

Summary
Background
In Phase 3 studies of ustekinumab, a fully human monoclonal IL‐12/23p40 antibody approved for moderate‐to‐severe Crohn's disease, patients entered a long‐term extension after completing 8 weeks of induction and 44 weeks of maintenance treatment. Efficacy through 92 weeks and safety through 96 weeks of IM‐UNITI maintenance are reported.
Methods
UNITI‐1 (TNF‐antagonist failures) and UNITI‐2 (conventional therapy failures) patients (N = 1281) entered IM‐UNITI, including 397 ustekinumab intravenous induction responders randomised to subcutaneous ustekinumab 90 mg every 12 weeks, every 8 weeks, or placebo and 884 nonrandomised patients. Dose‐adjustment to 90 mg every 8 weeks occurred in patients randomised to 90 mg every 12 weeks and placebo patients with loss of response (Weeks 8–32). All Week 44 completers could enter the long‐term extension without further dose adjustment. Placebo patients discontinued following study unblinding.
Results
A total of 718 patients (all treated) entered the long‐term extension (298 randomised and 420 not randomised). Overall, 86.5% (621/718) completed Week 96. The proportions of randomised patients in clinical remission were generally maintained from Week 44 through 92 in ustekinumab 90 mg every 12 weeks (77.4% to 72.6%), every 8 weeks (84.1% to 74.4%), and prior dose adjustment groups (63.4% to 53.5%). At Week 92, the proportions of patients in clinical remission were similar in the ustekinumab 90 mg every 12 weeks and every 8 weeks groups and lower in patients with prior dose adjustment. Proportions of patients in clinical remission at Week 92 for all treated every 8 weeks (64.4%) and every 12 weeks (64.3%) groups were lower than randomised every 8 weeks (74.4%) and every 12 weeks (72.6%) groups, but similarly maintained. Safety events (per hundred patient‐years) were similar among all placebo and ustekinumab patients (Week 0‐96), including adverse events (484.39 vs 447.76), serious adverse events (19.24 vs 18.82), and serious infections (4.09 vs 4.02). No dose effect was observed.
Conclusions
Subcutaneous ustekinumab maintained clinical response and remission through Week 92. No new safety signals were observed. ClinicalTrials.gov number NCT01369355.
1. INTRODUCTION
Crohn's disease is a chronic inflammatory bowel disease characterised by granulomatous inflammation, most commonly involving the terminal ileum and colon. The onset is most frequently in the second to fourth decade with characteristic symptoms of abdominal pain, diarrhoea, and weight loss and potential systemic and extraintestinal complications. Most patients have alternating periods of relapse and remission with half of patients requiring surgery within 10 years of diagnosis. The approach to treatment has evolved from control of symptoms to preventing progression of the disease. Therefore, treatments that safely maintain long‐term remission are essential.1
Treatment guidelines for Crohn's disease recommend maintenance therapy after remission is achieved, particularly for moderate‐to‐high risk patients.2 Potential benefits include reduction in hospitalisation and surgery and improved quality of life.3, 4 Long‐term efficacy has been demonstrated with azathioprine/mercaptopurine,5 methotrexate,6 tumour necrosis factor (TNF) antagonists,7, 8, 9, 10, 11 natalizumab,12 and vedolizumab.13 Although TNF antagonists have significantly advanced the care of Crohn's disease, their efficacy is limited14 and the development of anti‐drug antibodies is associated with loss of response.15, 16, 17 In addition, potential significant side effects of maintenance treatments include bone marrow suppression,18 malignancy,19, 20 and serious infections.21, 22 Therefore, a need exists for safer agents that have demonstrated improved long‐term maintenance efficacy.
Ustekinumab is a monoclonal antibody to the p40 subunit of interleukin‐12 and interleukin‐23 that has been approved for use in the treatment of psoriasis, psoriatic arthritis, and Crohn's disease.23 Both clinical trial and registry data have confirmed the positive long‐term efficacy and safety profile of ustekinumab in psoriasis.24, 25, 26 The efficacy and safety of ustekinumab in Crohn's disease through 1 year has been previously established in the UNITI‐1 and UNITI‐2 (8 weeks) and IM‐UNITI (44 weeks) studies in patients who have failed TNF antagonists or conventional therapies (predominantly regimens excluding anti‐TNF medications).27 Here, we report the IM‐UNITI long‐term extension data demonstrating the maintenance of response and remission (Week 92) with a positive safety profile (Week 96).
