

Abstract
Disruption of the genes encoding for the transcription coactivators, peroxisome proliferator-activated receptor (PPAR)-interacting protein (PRIP/ASC-2/RAP250/ TRBP/NRC) and PPAR-binding protein (PBP/TRAP220/ DRIP205/MED1), results in embryonic lethality by affecting placental and multiorgan development. Targeted deletion of coactivator PBP gene in liver parenchymal cells (PBPLiv−/−) results in the near abrogation of the induction of PPARα and CAR (constitutive androstane receptor)-regulated genes in liver. Here, we show that targeted deletion of coactivator PRIP gene in liver (PRIPLiv−/−) does not affect the induction of PPARα-regulated pleiotropic responses, including hepatomegaly, hepatic peroxisome proliferation, and induction of mRNAs of genes involved in fatty acid oxidation system, indicating that PRIP is not essential for PPARα-mediated transcriptional activity. We also provide additional data to show that liver-specific deletion of PRIP gene does not interfere with the induction of genes regulated by nuclear receptor CAR. Furthermore, disruption of PRIP gene in liver did not alter zoxazolamine-induced paralysis, and acetaminophen-induced hepatotoxicity. Studies with adenovirally driven EGFP-CAR expression in liver demonstrated that, unlike PBP, the absence of PRIP does not prevent phenobarbital-mediated nuclear translocation/retention of the receptor CAR in liver in vivo and cultured hepatocytes in vitro. These results show that PRIP deficiency in liver does not interfere with the function of nuclear receptors PPARα and CAR. The dependence of PPARα- and CAR-regulated gene transcription on coactivator PBP but not on PRIP attests to the existence of coactivator selectivity in nuclear receptor function.
Key words: Nuclear receptor coactivators, PRIP, PBP/TRAP220/MED1, CAR, PPARα
INTRODUCTION
Nuclear receptor coactivators enhance the transcriptional activation of nuclear receptor-regulated genes and thus function as positive regulators of transcription (2,8,11,32,35,46,48). Over 100 nuclear receptor cofactors (coactivators, coactivator-associated proteins, corepressors, and others) are known to be involved in nuclear receptor-directed transcription (32,35,46), but information on the selectivity and specificity of cofactor requirement for a given nuclear receptor in cell and gene-specific transcription is rudimentary at best (35,62). Nuclear receptors, such as constitutive androstane receptor (CAR), pregnane X receptor (PXR), steroid and xenobiotic receptor (SXR), estrogen receptor α and β, and glucocorticoid receptor belong to the type I nuclear receptor category because they are localized in the cytoplasm and require ligand activation to facilitate their trans-location into the nuclear compartment (12,21,22,51). These ligand-activated type I nuclear receptors form homodimers or heterodimerize with retinoid X receptor to bind to receptor-specific sites (cognate-responsive elements) in the promoter of target genes (8,35). Nuclear receptors belonging to the type II category, such as thyroid hormone receptor (TR)-α and TRβ, and members of peroxisome proliferator-activated receptor (PPAR) subfamily, reside in the nucleus in the absence of ligand as heterodimers with retinoid X receptor (24). These heterodimerized receptors bind to DNA and are maintained in a repressed state by nuclear receptor corepressors in the absence of ligand activation (32,48). Following ligand activation, both type I and type II nuclear receptors undergo conformational changes to optimize interaction with LXXLL (L, leucine; X, any amino acid) nuclear receptor box motif(s) of transcription coactivators (8,35,48). Ligand activation of a nuclear receptor initiates an orchestrated recruitment of several transcriptional coactivators and coactivator-associated proteins that form multisubunit protein complexes to facilitate nucleosome remodeling and nuclear receptor linking to the basal transcription machinery (8,11,44,46,48,49).
Coactivators, such as p160/SRC-1 family members (SRC-1/NCoA1/p160; NCoA2/TIF2/GRIP1/SRC-2; and NCoA3/pCIP/ACTR/SRC-3), in cooperation with CBP/p300 and pCAF, form a complex with his-tone acetyltransferase activity (HAT complex) to remodel chromatin (7–9,19,35,39,42). Additional groups of coactivators and coactivator-associated proteins such as CARM1 and PIMT are then recruited sequentially, combinatorially, or by some other mechanism (3,35,37,52,70). Several of these proteins belong to the TRAP/DRIP/ARC/MED complex (44,48,49,63), with coactivator PBP/TRAP220/DRIP205/MED1 serving as core protein for this complex (44,63,68). Coactivator PRIP (also known as ASC-2/RAP250/ TRBP/NRC) forms another large steady-state protein complex [ASC-2 complex (ASCOM)] with retinoblastoma-binding protein RBQ-3, α/β-tubulins, and trithorax group of proteins ALR-1, ALR-2, HALR, and ASH (1,9,25,30,33,67). PRIP-interacting protein PIMT binds to PRIP, CBP/p300, and some members of TRAP/MED complex to function as a linker between key proteins of HAT, TRAP, and ASCOM complexes during transcription (9,37,70).
In order to understand the in vivo biological functions of these coactivators, molecular genetic approaches are being utilized to systematically explore their role in development, metabolism, and regulation of nuclear receptor-specific gene transcription (43,46, 54,55,59,60,62). Targeted gene knockout models reveal that some of the coactivators and coactivator-associated proteins are indispensable for embryonic development while others are considered dispensable (46). Deletion of coactivator PBP, PRIP, CBP, and p300 genes is embryonic lethal, implying that these coactivators are widely involved in transcription, possibly affecting the function of many transcription factors (1,15,28,34,66,69). Conditional deletion of PBP gene (PPARBP) in liver parenchymal cells (PBPLiv−/−) results in the abrogation of the effects of PPARα ligands, indicating that PBP is essential for PPARα signaling (17). Deletion of PBP gene in liver also abolishes hypertrophic and hyperplastic influences in liver mediated by CAR ligands, phenobarbital and 1,4-bis-2[-(3,5-dichloropyridyloxy)]benzene (TCPOBOP), and of acetaminophen-induced hepatotoxicity (16). CAR ligands such as phenobarbital and TCPOBOP facilitate the translocation of CAR into the hepatocyte nucleus, and this translocation fails to occur in PBP hepatocytes, accounting for the abrogation of CAR-mediated gene expression in liver (10,12,16,21). Here we report the generation of PRIP liver conditional null mice (PRIPLiv−/−) and present evidence to demonstrate that coactivator PRIP is not required for the functioning of nuclear receptors PPARα and CAR in liver.
MATERIALS AND METHODS
Generation of PRIP Conditional Null Mutation in Liver (PRIPLiv−/−)
Heterozygous LoxP-PRIP mice (41,66) were bred with albumin-Cre (AlbCre) transgenic mice (61) to delete the DNA fragment between LoxP1 and LoxP2 in the liver (66). The heterozygous mice with the expected deletion were interbred to generate homozygous mutants lacking PRIP in liver cells (PRIPLiv−/−). Southern blot analysis of genomic DNA isolated from PRIP+/+ and PRIPLiv−/− livers was performed to confirm deletion of targeted allele in PRIPLiv−/− livers. Genomic DNA was digested with SpeI and hybridized with a probe (probe 1) spanning part of intron 5 and exon 6 of the PRIP gene. To further confirm homozygosity of PRIP gene deletion, genomic DNA was hybridized with a probe (probe 2) designed within exon 7 of the PRIP gene. Generation of PBPLiv−/− has been described elsewhere (16–18). Mice were housed in a pathogen-free animal facility under standard 12-h light/12-h dark cycle and maintained on standard rodent chow and water ad libitum. All animal procedures used in this study were reviewed and preapproved by the Institutional Review Boards for Animal Research of the Northwestern University.
Treatment With PPARα and CAR Agonists
For assessing the role of PRIP in PPARα ligand-induced changes in liver, groups of PRIPLiv−/− mice (n = 5−8) aged 5−6 weeks were fed powdered diet containing PPARα-ligand, Wy-14,643 (0.125% w/w), for 2 weeks (47). Groups of mice were treated with CAR ligand, phenobarbital (PB; 100 mg/kg body weight), or TCPOBOP (3 mg/kg body weight) by IP injection daily for 3 days (5,16). A single IP injection of acetaminophen (APAP) (250 mg/kg body weight) was given to mice pretreated with corn oil, PB, or TCPOBOP as described above, 24 h before sacrifice (16). To assess cell proliferation in liver, mice treated with TCPOBOP or Wy-14,643 were given bromode-oxyuridine (BrdUrd) (1.0 mg/ml) in drinking water for 4 days and analyzed immunohistochemically for liver BrdUrd nuclear-labeling indices.
Zoxazolamine Paralysis Test
Mice pretreated for 3 days with either corn oil or PB, as described above, were given a single IP injection of zoxazolamine (300 mg/kg body weight, Sigma), 24 h after the last dose of PB. Mice were placed on their backs and paralysis time was monitored (the time required for the animal to regain sufficient consciousness to right itself repeatedly) as described (16,53,56,65).
Adenovirally Driven Expression of EGFP-CAR
Mouse CAR cDNA was cloned into pEGFP-C1 vector (Clontech) and EGFP-CAR fragment was obtained by PCR as described (10). EGFP-CAR was cloned into pShuttle-CMV expression vector (Quantum Biotechnologies, Inc.) at XhoI and HindIII sites. The linearized shuttle vector and AdEasy vector (Quantum Biotechnologies, Inc.) were then cotransformed into Escherichia coli strain BJ5183. Positive recombinant plasmid Ad/EGFP-CAR was selected and the Ad/EGFP-CAR virus was then generated as described for Ad/PBP virus (16). For the expression of adenovirally driven EGFP-CAR, wild-type (C57BL/6J) and PRIPLiv−/− mice were injected intravenously (via tail vein) with 4 × 1011 recombinant adenovirus particles in a volume of 200 μl and sacrificed 5 days after injection. Phenobarbital (100 mg/kg body weight) or the solvent was injected IP 3 h prior to sacrifice. Small pieces of liver were used for cutting frozen sections, ∼8 Jim thick, fixed in 4% para-formaldehyde, and examined for fluorescence. Nuclear DNA was visualized by DAPI staining (10).
