

Abstract
Kidney disease is an increasingly common comorbidity that alters the pharmacokinetics of many drugs. Prescribing to patients with kidney disease requires knowledge about the drug, the extent of the patient’s altered physiology, and pharmacokinetic principles that influence the design of dosing regimens. There are multiple physiologic effects of impaired kidney function, and the extent to which they occur in an individual at any given time can be difficult to define. Although some guidelines are available for dosing in kidney disease, they may be on the basis of limited data or not widely applicable, and therefore, an understanding of pharmacokinetic principles and how to apply them is important to the practicing clinician. Whether kidney disease is acute or chronic, drug clearance decreases, and the volume of distribution may remain the same or increase. Although in CKD, these changes progress relatively slowly, they are dynamic in AKI, and recovery is possible depending on the etiology and treatments. This, and the use of kidney replacement therapies further complicate attempts to quantify drug clearance at the time of prescribing and dosing in AKI. The required change in the dosing regimen can be estimated or even quantitated in certain instances through the application of pharmacokinetic principles to guide rational drug dosing. This offers an opportunity to provide personalized medical care and minimizes adverse drug events from either under- or overdosing. We discuss the principles of pharmacokinetics that are fundamental for the design of an appropriate dosing regimen in this review.
Keywords: pharmacokinetics, kidney disease, chronic kidney disease, acute kidney injury, dialysis, clearance, volume of distribution
Introduction
Drugs are an important and frequently used treatment for patients with kidney disease. Knowledge of basic pharmacokinetic principles is important for all prescribers, but it is particularly important for nephrologists and other physicians who routinely see patients with organ dysfunction that affects drug handling.
Prescribing to patients with kidney disease is complicated, because kidney disease has multiple effects on pharmacokinetics, and these effects are dependent on both the drug and the clinical context. For example, kidney disease may be chronic (slowly progressive over months or years) or acute (rapidly evolving), and each scenario requires a different approach to drug dosing. Understanding how changes to physiology affect the pharmacokinetics of a given drug is essential to rational drug use and the optimization of treatment regimens.
Failure to properly account for the effect of kidney disease when designing appropriate drug-dosing regimens can predispose an individual to treatment failure or adverse drug events. Guidelines for adjustment of the dosing regimen in varying stages of CKD are provided by the manufacturer.
Furthermore, dose recommendations in the setting of kidney disease are frequently on the basis of limited data, and they may not adequately account for interindividual variability or acute changes, such as during AKI. For example, between 2002 and 2007, only 57% of new drug applications to the Food and Drug Administration (FDA) examined pharmacokinetics in kidney impairment, and only 44% of those with data in kidney impairment evaluated patients on hemodialysis (1). This reflects the FDA policy that manufacturers are not required to determine the effect of kidney disease on drug dosing (2).
In many cases, it is reasonable to simply prescribe the dose recommended by the manufacturer, particularly if the drug has a wide therapeutic index, the duration of therapy is short, the dose is low (e.g., prophylaxis dosing), and/or there is the ability to titrate the dose on the basis of clinical and laboratory progression. Other dosing guidance is available through textbooks, online references, and local procedures for many drugs but not all, and there may be significant differences in the suggested change in dose between different resources (3). Unfortunately, limited data or other safety concerns may simply lead the manufacturer to declare that the drug is contraindicated in patients with advanced kidney disease, which can deprive patients with kidney disease of important drug options.
In recent years, the application of pharmacokinetic principles to drugs in the postmarketing phase has raised the prospect of using drugs that were previously considered contraindicated in patients with eGFR<30 ml/min per 1.73 m2, including metformin (4) and novel-acting oral anticoagulants (NOACs) or direct-acting oral anticoagulants (DOACs; for example, apixaban) (5). There may also be circumstances when additional adjustments to the dosing regimen may be required in a patient (for example, a change in clearance due to a coprescribed drug that induces or inhibits elimination pathways of the index drug).
Therefore, it is necessary to have a rational approach to prescribing in patients with kidney disease. This requires knowledge about pharmacokinetic principles, properties of the drug, and how the drug will be handled by an individual patient. The purpose of this review is to provide an overview of pharmacokinetic principles that affect the design of a dosing regimen and provide the basis for discussions regarding the delivery of personalized medicine to those with kidney disease.
Relationship between Dosing Regimen and the Effect of a Drug
An individual’s response to a drug is determined by both the pharmacokinetics and pharmacodynamics of that drug. Pharmacodynamics is concerned with the effect of the drug on the body, including interactions between the drug, its target, and downstream biochemical effects. Pharmacokinetics describes the effect of the body on a drug and reflects the physiologic processes of absorption, distribution, metabolism, and excretion. Each of these processes may be altered in patients with kidney disease and affect therapeutic outcomes.
The concentration-time profile of a drug reflects the net effects of these pharmacokinetic processes after drug administration (Figure 1). The concentration-time profile approximates the clinical effect of most drugs, and drug exposure relates to the maximum plasma concentration (Cmax) and/or the area under the concentration-time curve (AUC). In general, high drug exposures increase the risk of adverse drug reactions, and low drug exposures are ineffective.
Figure 1.

Plasma concentration-time profile after oral administration of a single dose. The components relevant to the pharmacokinetics of a drug’s concentration-time profile are the peak or maximum plasma concentration (Cmax) and the time when it occurs (Tmax), the area under the concentration-time curve (represented as shaded area), and the elimination t1/2 (determined using the blue lines).
When the changes in pharmacokinetics due to kidney disease and other conditions are understood, the dosing regimen can be adjusted so that the concentration-time profile is optimized for the individual.
Reasons to Optimize Dosing Regimens
Either sub- or supratherapeutic dosing can occur when appropriate dose adjustments are not made in patients with kidney disease, and both have negative effects on patient outcomes, including morbidity, prolonged hospital admissions, and potentially, death. Subtherapeutic dosing increases the risk of treatment failure, which may be life threatening (e.g., anti-infectives) or organ threatening (e.g., immunosuppressive drugs). The risk of supratherapeutic exposure from drugs (or their active or toxic metabolites) that rely on kidney elimination is amplified when the drug has a narrow therapeutic index, such as digoxin or lithium. In many cases, accumulation develops over weeks, and the onset of drug toxicity is insidious. These principles are reflected in the examples below.