2. METHODS
2.1. Study design and endpoints
IM‐UNITI is a continuing Phase 3 ustekinumab maintenance study in patients with Crohn's disease. The Phase 3 programme included two 8‐week, identically designed, induction studies (UNITI‐1 and UNITI‐2) with distinct populations and one maintenance study (Figure 1). The detailed study designs, eligibility criteria, and results for the UNITI and IM‐UNITI studies through Week 44 have been previously published.27 Briefly, UNITI‐1 patients were required to have received one or more TNF antagonists at approved doses and to have met the criteria for primary nonresponse (the absence of a response), secondary nonresponse (a response that was not maintained), or to have had unacceptable side effects to TNF antagonists. UNITI‐2 patients were required to have failed (including unacceptable side effects) conventional therapy (steroids or immunomodulators [mercaptopurine, azathioprine or methotrexate]), but had not previously failed TNF antagonists. Induction study patients were eligible to enrol in IM‐UNITI.
Figure 1.
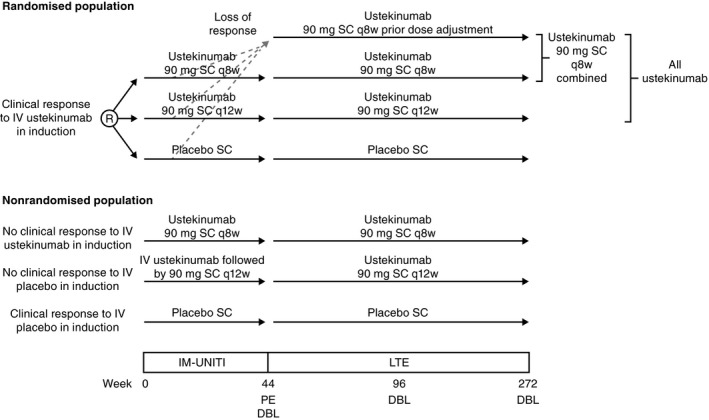
Study design for IM‐UNITI phase 3 Crohn's disease program*. R, randomisation; IV, intravenous; SC, subcutaneous; q8w, every 8 weeks; q12w, every 12 weeks; PE, primary endpoint; LTE, long‐term extension; DBL, database lock. *Unblinding occurred when the final patients reached Week 44
The primary population (randomised) for IM‐UNITI comprised 397 patients from UNITI‐1 or UNITI‐2 who were in clinical response at Week 8 after ustekinumab intravenous induction, defined as a reduction from baseline in Crohn's Disease Activity Index (CDAI) of ≥100 points or being in remission. These patients were randomly assigned, in a 1:1:1 ratio, to receive subcutaneous injections of 90 mg of ustekinumab every 8 weeks, 90 mg of ustekinumab every 12 weeks, or placebo through Week 40 with a Week 44 primary endpoint of clinical remission, defined as a CDAI score of <150 points. Randomised patients who subsequently met loss of response criteria at any time between Week 8 and Week 32 were eligible to have a single dose adjustment to ustekinumab 90 mg every 8 weeks; these patients were evaluated 16 weeks after dose adjustment and were discontinued from ustekinumab if not clinically improved.
The remainder of UNITI‐1 or UNITI‐2 patients could continue in IM‐UNITI but were not randomised and were treated as follows: Placebo induction responders continued placebo and placebo nonresponders received intravenous ustekinumab 130 mg at IM‐UNITI Week 0, and continued subcutaneous ustekinumab 90 mg every 12 weeks if in clinical response at Week 8. Intravenous ustekinumab induction nonresponders received subcutaneous ustekinumab 90 mg at IM‐UNITI Week 0 and continued ustekinumab 90 mg every 8 weeks if in clinical response at Week 8.
All patients (randomised and nonrandomised) who completed the safety and efficacy evaluation at Week 44 were eligible to participate in the IM‐UNITI extension, based upon the investigator's opinion that they would benefit from continued treatment. Throughout the extension, patients continued to receive the same treatment regimen that they were receiving at Week 44, without any further dose adjustment. The IM‐UNITI study was unblinded when the last patient in the main study completed the Week 44 evaluations and the Week 44 analyses were completed.
Efficacy was assessed through Week 92, with clinical remission defined as a CDAI score of <150 points and clinical response defined as a reduction from Week 0 of UNITI‐1 or UNITI‐2 in the CDAI score of ≥100 points (or if a CDAI score of <150 was attained). Corticosteroid‐free remission was defined as a CDAI score of <150 points without receiving corticosteroids at Week 92. Additional efficacy evaluations in the IM‐UNITI long‐term extension included CDAI and serum C‐reactive protein (CRP) every 12 weeks until Week 44 unblinding and then at every dosing visit, and the Inflammatory Bowel Disease questionnaire (IBDQ) and the 36‐Item Short Form Survey (SF‐36) every 6 months. Safety was assessed every 4 weeks through Week 96.
