during mitosis. J. Cell Sci. 116(Pt. 16):3399–3411; 2003. [DOI] [PubMed] [Google Scholar]
- 16. Digilio M. C.; Angioni A.; De Santis M.; Lombardo A.; Giannotti A.; Dallapiccola B.; Marino B. Spectrum of clinical variability in familial deletion 22q11.2: From full manifestation to extremely mild clinical anomalies. Clin. Genet. 63(4):308–313; 2003. [DOI] [PubMed] [Google Scholar]
- 17. Dolk H.; Loane M.; Garne E.; De Walle H.; Queisser-Luft A.; De Vigan C.; Addor M. C.; Gener B.; Haeusler M.; Jordan H.; Tucker D.; Stoll C.; Feijoo M.; Lillis D.; Bianchi F. Trends and geographic inequalities in the prevalence of Down syndrome in Europe, 1980–1999. Rev. Epidemiol. Sante Publique 53(Spec No. 2):2S87–95; 2005. [PubMed] [Google Scholar]
- 18. Dunham I.; Shimizu N.; Roe B. A.; Chissoe S.; Hunt A. R.; Collins J. E.; Bruskiewich R.; Beare D. M.; Clamp M.; Smink L. J.; Ainscough R.; Almeida J. P.; Babbage A.; Bagguley C.; Bailey J.; Barlow K.; Bates K. N.; Beasley O.; Bird C. P.; Blakey S.; Bridgeman A. M.; Buck D.; Burgess J.; Burrill W. D.; O’Brien K. P.; et al. The DNA sequence of human chromosome 22. Nature 402(6761):489–495; 1999. [DOI] [PubMed] [Google Scholar]
- 19. Egan M. F.; Goldberg T. E.; Kolachana B. S.; Callicott J. H.; Mazzanti C. M.; Straub R. E.; Goldman D.; Weinberger D. R. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc. Natl. Acad. Sci. USA 98(12):6917–6922; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Farrell M. J.; Stadt H.; Wallis K. T.; Scambler P.; Hixon R. L.; Wolfe R.; Leatherbury L.; Kirby M. L. HIRA, a DiGeorge syndrome candidate gene, is required for cardiac outflow tract septation. Circ. Res. 84(2):127–135; 1999. [DOI] [PubMed] [Google Scholar]
- 21. Fibison W. J.; Budarf M.; McDermid H.; Greenberg F.; Emanuel B. S. Molecular studies of DiGeorge syndrome. Am. J. Hum. Genet. 46(5):888–895; 1990. [PMC free article] [PubMed] [Google Scholar]
- 22. Funke B.; Puech A.; Saint-Jore B.; Pandita R.; Skoultchi A.; Morrow B. Isolation and characterization of a human gene containing a nuclear localization signal from the critical region for velo-cardio-facial syndrome on 22q11. Genomics 53(2):146–154; 1998. [DOI] [PubMed] [Google Scholar]
- 23. Galili N.; Baldwin H. S.; Lund J.; Reeves R.; Gong W.; Wang Z.; Roe B. A.; Emanuel B. S.; Nayak S.; Mickanin C.; Budarf M. L.; Buck C. A. A region of mouse chromosome 16 is syntenic to the DiGeorge, velocardiofacial syndrome minimal critical region. Genome Res. 7(1):17–26; 1997. [DOI] [PubMed] [Google Scholar]
- 24. Garg V.; Yamagishi C.; Hu T.; Kathiriya I. S.; Yamagishi H.; Srivastava D. Tbx1, a DiGeorge syndrome candidate gene, is regulated by sonic hedgehog during pharyngeal arch development. Dev. Biol. 235(1):62–73; 2001. [DOI] [PubMed] [Google Scholar]
- 25. Glaser B.; Moskvina V.; Kirov G.; Murphy K. C.; Williams H.; Williams N.; Owen M. J.; O’Donovan M. C. Analysis of ProDH, COMT and ZDHHC8 risk variants does not support individual or interactive effects on schizophrenia susceptibility. Schizophr. Res. 87(1–3):21–27; 2006. [DOI] [PubMed] [Google Scholar]