Mouse Primary Hepatocyte Culture and Adenovirus Infection
Hepatocytes were isolated from mouse livers using collagenase perfusion method as described previously (23). Cells were plated on collagen-coated 12-well plates containing William’s E medium at a cell density of 2.5−3.0 × 105 cells/well. After 8-h or overnight culture, the cells were infected with Ad/EGFP-CAR at a multiplicity of infection of ∼40. Twenty-four hours postinfection, cells were treated with or without ligand (1 mM phenobarbital). Cellular localization of CAR was assessed by fluorescence microscopy 2 h after the addition of CAR ligand penobarbital.
Northern and Western Blot Analyses
Total RNA isolated using TRIzol reagent (In-vitrogen) was glyoxylated, separated on an 0.8% agarose gel, transferred to nylon membrane, and probed with selected 32P-labeled cDNAs (16,43). Equal loading of total RNA in the gel was verified by measuring the intensity of 18S and 28S rRNA bands stained with Fast RNA Stain (HealthGene Corp). Immunoblotting of liver homogenates was performed using antibodies against CAR (Santa Cruz Biotechnology and M. Negishi, National Institute of Environmental Health Sciences, Research Triangle Park, NC), GSTπ (Dako), GFP (Invitrogen), and catalase (16,43).
RT-PCR and Quantitative PCR
RT-PCR was performed with the SuperScript one-step RT-PCR kit from Invitrogen as instructed. Primers 5′-CCC AGG CCA GTT CAC AGC-3′ and 5′-GGC CTG TGG TGT CCA A-3′ were used to amplify the region flanking exon 7 in the PRIP homozygous mutant. Total RNA (1 μg) was reverse transcribed with SuperScript II reverse transcriptase followed by PCR amplification consisting of 35 cycles of denaturing at 94°C for 15 s, annealing at 55°C for 30 s, and extension at 68°C for 1 min using a One-step RT-PCR kit (Invitrogen). For quantitative PCR, cDNA was synthesized from 2 μg total RNA using a Superscript III First Strand Synthesis System (Invitrogen) as per kit instructions. The PCR primers 5′-GCA AAT GCT GCC ATA GCC TCT GG- 3′ and 5′-GCC GAA GCA GGT CGA GAG GAT C- 3′ were used to detect PRIP mRNA expression. qPCR was carried out in triplicates for the amplification of PRIP gene and expression values were normalized with 18S ribosomal rRNA levels. The PCR reaction consisted of 1 μl (100 μmol) of sense and antisense primers and 10 μl of 2× SYBR Green denaturation at 95°C for 10 min and 40 cycles of 95°C for 15 s, 60°C and 95°C for 15 s. The generation of specific PCR products was confirmed by melting curve analysis and relative gene expression changes were measured using the comparative CT method, X = 2-ΔΔCT.
Immunohistochemical Analyses
Livers were fixed in either 10% formalin or 4% paraformaldehyde, and 4-μm-thick paraffin sections were cut and stained with hematoxylin and eosin. For immunohistochemical analyses, sections were deparaffinized and rehydrated. After inactivation of endogenous peroxidase activity and antigen retrieval, sections were blocked with 10% normal bovine serum in phosphate-buffered saline, followed by sequential incubation at room temperature with antibodies against CAR (M-150, SC-13065; Lot I2404, Santa Cruz Biotechnology) or PRIP for 3 h, biotinylated anti-rabbit IgG for 1 h, streptavidin-linked horseradish peroxidase (Vector laboratories) for 30 min, and finally 3,3′-diaminobenzidine tetrahydrochloride solution (Biogenex Inc.) for 4 min (16). BrdUrd incorporation was determined using a mouse anti-BrdUrd monoclonal antibody (DAKO) and Vectastain ABC Kit (Vector Laboratories).
Serum Alanine Aminotransferase (ALT) Assay
Blood collected from inferior vena cava was used for determination of serum (ALT) activity by using an ALT assay kit (Thermo Electron Corp.). Liver homogenates were used for GSH assay (Sigma) (16).
RESULTS
PRIP Liver Conditional (PRIPLiv−/−) Nulls
PRIP null mutation results in embryonic lethality that precludes investigations on the role of this coactivator in the adult liver (1,28,34,66). To circumvent this, mice with the LoxP integrated recombinant PRIP gene (LoxP-PRIP) were generated (41,66) and crossed with transgenic mice with the Cre transgene driven by the albumin promoter (Alb-Cre) (61). Alb-Cre transgenic mouse expresses Cre recombinase specifically in hepatic parenchymal cells but not in other cells in liver (16,17,61). Expression in liver of albumin promoter-driven Cre recombinase, commencing during the immediate postpartum stage with maximal expression occurring at around 2 weeks postnatally, results in the deletion of the exon 7 of the PRIP gene flanked by two LoxP sites (Fig. 1A). Deletion of exon 7 leads to a reading frame-shift resulting in the generation of a stop codon right after the fusion between exon 6 and exon 8. Deletion of PRIP gene in hepatocytes was confirmed by Southern blotting (Fig. 1B), RT-PCR (Fig. 1C), quantitative PCR (Fig. 1D), and immunohistochemical staining of liver sections for Cre and PRIP expression (Fig. 1E). As expected, Cre protein is absent in wild-type (PRIP+/+) livers but is expressed in PRIPLiv−/−(Fig. 1E). On the other hand, PRIP nuclear staining is seen in PRIP+/+ wild-type livers but not in Cre expressing PRIPLiv−/−, thus visually confirming that the deleted PRIP gene did not express the PRIP protein in liver (Fig. 1E).
Figure 1.
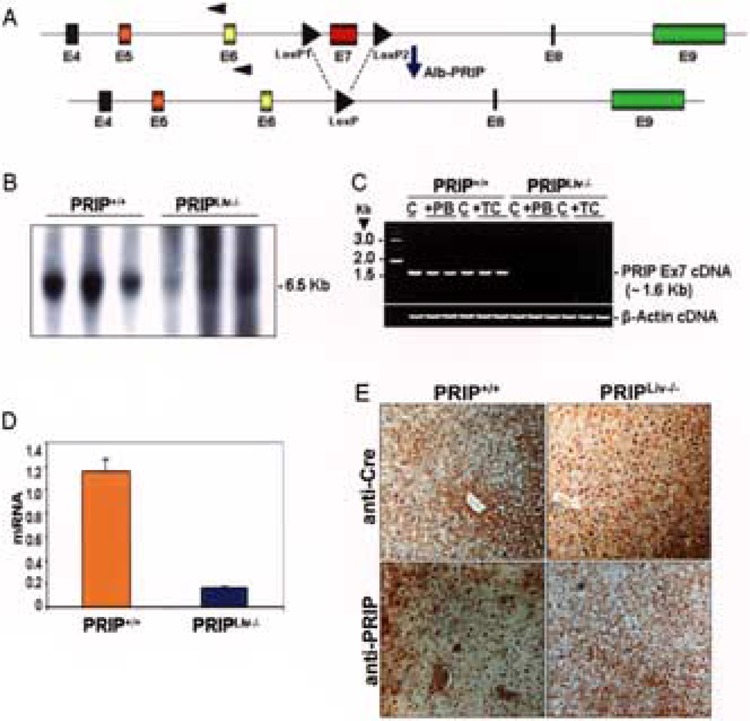
Conditional disruption of the PRIP gene in mouse liver. (A) Schematic diagram of LoxP-PRIP gene, with exon 7 flanked by LoxP sites and the resulting Cre mediated deletion of exon 7. LoxP-PRIP mouse was crossed with an Alb-Cre mouse to produce LoxP-PRIP mice expressing Alb-Cre. Expression in liver of Cre recombinase is necessary for the deletion of the exon 7. (B) Genomic DNA from liver of wild-type (PRIP+/+) and PRIPLiv−/− mice (3 each group) digested with SpeI and hybridized with exon 7 specific probe. A 6.5-kb band seen in PRIP+/+ mice is not apparent in PRIPLiv−/− mice. (C) Total RNA isolated from liver of PRIP+/+ or PRIPLiv−/− (C, control; PB, phenobarbital treated; TC, TCPOBOP treated) was used for RT-PCR amplification with primers specific for the LoxP-PRIP transgene and the deleted PRIP gene. An expected band was observed in wild-type PRIP+/+ mouse livers but not in PRIPLiv−/− livers. β-Actin served as a loading control. (D) Expression levels of PRIP mRNA in PRIP+/+ or PRIPLiv−/− livers as assessed by qPCR. mRNA levels were normalized to 18S rRNA levels. A mean of triplicates is shown (mean ± SE; n = 3). (E) Immunohistochemical staining of liver sections from PRIP+/+ and PRIPLiv−/− mice with anti-PRIP and anti-Cre antibodies. Cre staining of nuclei is seen in PRIPLiv−/− livers but not in wild-type PRIP+/+ livers, whereas PRIP nuclear staining is absent in Cre-expressing livers but present in the PRIP+/+ livers.