Patients were evaluated for antibodies to ustekinumab every 6 months. Analyses of antibodies to ustekinumab were performed using a validated, drug‐tolerant electro‐chemiluminescence immunoassay, in which ustekinumab was used to capture and detect induced immune responses to ustekinumab. Janssen or its designee assayed these samples, under conditions in which the identification of the patient and treatment assignment was blinded.
2.2. Statistical analysis
Because placebo patients discontinued the study after treatment assignments were unblinded to investigative sites (after the last patient completed Week 44 and database lock and subsequent analyses were completed), direct comparisons and/or statistical comparisons of efficacy results between placebo and ustekinumab treatment groups throughout the extension were not possible.
Demographic and baseline disease characteristics, efficacy and safety analyses were based on all patients treated with at least 1 administration of study agent during the long‐term extension. Descriptive statistics (eg, mean, median, standard deviation, interquartile range, minimum and maximum) were used to summarise continuous variables. Counts and percentages were used to summarise categorical variables.
For the IM‐UNITI long‐term extension, three types of analyses were conducted for remission data. Details of these analyses and the populations included are described in Table 1. In brief, they include (1) Analysis of patients who entered the long‐term extension, (2) Observed case analysis with inclusion only of patients who entered the long‐term extension who have data at that visit, (3) Analysis of all randomised patients in maintenance, regardless of whether they entered the long‐term extension.
Table 1.
Analyses for IM‐UNITI Long‐term Extension
| Analysis type | Population included |
|---|---|
|
|
|
|
|
|
ITT, intent‐to‐treat; CDAI, Crohn's disease activity index.
The efficacy population included 388 patients (excluding 9 patients enrolled prior to study restart who had received a prior investigational intravenous formulation in induction)
For all efficacy analyses other than the observed case analysis, patients who had a Crohn's disease‐related surgery or discontinued study agent due to lack of efficacy or an adverse event indicated to be worsening Crohn's disease after Week 44 and prior to the designated analysis time point were considered not to be in clinical remission/response, regardless of their CDAI score. Patients with insufficient data to calculate the CDAI score at the designated analysis time point were considered not to be in clinical remission/response. Other dichotomous endpoints followed the same treatment failure and missing data rules.
For continuous endpoints, patients meeting these treatment failure rules prior to the designated analysis time point had their induction baseline value carried forward and if there was insufficient data to calculate the variable at the designated analysis time points their last value was carried forward.
The incidence of antibodies to ustekinumab were summarised for all treated patients who entered the long‐term extension and received at least 1 administration of ustekinumab, either in the induction studies or in this study, and had appropriate samples for detection of antibodies to ustekinumab (ie, patients with at least 1 sample obtained after their first dose of ustekinumab). Patients were considered positive even if antibodies were detected at any single timepoint. The relationships between antibody to ustekinumab status at Week 92 and clinical response and remission status, as well as injection‐site reactions at Week 92 were explored.
3. RESULTS
A total of 718 patients continued into the IM‐UNITI long‐term extension, of which 567 patients were treated with subcutaneous ustekinumab 90 mg every 12 weeks (n = 213) or every 8 weeks (n = 354). The other 151 patients received subcutaneous placebo (Figure S1). Of the patients continuing to the long‐term extension, 84, 153 (71 with prior dose adjustment), and 61 patients on every 12 weeks, every 8 weeks, and placebo, respectively, had been in the primary population, representing approximately 75% (298/397) of the original randomised, primary population patients.
The clinical disease characteristics at Week 0 of UNITI‐1 and UNITI‐2 and at Week 0 of IM‐UNITI for randomised patients in the long‐term extension were generally similar among treatment groups and representative of a population with moderately to severely active Crohn's disease.27 As they entered the long‐term extension (Week 44 of IM‐UNITI), clinical disease characteristics of randomised patients were generally similar for ustekinumab 90 mg subcutaneous every 12 weeks and every 8 weeks groups and numerically higher (eg, CDAI scores, CRP concentrations) or lower (eg, IBDQ, patients in remission) for patients with prior dose adjustment (Table 2).
Table 2.
Disease characteristics of randomised patients who entered long‐term extension (at Week 44)
| Placebo | Continuous UST SC 90 mg q12w | Continuous UST SC 90 mg q8w | Patients with Prior Dose Adjustmenta | |
|---|---|---|---|---|
| N | 61 | 84 | 82 | 71 |
| Median CDAI | 96.0 | 95.5 | 70.5 | 130.0 |
| Median IBDQ | 180.5 | 189.0 | 185.5 | 171.0 |
| Median CRP (mg/L) | 6.7 | 3.5 | 3.7 | 4.0 |
| Patients in clinical remission | 77.0% | 77.4% | 84.1% | 63.4% |
SC, subcutaneous; UST, ustekinumab; CDAI, Crohn's disease activity index; IBDQ, inflammatory bowel disease questionnaire; CRP, C‐reactive protein; q12w, every 12 weeks; q8w, every 8 weeks.