- 26. Glaser B.; Schumacher J.; Williams H. J.; Jamra R. A.; Ianakiev N.; Milev R.; Ohlraun S.; Schulze T. G.; Czerski P. M.; Hauser J.; Jonsson E. G.; Sedvall G. C.; Klopp N.; Illig T.; Becker T.; Propping P.; Williams N. M.; Cichon S.; Kirov G.; Rietschel M.; Murphy K. C.; O’Donovan M. C.; Nothen M. M.; Owen M. J. No association between the putative functional ZDHHC8 single nucleotide polymorphism rs175174 and schizophrenia in large European samples. Biol. Psychiatry 58(1):78–80; 2005. [DOI] [PubMed] [Google Scholar]
- 27. Gogos J. A.; Morgan M.; Luine V.; Santha M.; Ogawa S.; Pfaff D.; Karayiorgou M. Catechol-O-methyltransferase-deficient mice exhibit sexually dimorphic changes in catecholamine levels and behavior. Proc. Natl. Acad. Sci. USA 95(17):9991–9996; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gogos J. A.; Santha M.; Takacs Z.; Beck K. D.; Luine V.; Lucas L. R.; Nadler J. V.; Karayiorgou M. The gene encoding proline dehydrogenase modulates sensorimotor gating in mice. Nat. Genet. 21(4):434–439; 1999. [DOI] [PubMed] [Google Scholar]
- 29. Gong W.; Gottlieb S.; Collins J.; Blescia A.; Dietz H.; Goldmuntz E.; McDonald-McGinn D. M.; Zackai E. H.; Emanuel B. S.; Driscoll D. A.; Budarf M. L. Mutation analysis of TBX1 in non-deleted patients with features of DGS/VCFS or isolated cardiovascular defects. J. Med. Genet. 38(12):E45; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guy J. D.; Majorski L. V.; Wallace C. J.; Guy M. P. The incidence of minor physical anomalies in adult male schizophrenics. Schizophr. Bull. 9(4):571–582; 1983. [DOI] [PubMed] [Google Scholar]
- 31. Hall C.; Nelson D. M.; Ye X.; Baker K.; DeCaprio J. A.; Seeholzer S.; Lipinski M.; Adams P. D. HIRA, the human homologue of yeast Hir1p and Hir2p, is a novel cyclin-cdk2 substrate whose expression blocks S-phase progression. Mol. Cell. Biol. 21(5):1854–1865; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hierck B. P.; Molin D. G.; Boot M. J.; Poelmann R. E.; Gittenbergerde Groot A. C. A chicken model for DGCR6 as a modifier gene in the DiGeorge critical region. Pediatr. Res. 56(3):440–448; 2004. [DOI] [PubMed] [Google Scholar]
- 33. Hopwood B.; Dalton S. Cdc45p assembles into a complex with Cdc46p/Mcm5p, is required for minichromosome maintenance, and is essential for chromosomal DNA replication. Proc. Natl. Acad. Sci. USA 93(22):12309–12314; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Iwamoto K.; Bundo M.; Kato T. Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum. Mol. Genet. 14(2):241–253; 2005. [DOI] [PubMed] [Google Scholar]
- 35. Jacquet H.; Raux G.; Thibaut F.; Hecketsweiler B.; Houy E.; Demilly C.; Haouzir S.; Allio G.; Fouldrin G.; Drouin V.; Bou J.; Petit M.; Campion D.; Frebourg T. PRODH mutations and hyperprolinemia in a subset of schizophrenic patients. Hum. Mol. Genet. 11(19):2243–2249; 2002. [DOI] [PubMed] [Google Scholar]
- 36. Jerome L. A.; Papaioannou V. E. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat. Genet. 27(3):286–291; 2001. [DOI] [PubMed] [Google Scholar]