PPARα and CAR Ligand-Induced Hepatocellular Proliferation
To evaluate the role of coactivator PRIP in PPARα and CAR ligand-induced liver cell proliferation, PRIPLiv−/− mice were treated with either Wy-14,643, a potent PPARα ligand, or with CAR ligands, phenobarbital or TCPOBOP (Fig. 2). Short-term treatment with either PPARα or CAR ligands increased liver weight/body weight ratios in both PRIP+/+ and PRIPLiv−/− mice (Fig. 2A). The hepatomegalic effect was more pronounced in both PRIP+/+ and PRIPLiv−/− mice treated with TCPOBOP and Wy-14,643, compared to phenobarbital treatment. Hepatocellular proliferation, as assessed by BrdUrd labeling indices, was prominent in the livers of both PRIP+/+ and PRIPLiv−/− following TCPOBOP and Wy-14,643 treatment. There was no difference in the extent of liver cell proliferation with either ligand in the presence or absence of PRIP expression (Fig. 2B, C). Figure 2C illustrates representative photomicrographs of liver sections immunohistochemically analyzed for BrdUrd incorporation. In untreated livers, BrdUrd labeling was seen in an occasional hepatocyte nucleus but the number of labeled hepatocytes was markedly increased throughout the liver lobule in PRIP+/+ and PRIPLiv−/− mice.
Figure 2.
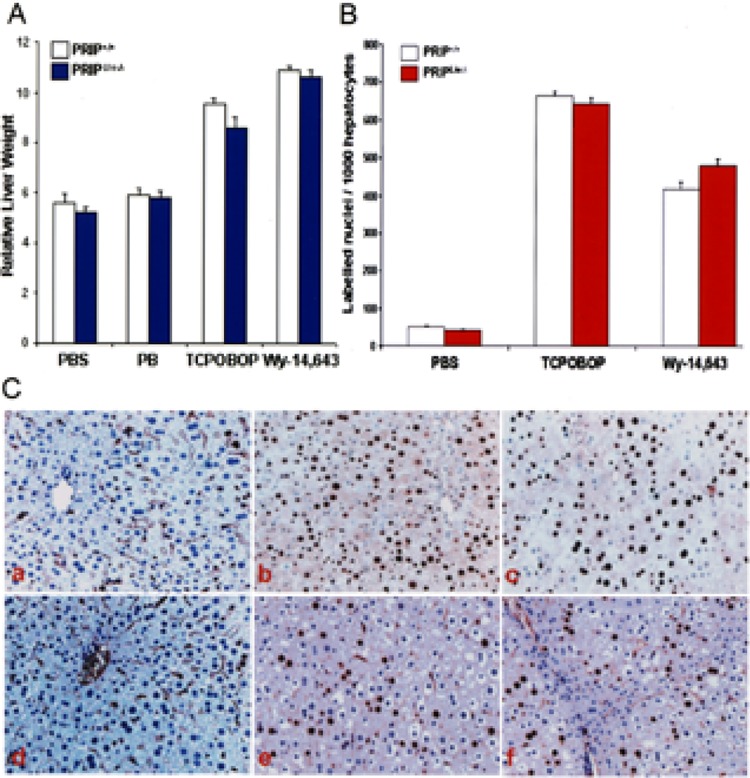
Changes in liver weight and hepatocellular proliferation. (A) Liver weight/body weight ratios in PRIP+/+ and PRIPLiv−/− mice treated IP with PB or TCPOBOP (TC) daily for 3 days, or fed Wy-14,643 (0.125%) in the diet for 4 days compared with PBS injected controls. (B) Hepatocellular BrdUrd labeling indices in TCPOBOP- and Wy-14,643-treated mice. BrdUrd was given in drinking water for 3 days (1.0 mg/ml) to mice given TCPOBOP for 3 days, or fed a diet containing Wy-14,643 for 4 days. Liver cell proliferation is similar for each compound in both PRIP+/+ and PRIPLiv−/−mice. (C) Representative photomicrographs to illustrate BrdUrd-labeled nuclei in the livers of untreated PRIP+/+ (a) and PRIPLiv−/− (d) controls, and TCPOBOP (b, c) and Wy-14,643 (e, f) treated PRIP+/+ (b, e) and PRIPLiv−/− (c, f) mice.
Induction of Peroxisome Proliferation and PPARα-Regulated Genes in Liver
The highly reproducible pleiotropic responses in the rat and mouse liver induced by peroxisome proliferators have been well documented (45). These structurally diverse chemicals act by functioning as ligands for the nuclear receptor PPARα and disruption of PPARα gene abrogates these responses (14,29). To assess the role of PRIP in PPARα ligand-induced peroxisome proliferation and gene expression in liver, diet containing Wy-14,643 was fed for 2 weeks and livers were analyzed for morphological alterations and for the expression of selected PPARα target genes. Light microscopy of hematoxylin and eosin (H&E)-stained liver sections revealed hypertrophy of all hepatocytes in the liver lobule with granular eosinophilic cytoplasm in Wy-14,643-treated PRIP+/+ and PRIPLiv−/− mice compared to untreated controls (Fig. 3A). Evaluation of 0.5-μ-thick sections of liver that were processed for visualizing peroxisomal catalase showed a profound increase in the number of these catalase-positive organelles in all hepatocytes in both PRIP+/+ and PRIPLiv−/− mice (Fig. 3B). The degree of peroxisome proliferation in liver cells, as assessed by light and electron microscopy (not illustrated), was essentially similar in both PRIP+/+ and PRIPLiv−/− mice. These observations suggest that the presence of coactivator PRIP is not essential for PPARa ligand-induced peroxisome proliferation in liver.
Figure 3.
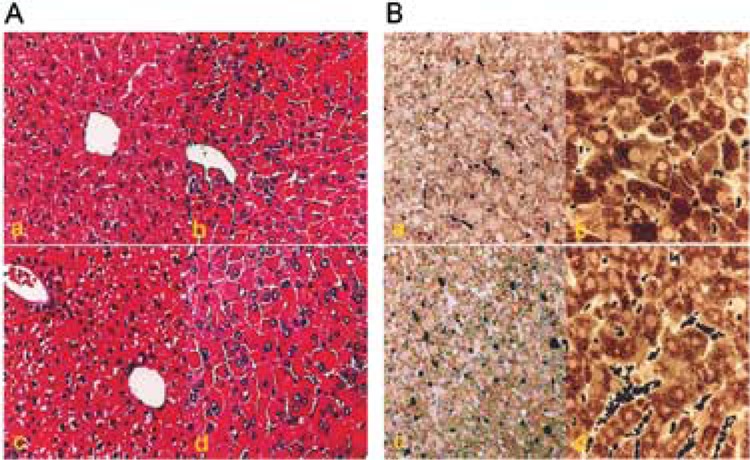
Comparative changes in liver morphology and cytochemical localization of peroxisomal catalase. (A) Liver morphology of PRIP+/+ (a, b) and PRIPLiv−/−(c, d) mice treated with Wy-14,643 (b, d) for 7 days (H&E stained). (B) Peroxisome proliferation in hepatocytes of wild-type and PRIPLiv−/− mice treated with Wy-14,643 for 7 days. A massive increases increase in peroxisomes is evident in the livers of wild-type and PRIPLiv−/− mice treated with Wy-14,643 (b, d).
Analysis of RNA isolated from livers of untreated and peroxisome proliferator-treated mice for PPARα-regulated gene expression revealed no differences in the expression of key genes involved in peroxisomal β-oxidation and microsomal ω-oxidation (Fig. 4A). The hepatic mRNA levels of all three P-oxidation system genes, namely fatty acyl-CoA oxidase (AOX), enoyl-CoA hydratase/L-3-hydroxyacyl-CoA dehydrogenase (L-bifunctional enzyme; L-PBE), and peroxisomal thiolase (PTL), increased markedly in Wy-14,643-treated PRIP+/+ and PRIPLiv−/− mice. The mRNA level of CYP4A1, which is responsible for microsomal cytochrome p450 fatty acid cohydroxylation, is also increased in these livers following treatment with a PPARα ligand (Fig. 4A). PEX-11, which is involved in peroxisome biogenesis, is also upregulated in livers with peroxisome proliferation. Catalase is a peroxisomal marker enzyme and treatment with peroxisome proliferators causes only a mild increase in its mRNA level. PPARα mRNA levels remain essentially similar in control and Wy-14,643-treated mice. Immunoblotting data confirm similar levels of increases in the amount of AOX, L-PBE, and PTL proteins in PRIP+/+ and PRIPLiv−/− mouse livers following Wy-14,643 treatment (Fig. 4B). The levels of D-bifunctional enzyme (D-PBE), and sterol carrier protein-x (SCP-x), the two enzymes involved in the branched chain fatty acid β-oxidation, increased only slightly in both types of mice. Immunoblotting also showed a visible but slight increase in PPARα and catalase proteins with Wy-14,643 treatment in PRIP+/+ and PRIPLiv−/− mouse livers. These data further confirm that PRIP gene is not involved in the regulation of these genes.
Figure 4.