Patients who were in clinical response to UST induction dosing, were randomised, met loss of clinical response criteria from Week 8 through Week 32, and received UST 90 mg SC q8w (this includes patients who were randomised to 90 mg SC q8w who had a sham dose adjustment).
Of the 298 randomised patients who were treated in the long‐term extension, 76/298 (25.5%) discontinued study agent from Week 44 through Week 96 (35/61 [57%] and 41/237 [17.3%] in the placebo and all ustekinumab 90 mg subcutaneous groups, respectively).
All patients remained blinded from Week 44 through Week 96 in the long‐term extension until the study unblinding, which occurred after completion of the Week 44 evaluations and analyses.
3.1. Randomised patients entering long‐term extension
3.1.1. Clinical remission Weeks 44‐92
The proportions of patients in clinical remission were generally maintained from Week 44 through Week 92 in the ustekinumab 90 mg every 12 weeks group (65/84 [77.4%] to 61/84 [72.6%]), and in the every 8 weeks group (69/82 [84.1%] to 61/82 [74.4%], respectively; Figure 2A). At Week 92, the proportions of patients in clinical remission were similar in the ustekinumab 90 mg every 12 weeks (61/84 [72.6%]) and every 8 weeks (61/82 [74.4%]) groups and lower in patients with prior dose adjustment (38/71 [53.5%]; Table 3).
Figure 2.
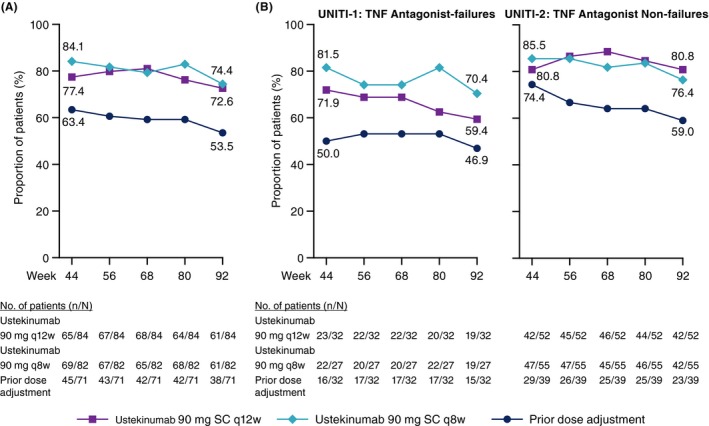
Clinical remission from Week 44 through Week 92 of treatment among (A) all randomised patients entering the long‐term extension and (B) by induction study subgroups. SC, subcutaneous; TNF, tumour necrosis factor; q8w, every 8 weeks; q12w, every 12 weeks
Table 3.
IM‐UNITI efficacy assessments at Week 92 among randomised patients who entered long‐term extensiona
| All Patients | UNITI‐1 | UNITI‐2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Continuous SC UST 90 mg q12w | Continuous SC UST 90 mg q8w | Pts with Prior Dose Adjustmentb | Continuous SC UST 90 mg q12w | Continuous SC UST 90 mg q8w | Pts with Prior Dose Adjustmentb | Combined | Continuous SC UST 90 mg q12w | Continuous SC UST 90 mg q8w | Pts with Prior Dose Adjustmentb | Combined | |
| N | 84 | 82 | 71 | 32 | 27 | 32 | 59 | 52 | 55 | 39 | 94 |
| Clinical Remission (%) | 72.6 | 74.4 | 53.5 | 59.4 | 70.4 | 46.9 | 57.6 | 80.8 | 76.4 | 59.0 | 69.1 |
| Clinical response (%) | 83.3 | 80.5 | 67.6 | 68.8 | 81.5 | 56.3 | 67.8 | 92.3 | 80.0 | 76.9 | 78.7 |
| Clinical remission and not receiving corticosteroids (%) | 67.9 | 63.4 | 42.3 | 56.3 | 59.3 | 31.3 | 44.1 | 75.0 | 65.5 | 51.3 | 59.6 |
|
Clinical remission in UNITI‐1 subset N (%)c |
19/32 (59.4) | 19/27 (70.4) | 15/32 (46.9) | 19/32 (59.4) | 19/27 (70.4) | 15/32 (46.9) | 34/59 (57.6) | ||||
|
Clinical remission in the UNITI‐2 subset patients naive to TNF antagonistsd
N (%) |
29/38 (76.3) | 29/39 (74.4) | 18/28 (64.3) | 29/38 (76.3) | 29/39 (74.4) | 18/28 (64.3) | 47/67 (70.1) | ||||
| Median change in CDAI from maintenance baselinee | −34.0 | −40.0 | −24.0 | −21.0 | −49.0 | 11.0 | −33.0 | −51.5 | −40.0 | −27.0 | −37.5 |
SC, subcutaneous; UST, ustekinumab; CDAI, Crohn's disease activity index; q12w, every 12 weeks; q8w, every 8 weeks; Pts, patients; TNF, tumour necrosis factor.