Selected Examples of Drugs That Require Special Consideration When Prescribing to Patients with Kidney Disease
Antibiotics
The efficacy of antibiotics depends on their concentration relative to the minimum inhibitory concentration (MIC) of the culprit bacteria. Three pharmacokinetic-pharmacodynamic targets describe features of the concentration-time profile that maximize antibiotic efficacy.
(1) The ratio of maximum free drug plasma concentration to MIC (concentration dependent; e.g., aminoglycosides)
(2) The ratio of the AUC to MIC (AUC:MIC; e.g., vancomycin)
(3) The proportion of time that the plasma concentration exceeds the MIC (time-dependent killing; e.g., β-lactam antibiotics)
Therefore, the actual target depends on the specific antibiotic and the MIC of the culprit bacteria (6). Plasma concentrations below the target concentration predispose to therapeutic failure and development of multiresistant organisms. Although prescribing higher doses increases the likelihood of achieving pharmacokinetic-pharmacodynamic targets, it also increases the risk of adverse events, including in drugs considered to have a wide therapeutic index, like β-lactam antibiotics (7).
Lithium and Digoxin
Lithium excretion depends on kidney function. The most common cause of adverse reactions to lithium is chronic poisoning, which typically occurs in the setting of reduced kidney function as a result of dehydration or dose adjustment with inadequate monitoring (8,9). The resultant neurotoxicity can be severe and persist for days or weeks, and in rare instances, it can be irreversible (9).
Similarly, digoxin has a narrow therapeutic index and accumulates when there is impaired kidney function if the dose is not decreased (10). Digoxin poisoning is reasonably common, being associated with prolonged hospital admissions and high resource utilization, including antidigoxin Fab (11).
Both agents commonly undergo therapeutic drug monitoring, and the frequency at which this occurs should be increased in settings where the drug clearance (CL) is significant reduced or where this fluctuates, as in AKI.
Cyclophosphamide
Cyclophosphamide is used to treat various autoimmune diseases and malignancies, and much of the effect of cyclophosphamide occurs through CYP450-mediated formation of active metabolites, which are eliminated by the kidney. Thus, active dose reductions are performed in patients with impaired kidney function (e.g., plasma creatinine concentration >300 μmol/L [12]) in autoimmune disease and oncology (13) to limit the accumulation of cyclophosphamide and active metabolites (13,14). Cyclophosphamide bioactivation may increase in patients with GN compared with those with other types of kidney disease, which may prompt different approaches to dose adjustment (15). Inadequate dose reductions of cyclophosphamide in CKD may contribute to the increased adverse events and death in patients with systemic vasculitis in the first 12 months of treatment (16). However, studies have also highlighted that low-dose cyclophosphamide reduces treatment efficacy in, for example, the treatment of lupus nephritis (17). Therefore, more research is required to determine how to optimize cyclophosphamide therapy in patients with CKD, which ideally incorporates both pharmacokinetic and pharmacodynamic measures of effect.
Metformin
Metformin is the first-line oral antihyperglycemic drug for type 2 diabetes mellitus. However, its use was formerly considered to be contraindicated in patients with CKD due to concerns around metformin-associated lactic acidosis. Metformin-associated lactic acidosis is attributed to metformin accumulation in the context of impaired kidney function, and it is characterized by severe lactic acidosis (18,19). Regardless, preliminary studies have shown that metformin can be safely prescribed to patients with advanced CKD after appropriate dose reduction (4,20), increasing the treatment options for these patients.
Novel-acting/Direct-acting Oral Anticoagulants
The increasing use of NOACs/DOACs has presented issues for patients with both acute kidney disease and CKD, and NOACs/DOACs were previously contraindicated in advanced CKD. There is variability in the extent to which CL of these drugs depends on kidney function, such that kidney disease has differing effects on drug exposure and the risk of adverse events. For example, dabigatran pharmacokinetics is largely dependent on kidney CL and P-glycoprotein transporters, and very significant increases in AUC can occur with progressive decline in kidney function (21), predisposing to adverse events. In comparison, there is less of a decrease in the CL of apixaban from advanced kidney disease, and after studies on the basis of core pharmacokinetic principles, an appropriate dose reduction was determined and tested (5), providing guidance for its use in patients who are dialysis dependent (22). However, data about interindividual variability are still limited for these drugs, and therefore, there may be circumstances where therapeutic drug monitoring may be beneficial.
Pharmacokinetic Principles and Parameters
Quantifying changes in pharmacokinetics allows the dosing regimen to be adjusted with some precision to maximize the likelihood that the desired drug concentration-time profile is achieved. CL and volume of distribution (Vd) are the primary pharmacokinetic parameters that determine the concentration-time profile (drug exposure) and therefore, the appropriate dosing regimen (more discussion is in part 2 [23]). Patients with kidney disease are particularly susceptible to changes in both CL and Vd in both chronic and acute conditions. Half-life (t1/2) is a widely used pharmacokinetic parameter, which depends on both CL and Vd, and therefore, it is referred to as a secondary parameter (Equation 5).
Absolute Bioavailability
Absolute bioavailability is the fraction of drug that reaches the systemic circulation after administration, and it is calculated by comparing the AUC of an administered dose with the AUC achieved after rapid intravenous infusion (Equation 1). This is most commonly thought of in terms of oral bioavailability, where an orally administered drug’s bioavailability depends on the extent of gastrointestinal absorption and hepatic first-pass elimination:
| (1) |
where F is the absolute bioavailability, AUCpo is the AUC with oral dosing, AUCiv is the AUC with intravenous dosing, Dpo is the oral dose administered, and Div is the intravenous dose administered.
