

Abstract
Animal models suggest that a deficiency in myeloperoxidase (MPO; EC 1.11.1.7), a lysosomal hemoprotein involved in host defense, may be associated with a decreased level of immunity. A nonsynonymous mutation, resulting in an arginine to cysteine substitution (Arg499Cys or R499C), has been identified in the exon 9 genetic coding region of a Japanese patient with complete MPO deficiency. Genetic analysis revealed that the mRNA of the patient could be correctly transcribed then further translated into a peptide sequence. However, the Western blot analysis confirmed the absence of MPO peptides. An initial screening assay of the patient’s blood exhibited an abnormal hematograph, and no MPO activity was detected. To determine if this mutation might be associated with MPO deficiency, DNA samples for 387 controls were examined. Genetic analysis was performed using standard PCR techniques for amplification and sequencing. None of the control samples possessed the R499C substitution. This mutation is in close proximity to a different mutation (G501S) previously found in another Japanese MPO-deficient patient, and the amino acid, H502, which is strongly involved in heme binding, leading to the speculation that heme binding may play a role in complete MPO deficiency.
Key words: Myeloperoxidase (MPO), Myeloperoxidase deficiency, Arg499Cys, R499C
INTRODUCTION
Upon comparison with the United States and Europe, complete myeloperoxidase deficiency [Mendelian inheritance in Man (MIM) #254600] is considered a rare occurrence in the Japanese population with a prevalence of 1.75/100,000 (15). Located in neutrophils and monocytes, myeloperoxidase (MPO) plays a role in host defense against a wide range of organisms. Mouse studies by Aratani et al. (1,2) suggest a link between MPO deficiency and an increased occurrence of infections.
In the biosynthesis of MPO, the translational product is first converted into the enzymatically inactive apoproMPO. Heme binding transforms this protein to an active proMPO form and subsequently mature MPO. It has been postulated that MPO deficiency may be a result of its inability to undergo posttranslational processing (11,12). Though the translational process leaves a margin for the development of MPO deficiency, genetic anomalies in the coding region also contribute to the inability to produce this enzyme.
To date, there have been four allelic mutations related to MPO deficiency. These include: arginine to tryptophan substitution (R569W) (11,12), tyrosine to cysteine conversion (Y173C) (4), methionine to threonine substitution (M251T) (14), and glycine to serine conversion (G501S) (13). In addition to nonsynonymous mutations, studies have found enzyme deficiency due to alternative splicing and deletions in the genetic sequence for MPO (6,14).
This study presents a novel nonsynonymous mutation found in a Japanese patient with complete MPO deficiency. This point mutation is in close proximity to the G501S mutation, also identified in the Japanese population, as well as the amino acid histidine, at position 502 (H502), that is involved in heme binding. DNA samples from the indigenous population were analyzed to determine the probable prevalence of the mutation.
MATERIALS AND METHODS
Subjects
A 28-year-old Japanese female presented with complete MPO deficiency as identified by Bayer-Technicon automated hematography (15). Blood from one healthy control was sampled along with the patient for simultaneous measurement of enzymatic activity and superoxide anion production. DNA samples from 387 individuals were utilized in the genetic analysis. These genetic samples included rheumatoid arthritis patients (21%), hepatitis C patients (41%), and healthy volunteers (38%). No information on the activity of MPO or superoxide dismutase was available for these donors. Informed consent was obtained from all subjects. The low prevalence of both complete and partial MPO deficiency was used to justify the unlikelihood of these DNA samples being from MPO-deficient donors.
MPO Activity and Superoxide Production
MPO release and superoxide anion production from neutrophils were assayed as described previously (8). Briefly, neutrophils were prewarmed at 37°C then transferred onto plates containing cytochalsin B (CB) and fMet-Leu-Phe (FMLP) for 10 min. The cell suspension was then centrifuged at 400 × g for 5 min. MPO activities in the supernatant and in the homogenate of the cell pellet were measured spectrophotometrically using 3,3′,5,5′-tetramethylbenzidine as a substrate (16,17). For elucidating superoxide anion production, neutrophils were mixed with ferricytochrome C for 2 min at 37°C prior to the addition of CB and FMLP. Total MPO activity was calculated as the sum of the enzyme activity in supernatants and cell pellet homogenates after treatment with CB and FMLP. MPO release was expressed as the percentage of total enzyme activity (MPO activity in supernatant ÷ total enzyme activity). Superoxide anion production was determined by measuring the increase in absorbance at 550 nm in 1-min intervals using a microplate reader.