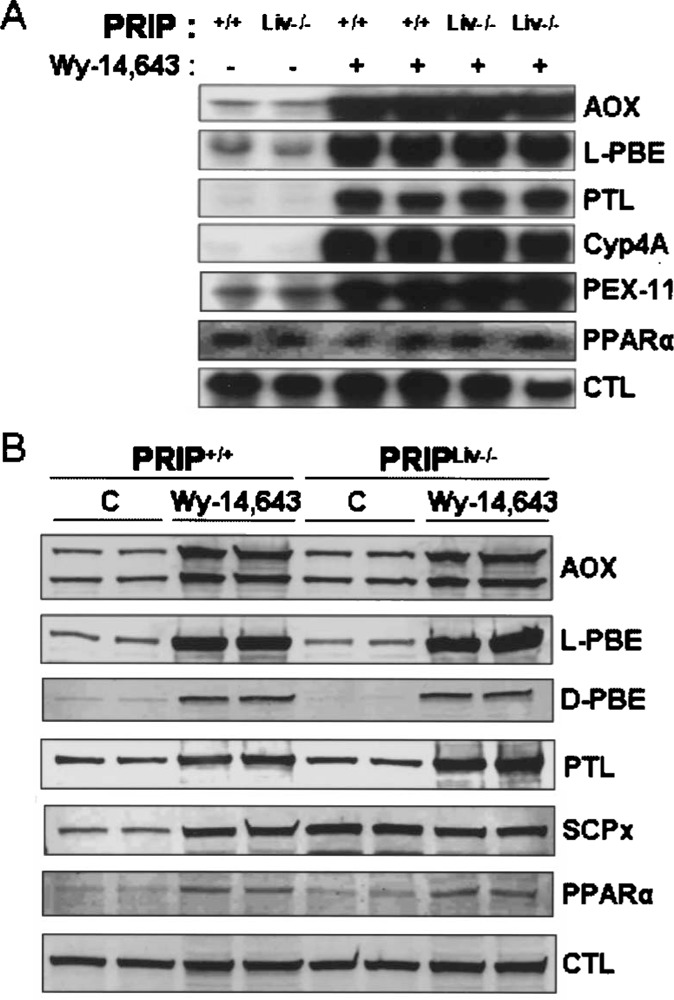
Expression of selected PPARα regulated genes in Wy-14,643-treated PRIP+/+ and PRIPLiv−/− mouse liver. (A) Northern blot analysis of total liver RNA from Wy-14,643-treated and untreated PRIPLiv−/− and wild-type (PRIP+/+) mice for peroxisomal P-oxidation system genes (AOX, L-PBE, PTL), microsomal co-oxidation (CYP4A3) gene, and others. Peroxisomal marker enzyme catalase (CTL) is also included. (B) Immunoblot analysis of liver proteins of untreated and Wy-14,643-treated wild-type (+/+) and PRIPLiv−/− (Liv−/−) mice for AOX (two bands represent AOX components A (72 kDa) and B (51 kDa)), L-PBE, D-PBE, PTL, SCPx, PPARa, and CTL.
PRIP Is Redundant for CAR-Regulated Gene Expression
In a previous brief report, we reported that induction of CYP genes in liver by CAR ligands, phenobarbital and TCPOBOP, is not dependent on coactivator PRIP. We now show that both these ligands induce CYP2B10, CYP3A11, CYP2C29, and CYP1A2 mRNA levels to a similar extent in both PRIP+/+ and PRIPLiv−/− mouse livers (Fig. 5A, B) and also include functional data showing that PRIP deletion does not affect zoxazolamine-induced paralysis (Table 1). PRIP deficiency does not affect the levels of expression of many genes analyzed by Northern blotting (Fig. 5A) and these include not only CYP genes but also three nuclear receptors, namely CAR, pregnane X receptor (PXR), and farnesyl X receptor (FXR) (Fig. 5A). The mRNA expression of cytoplasmic CAR retention protein (CCRP) in liver appeared similar in treated and untreated groups (26). We also compared the mRNA expression levels of some key genes (CYP2B10, CYP3A11, CYP1A2) in the liver of PRIPLiv−/− and PBPLiv−/− mice treated with CAR ligands phenobarbital and TCPOBOP (Fig. 5B). The induction, for example, of CAR-regulated gene CYP2B10 was robust in PRIP-null livers but minimal in PBP-null livers (Fig. 5B). The slight increase in CYP2B10 mRNA in PBP livers following TCPOBOP treatment is due to residual presence of a small fraction of hepatocytes that escaped Cre-mediated gene deletion. Immunoblotting for CAR, GSTPi, and catalase showed similar levels of expression in all groups (Fig. 5C).
Figure 5.
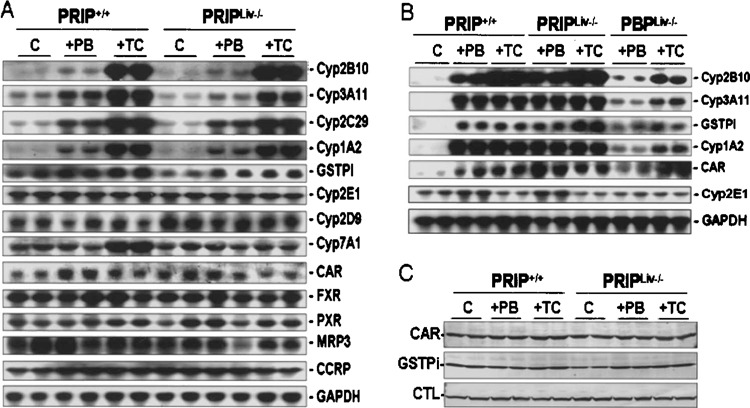
Changes in PB- and TCPOBOP-induced CAR target gene expression in liver. (A) Northern blot analysis of liver RNA from wild-type (PRIP+/+) and PRIPLiv−/− mice given saline (con), phenobarbital (PB), or TCPOBOP (TC) injections for 3 days. The cDNA probes used for the analysis are indicated. Increases in Cyp2B10, Cyp3A11, Cyp2C29, and Cyp1A2 mRNA levels in wild-type and PRIP-null livers by PB or TCPOBOP appear similar. No differences in the expression levels of CAR, FXR, PXR, and CCRP are seen among different groups. GAPDH is used for assessing RNA loading. (B) Northern blot analysis for the changes in the expression of selected genes in PRIPLiv−/− and PBPLiv−/− mouse livers. Robust induction of CYP2B10 is seen in PRIPLiv−/− but not in PBPLiv−/− livers. (C) Immunoblot analysis of liver proteins for CAR, GSTPi, and catalase (CTL) in PB- and TCPOBOP (TC)-treated PRIP+/+ and PRIPLiv−/− mice.
TABLE 1.
ZOXAZOLAMINE PARALYSIS TEST
| PRIP+/+ | PRIPLiv−/− | |
|---|---|---|
| Control | 4/13: 2-h paralysis; recovered | 1/11: 2-h paralysis; recovered |
| 5/13: 3-h paralysis; recovered | 6/11: 3-h paralysis; recovered | |
| 1/13: 4-h paralysis; recovered | 1/11: 4-h paralysis; dead | |
| 1/13: 6-h paralysis; recovered | 1/11: 8-h paralysis; dead | |
| 1/13: 6-h paralysis; dead | 2/11: 12-h paralysis; recovered | |
| 1/13: 9-h paralysis; dead | ||
| PB (3 days) | 8/8: not paralyzed | 8/8: not paralyzed |
Control and 3-day PB-treated male mice (5–6 weeks old) were given a single IP injection of zoxazolamine (300 mg/kg of body weight), and paralysis duration was recorded as the time when the mice were able to right themselves repeatedly. Very similar results were observed in both wild-type (PRIP+/+) and knockout (PRIP−/−) mouse groups.
PRIP Deficiency Does Not Influence Zoxazolamine Paralysis and APAP-Induced Hepatic Necrosis
The functional significance of the induction of CAR-mediated transcription of CYP genes was examined by the assessment of paralysis induced by the muscle relaxant zoxazolamine and acetaminophen (APAP) hepatotoxicity (16,56). Induction of CYP1A2 by certain xenobiotics, including CAR ligand phenobarbital, leads to rapid metabolic inactivation of zoxazolamine, resulting in significant decrease in the duration of paralysis (16,56). We have previously reported that PBPLiv−/− mice exhibited significantly enhanced zoxazolamine sensitivity and phenobarbital pretreatment had no effect on zoxazolamine paralysis, confirming the functional significance of the failure of CYP gene induction in PBP-null livers (16). In the present study, PRIP gene deletion had no effect on the inducibility of CYP genes in liver and as a corollary the zoxazolamine paralysis was similar to that noted in wild-type (PRIP+/+) mice (Table 1). We further examined the role of PRIP in APAP-induced hepatotoxicity. APAP hepatotoxic effects are influenced by a variety of factors that culminate in the conversion of APAP to its highly toxic intermediate metabolite, N-acetyl-p-benzoquinone imine (16,56, 65). It is well known that induction of CYP isoforms by activators of CAR or PXR enhances APAP hepatotoxicity whereas the disruption of CAR and PBP genes has an abrogating effect on this toxicity (6, 56,65). We now show that PRIP gene deletion had no abrogating effect on APAP hepatotoxicity (Fig. 6). Phenobarbital or TCPOBOP pretreatment resulted in the induction of expression of CYP genes in PRIPLiv−/− mouse liver and as a consequence developed centrilobular hepatic necrosis (Fig. 6A) due to rapid and efficient APAP metabolic conversion to the toxic metabolite, N-acetyl-p-benzoquinone imine (56). Serum ALT levels were high in both PRIP+/+ and PRIPLiv−/− mice pretreated with phenobarbital or TCPOBOP prior to APAP exposure (Fig. 6B). Administration of APAP alone without prior treatment with a CAR ligand did not result in hepatic necrosis. We also measured GSH content in liver to ascertain if hepatotoxicity correlates with diminished GSH content. Decrease in GSH level occurred in both PRIP+/+ and PRIPLiv−/− mouse livers with APAP-induced hepatic necrosis (Fig. 6C).
Figure 6.