The principles can also be used to quantify the effect of kidney disease on drug exposure. Several processes involved in drug absorption and hepatic metabolism are affected by kidney disease (Table 1), but the significance of these changes for a given drug is not well defined. Indeed, the relative influence of bioavailability and/or CL on a change in the AUC cannot be readily differentiated in many instances. However, if an increase in AUC is mostly due to an increase in bioavailable dose, then the Cmax and AUC would be expected to increase to a similar extent (Equation 2). For example, in Figure 5A, the effect of a 50% decrease in CL is that the Cmax increased 20% and the AUC increased 100%, which is consistent with the relationship shown in Equation 6 rather than an increase in bioavailable dose (Equation 2). Clinical applications of this in patients with kidney disease are discussed in part 2 of this series (23).
Table 1.
| Process | Example Drugs | Anatomic Location | Alteration to This Process in CKD | Potential Change in Kinetics with CKD | Potential Implications for Dosing Regimen |
|---|---|---|---|---|---|
| Absorption and bioavailability | |||||
| Passive: concentration-dependent absorption | Multiple | Enterocytes | Decrease or increase | Decreased or increased bioavailability | Increased or decreased dose |
| Enzymatic metabolism (multiple; in particular, CYP3A4) | See below | Enterocytes | Decreased | Increased bioavailability | Decrease in dose |
| Active: P-glycoprotein (ABCB1) | Calcineurin inhibitors, digoxin, methotrexate | Enterocytes | Decreased | Increased bioavailability | Decrease in dose |
| Distribution | |||||
| Passive: concentration-dependent diffusion | Multiple | Systemic | No change or increased | No change or increased | No change or increase in initial dose |
| Protein binding | Multiple | Systemic | Decrease in protein concentration or protein binding | Increase in free (unbound) fraction, which can increase clearance and distribution | Potential increase in dose and either increase or decrease in frequency depending on change in Vd relative to CL |
| Active transporters (P-glycoprotein; ABCB1) | See above | Liver, brain, elsewhere | Unknown | Decreased activity: increased Vd | No change or increase in initial dose |
| Drug Clearance | |||||
| Passive: glomerular filtration | Multiple, including methotrexate | Glomerulus | Decreased | Decreased clearance | Decrease maintenance dose or frequency of dosing |
| Active: organic anion transporting polypeptide | β-Lactam antibiotics, methotrexate, atorvastatin, imatinib, rosuvastatin | Brain, liver, kidneys, intestine | Decreased | Decreased clearance | Decrease maintenance dose or frequency of dosing |
| Active: organic cation transporter | Metformin | Liver, kidney, brain, lung, etc. | Decreased | Decreased clearance | Decrease maintenance dose or frequency of dosing |
| Active: P-glycoprotein (ABCB1) | See above | Liver, kidney | Unknown (decreased in rats) | Decreased clearance | Decrease maintenance dose or frequency of dosing |
| Enzymatic: CYP2C8/9a | S-Warfarin, fluoxetine, tamoxifen, glipizide | Liver | Decreased or no change | Decreased clearance | Decrease maintenance dose or frequency of dosing |
| Enzymatic: CYP2C19a | Citalopram, cyclophosphamide, warfarin, diazepam | Liver | Decreased or no change | Decreased clearance | Decrease maintenance dose or frequency of dosing |
| Enzymatic: CYP2D6a | Carvedilol, metoprolol, tramadol, tamoxifen, codeine | Liver | Decreased | Decreased clearance | Decrease maintenance dose or frequency of dosing |
| Enzymatic: CYP3A4/5a | Atorvastatin, verapamil, tacrolimus, fluconazole, cyclophosphamide, carbamazepine, tolvaptan | Liver, enterocytes, kidneys (CYP3A5) | Decreased or no change | Decreased clearance | Decrease maintenance dose or frequency of dosing |
| Enzymatic: CYP1Aa | Caffeine, theophylline, warfarin | Liver | Decreased or no change | Decreased clearance | Decrease maintenance dose or frequency of dosing |
| Enzymatic: CYP2B6a | Cyclophosphamide, bupropion, methadone | Liver, kidney | Increased or decreased | Increased or decreased clearance | Increase or decrease maintenance dose or frequency of dosing |
Vd, volume of distribution;
The effect of CKD on the expression and activity of some cytochrome P450 isoenzymes is controversial and may instead reflect changes in transporter function as discussed in the text. Rowland Yeo et al. (45) found a reduction in cytochrome P450 activity across a range of isoenzymes. However, although some studies have identified progressive reductions in clearance by individual isoenzymes (for example, CYP2D6 [46]), others have found no difference in enzyme activity in advanced CKD for CYP3A4/5 (16,46) and CYP2C9 (47–50). Additional studies in human subjects are required to clarify the extent of any effect.
Figure 5.

A change in either volume of distribution or clearance has differing effects on the concentration-time profile. Graphs are drawn to scale for ready comparison. (A) A doubling in volume of distribution (Vd) and a halving of clearance have the same effect on the elimination t1/2, but they incur substantially different concentration-time profiles. Halving clearance leads to a doubling of the area under the concentration-time curve (Equation 6). The doubling in Vd leads to a reduction in maximum plasma concentration (Equation 2) but no change in the area under the concentration-time curve, despite the change in the concentration-time profile. (B) In patients with altered kinetics, continuous dosing will lead to drug accumulation if the regimen is not adjusted. Onset of toxicity will occur earlier from a decrease in clearance. (C) In patients with altered kinetics, increasing the dosing interval will prevent drug accumulation. Here, because the t1/2 was doubled in both cases, the dosing interval was also doubled. Although the trough concentrations are similar after the decrease in dosing frequency, the maximum plasma concentration and average concentration are lower when Vd is doubled, which may decrease the effectiveness of this regimen compared with in a patient with normal kinetics.