Patients who had insufficient data at Week 92 are considered not to be in clinical remission or response.
Patients who were in clinical response to ustekinumab induction dosing, were randomised, met loss of response criteria from Week 8 through Week 32 and received 90 mg SC q8w (this includes patients who were randomised to 90 mg SC q8w who had a sham dose adjustment).
Patients refractory or intolerant to TNF antagonists (UNITI‐1).
Patients naive to TNF antagonists from UNITI‐2.
Patients who had insufficient data to calculate the CDAI score at Week 92 had their last value carried forward.
3.1.2. Clinical remission by induction study, Weeks 44‐92
The proportions of patients in clinical remission were generally maintained from Week 44 through Week 92 for patients from both the UNITI‐1 and UNITI‐2 populations (Figure 2B).
The proportions of UNITI‐1 patients in clinical remission at Week 92 were 19/32 (59.4%) in the ustekinumab 90 mg every 12 weeks group, 19/27 (70.4%) in the every 8 weeks group and 15/32 (46.9%) for patients with prior dose adjustment (Table 3).
At Week 92, the proportions of UNITI‐2 patients in clinical remission were 42/52 (80.8%) in the ustekinumab 90 mg every 12 weeks group, 42/55 (76.4%) in the every 8 weeks group and 23/39 (59.0%) in patients with prior dose adjustment. In the subset of patients who were TNF antagonist‐naïve, data were generally consistent with the overall UNITI‐2 population (Table 3).
3.1.3. Corticosteroid‐free remission
The proportions of patients in clinical remission at Week 92 and not receiving corticosteroids at Week 92 were similar in the ustekinumab 90 mg every 12 weeks (57/84; 67.9%) and every 8 weeks (52/82; 63.4%) groups and were lower in patients with prior dose adjustment (30/71; 42.3%; Table 3).
3.1.4. Clinical response
At Week 92, the proportions of patients in clinical response were similar for patients in the ustekinumab 90 mg every 12 weeks (83.3%) and every 8 weeks (80.5%) groups and lower in patients with prior dose‐adjustment (67.6%) (Table 3).
3.1.5. Change in CDAI over time from maintenance baseline
The median change from maintenance baseline in CDAI scores for randomised patients at Week 92 was −34.0 in the ustekinumab 90 mg every 12 weeks group, −40.0 in the every 8 weeks group, and −24.0 in patients with prior dose adjustment (Table 3).
3.1.6. Patients with normalised CRP
The proportions of randomised patients with normalised CRP levels were generally stable from Week 44 through Week 92 in both the ustekinumab 90 mg every 12 weeks (36.1% to 29.5%) and every 8 weeks (30.4% to 26.8%) groups (Figure 3).
Figure 3.
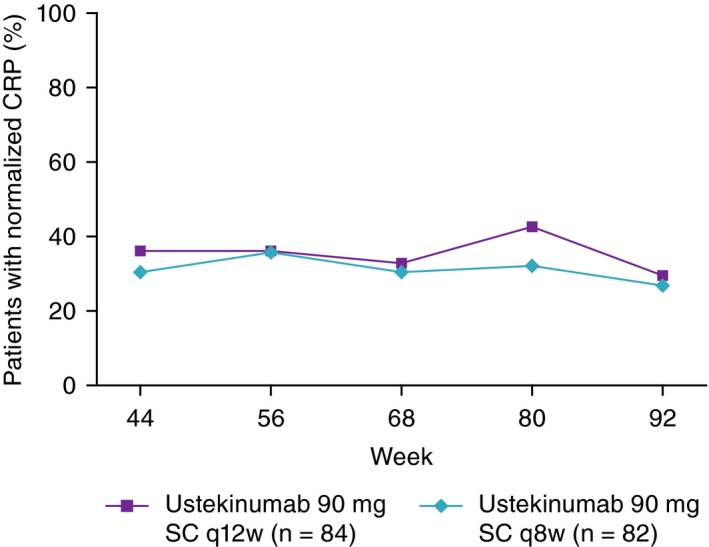
Proportions of patients with normalised CRP (≥3 mg/L) from Week 44 through Week 92; randomised patients who entered the long‐term extension. CRP, C‐reactive protein; SC, subcutaneous; q8w, every 8 weeks; q12w, every 12 weeks
3.1.7. Patient reported outcomes
IBDQ
At Week 0 of IM‐UNITI, the median IBDQ total score, IBDQ dimension (bowel, emotional, systemic, and social) scores were similar across treatment groups (Table 4). The proportions of patients with at least a 16‐point improvement in IBDQ score from baseline of the induction study were similar at Week 92 for patients in the ustekinumab 90 mg every 12 weeks (73.8%) and every 8 weeks group (76.8%), and lower in patients with prior dose adjustment (62.0%; Table 4).