- 37. Jiang R.; Lan Y.; Norton C. R.; Sundberg J. P.; Gridley T. The Slug gene is not essential for mesoderm or neural crest development in mice. Dev. Biol. 198(2):277–285; 1998. [PubMed] [Google Scholar]
- 38. Johnson E. S.; Ma P. C.; Ota I. M.; Varshavsky A. A proteolytic pathway that recognizes ubiquitin as a degradation signal. J. Biol. Chem. 270(29):17442–17456; 1995. [DOI] [PubMed] [Google Scholar]
- 39. Kimber W. L.; Hsieh P.; Hirotsune S.; Yuva-Paylor L.; Sutherland H. F.; Chen A.; Ruiz-Lozano P.; Hoogstraten-Miller S. L.; Chien K. R.; Paylor R.; Scambler P. J.; Wynshaw-Boris A. Deletion of 150 kb in the minimal DiGeorge/velocardiofacial syndrome critical region in mouse. Hum. Mol. Genet. 8(12):2229–2237; 1999. [DOI] [PubMed] [Google Scholar]
- 40. LaMantia A. S. Forebrain induction, retinoic acid, and vulnerability to schizophrenia: Insights from molecular and genetic analysis in developing mice. Biol. Psychiatry 46(1):19–30; 1999. [DOI] [PubMed] [Google Scholar]
- 41. Li T.; Ma X.; Sham P. C.; Sun X.; Hu X.; Wang Q.; Meng H.; Deng W.; Liu X.; Murray R. M.; Collier D. A. Evidence for association between novel polymorphisms in the PRODH gene and schizophrenia in a Chinese population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 129(1):13–15; 2004. [DOI] [PubMed] [Google Scholar]
- 42. Lindsay E. A.; Botta A.; Jurecic V.; Carattini-Rivera S.; Cheah Y. C.; Rosenblatt H. M.; Bradley A.; Baldini A. Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature 401(6751):379–383; 1999. [DOI] [PubMed] [Google Scholar]
- 43. Lindsay E. A.; Goldberg R.; Jurecic V.; Morrow B.; Carlson C.; Kucherlapati R. S.; Shprintzen R. J.; Baldini A. Velo-cardio-facial syndrome: frequency and extent of 22q11 deletions. Am. J. Med. Genet. 57(3):514–522; 1995. [DOI] [PubMed] [Google Scholar]
- 44. Lindsay E. A.; Vitelli F.; Su H.; Morishima M.; Huynh T.; Pramparo T.; Jurecic V.; Ogunrinu G.; Sutherland H. F.; Scambler P. J.; Bradley A.; Baldini A. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 410(6824):97–101; 2001. [DOI] [PubMed] [Google Scholar]
- 45. Liu H.; Heath S. C.; Sobin C.; Roos J. L.; Galke B. L.; Blundell M. L.; Lenane M.; Robertson B.; Wijsman E. M.; Rapoport J. L.; Gogos J. A.; Karayiorgou M. Genetic variation at the 22q11 PRODH2/DGCR6 locus presents an unusual pattern and increases susceptibility to schizophrenia. Proc. Natl. Acad. Sci. USA 99(6):3717–3722; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Long J. M.; Laporte P.; Merscher S.; Funke B.; Saint-Jore B.; Puech A.; Kucherlapati R.; Morrow B. E.; Skoultchi A. I.; Wynshaw-Boris A. Behavior of mice with mutations in the conserved region deleted in velocardiofacial/DiGeorge syndrome. Neurogenetics 7(4):247–257; 2006. [DOI] [PubMed] [Google Scholar]
- 47. Lund J.; Chen F.; Hua A.; Roe B.; Budarf M.; Emanuel B. S.; Reeves R. H. Comparative sequence analysis of 634 kb of the mouse chromosome 16 region of conserved synteny with the human velocardiofacial syndrome region on chromosome 22q11.2. Genomics 63(3):374–383; 2000. [DOI] [PubMed] [Google Scholar]