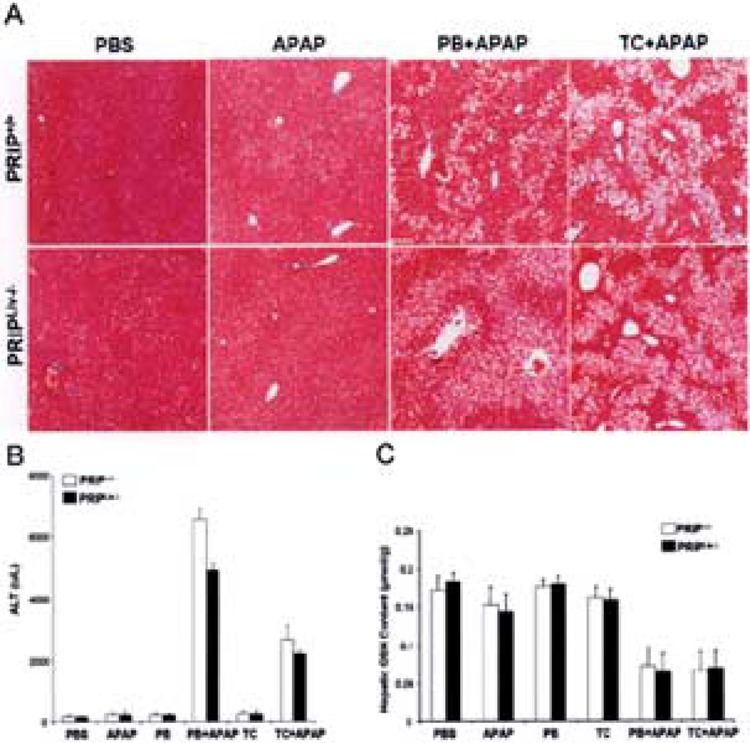
PRIP and CAR in APAP toxicity. (A) Histological appearance of livers from wild-type (PRIP+/+) and PRIPLiv−/− mice treated with PBS, APAP, PB plus APAP, or TCPOBOP (TC) plus APAP (H&E staining). Centrilobular hepatic necrosis is prominent in wild-type as well as PRIPLiv−/− mice pretreated with PB or TCPOBOP (TC) prior to APAP administration. Administration of PBS or APAP alone showed no hepatic necrosis. (B) Serum ALT levels in mice. ALT levels increased significantly in PB- or TCPOBOP-pretreated PRIP+/+ and PRIPLiv−/− mice given APAP. (C) Hepatic GSH levels. Both PRIP+/+ and PRIPLiv−/− mice treated for 3 days with PB or TCPOBOP (TC) prior to APAP showed significantly reduced levels of hepatic GSH compared to GSH levels in PBS, APAP, PB, or TCPOBOP treatment.
PRIP Is Not Needed for Phenobarbital-Mediated Nuclear Translocation of CAR
Nuclear receptor CAR is classified as a type I nuclear receptor, because it is cytoplasmic in location and undergoes nuclear translocation after ligand binding (13,20–22,27,31,64). Upon entering the nucleus, the agonist bound receptor interacts with the LXXLL (L, leucine; X, any amino acid) nuclear receptor (NR) box of coactivators to assemble the transcriptional complex for initiation of transcription (11,48). Immunohistochemical staining for CAR in liver sections of wild-type (PRIP+/+) and PRIPLiv−/− mice treated with phenobarbital revealed translocation of endogenous CAR into the nucleus of hepatocytes in contrast to the absence of CAR nuclear staining in untreated controls (Fig. 7A). Recently, we generated adenovirally driven EGFP-CAR vector for expression of CAR in liver in vivo and in hepatocytes in vitro. Expression of exogenous Ad/EGFP-CAR in liver was prominent in liver cells and, in the absence of CAR ligand, the EGFP-CAR was in the cytoplasm (Fig. 7B,D). Rapid translocation of CAR into the nucleus occurred within a short time after phenobarbital administration in both PRIP+/+ and PRIPLiv−/− livers (Fig. 7B-E). We also studied nuclear translocation of Ad/ EGFP-CAR in primary hepatocyte cultures (Fig. 8). EGFP-CAR appeared cytoplasmic in wild-type (PRIP+/+) and PRIPLiv−/− mouse hepatocytes in culture in the absence of CAR ligand, and with phenobarbital CAR translocated to the nucleus irrespective of the PRIP status.
Figure 7.
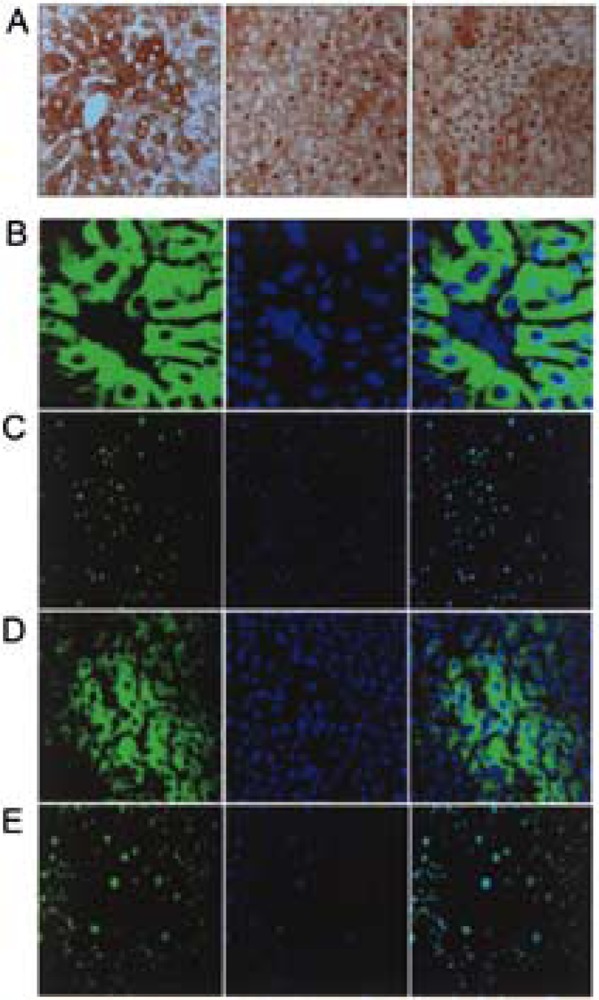
Absence of coactivator PRIP does not influence pheno-barbital-mediated nuclear translocation of CAR in mouse liver. (A) Immunoperoxidase staining of liver sections of wild-type control mouse (left panel), wild-type mouse treated with PB (middle panel), and PRIPLiv−/− mouse (right panel) treated with PB for localization of CAR. CAR was cytoplasmic in the untreated wild-type mouse whereas PB treatment caused nuclear translocation of CAR in wild-type as well as PRIPLiv−/− mouse livers. (B–E) Localization of exogenous Ad/EGFP-CAR in liver. EGFP fluorescence (left panels), DAPI staining (middle panels), and merged images (right panels) of wild-type (B, C) and PRIPLiv−/− (D, E) mice without (B, D) and with PB (C, E) treatment. PB induced nuclear translocation of Ad/EGFP-CAR in wild-type (C) and PRIPLiv−/− (E) mouse livers.
Figure 8.
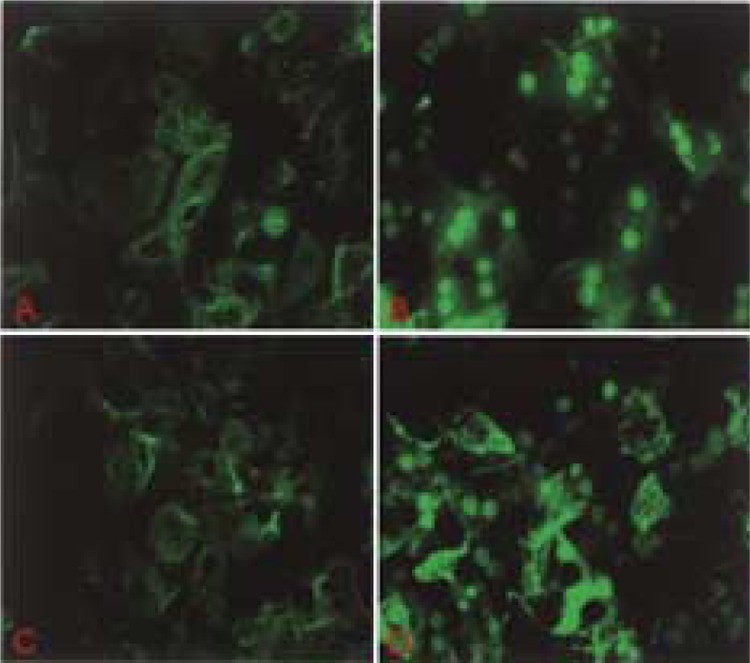
Nuclear translocation of Ad/EGFP-CAR in primary hepatocyte cultures. (A, B) Wild-type mouse hepatocytes in culture without (A) and with phenobarbital (B) in the medium. (C, D) PRIPLiv−/− mouse hepatocytes without (C) and with (D) PB in culture medium. CAR is mostly cytoplasmic in the absence of PB (A, C) and nuclear (B, D) following PB.