Table 4.
Inflammatory bowel disease questionnaire (IBDQ) and 36‐item short form survey (SF‐36) scores at baseline, Week 44, and Week 92
| Continuous SC UST 90 mg q12w | Continuous SC UST 90 mg q8w | Patients with Prior Dose Adjustmenta | All UST treated | |
|---|---|---|---|---|
| IBDQ | ||||
| N | 84 | 82 | 71 | 237 |
| Median baseline score | 175.0 | 179.0 | 175.0 | 178.0 |
| Median change from baseline at Week 44 | 8.0 | 5.0 | −1.0 | 5.0 |
| Median change from baseline at Week 92 | 3.5 | 9.0 | 6.0 | 6.0 |
| Patients with ≥16‐point improvement at Week 92 | 73.8% | 76.8% | 62.0% | 71.3% |
| Bowel | ||||
| N | 84 | 82 | 71 | 237 |
| Median baseline score | 57.0 | 57.5 | 54.0 | 56.0 |
| Median change from baseline at Week 92 | 1.0 | 3.0 | 2.0 | 2.0 |
| Emotional | ||||
| N | 84 | 82 | 71 | 237 |
| Median baseline score | 65.5 | 66.0 | 64.0 | 66.0 |
| Median change from baseline at Week 92 | 1.5 | 4.0 | 1.0 | 2.0 |
| Systemic | ||||
| N | 84 | 82 | 71 | 237 |
| Median baseline score | 24.0 | 25.0 | 25.0 | 25.0 |
| Median change from baseline at Week 92 | 1.0 | 2.0 | 0.0 | 1.0 |
| Social | ||||
| N | 84 | 81 | 71 | 236 |
| Median baseline score | 31.0 | 31.0 | 30.0 | 31.0 |
| Median change from baseline at Week 92 | 1.0 | 1.0 | 0.0 | 1.0 |
| SF‐36 | ||||
| PCS | ||||
| N | 76 | 75 | 69 | 220 |
| Median baseline score | 49.50 | 50.12 | 48.36 | 49.54 |
| Median change from baseline at Week 44 | 1.92 | 0.88 | 0.31 | 0.96 |
| Median change from baseline at Week 92 | 1.21 | 2.93 | 0.81 | 2.02 |
| Patients with a 5‐point improvement at Week 92 | 59.2% | 69.3% | 59.4% | 62.7% |
| MCS | ||||
| N | 76 | 75 | 69 | 220 |
| Median baseline score | 48.69 | 49.43 | 49.95 | 49.38 |
| Median change from baseline at Week 44 | 3.34 | 2.00 | −0.04 | 1.79 |
| Median change from baseline at Week 92 | 2.80 | 2.51 | −1.68 | 0.89 |
| Patients with a 5‐point improvement at Week 92 | 65.8% | 62.7% | 49.3% | 59.5% |
SC, subcutaneous; UST, ustekinumab; q12w, every 12 weeks; q8w, every 8 weeks; PCS, Physical Component score; MCS, Mental Component score.
Patients who were in clinical response to ustekinumab (UST) induction dosing, were randomised, met loss of response criteria from Week 8 through Week 32 and received 90 mg SC q8w (this includes patients who were randomised to 90 mg SC q8w who had a sham dose adjustment).
The median change from maintenance baseline in IBDQ scores was higher at Week 92 for patients in the ustekinumab 90 mg every 8 weeks group (9.0) as compared with patients in the every 12 weeks group (3.5), and those patients with prior dose adjustment (6.0). The median changes from maintenance baseline in IBDQ dimension scores (eg, bowel, emotional, systemic, and social) were generally higher for the ustekinumab 90 mg every 12 weeks and every 8 weeks groups as compared with the prior dose adjustment group.
SF‐36
The median SF‐36 scores at Week 0 of IM‐UNITI were similar across treatment groups (Table 4). At Week 92, comparable results were observed among these groups in the proportion of patients with at least a five‐point improvement in both the SF‐36 physical and mental component scores (Table 4).