- 48. Maldonado-Saldivia J.; Funke B.; Pandita R. K.; Schuler T.; Morrow B. E.; Schorle H. Expression of Cdcrel-1 (Pnutl1), a gene frequently deleted in velo-cardio-facial syndrome/DiGeorge syndrome. Mech. Dev. 96(1):121–124; 2000. [DOI] [PubMed] [Google Scholar]
- 49. Maynard T. M.; Haskell G. T.; Bhasin N.; Lee J. M.; Gassman A. A.; Lieberman J. A.; LaMantia A. S. RanBP1, a velocardiofacial/DiGeorge syndrome candidate gene, is expressed at sites of mesenchymal/epithelial induction. Mech. Dev. 111(1–2):177–180; 2002. [DOI] [PubMed] [Google Scholar]
- 50. Maynard T. M.; Haskell G. T.; Lieberman J. A.; LaMantia A. S. 22q11 DS: Genomic mechanisms and gene function in DiGeorge/velocardiofacial syndrome. Int. J. Dev. Neurosci. 20(3–5):407–419; 2002. [DOI] [PubMed] [Google Scholar]
- 51. Maynard T. M.; Haskell G. T.; Peters A. Z.; Sikich L.; Lieberman J. A.; LaMantia A. S. A comprehensive analysis of 22q11 gene expression in the developing and adult brain. Proc. Natl. Acad. Sci. USA 100(24):14433–14438; 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McConnell S. K.; Kaznowski C. E. Cell cycle dependence of laminar determination in developing neocortex. Science 254(5029):282–285; 1991. [DOI] [PubMed] [Google Scholar]
- 53. McDermid H. E.; Morrow B. E. Genomic disorders on 22q11. Am. J. Hum. Genet. 70(5):1077–1088; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McKie J. M.; Sutherland H. F.; Harvey E.; Kim U. J.; Scambler P. J. A human gene similar to Drosophila melanogaster peanut maps to the DiGeorge syndrome region of 22q11. Hum. Genet. 101(1):6–12; 1997. [DOI] [PubMed] [Google Scholar]
- 55. McLean S. D.; Saal H. M.; Spinner N. B.; Emanuel B. S.; Driscoll D. A. Velo-cardio-facial syndrome. Intrafamilial variability of the phenotype. Am. J. Dis. Child. 147(11):1212–1216; 1993. [DOI] [PubMed] [Google Scholar]
- 56. Meechan D. W.; Maynard T. M.; Wu Y.; Gopalakrishna D.; Lieberman J. A.; LaMantia A.-S. Gene dosage in the developing and adult brain in a mouse model of 22q11 deletion syndrome. Mol. Cell. Neurosci. 33(4):412–428; 2006. [DOI] [PubMed] [Google Scholar]
- 57. Merscher S.; Funke B.; Epstein J. A.; Heyer J.; Puech A.; Lu M. M.; Xavier R. J.; Demay M. B.; Russell R. G.; Factor S.; Tokooya K.; Jore B. S.; Lopez M.; Pandita R. K.; Lia M.; Carrion D.; Xu H.; Schorle H.; Kobler J. B.; Scambler P.; Wynshaw-Boris A.; Skoultchi A. I.; Morrow B. E.; Kucherlaspati R. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 104(4):619–629; 2001. [DOI] [PubMed] [Google Scholar]
- 58. Mirnics K.; Middleton F. A.; Lewis D. A.; Levitt P. Analysis of complex brain disorders with gene expression microarrays: Schizophrenia as a disease of the synapse. Trends Neurosci. 24(8):479–486; 2001. [DOI] [PubMed] [Google Scholar]
- 59. Morris C. A.; Mervis C. B. Williams syndrome and related disorders. Annu. Rev. Genomics Hum. Genet. 1:461–484; 2000. [DOI] [PubMed] [Google Scholar]
- 60. Mukai J.; Liu H.; Burt R. A.; Swor D. E.; Lai W. S.; Karayiorgou M.; Gogos J. A. Evidence that the gene encoding ZDHHC8 contributes to the risk of schizophrenia. Nat. Genet. 36(7):725–731; 2004. [DOI] [PubMed] [Google Scholar]