DISCUSSION
In this study, we report the generation of PRIP liver conditional null mice (PRIPLiv−/−) to study the role of this coactivator in the transcriptional regulation of genes controlled by nuclear receptors PPARα and CAR. We demonstrate that coactivator PRIP is not essential for the function of these two important xenobiotic sensing nuclear receptors. First, absence of PRIP in liver had no effect on PPARα ligand-induced hepatomegaly, hepatic peroxisome proliferation, and the induction of genes regulated by PPARα that participate in peroxisomal and microsomal fatty acid oxidation in liver. Second, the absence of PRIP in liver failed to abrogate CAR ligand-induced induction of certain CYP family genes involved in xenobiotic metabolism (53,56,57). Consequently, PRIP-null livers pretreated with CAR ligand phenobarbital showed hepatocellular necrosis when exposed to acetaminophen. In an earlier study, we demonstrated that steroid receptor coactivator-1 (SRC-1), a member of the p160/SRC-1 family of coactivators, is not essential for PPARα-regulated gene expression and induction of peroxisome proliferator-induced pleiotropic responses in mouse liver (43). SRC-1 gene deletion also exerted no effect on phenobarbital-mediated nuclear translocation of CAR and the induction of CAR-regulated genes in liver (D. Guo unpublished). Thus, it appears that coactivators SRC-1 and PRIP are redundant for the transcriptional activity of both PPARα and CAR, despite the fact that SRC-1 has intrinsic histone acetyltransferase activity needed for chromatin modification (8,48). In contrast, studies with PBP liver conditional null mice (PBPLiv−/−) established that coactivator PBP/MED1 deficiency in vivo mimics the absence of PPARα, implying that PBP is required for PPARα-regulated gene expression in liver (17). Furthermore, PBP gene deletion in liver abrogated acetaminophen hepatotoxicity similar to that observed in CAR-null mouse liver (65). Accordingly, while PBP is an essential coactivator for PPARα and CAR function, both PRIP and SRC-1 are considered redundant for the transcriptional activation of these two nuclear receptors.
The mechanism by which PBP/TRAP220 affects PPARα- and CAR-mediated transcriptional activation of their respective target genes is not fully known, but it appears that absence of PBP in liver cells diminishes the recruitment or association of other cofactors (16,17). The finding that both PPARα and CAR target gene expression mediated by cognate ligands in PRIP-null livers is similar to that noted in the wild-type mouse livers implies that coactivator assembly necessary for the function of these two nuclear receptors proceeds properly in the absence of PRIP. Furthermore, CAR is a cytoplasmic protein and it has to be translocated to the nucleus for transcriptional activity (26,36,38,40,50,53,58). We noted that the absence of PBP hinders the phenobarbital-mediated nuclear translocation of CAR or that PBP is required for retention of CAR in the nucleus (10,16). In an unliganded state, CAR is retained in the cytoplasm by complexing with Hsp90, and cytoplasmic CAR-retention protein (CCRP) (26,27,38). Phenobarbital-like CAR activators facilitate the recruitment of an okadaic acid-sensitive phosphatase to the CAR-Hsp90-CCRP complex, implying that CAR translocation may be phosphorylation dependent (53). Our results with PBP-null livers suggest that within the nucleus, imported CAR most likely binds to PBP and that PBP functions as a scaffolding protein to retain CAR. Robust binding of CAR to PBP containing two LXXLL motifs has been demonstrated, indicating a strong affinity between these two molecules (10). In the absence of this PBP scaffold, the influxed CAR is unlikely to be retained and concentrated within the nucleus (10,58). The fact that CAR is translocated to the nucleus and upregulates the expression of its target gene in the absence of coactivator PRIP is a clear indication that PRIP is not essential for the function of CAR. These observations clearly establish that coactivator PBP is required for CAR and PPARα function and induction of genes responsible for xenobiotic metabolism, whereas SRC-1 and PRIP appear not essential for the functioning of PPARα and CAR (16). SRC-1 and PRIP may be indirectly associated with PBP scaffold but the signal for PPARα- and CAR-driven transcription does not involve these two coactivators. Ligand-independent translocation of CAR into the nucleus was shown to occur in vivo in response to SRC-2/GRIP1 and expression of GRIP1 increased CAR-mediated transactivation about twofold (36,58). Recent work with SCR-1-null livers showed that it is not required for CAR nuclear translocation and it would be important to examine the role of SRC-2 and SRC-3 and combinations of these molecules using single and double knockout mice (43,55,59,60).
PPARα and CAR ligands induce hepatocellular proliferation during the acute exposure phase (5,45, 47). Wy-14,643, a potent PPARα ligand, exerts a profound primary liver cell proliferating effect in mice (49) and this effect is abolished in mice deficient in PPARα (29) or coactivator PBP/MED1 (17). Likewise, CAR ligand TCPOBOP is a potent hepatomitogen and its mitogenic potential is severely curtailed in mice with conditional deletion of coactivator PBP (5,16). In the present study, conditional deletion of PRIP gene in liver had no adverse effect on hepatocellular proliferation induced by PPAR and CAR ligands. Although lack of PPARα completely abrogates the hepatocyte proliferation in response to peroxisome proliferators, the absence of PPARα enhanced hepatocyte proliferation in response to CAR ligand TCPOBOP (5). These observations suggest a possible role for PPARa in CAR signaling (5). It is possible that absence of PPARa increases the RXR pool for CAR/RXR heterodimerization, resulting in enhanced cell proliferation (5). Both Wy-14,643 and TCPOBOP increased hepatocellular proliferation in PRIP+/+ and PRIPLiv−/− mice unlike that noted in the livers of mice deficient in PBP (16). These results show that PRIP does not play a role in the mitogenic effects mediated by PPARα and CAR ligands.
PRIP has been shown to enhance the transcriptional activation by CAR in the transfection assays in vitro, implying that PRIP serves as a coactivator for this xenobiotic receptor (4). PRIP also binds to CAR and this binding encompasses the first LXXLL motif of PRIP (4). These in vitro observations clearly imply that PRIP is involved in CAR-mediated gene transcription. However, the in vivo data obtained with PRIPLiv−/− mice in the present study failed to support the notion that PRIP is needed for the function of CAR. While PRIP amplifies CAR-mediated transcription in vitro, it appears that other factors may dominate in vivo, and supersede PRIP, making it redundant for CAR function. PRIP deficiency in liver failed to alter acetaminophen-induced hepatotoxicity in mice pretreated with CAR ligands PB or TCPOBOP. No differences in the upregulation of cytochrome P450 gene expression (CYP1A2, CYP2B10, CYP3A11, and CYP2E1) were seen following PB or TCPOBOP treatment between PRIP+/+ and PRIPLiv−/− mice. Moreover, CAR mRNA and protein levels were unchanged in PRIPLiv−/− mouse liver, suggesting that PRIP does not alter hepatic CAR expression. The mRNA levels of CCRP were also unchanged in PRIP-null livers. Functional analysis revealed that paralysis induced by zoxazolamine and hepatic necrosis induced by APAP were similar in PRIPLiv−/− and PRIP+/+ mice. These data firmly establish that PRIP deficiency in the liver does not protect the animal from the hepatotoxic effects of acetaminophen, unlike that seen with PBP deficiency (16). However, these findings are somewhat contradictory to the observations in transgenic mice expressing DN1, a fragment of PRIP/ ASC-2 containing the first LXXLL motif (4). These mice failed to show acetaminophen-induced hepatic necrosis, suggesting that PRIP/ASC-2 is a bona fide coactivator for CAR. These differences are attributes to DN1 containing a functional LXXLL motif and this motif conceivably suppressed the function of other critical coactivators for CAR function such as PBP (16). Our studies on liver-specific PRIP-null mice demonstrate a more specific and reliable approach wherein the function of only one gene (PRIP) is disrupted as opposed to the dominant negative approach, which might have a more general effect and exert a broader dysfunction. Our findings with PRIP deficiency are unlike those obtained with PBP deficiency with regard to transcriptional activation of PPARα- and CAR-regulated genes (16,17). The reason for this selective dependence of PPARα- and CAR-regulated gene transcription on coactivator PBP but not on PRIP may be due to the existence of coactivator selectivity in nuclear receptor function (2,3,8, 11,35).
We thus conclude that transcription coactivator PRIP is not essential for induction of the hepatic effects of xenobiotic receptors CAR and PPARα. Nonetheless, whole body knockout of this gene is embryonic lethal, implying that this is an essential coactivator and that it may be involved in the function of nuclear receptors and transcription factors other than CAR and PPARα. Additional studies are necessary to explore in detail the selectivity and specificity of coactivator requirement for transcription of a given nuclear receptor.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (NIH) Grants GM23750 (to J.K.R.) and CA104578 (to J.K.R.).