Volume of Distribution (Vd)
Vd is an apparent (theoretical) volume rather than being a true entity. It is the parameter relating the concentration of a drug in the plasma to the total amount of the drug in the body. It is quantified as liters per kilogram body weight, and it is mostly determined by the distribution and binding of the drug to extravascular tissues compared with plasma proteins. Vd is also used to estimate the Cmax (Figure 1) after a single dose, and it influences the loading dose (equation 1 in part 2 of this series in ref. 23) and t1/2 (Equation 5).
After a rapid bolus dose, the Cmax is predicted by Equation 2, where F is bioavailability (F=1 after intravenous administration; discussed above and in Equation 1):
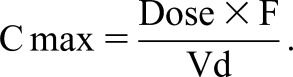 |
(2) |
Vd is highly dependent on not only body mass but also, body composition, notably the absolute and relative amounts of body water and adipose tissue. In the clinical circumstance where there is an increase in Vd (e.g., severe nephrotic syndrome), this can require a proportional increase in the dose to achieve the same Cmax. Conversely, changes in drug bioavailability may require a change in the dose, and bioavailability can increase or decrease in kidney disease, which is discussed later and in Table 1. Clinical applications of this are discussed in part 2 of this series (23).
Clearance
CL is the volume of blood cleared of a drug in a period of time usually measured in units of liters per hour or milliliters per minute, and it is the parameter that most closely describes drug elimination. CL determines the maintenance dose rate of a drug required to achieve a target plasma concentration (and therefore, effect) at steady state.
CL can be referred to by a particular organ (e.g., liver or kidney), a particular metabolic pathway, or the whole body. The total or systemic CL is the sum of the CL by individual organs, which incorporates both active (e.g., metabolism or active secretion) and passive (e.g., glomerular filtration) processes, as follows:
| (3) |
where CLK is kidney clearance, CLH is hepatic clearance, and CLother accounts for other routes of drug elimination (for example, kidney replacement therapy or metabolism by circulating esterases). The sum of CLH and CLother is sometimes referred to as nonrenal CL. The relationship between different routes of CL is shown graphically in Figure 2, where the anticipated change in total CL is related to GFR. Although this is a convenient way to think about changes in pharmacokinetics in the setting of kidney dysfunction, it can be an oversimplification for some drugs, because changes in nonrenal drug CL occur at the same time, which is discussed in detail below and represented in Figure 3.
Figure 2.

Changes in total drug clearance with declining kidney function relates to the extent of drug clearance by the kidney. Representation on the basis of Equation 3 for drugs with three different pharmacokinetic profiles. Here, Drug A is 100% cleared by the kidney, and therefore, it is predicted that a 50% decrease in GFR will correlate with a 50% decrease in total clearance, prompting a 50% decrease in dose or doubling of the dosing interval to maintain the same mean plasma concentration. Drugs from many classes can be represented: for example, antibiotics (A: β-lactams or aminoglycosides, B: macrolides, and C: fluoroquinolones), anticoagulants (A: dabigatran, B: warfarin, and C: rivaroxaban), and β-blockers (A: atenolol, B: metoprolol, and C: bisoprolol). Unfortunately, this representation is an oversimplification, because it does not consider changes to nonrenal clearance in kidney disease that occur with some drugs as discussed in the text.
Figure 3.

Drug clearance by metabolism can also decrease with declining kidney function. Drawn from data presented by Rowland Yeo et al. (45), the analyses are of clearance data in clinical studies after correcting for differences in protein binding and blood to plasma partitioning. The drugs were chosen as a probe of different CYP450s (theophylline for 1A2, rosiglitazone for 2C8, bosentan for 2C9, omeprazole for 2C19, bufuralol for 2D6, and midazolam for 3A4). Although these data are illustrative, the effect on expression and activity of some cytochrome P450 isoenzymes is controversial. For example, some studies have identified progressive reductions in clearance by CYP2D6 (46), whereas others have found no difference in enzyme activity in advanced CKD for CYP3A4/5 (16,46) and CYP2C9 (47). Instead, the changes in metabolic clearances noted in CKD may also relate to changes in expression or function of drug transporters (for example, those on the hepatocyte cell membrane). Additional studies in human subjects are required to further clarify the extent of any effect.
Kidney Clearance.
The traditional way to determine kidney CL is to measure the rate of excretion of the drug in urine and changes in the drug plasma concentration at the same time. Kidney CL is the net result of three different processes: filtration at the glomerulus, active secretion in the proximal tubule, and passive reabsorption along the kidney tubules:
| (4) |
where Fu is the fraction of the total drug concentration that is unbound to plasma proteins (free), CLsecretion is due to active secretion in the kidney tubules, and CLreabsorption refers to reabsorption from the glomerular filtrate back to the circulation.
Glomerular filtration varies with kidney blood flow, which can decrease when there is a reduced cardiac output or volume depletion. However, for some drugs, active secretion is significant, and therefore, the kidney CL exceeds GFR (for example, metformin, meropenem, amoxycillin, cefalexin, ampicillin, and piperacillin). The relative contributions of the processes shown in Equation 4 are illustrated in Figure 4, and Table 1 summarizes the more common drug transporters that contribute to this phenomenon.
Figure 4.

Total kidney clearance is dependent on the contributions of each of glomerular filtration, secretion in the proximal tubule, and reabsorption in the distal tubule. OATP, organic anion transporting polypeptide; OCT, organic cation transporter.
Furthermore, as GFR declines, the extent to which total kidney CL of a drug depends on active secretion can increase. Active transporters are also important, because drug-drug interactions may decrease CL due to competitive binding and being a saturable process. The clinical implication of this for drugs that are substrates of drug transporters in the kidney is that greater dose reductions are required in patients with kidney tubulopathy compared with those with a similarly reduced GFR due solely to glomerulopathy (24). In the case of glomerulopathy, drug CL may be preserved relative to the reduced GFR by preservation of active tubular secretion of drugs and/or metabolites (25,26). This is contrary to the intact nephron hypothesis that assumes that drug CL has a linear relationship to GFR, because reductions in kidney function are caused by a reduction in the number of intact (complete) nephrons. Furthermore, drug transporter activity can be pH dependent (for example, the active secretion of metformin is reduced when the filtrate in the tubular lumen is alkaline [27,28], which has the potential to decrease kidney CL). This challenges the use of GFR as the sole criterion for estimating kidney CL of drugs.