- 61. Murray S. A.; Gridley T. Snail family genes are required for left-right asymmetry determination, but not neural crest formation, in mice. Proc. Natl. Acad. Sci. USA 103(27):10300–10304; 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nieto M. A.; Sargent M. G.; Wilkinson D. G.; Cooke J. Control of cell behavior during vertebrate development by Slug, a zinc finger gene. Science 264(5160):835–839; 1994. [DOI] [PubMed] [Google Scholar]
- 63. Palmieri F. The mitochondrial transporter family (SLC25): Physiological and pathological implications. Pflugers Arch. 447(5):689–709; 2004. [DOI] [PubMed] [Google Scholar]
- 64. Papaioannou V. E.; Silver L. M. The T-box gene family. Bioessays 20(1):9–19; 1998. [DOI] [PubMed] [Google Scholar]
- 65. Paterlini M.; Zakharenko S. S.; Lai W. S.; Qin J.; Zhang H.; Mukai J.; Westphal K. G.; Olivier B.; Sulzer D.; Pavlidis P.; Siegelbaum S. A.; Karayiorgou M.; Gogos J. A. Transcriptional and behavioral interaction between 22q11.2 orthologs modulates schizophrenia-related phenotypes in mice. Nat. Neurosci. 8(11):1586–1594; 2005. [DOI] [PubMed] [Google Scholar]
- 66. Paylor R.; Glaser B.; Mupo A.; Ataliotis P.; Spencer C.; Sobotka A.; Sparks C.; Choi C. H.; Oghalai J.; Curran S.; Murphy K. C.; Monks S.; Williams N.; O’Donovan M. C.; Owen M. J.; Scambler P. J.; Lindsay E. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: Implications for 22q11 deletion syndrome. Proc. Natl. Acad. Sci. USA 103(20):7729–7734; 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Peng X. R.; Jia Z.; Zhang Y.; Ware J.; Trimble W. S. The septin CDCrel-1 is dispensable for normal development and neurotransmitter release. Mol. Cell. Biol. 22(1):378–387; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Plageman T. F. Jr.; Yutzey K. E. T-box genes and heart development: Putting the “T” in heart. Dev. Dyn. 232(1):11–20; 2005. [DOI] [PubMed] [Google Scholar]
- 69. Prescott K.; Ivins S.; Hubank M.; Lindsay E.; Baldini A.; Scambler P. Microarray analysis of the Df1 mouse model of the 22q11 deletion syndrome. Hum. Genet. 116(6):486–496; 2005. [DOI] [PubMed] [Google Scholar]
- 70. Puech A.; Saint-Jore B.; Funke B.; Gilbert D. J.; Sirotkin H.; Copeland N. G.; Jenkins N. A.; Kucherlapati R.; Morrow B.; Skoultchi A. I. Comparative mapping of the human 22q11 chromosomal region and the orthologous region in mice reveals complex changes in gene organization. Proc. Natl. Acad. Sci. USA 94(26):14608–14613; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rakic P. A century of progress in corticoneurogenesis: From silver impregnation to genetic engineering. Cereb. Cortex 16(Suppl. 1):i3–17; 2006. [DOI] [PubMed] [Google Scholar]
- 72. Rauch A.; Devriendt K.; Koch A.; Rauch R.; Gewillig M.; Kraus C.; Weyand M.; Singer H.; Reis A.; Hofbeck M. Assessment of association between variants and haplotypes of the remaining TBX1 gene and manifestations of congenital heart defects in 22q11.2 deletion patients. J. Med. Genet. 41(4):e40; 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ravnan J. B.; Chen E.; Golabi M.; Lebo R. V. Chromosome 22q11.2 microdeletions in velocardiofacial syndrome patients with widely variable manifestations. Am. J. Med. Genet. 66(3):250–256; 1996. [DOI] [PubMed] [Google Scholar]