REFERENCES
- 1. Antonson P.; Schuster G. U.; Wang L.; Rozell B.; Holter E.; Flodby P.; Treuter E.; Holmgren L.; Gustafsson J. A. Inactivation of the nuclear receptor coactivator RAP250 in mice results in placental vascular dysfunction. Mol. Cell. Biol. 23:1260–1268; 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bookout A. L.; Jeong Y.; Downes M.; Yu R. T.; Evans R. M.; Mangelsdorf D. J. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcription network. Cell 126:789–799; 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen D.; Ma H.; Hong H.; Koh S. S.; Huang S. M.; Schurter B. T.; Aswad D. W.; Stallcup M. R. Regulation of transcription by a protein methyltransferase. Science 284:2174–2177; 1999. [DOI] [PubMed] [Google Scholar]
- 4. Choi E.; Lee S.; Yeom S.-Y.; Kim G. H.; Lee J. W.; Kim S.-W. Characterization of activating signal cointegrator-2 as a novel transcriptional coactivator of the xenobiotic nuclear receptor constitutive androstane receptor. Mol. Endocrinol. 19:1711–1719; 2005. [DOI] [PubMed] [Google Scholar]
- 5. Columbano A.; Ledda-Columbano G. M.; Pibri M.; Concas D.; Reddy J. K.; Rao M. S. Peroxisome proliferator-activated receptor-α−/− mice show enhanced hepatocyte proliferation in response to the hepatomitogen1,4-bis[2-(3,5-dichloropyridyloxy)] benzene, a ligand of constitutive androstane receptor. Hepatology 34:262–266; 2001. [DOI] [PubMed] [Google Scholar]
- 6. Faucette S. R.; Sueyoshi T.; Smith C. M.; Negishi M.; Lecluyse E. L.; Wang H. Differential regulation of hepatic CYP2B6 and CYP3A4 genes by constitutive androstane receptor but not pregnane X receptor. J. Pharmacol. Exp. Ther. 317:1200–1209; 2006. [DOI] [PubMed] [Google Scholar]
- 7. Géhin M.; Mark M.; Dennefeld C.; Dierich A.; Gronemeyer H.; Chambon P. The function of TIF2/ GRIP1 in mouse reproduction is distinct from those of SRC-1 and p/CIP. Mol. Cell. Biol. 22:5923–5937; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Glass C. K.; Rosenfeld M. G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 14:121–141; 2000. [PubMed] [Google Scholar]
- 9. Goo Y. H.; Sohn Y. C.; Kim D-H.; Kin S-W.; Kang M-J.; Jung D-J.; Kwak N. A.; Barlev N. A.; Berger S. L.; Chow V. T.; Roeder R. G.; Azorsa D. O.; Meltzer P. S.; Suh P-G.; Song E.; Lee K. J.; Lee Y. C.; Lee J. W. Activating signal cointegrator 2 belongs to a novel steady-state complex that contains a subset of Trithorax group proteins. Mol. Cell. Biol. 23:140–149; 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guo D.; Sarkar J.; Ahmed M. R.; Viswakarma N.; Jia Y.; Yu S.; Rao M. S.; Reddy J. K. Peroxisome proliferator-activated receptor (PPAR)-binding protein (PBP) but not PPAR-interacting protein (PRIP) is required for nuclear translocation of constitutive androstane receptor in mouse liver. Biochem. Biophys. Res. Commun. 347:485–495; 2006. [DOI] [PubMed] [Google Scholar]
- 11. Hermanson O.; Glass C. K.; Rosenfeld M. G. Nuclear receptor coregulators: Multiple modes of modification. Trends Endocrinol. Metabol. 13:55–60; 2002. [DOI] [PubMed] [Google Scholar]
- 12. Honkakoski P.; Sueyoshi T.; Negishi M. Drug-activated nuclear receptors CAR and PXR. Ann. Med. 35:172–182; 2003. [DOI] [PubMed] [Google Scholar]
- 13. Hosseinpour F.; Moore R.; Negishi M.; Sueyoshi T. Serine 202 regulates the nuclear translocation of constitutive active/androstane receptor. Mol. Pharmacol. 69:1095–1102; 2006. [DOI] [PubMed] [Google Scholar]
- 14. Issemann I.; Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 347:645–650; 1990. [DOI] [PubMed] [Google Scholar]
- 15. Ito M.; Yuan C. X.; Okano H. J.; Darnell R. B.; Roeder M. G. Involvement of the TRAP220 component of the TRAP/SMCC coactivator complex in embryonic development and thyroid hormone action. Mol. Cell 5:683–693; 2000. [DOI] [PubMed] [Google Scholar]
- 16. Jia Y.; Guo G. L.; Surapureddi S.; Sarkar J.; Qi C.; Guo D.; Xia J.; Kashireddi P.; Yu S.; Cho Y. W.; Rao M. S.; Kemper B.; Ge K.; Gonzalez F. J.; Reddy J. K. Transcription coactivator peroxisome proliferators-activated receptor binding protein/mediator 1 deficiency abrogates acetaminophen hepatotoxicity. Proc. Natl. Acad. Sci. USA 102:12531–12536; 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jia Y.; Qi C.; Kashireddi P.; Surapureddi S.; Zhu Y. J.; Rao M. S.; Le Roith D.; Chambon P.; Gonzalez F. J.; Reddy J. K. Transcription coactivator PBP, the peroxisome proliferators-activated receptor (PPAR)-binding protein, is required for PPARα-regulated gene expression in liver. J. Biol. Chem. 279:24427–24434; 2004. [DOI] [PubMed] [Google Scholar]
- 18. Jia Y.; Qi C.; Zhang Z.; Zhu Y. T.; Rao M. S.; Zhu Y-J. Peroxisome proliferator-activated receptor-binding protein null mutation results in defective mammary gland development. J. Biol. Chem. 280:10766–10773; 2005. [DOI] [PubMed] [Google Scholar]
- 19. Kamei Y.; Xu L.; Heinzel T.; Torchia J.; Kurokawa R.; Gloss B.; Lin S. C.; Heyman R. A.; Rose D. W.; Glass C. K.; Rosenfeld M. G. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell 85:403–414; 1996. [DOI] [PubMed] [Google Scholar]
- 20. Kanno Y.; Suzuki M.; Nakahama T.; Inouye Y. Characterization of nuclear localization signals and cytoplasmic retention region in nuclear receptor CAR. Biochim. Biophys. Acta 1745:215–222; 2005. [DOI] [PubMed] [Google Scholar]
- 21. Kawamoto T.; Sueyoshi T.; Zelko I.; Moore R.; Washburn K.; Negishi M. Phenobarbital-responsive nuclear translocation of the receptor CAR in induction of the CYP2B gene. Mol. Cell. Biol. 19:6318–6322; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kawana K.; Ikuta T.; Kobayashi Y.; Gotoh O.; Takeda K.; Kawajiri K. Molecular mechanisms of nuclear translocation of an orphan nuclear receptor, SXR. Mol. Pharmacol. 63:524–531; 2003. [DOI] [PubMed] [Google Scholar]
- 23. Klaunig J. E.; Goldblatt P. J.; Hinton D. E.; Lipsky M. M.; Chacko J.; Trump B. F. Mouse liver cell culture. I. Hepatocyte isolation. In Vitro 17:913–925; 1981. [DOI] [PubMed] [Google Scholar]
- 24. Kliewer S. A.; Umesono K.; Noonan D. J.; Heyman R. A.; Evans R. M. Convergence of 9-cis retinoic acid and peroxisome proliferator signaling pathways through heterodimer formation of their receptors. Nature 358:771–774; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ko L.; Cardona G. R.; Chin W. W. Thyroid hormone receptor-binding protein an LXXLL motif-containing protein, functions as a general coactivator. Proc. Natl. Acad. Sci. USA 97:6212–6217; 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kobayashi K.; Sueyoshi T.; Inoue K.; Morre R.; Negishi M. Cytoplasmic accumulation of the nuclear receptor CAR by a tetratricopeptide repeat protein in HepG2 cells. Mol. Pharmacol. 64:1069–1075; 2003. [DOI] [PubMed] [Google Scholar]
- 27. Koike C.; Moore R.; Negishi M. Localization of the nuclear receptor CAR at the cell membrane of mouse liver. FEBS Lett. 579:6733–6736; 2005. [DOI] [PubMed] [Google Scholar]
- 28. Kuang S. Q.; Liao L.; Zhang H.; Pereira F. A.; Yuan Y.; DeMayo F. J.; Ko L.; Xu J. Deletion of the cancer amplified coactivator AIB3 results in defective placentation and embryonic lethality. J. Biol. Chem. 277:45356–45360; 2002. [DOI] [PubMed] [Google Scholar]
- 29. Lee S. S. T.; Pineau T.; Drago J.; Lee E. J.; Owens J. W.; Kroetz D. L.; Fernandez-Salguero P. M.; Westphal H.; Gonzalez F. J. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol. Cell. Biol. 15:3012–3022; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee S. K.; Anzick S. L.; Choi J. E.; Bubendorf L.; Guan X-Y.; Jung Y. K.; Kallioniemi O. P.; Kononen J.; Trent J. M.; Azorsa D.; Jhun B. H.; Cheong J. H.; Lee Y. C.; Meltzer P. S.; Lee J. W. A nuclear factor, ASC-2, is a cancer-amplified transcriptional coactivators essential for ligand-dependent transactivation by nuclear receptors in vivo. J. Biol. Chem. 274:34283–34293; 1999. [DOI] [PubMed] [Google Scholar]
- 31. Leung Y-K.; Mak P.; Hassan S.; Ho S-M. Estrogen receptor (ER)-b isoforms: A key to understanding ER-signaling. Proc. Natl. Acad. Sci. USA 103:13162–13167; 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lewis B. A.; Reinberg D. The mediator coactivator complex: Functional and physical roles in transcriptional regulation. J. Cell Sci. 116:3667–3675; 2003. [DOI] [PubMed] [Google Scholar]
- 33. Mahajan M. A.; Samuels H. H. A new family of nuclear receptor coregulators that integrate nuclear receptor signaling through CREB-binding protein. Mol. Cell. Biol. 20:5048–5063; 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mahajan M. A.; Das S.; Zhu H.; Tomic-Canic M.; Samuels H. H. The nuclear hormone receptor coactivator NRC is a pleiotropic modulator affecting growth, development, apoptosis, reproduction, and wound repair. Mol. Cell. Biol. 24:4994–5004; 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McKenna N. J.; O’Malley B. W. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 108:465–474; 2002. [DOI] [PubMed] [Google Scholar]