Western Blot Analysis
Western blot analysis was performed on the patient and the healthy control using the method described previously (13). Briefly, neutrophils were dissolved in a loading buffer, consisting of 50 mM Tris-HCl, pH 6.8, 10% glycerol, 1% sodium dodecyl sulfate (SDS), 0.3 M β-mercaptoethanol, and 0.01% bromophenol blue, and incubated at 98°C for complete reduction. The samples were fractionated by electrophoresis on 8–12% SDS polyacrylamide gel and transferred onto a PVDF membrane (Millipore, Bedford, MA, USA). Antibodies used for immunodetection included 1:2000 anti-human MPO rabbit antibody (Dako, Glostrup, Denmark) at room temperature for 2 h, followed by an incubation with a 1:2000 alkaline phosphatase-conjugated anti-rabbit IgG goat antibody (Organon Teknika, Durham, NC, USA) for 2 h. The PVDF membrane was visualized using an AP conjugate substrate kit (Bio-Rad, Hercules, CA, USA).
cDNA and Genomic DNA Analysis
MPO cDNA was acquired from the mRNA of mononuclear cells (MNC) by polymerase chain reaction (PCR) after reverse transcription (RT) reaction with AMV reverse transcriptase, using the specific primer pairs for MPO mRNA as described in Ohashi et al. (13). Genomic fragments were also amplified by PCR. The obtained RT-PCR products were compared with a normal MPO cDNA sequence (7,9,18).
DNA segments obtained were purified with the QIAquick PCR purification system (Qiagen, Hilden, Germany) and served as templates in a PRISM Ready Reaction Dye Terminator Cycle Sequencing procedure utilizing an automatic sequencer (Applied Biosystems, CA, USA). Sequences were then analyzed using Genetyx-win software (Software Development Co., Ltd., Tokyo, Japan).
RESULTS
MPO Activity of Neutrophils
Diminished levels of MPO activity were detected in the patient (i.e., 0.9 units/106 cells), which is a level approximately one sixth that observed in the healthy control (Table 1). The patient’s elevated superoxide dismutase activity, compared to the healthy control, was reflective of MPO deficiency.
TABLE 1.
MPO AND SUPEROXIDE DISMUTASE ACTIVITY IN THE PATIENT AND HEALTHY CONTROL
| MPO Activity (Units/106 Cells) | Superoxide Anion Production (nmol/106 Cells) | |
|---|---|---|
| Patient | 0.9 | 10.5 |
| Control | 5.2 | 6.8 |
Western Blot Analysis
Western blot analysis of the neutrophils from the patient and the healthy control was performed alongside purified MPO, using anti-human MPO antibody. While the healthy control and purified MPO clearly showed 59 kDa of heavy chain and 14 kDa of light subunit, MPO peptides could not be confirmed in the patient (Fig. 1).
Figure 1.
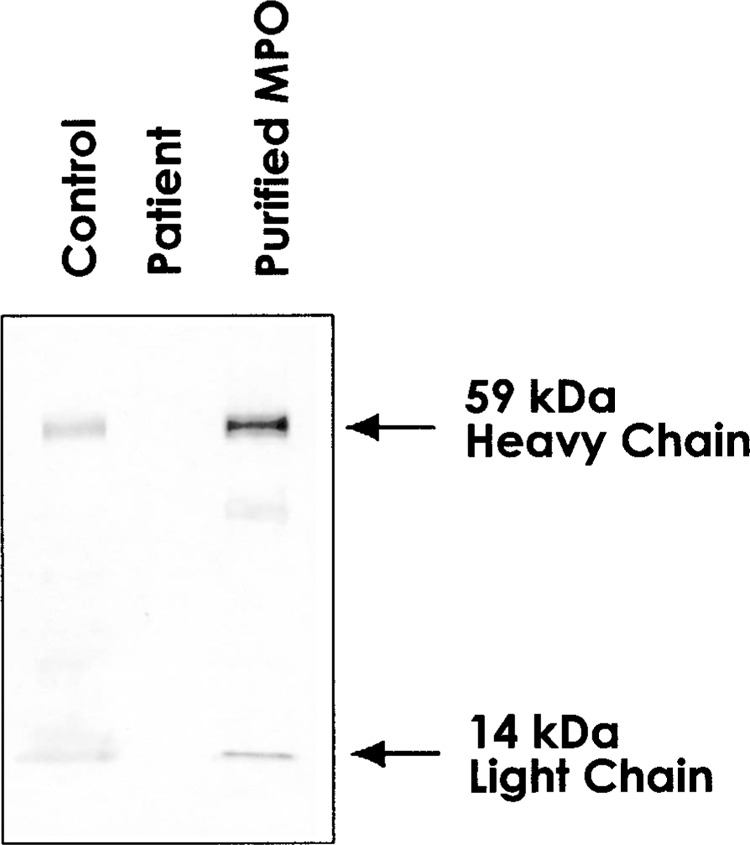
Western blot analysis. Proteins in neutrophils of the patient, normal control, and purified MPO protein were fractionated on 8–12% polyacrylamide gel, transferred on PVDF membrane, and detected by rabbit anti-human MPO antibody. MPO polypeptides are indicated on the right.