- 74. Roberts C.; Sutherland H. F.; Farmer H.; Kimber W.; Halford S.; Carey A.; Brickman J. M.; Wynshaw-Boris A.; Scambler P. J. Targeted mutagenesis of the Hira gene results in gastrulation defects and patterning abnormalities of mesoendodermal derivatives prior to early embryonic lethality. Mol. Cell. Biol. 22(7):2318–2328; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Roubertoux P. L.; Kerdelhue B. Trisomy 21: From chromosomes to mental retardation. Behav. Genet. 36(3):346–354; 2006. [DOI] [PubMed] [Google Scholar]
- 76. Saito S.; Ikeda M.; Iwata N.; Suzuki T.; Kitajima T.; Yamanouchi Y.; Kinoshita Y.; Takahashi N.; Inada T.; Ozaki N. No association was found between a functional SNP in ZDHHC8 and schizophrenia in a Japanese case-control population. Neurosci. Lett. 374(1):21–24; 2005. [DOI] [PubMed] [Google Scholar]
- 77. Saitta S. C.; McGrath J. M.; Mensch H.; Shaikh T. H.; Zackai E. H.; Emanuel B. S. A 22q11.2 deletion that excludes UFD1L and CDC45L in a patient with conotruncal and craniofacial defects. Am. J. Hum. Genet. 65(2):562–566; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sakai D.; Suzuki T.; Osumi N.; Wakamatsu Y. Cooperative action of Sox9, Snail2 and PKA signaling in early neural crest development. Development 133(7):1323–1333; 2006. [DOI] [PubMed] [Google Scholar]
- 79. Scambler P. J.; Carey A. H.; Wyse R. K.; Roach S.; Dumanski J. P.; Nordenskjold M.; Williamson R. Microdeletions within 22q11 associated with sporadic and familial DiGeorge syndrome. Genomics 10(1):201–206; 1991. [DOI] [PubMed] [Google Scholar]
- 80. Sefton M.; Sanchez S.; Nieto M. A. Conserved and divergent roles for members of the Snail family of transcription factors in the chick and mouse embryo. Development 125(16):3111–3121; 1998. [DOI] [PubMed] [Google Scholar]
- 81. Shaikh T. H.; Kurahashi H.; Emanuel B. S. Evolutionarily conserved low copy repeats (LCRs) in 22q11 mediate deletions, duplications, translocations, and genomic instability: An update and literature review. Genet. Med. 3(1):6–13; 2001. [DOI] [PubMed] [Google Scholar]
- 82. Shifman S.; Bronstein M.; Sternfeld M.; Pisante-Shalom A.; Lev-Lehman E.; Weizman A.; Reznik I.; Spivak B.; Grisaru N.; Karp L.; Schiffer R.; Kotler M.; Strous R. D.; Swartz-Vanetik M.; Knobler H. Y.; Shinar E.; Beckmann J. S.; Yakir B.; Risch N.; Zak N. B.; Darvasi A. A highly significant association between a COMT haplotype and schizophrenia. Am. J. Hum. Genet. 71(6):1296–1302; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Stegmann K.; Boecker J.; Kosan C.; Ermert A.; Kunz J.; Koch M. C. Human transcription factor SLUG: Mutation analysis in patients with neural tube defects and identification of a missense mutation (D119E) in the Slug subfamily-defining region. Mutat. Res. 406(2–4):63–69; 1999. [DOI] [PubMed] [Google Scholar]
- 84. Taylor C.; Wadey R.; O’Donnell H.; Roberts C.; Mattei M. G.; Kimber W. L.; Wynshaw-Boris A.; Scambler P. J. Cloning and mapping of murine Dgcr2 and its homology to the Sez-12 seizure-related protein. Mamm. Genome 8(5):371–375; 1997. [DOI] [PubMed] [Google Scholar]