Finally, some drugs are reabsorbed from the glomerular filtrate in the tubules, and the extent of reabsorption can vary with urine pH and flow (e.g., weak acids, such as salicylate or some herbicides), knowledge of which has been used in the treatment of poisoning (29). The effect of kidney disease on tubular reabsorption and the implications on drug dosing are poorly defined.
Nonrenal Clearance.
There can be an apparent increase in nonrenal CL in patients with kidney disease, which probably reflects increased opportunity for elimination by alternative CL mechanisms or possibly, upregulation in other CL processes. For example, lower proportions of the dose of meropenem and piperacillin are eliminated in urine in patients with CKD compared with that predicted from data in healthy subjects (30,31), which is not consistent with Equation 3 or Figure 2.
However, for some drugs, nonrenal CL is decreased in the context of kidney disease, although most of these data are in the setting of CKD rather than AKI. The proposed mechanism for decreased nonrenal CL is inhibition of enzymes and transporters by circulating uremic toxins, which can be reversed (corrected) with their removal by hemodialysis (32). Here, the t1/2 decreases (rectifies) when affected drugs are administered immediately after hemodialysis (33). This is supported by in vitro studies (34–36), with some exceptions (37), and therefore, other mechanisms may also contribute, such as changes in protein expression (36,38). Of note, inhibition of drug transporters may decrease nonrenal drug CL due to either decreased secretion (e.g., P-glycoprotein, organic anion transporting polypeptide, or organic cation transporter) or uptake into hepatocytes (e.g., organic anion transporting polypeptide or organic cation transporter). The extent to which kidney disease decreases the CL of selected drugs that are substrates of the cytochrome P450 isoenzyme system is shown in Figure 3 and Table 1, potentially reflecting changes in both enzyme and transporter activity.
Another factor to consider when interpreting nonrenal drug CL data is the decrease in protein binding that occurs in CKD and the limited data describing changes in free (unbound compared with total) drug CL. For example, research describing the effect of CKD on benzodiazepine hepatic CL noted a decrease in CL of the free fraction in only two of nine studies, whereas in some studies, there was an increase in CL (32).
Despite these complexities, a common method to estimate the change in total drug CL between specific patient populations is to quantify the change in the function of the organ that has a significant role in the total drug CL (for example, determining kidney function by estimating GFR for a drug that is predominantly cleared by the kidney). Subsequently, using Equation 3, one can estimate the percentage change in drug CL in those with kidney impairment relative to healthy subjects. Drug CL relative to kidney function can be found in some textbooks and reviews (for example, ref. 39 for antibiotics). Where possible, it is preferable to understand how total CL changes with decreasing kidney function or consider the change in t1/2, because this incorporates both CL and Vd (Equation 5). Another factor that may limit the precision with which GFR reliably estimates drug CL includes the interindividual variability in pharmacokinetics. The clinical applications of the changes in CL are discussed further in part 2 of this series (23).
Elimination t1/2
The elimination t1/2 is the time required for the plasma concentration to decrease by 50% (Figure 1). t1/2 is determined in an individual by measuring the rate of decrease in serial (a minimum of three where possible) plasma drug concentrations. Plasma sampling can occur soon after an intravenous dose or in the case of orally administered drugs, after completion of absorption (after Cmax or Tmax) (Figure 1).
t1/2 is a major determinant of the duration of action after a single dose, the time required to reach steady-state plasma concentrations (obtained approximately 4–5 t1/2 after drug initiation) with multiple doses, and the dosing frequency. It is important to recognize that the time to reach steady-state concentration will be delayed for drugs with relatively prolonged half-lives.
t1/2 incorporates both Vd and CL:
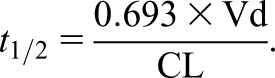 |
(5) |
t1/2 is prolonged in proportion to an increase in Vd or a decrease in CL. For example, the t1/2 will double after either a 50% decrease in CL or doubling of Vd (Figure 5A). Failure to dose adjust in the case of impaired kidney CL will lead to drug accumulation and risk of toxicity (Figure 5B), especially for chronic drug therapy. An example of this is the use of atenolol in patients with ESKD, in whom the t1/2 increases from 6 to 100 hours compared with in patients with preserved kidney function (40). A change in either CL or Vd has a very different effect on the concentration-time profile (Figure 5, A and B), but in each case, the dosing interval should be doubled (Figure 5C). However, Figure 5 is probably an oversimplification, because both CL and Vd can change in acute and chronic clinical situations, such as sepsis, kidney disease, and liver disease.
Finally, the t1/2 to consider is not only that of the parent drug, but also that of active or toxic metabolites. There are many cases of poisoning occurring due to accumulation of metabolites that are eliminated by the kidney, such as morphine causing coma, meperidine (pethidine) causing seizures, allopurinol causing toxic epidermal necrolysis, glyburide (glibenclamide) causing hypoglycemia, and cyclophosphamide causing immunosuppression. For example, relative to patients with normal kidney function, active cyclophosphamide metabolites had a significantly prolonged t1/2 and accumulated in a patient with a creatinine clearance of 18 ml/min, which contributed to prolonged bone marrow suppression (41) (discussed in Examples of Drugs That Require Special Consideration When Prescribing to Patients with Kidney Disease).
Area Under the Curve (AUC)
For a given dose, the AUC is proportional to the decrease in CL. This relationship between AUC and CL is expressed by Equation 6:
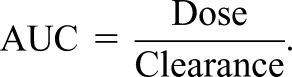 |
(6) |
Changes in drug CL as the result of kidney disease can, therefore, increase the AUC and overall drug exposure for a given dose, which in turn, increases the risk of adverse drug reactions. Numerically, this can be quantified using the equation
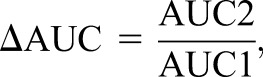 |
(7) |
where AUC1 is the initial or baseline AUC (e.g., with normal kidney function or before an intervention) and AUC2 is the observed AUC after the change (e.g., with kidney disease or after the intervention). For example, a 50% decrease in CL will double the AUC (ΔAUC=2, 100% increase, or twofold increase), which is shown graphically in Figure 5A.