Mutational Analysis
No alternative splice variations were found in the complementary DNA (cDNA) of the MPO mRNA, obtained from the patient’s monocytes. Notably, this observation was also recorded by Ohashi et al. (13). The sequenced cDNA fragment lengths were found to be the appropriate length for each region. This finding shows that the MPO gene in the patient was normally transcribed without any alternative transcription. Both the genomic and cDNA revealed a point mutation in the patient sample (Fig. 2): a non-synonymous homozygous mutation in the coding region of exon 9 with thymine (T) being substituted for cytosine (C) at the 1495th position from the adenine of the first AUG codon. This C to T substitution resulted in an arginine to cysteine substitution (R499C). None of the 387 DNA samples were found to possess the novel R499C mutation identified in the patient.
Figure 2.
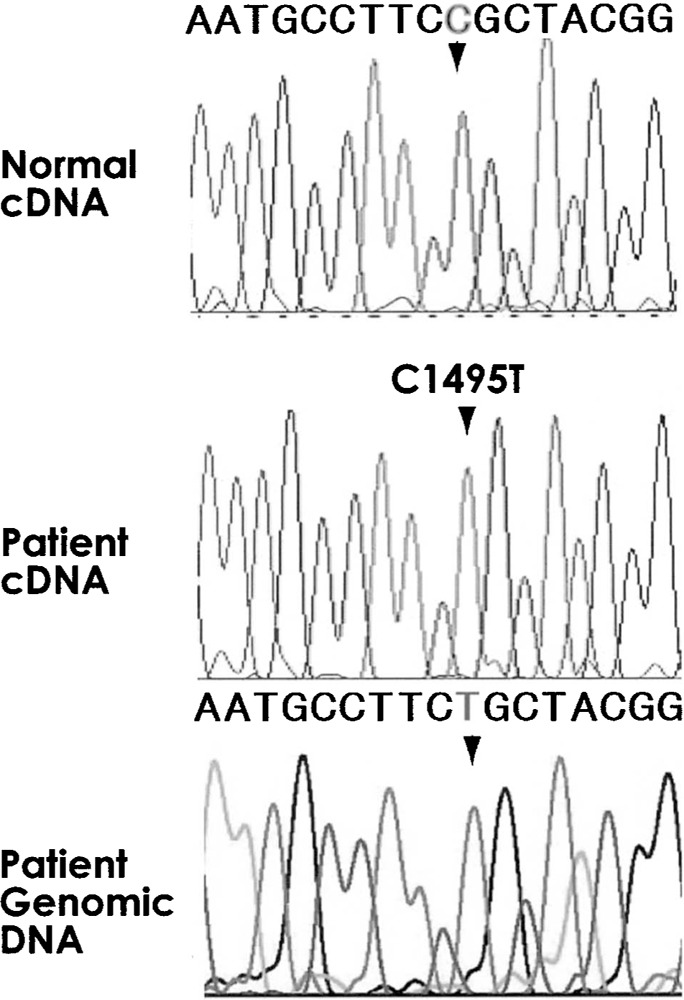
Sequence histogram around the mutation point of cDNA and genomic DNA of the patient. Top panel shows a histogram of the same region from normal control. In the middle and bottom are histograms of cDNA and genomic DNA from the patient. Arrowheads indicate the mutation position 1495.