3.2. All treated patients entering long‐term extension
3.2.1. Remission and response Weeks 44‐92
From Week 44 to Week 92, the proportions of patients in clinical remission remained similar in the every 12 weeks (68.5% to 64.3%; n = 213) and the every 8 weeks groups (68.9% to 64.4%; n = 354, data not shown).
In these same groups, from Week 44 to 92, the proportions of patients in clinical response remained similar in both the every 12 weeks (86.9% to 79.3%; n = 213) and the every 8 weeks groups (89.0% to 75.7%; n = 354).
3.3. Observed case analysis of randomised patients entering long‐term extension
Using observed data, clinical remission rates were also maintained from Week 44 through Week 92 in the ustekinumab 90 mg every 12 weeks group (77.4%‐79.2%), as well as in the every 8 weeks group (84.1%‐87.1%; Figure 4).
Figure 4.
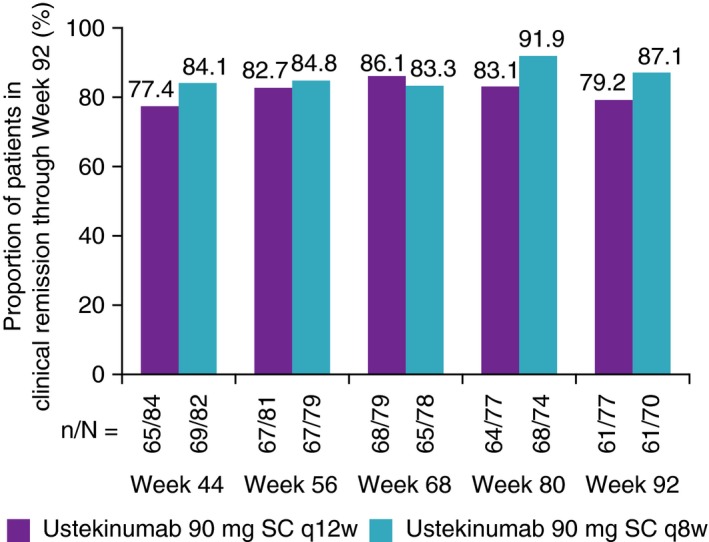
Patients in clinical remission from Week 44 through Week 92 of IM‐UNITI long‐term extension: observed data
3.4. Intent‐to‐treat analysis of randomised patients from Week 0 of maintenance
Of patients originally randomised to ustekinumab 90 mg every 12 weeks, 45.0% (58/129) were in remission at Week 92, and 46.9% (60/128) of patients originally randomised to 90 mg every 8 weeks were in remission at Week 92, including 55.8% (29/52) of patients in the TNF‐antagonist naïve subset. Consistent with other analyses, remission rates were sustained from Week 44 through Week 92 (Figure 5).
Figure 5.
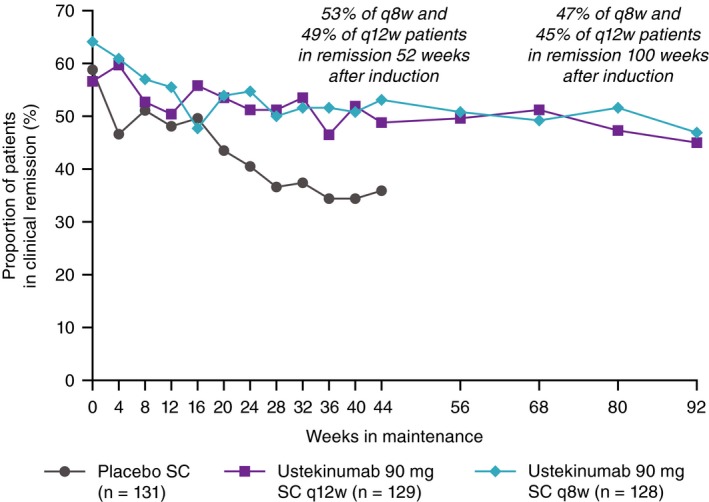
Long‐term rates of remission for patients responding to ustekinumab induction through Week 92 (intent‐to‐treat analysis of IM‐UNITI primary population from Week 0). SC, subcutaneous; q8w, every 8 weeks; q12w, every 12 weeks
3.5. Safety in all treated patients entering long‐term extension
3.5.1. Weeks 0‐96
Safety events (per hundred patient‐years) were similar among all ustekinumab‐treated patients compared to placebo, including overall adverse events (484.39 vs 447.76), serious adverse events (19.24 vs 18.82), and serious infections (4.09 vs 4.02), with 244.2 patient‐years of follow‐up among placebo‐treated patients and 1020.0 patient‐years years among all ustekinumab‐treated patients. The number of adverse events, serious adverse events, and serious infections per hundred patient‐years were generally comparable for the ustekinumab 90 mg every 12 weeks and every 8 weeks groups, without dose effect (Table 5).