When Should the Usual Dosing Regimen Be Adjusted?
The minimum change in kidney function that necessitates a change in dosing is not well defined. A long-standing rule of thumb is that dose adjustment is not required if a pharmacokinetic parameter changes by <30% (42), but this threshold is conservative. An FDA draft document recommends that detailed pharmacokinetic studies should be performed if kidney disease has a “substantial effect” on pharmacokinetics (for example, the drug exposure expressed as the AUC [Figure 1], increases by at least 50%–100%) or less effect if the drug has a narrow therapeutic range compared with in healthy subjects (2). When comparing the same dose, an increase in AUC is usually proportional to the decrease in CL (Equations 6 and 7). Therefore, a decrease in kidney function is unlikely to be clinically significant if drug clearance decreases by less than 50% (the “no effect boundary”).
The extent to which drugs (or their relevant metabolites) are excreted by the kidney are also important in determining whether dose adjustment is necessary in kidney disease. In general, dose adjustment is unlikely to be required when <30% of a dose is excreted by the kidneys (2). It is also important to consider instances where dose adjustments for primarily hepatically metabolized drugs may be required, because their pharmacologically active and/or toxic metabolites are primarily excreted by the kidney, which may increase the pharmacologic effect and/or risk of adverse events. For example, morphine is metabolized to morphine-6-glucuronide (up to 360 times more potent than the parent drug [43]), which is cleared by the kidney and accumulates in kidney failure. Mycophenolate is metabolized to mycophenolic acid glucuronide (inactive), which is cleared by the kidney, and it can accumulate in kidney impairment and may contribute to the gastrointestinal intolerance of this medication seen in severe CKD (44).
Other considerations include the risk of drug accumulation and the clinical manifestations when this occurs. For example, dose adjustments are less necessary for a low-toxicity drug being prescribed for a short course of treatment (e.g., an oral penicillin), where the risk of accumulation is mitigated by the short duration of therapy (for example, several days). In contrast, dose adjustments are required for drugs with a long treatment duration and a higher intrinsic toxicity (e.g., metformin, digoxin, lithium, or colchicine) (discussed in Examples of Drugs That Require Special Consideration When Prescribing to Patients with Kidney Disease), particularly if they have a long elimination t1/2 (e.g., >24 hours).
Therefore, dose adjustments may be required for selected drugs with pharmacokinetics that change significantly in kidney disease, particularly if there is a high risk of drug accumulation and severe and/or irreversible toxicity. Methods for dose adjusting in patients with kidney disease are discussed in detail in part 2 of this series (23).
Conclusions
Pharmacokinetic factors that inform the dosing of drugs are well described. However, limited data in patients with kidney disease, particularly for certain drugs, and marked interindividual variability complicate the development of dosing guidelines. Furthermore, kidney disease can cause wide-ranging changes in pharmacokinetics through derangement of not only kidney drug CL but also, nonrenal CL, Vd, and bioavailability. These considerations apply to both the parent drug and any active or toxic metabolites. Each requires a different approach to adjustment of the dosing regimen, and inappropriate adjustments, particularly with maintenance therapy, lead to drug concentrations that are too low or too high, predispose patients to harm due to therapeutic failure, or adverse drug reactions.
Drug dosing can be optimized on a case by case basis by the use of rational dose design grounded in an understanding of basic pharmacokinetic concepts and therapeutic drug monitoring, particularly for drugs that have a narrow therapeutic index. This is a key component in the development of personalized medical care for patients with kidney disease, and it is discussed further in part 2 of this series (23).
Disclosures
None.
Acknowledgments
D.M.R. is a recipient of the Jacquot Research Establishment Fellowship, Royal Australasian College of Physicians and the Clinician ‘Buy-Out’ Program, St. Vincent's Centre for Applied Medical Research.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
See related article, “Introduction to Nephropharmacology for the Clinician: A New CJASN Series,” on pages 1083–1084.