DISCUSSION
Complete deficiency of MPO, originating from a novel mutation, is a unique phenomenon, requiring two recessive alleles due to the bi-allelic manner of MPO gene expression. With rare migration rates throughout different areas of Japan, these homozygous patients may serve as a sentinel in identifying clusters with the same mutation. The biosynthesis of MPO includes N-linked glycosylation, heme insertion, proteolytic processing, and dimerization (5). Our findings reveal another mutation in the MPO gene that may be responsible for MPO deficiency. Both the R499C mutation and the previously published G501S mutation found in MPO-deficient Japanese individuals are proximal to the histidine in position 502 (H502) in the myeloperoxidase structure (3,19). This amino acid is strongly involved with heme binding and thus a mutation near this area might interfere with subsequent development of mature MPO (Fig. 3). Our findings also support the hypothesis that immature peptides are disrupted, as the Western blot analysis did not reveal any peptides with the same antigenic determinants of MPO. Lastly, among the almost 400 Japanese individuals tested no nonsynonymous mutations in exon 9 genetic coding region were identified.
Figure 3.
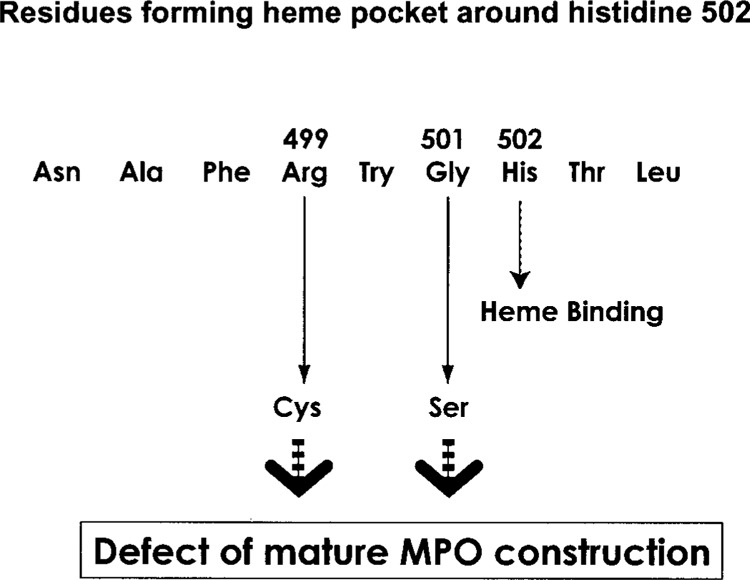
Schematic diagram of the amino acid residues around histidine 502. R499C substitution causes a similar effect on mature MPO construction with G501S.
ACKNOWLEDGMENTS
This study was supported partly by a grant from the Ministry of Health, Labour and Welfare and Human Science Foundation Japan.
REFERENCES
- 1. Aratani Y.; Kura F.; Watanabe H.; Akagawa H.; Takano Y.; Suzuki K.; Maeda N.; Koyama H. Differential host susceptibility to pulmonary infections with bacteria and fungi in mice deficient in myeloperoxidase. J. Infect. Dis. 182:1276–1279; 2000. [DOI] [PubMed] [Google Scholar]
- 2. Aratani Y.; Koyama H.; Nyui S.; Suzuki K.; Kura F.; Maeda N. Severe impairment in early host defense against Candida albicans in mice deficient in myeloperoxidase. Infect. Immun. 67:1828–1836; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Davey C. A.; Fenna R. E. 2.3 A resolution X-ray crystal structure of the bisubstrate analogue inhibitor salicylhydroxamic acid bound to human myeloperoxidase: A model for a prereaction complex with hydrogen peroxide. Biochemistry 35:10967–10973; 1996. [DOI] [PubMed] [Google Scholar]
- 4. DeLeo F. R.; Goedken M.; McCormick S. J.; Nauseef W. M. A novel form of hereditary myeloperoxidase deficiency linked to endoplasmic reticulum/proteasome degradation. J. Clin. Invest. 101:2900–2909; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gullberg U.; Andersson E.; Garwicz D.; Lindmark A.; Olsson I. Biosynthesis, processing and sorting of neutrophil proteins: Insight into neutrophil granule development. Eur. J. Haematol. 58:137–153; 1997. [DOI] [PubMed] [Google Scholar]
- 6. Hashinaka K.; Nishio C.; Hur S. J.; Sakiyama F.; Tsunasawa S.; Yamada M. Multiple species of myeloperoxidase messenger RNAs produced by alternative splicing and differential polyadenylation. Biochemistry 27:5906–5914; 1988. [DOI] [PubMed] [Google Scholar]