- 36. Min G.; Kemper J. K.; Kemper B. Glucocorticoid receptor-interacting protein 1 mediates ligand independent nuclear translocation and activation of constitutive androstane receptor in vivo. J. Biol. Chem. 277:26356–26363; 2002. [DOI] [PubMed] [Google Scholar]
- 37. Misra P.; Qi C.; Yu S.; Shah S. H.; Cao W-Q.; Rao M. S.; Thimmapaya B.; Zhu Y.; Reddy J. K. Interaction of PIMT with transcription coactivators CBP, p300, and PBP differential role in transcriptional regulation. J. Biol. Chem. 277:20011–20019; 2002. [DOI] [PubMed] [Google Scholar]
- 38. Numazawa S.; Shindo S.; Maruyama K.; Chibana F.; Kawahara Y.; Ashino T.; Tanaka S.; Yoshida T. Impaired nuclear translocation of CAR in hepatic pre-neoplastic lesions: Association with an attenuated CYP2B induction by phenobarbital. FEBS Lett. 579:3560–3564; 2005. [DOI] [PubMed] [Google Scholar]
- 39. Onate S. A.; Tsai S. Y.; Tsai M.-J.; O’Malley B. W. Sequence and characterization of coactivator of the steroid hormone receptor superfamily. Science 270:1354–1357; 1995. [DOI] [PubMed] [Google Scholar]
- 40. Pascussi J-M.; Gerbal-Chaloin S.; Fabre J-M.; Muarel P.; Vilarem M-J. Dexamethasone enhances constitutive androstane receptor expression in human hepatocytes: Consequences on cytochrome p450 gene regulation. Mol. Pharmacol. 58:1441–1450; 2000. [DOI] [PubMed] [Google Scholar]
- 41. Qi C.; Kashireddy P.; Zhu Y. T.; Rao S. M.; Zhu Y.-J. Null mutation of peroxisome proliferator-activated receptor-interacting protein in mammary glands causes defective mammopoiesis. J. Biol. Chem. 279:33696–33701; 2004. [DOI] [PubMed] [Google Scholar]
- 42. Qi C.; Zhu Y.; Reddy J. K. Peroxisome proliferator-activated receptors, coactivators, and downstream targets. Cell. Biochem. Biophys. 32:187–204; 2000. [DOI] [PubMed] [Google Scholar]
- 43. Qi C.; Zhu Y.; Pan J.; Yeldandi A. V.; Rao M. S.; Maeda N.; Subbarao V.; Pulikuri S.; Hashimoto T.; Reddy J. K. Mouse steroid receptor coactivator-1 is not essential for peroxisome proliferator activated receptor α-regulated gene expression. Proc. Natl. Acad. Sci. USA 96:1585–1590; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rachez C.; Lemon B. D.; Suldan Z.; Bromleigh V.; Gamble M.; Naar A. M.; Erdjument-Bromage H.; Tempst P.; Freedman L. P. Ligand-dependent transcription activation by nuclear receptors requires the DRIP complex. Nature 398:824–828; 1999. [DOI] [PubMed] [Google Scholar]
- 45. Reddy J. K. Peroxisome proliferators and peroxisome proliferators-activated receptor α. Biotic and xenobiotic sensing. Am. J. Pathol. 164:2305–2321; 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Reddy J. K.; Guo D.; Jia Y.; Yu S.; Rao M. S. Nuclear receptor transcriptional coactivators in development and metabolism. Adv. Dev. Biol. 16:389–420; 2006. [Google Scholar]
- 47. Reddy J. K.; Rao M. S.; Azarnoff D. L.; Sell S. Mitogenic and carcinogenic effects of a hypolipidemic peroxisome proliferator, [4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio]acetic acid (Wy-14,643), in rat and mouse liver. Cancer Res. 39:152–161; 1979. [PubMed] [Google Scholar]
- 48. Roeder R. G. Transcriptional regulation and the role of diverse coactivators in animal cells. FEBS Lett. 579:909–915; 2005. [DOI] [PubMed] [Google Scholar]
- 49. Ryu S.; Zhou S.; Ladurner A. G.; Tjian R. The transcriptional cofactor complex CRSP is required for activity of the enhancer-binding protein Spl. Nature 397:446–450; 1999. [DOI] [PubMed] [Google Scholar]
- 50. Shiraki T.; Sakai N.; Kanaya E.; Jingami H. Activation of orphan nuclear constitutive androstane receptor requires subnuclear targeting by peroxisome proliferator-activated receptor γ coactivators-1α. A possible link between xenobiotic response and nutritional state. J. Biol. Chem. 278:11344–11350; 2003. [DOI] [PubMed] [Google Scholar]
- 51. Squires E. J.; Sueyoshi T.; Negishi M. Cytoplasmic localization of pregnane X receptor and ligand-dependent nuclear translocation in mouse liver. J. Biol. Chem. 279:49307–49314; 2004. [DOI] [PubMed] [Google Scholar]
- 52. Surapureddi S.; Yu S.; Bu H.; Hashimoto T.; Yeldandi A. V.; Kashireddy P.; Cherkaoui-Malaki M.; Qi C.; Zhu Y.-J.; Rao M. S.; Reddy J. K. Identification of a transcriptionally active peroxisome proliferator-activated receptor alpha-interacting cofactor complex in rat liver and characterization of PRIC285 as a coactivator. Proc. Natl. Acad. Sci. USA 99:11836–11841; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Swales K.; Negishi M. CAR, driving into the future. Mol. Endocrinol. 18:1589–1598; 2004. [DOI] [PubMed] [Google Scholar]
- 54. Wang Z.; Rose D. W.; Hermanson O.; Liu F.; Herman T.; Wu W.; Szeto D.; Gleiberman A.; Krones A.; Pratt K.; Rosenfeld R.; Glass C. K.; Rosenfeld M. G. Regulation of somatic growth by the p160 coactivator p/CIP. Proc. Natl. Acad. Sci. USA 97:13549–13554; 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang Z.; Qi C.; Krones A.; Woodring P.; Zhu X.; Reddy J. K.; Evans R. M.; Rosenfeld M. G.; Hunter T. Critical roles of the p160 transcriptional coactivatros p/CIP and SRC-1 in energy balance. Cell Metabol. 3:111–122; 2006. [DOI] [PubMed] [Google Scholar]
- 56. Wei P.; Zhang J.; Egan-Hafley M.; Liang S.; Moore D. D. The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature 407:920–923; 2000. [DOI] [PubMed] [Google Scholar]
- 57. Wyde M. E.; Bartolucci E.; Ueda A.; Zhang H.; Yan B.; Negishi M.; You L. The environmental pollutant 1,1,-dichloro-2,2-bis(p-chlorophrnyl)ethylene induces rat hepatic cytochrome P450 2B and 3A expression through the constitutive androstane receptor and pregnane X receptor. Mol. Pharmacol. 64:474–481; 2003. [DOI] [PubMed] [Google Scholar]
- 58. Xia J.; Kemper B. Structural determinant of constitutive androstane receptor required for its glucocorticoid receptor interacting protein-1-mediated nuclear accumulation. J. Biol. Chem. 280:7285–7293; 2005. [DOI] [PubMed] [Google Scholar]
- 59. Xu J.; Liao L.; Ning G.; Yoshida-Komiya H.; Deng C.; O’Malley B. W. The steroid receptor coactivators SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc. Natl. Acad. Sci. USA 97:6379–6384; 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xu J.; Qiu Y.; DeMayo F. J.; Tsai S. Y.; Tsai M. J.; O’Malley B. W. Partial hormone resistance in mice with disruption of the steroid receptor coactivator-1 (SRC-1) gene. Science 279:1922–1925; 1998. [DOI] [PubMed] [Google Scholar]
- 61. Yakar S.; Liu J.-L.; Stannard B.; Butler A.; Accili D.; Sauer B.; Le Roith D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc. Natl. Acad. Sci. USA 96:7342–7329; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yu S.; Reddy J. K. Transcription coactivators for peroxisome proliferator-activated receptors. Biochim. Biophys. Acta; in press. [DOI] [PubMed]
- 63. Yuan C. X.; Ito M.; Fondell J. D.; Fu Z. Y.; Roeder R. G. The TRAP220 component of a thyroid hormone receptor-associated protein (TRAP) coactivator complex interacts directly with nuclear receptors in a ligand-dependent fashion. Proc. Natl. Acad. Sci. USA 95:7939–7944; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zelko I.; Sueyoshi T.; Kawamoto T.; Moore R.; Negishi M. The peptide near the C-terminus regulates receptor CAR nuclear translocation induced by xenochemicals in mouse liver. Mol. Cell. Biol. 21:2838–2846; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang J.; Huang W.; Chua S. S.; Wei P.; Moore D. D. Modulation of acetaminophen-induced hepatotoxicity by the xenobiotic receptor CAR. Science 298:422–424; 2002. [DOI] [PubMed] [Google Scholar]
- 66. Zhu Y. J.; Crawford S. E.; Stellmach V.; Dwivedi R. S.; Rao M. S.; Gonzalez F. J.; Qi C.; Reddy J. K. Coactivator PRIP, the peroxisome proliferator-activated receptor-interacting protein, is a modulator of placental, cardiac, hepatic and embryonic development. J. Biol. Chem. 278:1986–1990; 2003. [DOI] [PubMed] [Google Scholar]
- 67. Zhu Y.; Kan L.; Qi C.; Kanwar Y. S.; Yeldandi A. Y.; Rao M. S.; Reddy J. K. Isolation and characterization of peroxisome proliferator-activated receptor (PPAR) interacting protein (PRIP) as a coactivator for PPAR. J. Biol. Chem. 275:13510–13516; 2000. [DOI] [PubMed] [Google Scholar]
- 68. Zhu Y.; Qi C.; Jain S.; Rao M. S.; Reddy J. K. Isolation and characterization of PBP, a protein that interacts with peroxisome proliferator-activated receptor. J. Biol. Chem. 272:25500–25506; 1997. [DOI] [PubMed] [Google Scholar]
- 69. Zhu Y.; Qi C.; Jia Y.; Nye J. S.; Rao M. S.; Reddy J. K. Deletion of PBP/PPARBP, the gene for nuclear receptor coactivator peroxisome proliferator-activated receptor-binding protein, results in embryonic lethality. J. Biol. Chem. 275:14779–14782; 2000. [DOI] [PubMed] [Google Scholar]
- 70. Zhu Y.; Qi C.; Cao W-Q.; Yeldandi A. V.; Rao M. S.; Reddy J. K. Cloning and characterization of PIMT, a protein with methyltransferase domain, which interacts with and enhances nuclear receptor coactivator PRIP function. Proc. Natl. Acad. Sci. USA 98:10380–10385; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]