References
- 1.Zhang Y, Zhang L, Abraham S, Apparaju S, Wu TC, Strong JM, Xiao S, Atkinson AJ Jr, Thummel KE, Leeder JS, Lee C, Burckart GJ, Lesko LJ, Huang SM: Assessment of the impact of renal impairment on systemic exposure of new molecular entities: Evaluation of recent new drug applications. Clin Pharmacol Ther 85: 305–311, 2009 [DOI] [PubMed] [Google Scholar]
- 2.FDA: Pharmacokinetics in Patients with Impaired Renal Function—Study Design, Data Analysis, and Impact on Dosing and Labeling, 2010. Available at: https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm204959.pdf. Accessed December 24, 2017
- 3.Khanal A, Castelino RL, Peterson GM, Jose MD: Dose adjustment guidelines for medications in patients with renal impairment: How consistent are drug information sources? Intern Med J 44: 77–85, 2014 [DOI] [PubMed] [Google Scholar]
- 4.Duong JK, Roberts DM, Furlong TJ, Kumar SS, Greenfield JR, Kirkpatrick CM, Graham GG, Williams KM, Day RO: Metformin therapy in patients with chronic kidney disease. Diabetes Obes Metab 14: 963–965, 2012 [DOI] [PubMed] [Google Scholar]
- 5.Leil TA, Feng Y, Zhang L, Paccaly A, Mohan P, Pfister M: Quantification of apixaban’s therapeutic utility in prevention of venous thromboembolism: Selection of phase III trial dose. Clin Pharmacol Ther 88: 375–382, 2010 [DOI] [PubMed] [Google Scholar]
- 6.Blot S, Lipman J, Roberts DM, Roberts JA: The influence of acute kidney injury on antimicrobial dosing in critically ill patients: Are dose reductions always necessary? Diagn Microbiol Infect Dis 79: 77–84, 2014 [DOI] [PubMed] [Google Scholar]
- 7.Imani S, Buscher H, Marriott D, Gentili S, Sandaradura I: Too much of a good thing: A retrospective study of β-lactam concentration-toxicity relationships. J Antimicrob Chemother 72: 2891–2897, 2017 [DOI] [PubMed] [Google Scholar]
- 8.Oakley PW, Whyte IM, Carter GL: Lithium toxicity: An iatrogenic problem in susceptible individuals. Aust N Z J Psychiatry 35: 833–840, 2001 [DOI] [PubMed] [Google Scholar]
- 9.Baird-Gunning J, Lea-Henry T, Hoegberg LCG, Gosselin S, Roberts DM: Lithium poisoning. J Intensive Care Med 32: 249–263, 2017 [DOI] [PubMed] [Google Scholar]
- 10.Roberts DM, Gallapatthy G, Dunuwille A, Chan BS: Pharmacological treatment of cardiac glycoside poisoning. Br J Clin Pharmacol 81: 488–495, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan BS, Isbister GK, O’Leary M, Chiew A, Buckley NA: Efficacy and effectiveness of anti-digoxin antibodies in chronic digoxin poisonings from the DORA study (ATOM-1). Clin Toxicol (Phila) 54: 488–494, 2016 [DOI] [PubMed] [Google Scholar]
- 12.de Groot K, Harper L, Jayne DR, Flores Suarez LF, Gregorini G, Gross WL, Luqmani R, Pusey CD, Rasmussen N, Sinico RA, Tesar V, Vanhille P, Westman K, Savage CO; EUVAS (European Vasculitis Study Group): Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: A randomized trial. Ann Intern Med 150: 670–680, 2009 [DOI] [PubMed] [Google Scholar]
- 13. Li YF, Fu S, Hu W, Liu JH, Finkel KW, Gershenson DM, Kavanagh JJ: Systemic anticancer therapy in gynecological cancer patients with renal dysfunction. Int J Gynecol Cancer 17: 739–763, 2007. [DOI] [PubMed]
- 14.Haubitz M, Bohnenstengel F, Brunkhorst R, Schwab M, Hofmann U, Busse D: Cyclophosphamide pharmacokinetics and dose requirements in patients with renal insufficiency. Kidney Int 61: 1495–1501, 2002 [DOI] [PubMed] [Google Scholar]
- 15.Joy MS: Impact of glomerular kidney diseases on the clearance of drugs. J Clin Pharmacol 52[Suppl]: 23S–34S, 2012 [DOI] [PubMed] [Google Scholar]
- 16.Little MA, Nightingale P, Verburgh CA, Hauser T, De Groot K, Savage C, Jayne D, Harper L; European Vasculitis Study (EUVAS) Group: Early mortality in systemic vasculitis: Relative contribution of adverse events and active vasculitis. Ann Rheum Dis 69: 1036–1043, 2010 [DOI] [PubMed] [Google Scholar]
- 17.Singh JA, Hossain A, Kotb A, Wells GA: Comparative effectiveness of immunosuppressive drugs and corticosteroids for lupus nephritis: A systematic review and network meta-analysis. Syst Rev 5: 155, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duong JK, Furlong TJ, Roberts DM, Graham GG, Greenfield JR, Williams KM, Day RO: The role of Metformin in Metformin-Associated Lactic Acidosis (MALA): Case series and formulation of a model of pathogenesis. Drug Saf 36: 733–746, 2013 [DOI] [PubMed] [Google Scholar]
- 19.Juurlink DN, Roberts DM: The enigma of metformin-associated lactic acidosis. Clin Toxicol (Phila) 52: 85–87, 2014 [DOI] [PubMed] [Google Scholar]
- 20.Duong JK, Kroonen MYAM, Kumar SS, Heerspink HL, Kirkpatrick CM, Graham GG, Williams KM, Day RO: A dosing algorithm for metformin based on the relationships between exposure and renal clearance of metformin in patients with varying degrees of kidney function. Eur J Clin Pharmacol 73: 981–990, 2017 [DOI] [PubMed] [Google Scholar]
- 21.Stangier J, Rathgen K, Stähle H, Mazur D: Influence of renal impairment on the pharmacokinetics and pharmacodynamics of oral dabigatran etexilate: An open-label, parallel-group, single-centre study. Clin Pharmacokinet 49: 259–268, 2010 [DOI] [PubMed] [Google Scholar]
- 22.Mavrakanas TA, Samer CF, Nessim SJ, Frisch G, Lipman ML: Apixaban pharmacokinetics at steady state in hemodialysis patients. J Am Soc Nephrol 28: 2241–2248, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roberts DM, Sevastos J, Carland JE, Stocker SL, Lea-Henry TN: Clinical pharmacokinetics in kidney disease (part 2): Application to rational design of dosing regimens. Clin J Am Soc Nephrol 2018, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hori R, Okumura K, Kamiya A, Nihira H, Nakano H: Ampicillin and cephalexin in renal insufficiency. Clin Pharmacol Ther 34: 792–798, 1983 [DOI] [PubMed] [Google Scholar]