- 7. Johnson K. R.; Nauseef W. M.; Care A.; Wheelock M. J.; Shane S.; Hudson S.; Koeffler H. P.; Selsted M.; Miller C.; Rovera G. Characterization of cDNA clones for human myeloperoxidase: predicted amino acid sequence and evidence for multiple mRNA species. Nucleic Acids Res. 15:2013–2028; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kawai Y.; Okawarab A. I.; Okuyama H.; Kura F.; Suzuki K. Modulation of chemotaxis, O2- production and myeloperoxidase release from human polymorphonuclear leukocytes by the ornithine-containing lipid and serineglycine-containing lipid of Flavobacterium. FEMS Immunol. Med. Microbiol. 28:205–209; 2000. [DOI] [PubMed] [Google Scholar]
- 9. Morishita K.; Kubota N.; Asano S.; Kaziro Y.; Nagata S. Molecular cloning and characterization of cDNA for human myeloperoxidase. J. Biol. Chem. 262:3844–3851; 1987. [PubMed] [Google Scholar]
- 10. Nauseef W. M.; Cogley M.; Bock S.; Petrides P. E. Pattern of inheritance in hereditary myeloperoxidase deficiency associated with the R569W missense mutation. J. Leukoc. Biol. 63:264–269; 1998. [DOI] [PubMed] [Google Scholar]
- 11. Nauseef W. M.; Cogley M.; McCormick S. Effect of the R569W missense mutation on the biosynthesis of myeloperoxidase. J. Biol. Chem. 271:9546–9549; 1996. [DOI] [PubMed] [Google Scholar]
- 12. Nauseef W. M.; Brigham S.; Cogley M. Hereditary myeloperoxidase deficiency due to a missense mutation of arginine 569 to tryptophan. J. Biol. Chem. 269:1212–1216; 1994. [PubMed] [Google Scholar]
- 13. Ohashi Y. Y.; Kameoka Y.; Persad A. S.; Koi F.; Yamagoe S.; Hashimoto K.; Suzuki K. Novel missense mutation found in a Japanese patient with myeloperoxidase deficiency. Gene 327:195–200; 2004. [DOI] [PubMed] [Google Scholar]
- 14. Romano M.; Dri P.; Dadalt L.; Patriarca P.; Baralle F. E. Biochemical and molecular characterization of hereditary myeloproliferative deficiency. Blood 90:4126–4134; 1997. [PubMed] [Google Scholar]
- 15. Suzuki K.; Nunoi H.; Miyazaki M.; Koi F. Prevalence of inherited myeloperoxidase deficiency in Japan. In: Petrides P. E.; Nauseef W. M., eds. The peroxidase multigene family of enzymes. Berlin: Springer-Verlag; 2000:45–149. [Google Scholar]
- 16. Suzuki K.; Yamada M.; Akashi K.; Fujikura T. Similarity of kinetics of three types of myeloperoxidase from human leukocytes and four types from HL-60 cells. Arch. Biochem. Biophys. 245:167–173; 1986. [DOI] [PubMed] [Google Scholar]
- 17. Suzuki K.; Ota H.; Sasagawa S.; Sakatani T.; Fujikura T. Assay method for myeloperoxidase in human polymorphonuclear leukocytes. Anal. Biochem. 132:345–352; 1983. [DOI] [PubMed] [Google Scholar]
- 18. Yamada M.; Hur S. J.; Hashinaka K.; Tsuneoka K.; Saeki T.; Nishio C.; Sakiyama F.; Tsunasawa S. Isolation and characterization of a cDNA coding for human myeloperoxidase. Arch. Biochem. Biophys. 255:147–155; 1987. [DOI] [PubMed] [Google Scholar]
- 19. Zeng J.; Fenna R. E. X-ray crystal structure of canine myeloperoxidase at 3 A resolution. J. Mol. Biol. 226:185–207; 1992. [DOI] [PubMed] [Google Scholar]