- 85. Twigg S. R.; Wilkie A. O. Characterisation of the human snail (SNAI1) gene and exclusion as a major disease gene in craniosynostosis. Hum. Genet. 105(4):320–326; 1999. [DOI] [PubMed] [Google Scholar]
- 86. Vermot J.; Niederreither K.; Garnier J. M.; Chambon P.; Dolle P. Decreased embryonic retinoic acid synthesis results in a DiGeorge syndrome phenotype in newborn mice. Proc. Natl. Acad. Sci. USA 100(4):1763–1768; 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Vitelli F.; Taddei I.; Morishima M.; Meyers E. N.; Lindsay E. A.; Baldini A. A genetic link between Tbx1 and fibroblast growth factor signaling. Development 129(19):4605–4611; 2002. [DOI] [PubMed] [Google Scholar]
- 88. Voelckel M. A.; Girardot L.; Giusiano B.; Levy N.; Philip N. Allelic variations at the haploid TBX1 locus do not influence the cardiac phenotype in cases of 22q11 microdeletion. Ann. Genet. 47(3):235–240; 2004. [DOI] [PubMed] [Google Scholar]
- 89. Waddington J. L.; Lane A.; Larkin C.; O’Callaghan E. The neurodevelopmental basis of schizophrenia: clinical clues from cerebro-craniofacial dysmorphogenesis, and the roots of a lifetime trajectory of disease. Biol. Psychiatry 46(1):31–39; 1999. [DOI] [PubMed] [Google Scholar]
- 90. Wadey R.; McKie J.; Papapetrou C.; Sutherland H.; Lohman F.; Osinga J.; Frohn I.; Hofstra R.; Meijers C.; Amati F.; Conti E.; Pizzuti A.; Dallapiccola B.; Novelli G.; Scambler P. Mutations of UFD1L are not responsible for the majority of cases of DiGeorge Syndrome/velocardiofacial syndrome without deletions within chromosome 22q11. Am. J. Hum. Genet. 65(1):247–249; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Williams H. J.; Glaser B.; Williams N. M.; Norton N.; Zammit S.; MacGregor S.; Kirov G. K.; Owen M. J.; O’Donovan M. C. No association between schizophrenia and polymorphisms in COMT in two large samples. Am. J. Psychiatry 162(9):1736–1738; 2005. [DOI] [PubMed] [Google Scholar]
- 92. Wilming L. G.; Snoeren C. A.; van Rijswijk A.; Grosveld F.; Meijers C. The murine homologue of HIRA, a DiGeorge syndrome candidate gene, is expressed in embryonic structures affected in human CATCH22 patients. Hum. Mol. Genet. 6(2):247–258; 1997. [DOI] [PubMed] [Google Scholar]
- 93. Yagi H.; Furutani Y.; Hamada H.; Sasaki T.; Asakawa S.; Minoshima S.; Ichida F.; Joo K.; Kimura M.; Imamura S.; Kamatani N.; Momma K.; Takao A.; Nakazawa M.; Shimizu N.; Matsuoka R. Role of TBX1 in human del22q11.2 syndrome. Lancet 362(9393):1366–1373; 2003. [DOI] [PubMed] [Google Scholar]
- 94. Yamagishi C.; Hierck B. P.; Gittenberger-De Groot A. C.; Yamagishi H.; Srivastava D. Functional attenuation of UFD1l, a 22q11.2 deletion syndrome candidate gene, leads to cardiac outflow septation defects in chicken embryos. Pediatr. Res. 53(4):546–553; 2003. [DOI] [PubMed] [Google Scholar]
- 95. Yamagishi H.; Garg V.; Matsuoka R.; Thomas T.; Srivastava D. A molecular pathway revealing a genetic basis for human cardiac and craniofacial defects. Science 283(5405):1158–1161; 1999. [DOI] [PubMed] [Google Scholar]