- 25.Chapron A, Shen DD, Kestenbaum BR, Robinson-Cohen C, Himmelfarb J, Yeung CK: Does secretory clearance follow glomerular filtration rate in chronic kidney diseases? Reconsidering the intact nephron hypothesis. Clin Transl Sci 10: 395–403, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Putt TL, Duffull SB, Schollum JB, Walker RJ: GFR may not accurately predict aspects of proximal tubule drug handling. Eur J Clin Pharmacol 70: 1221–1226, 2014 [DOI] [PubMed] [Google Scholar]
- 27.König J, Zolk O, Singer K, Hoffmann C, Fromm MF: Double-transfected MDCK cells expressing human OCT1/MATE1 or OCT2/MATE1: Determinants of uptake and transcellular translocation of organic cations. Br J Pharmacol 163: 546–555, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Müller F, König J, Glaeser H, Schmidt I, Zolk O, Fromm MF, Maas R: Molecular mechanism of renal tubular secretion of the antimalarial drug chloroquine. Antimicrob Agents Chemother 55: 3091–3098, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts DM, Buckley NA: Pharmacokinetic considerations in clinical toxicology: Clinical applications. Clin Pharmacokinet 46: 897–939, 2007 [DOI] [PubMed] [Google Scholar]
- 30.Brogard JM, Jehl F, Blickle JF, Dorner M, Arnaud JP, Monteil H: Biliary pharmacokinetic profile of piperacillin: Experimental data and evaluation in man. Int J Clin Pharmacol Ther Toxicol 28: 462–470, 1990 [PubMed] [Google Scholar]
- 31.Ljungberg B, Nilsson-Ehle I: Pharmacokinetics of meropenem and its metabolite in young and elderly healthy men. Antimicrob Agents Chemother 36: 1437–1440, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greenblatt DJ: Hepatic clearance of drugs in patients with renal insufficiency. Clin Pharmacol Drug Dev 2: 295–297, 2013 [DOI] [PubMed] [Google Scholar]
- 33.Nolin TD, Appiah K, Kendrick SA, Le P, McMonagle E, Himmelfarb J: Hemodialysis acutely improves hepatic CYP3A4 metabolic activity. J Am Soc Nephrol 17: 2363–2367, 2006 [DOI] [PubMed] [Google Scholar]
- 34.Nolin TD, Frye RF, Le P, Sadr H, Naud J, Leblond FA, Pichette V, Himmelfarb J: ESRD impairs nonrenal clearance of fexofenadine but not midazolam. J Am Soc Nephrol 20: 2269–2276, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Volpe DA, Tobin GA, Tavakkoli F, Dowling TC, Light PD, Parker RJ: Effect of uremic serum and uremic toxins on drug metabolism in human microsomes. Regul Toxicol Pharmacol 68: 297–303, 2014 [DOI] [PubMed] [Google Scholar]
- 36.Sun H, Frassetto L, Benet LZ: Effects of renal failure on drug transport and metabolism. Pharmacol Ther 109: 1–11, 2006 [DOI] [PubMed] [Google Scholar]
- 37.De Martin S, Orlando R, Bertoli M, Pegoraro P, Palatini P: Differential effect of chronic renal failure on the pharmacokinetics of lidocaine in patients receiving and not receiving hemodialysis. Clin Pharmacol Ther 80: 597–606, 2006 [DOI] [PubMed] [Google Scholar]
- 38.Yeung CK, Shen DD, Thummel KE, Himmelfarb J: Effects of chronic kidney disease and uremia on hepatic drug metabolism and transport. Kidney Int 85: 522–528, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roberts DM: The relevance of drug clearance to antibiotic dosing in critically ill patients. Curr Pharm Biotechnol 12: 2002–2014, 2011 [DOI] [PubMed] [Google Scholar]
- 40.McAinsh J, Holmes BF, Smith S, Hood D, Warren D: Atenolol kinetics in renal failure. Clin Pharmacol Ther 28: 302–309, 1980 [DOI] [PubMed] [Google Scholar]
- 41.Bagley CM Jr, Bostick FW, DeVita VT Jr: Clinical pharmacology of cyclophosphamide. Cancer Res 33: 226–233, 1973 [PubMed] [Google Scholar]
- 42.Levy G: Pharmacokinetics in renal disease. Am J Med 62: 461–465, 1977 [DOI] [PubMed] [Google Scholar]
- 43.Frances B, Gout R, Monsarrat B, Cros J, Zajac JM: Further evidence that morphine-6 beta-glucuronide is a more potent opioid agonist than morphine. J Pharmacol Exp Ther 262: 25–31, 1992 [PubMed] [Google Scholar]
- 44.MacPhee IA, Spreafico S, Bewick M, Davis C, Eastwood JB, Johnston A, Lee T, Holt DW: Pharmacokinetics of mycophenolate mofetil in patients with end-stage renal failure. Kidney Int 57: 1164–1168, 2000 [DOI] [PubMed] [Google Scholar]
- 45.Rowland Yeo K, Aarabi M, Jamei M, Rostami-Hodjegan A: Modeling and predicting drug pharmacokinetics in patients with renal impairment. Expert Rev Clin Pharmacol 4: 261–274, 2011 [DOI] [PubMed] [Google Scholar]
- 46.Yoshida K, Sun B, Zhang L, Zhao P, Abernethy DR, Nolin TD, Rostami-Hodjegan A, Zineh I, Huang SM: Systematic and quantitative assessment of the effect of chronic kidney disease on CYP2D6 and CYP3A4/5. Clin Pharmacol Ther 100: 75–87, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Joy MS, Frye RF, Nolin TD, Roberts BV, La MK, Wang J, Brouwer KL, Dooley MA, Falk RJ: In vivo alterations in drug metabolism and transport pathways in patients with chronic kidney diseases. Pharmacotherapy 34: 114–122, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bergeron MG, Gennari FJ, Barza M, Weinstein L, Cortell S: Renal tubular transport of penicillin G and carbenicillin in the rat. J Infect Dis 132: 374–383, 1975 [DOI] [PubMed] [Google Scholar]
- 49.Graham GG, Punt J, Arora M, Day RO, Doogue MP, Duong JK, Furlong TJ, Greenfield JR, Greenup LC, Kirkpatrick CM, Ray JE, Timmins P, Williams KM: Clinical pharmacokinetics of metformin. Clin Pharmacokinet 50: 81–98, 2011 [DOI] [PubMed] [Google Scholar]
- 50.Debruyne D, Ryckelynck JP: Clinical pharmacokinetics of fluconazole. Clin Pharmacokinet 24: 10–27, 1993 [DOI] [PubMed] [Google Scholar]