Table 5.
Summary of key safety finding per hundred patient‐years of follow‐up from Week 0 through Week 96: all patients who entered long‐term extensiona
| Placebo | SC UST 90 mg q12w | SC UST 90 mg q8w | All UST | |
|---|---|---|---|---|
| N | 151 | 213 | 354 | 567 |
| Average duration of follow‐up (weeks) | 84.1 | 93.8 | 93.4 | 93.5 |
| Total patient‐years of follow‐up | 244.2 | 384.3 | 635.6 | 1020.0 |
| Deaths | 0 | 1b | 2 | 3 |
| Number of specified events per hundred patient‐years of follow‐up (95% confidence interval) | ||||
| Adverse events | 484.39 (457.18, 512.80) | 413.70 (393.62, 434.55) | 468.36 (451.68, 485.49) | 447.76 (434.87, 460.94) |
| Serious adverse events | 19.24 (14.14, 25.59) | 19.51 (15.35, 24.46) | 18.41 (15.22, 22.06) | 18.82 (16.26, 21.68) |
| Infections | 105.64 (93.14, 119.35) | 112.66 (102.30, 123.79) | 120.51 (112.13, 129.36) | 117.55 (110.99, 124.40) |
| Serious infections | 4.09 (1.96, 7.53) | 5.72 (3.59, 8.67) | 2.99 (1.80, 4.67) | 4.02 (2.88, 5.45) |
SC, subcutaneous; UST, ustekinumab; q12w, every 12 weeks; q8w, every 8 weeks
Data are based on all randomised and nonrandomised patients combined by regimen who entered the long‐term extension
Death occurred in a nonrandomised patient on q12w UST after withdrawal of consent
3.5.2. Weeks 44‐96
Safety events (per hundred patient‐years) were similar among all ustekinumab‐treated patients compared to placebo, including overall adverse events (401.27 vs 371.84), serious adverse events (24.27 vs 19.56), and serious infections (5.20 vs 3.73), with 115.4 patient‐years of follow‐up among placebo‐treated patients and 536.8 patient‐years years among all ustekinumab‐treated patients. No dose effect was seen.
One case of tuberculosis deemed unrelated to study drug by the investigator was reported in a 45‐year‐old male from South Africa who received ustekinumab every 8 weeks during the long‐term extension. Although negative for tuberculosis during screening at study entry and asymptomatic, the patient had a positive QuantiFERON‐TB Gold test on routine annual screening and had bronchial brushings that were positive for mycobacterium tuberculosis. The patient completed 6 months of therapy for tuberculosis with complete resolution. A single, serious opportunistic infection of cytomegalovirus colitis occurred in a 32‐year old who received ustekinumab induction followed by placebo maintenance (440 days after last exposure to ustekinumab). The patient was treated with ganciclovir with resolution of the infection.
The number of treatment‐emergent malignancies per hundred patient‐years of follow‐up was 2.60 for placebo patients, and 0.37 for ustekinumab patients. Five malignancies were reported in patients during the long‐term extension. One squamous cell cutaneous carcinoma in a patient randomised in maintenance to placebo and who continued to receive placebo in the long‐term extension; one testicular seminoma in a 31‐year‐old male randomised in maintenance to ustekinumab 90 mg subcutaneous every 8 weeks; and one basal cell carcinoma in a patient who was not randomised but received ustekinumab 90 mg subcutaneous every 12 weeks during the long‐term extension. Two malignancies (papillary thyroid cancer and squamous cell cutaneous carcinoma) were reported in two patients who were not randomised and did not receive ustekinumab throughout the entire study.
Three deaths were reported, two of presumed cardiovascular causes and one suicide. Two of the three patients received ustekinumab 90 mg every 8 weeks and one patient received ustekinumab 90 mg every 12 weeks.
The proportions of patients with 1 or more injection‐site reactions was 2.0% (n = 11) for ustekinumab patients and 0.6% (n = 6) for placebo patients with no reports of serious reactions, anaphylaxis or serum sickness‐like reactions.
3.6. Immunogenicity in randomised patients entering long‐term extension
A total of 237 randomised patients who received ustekinumab during the long‐term extension had appropriate samples for antibody to ustekinumab status through Week 96. Across all randomised treatment groups, 4.2% (10/237) of ustekinumab subcutaneous‐treated patients were positive for antibodies to ustekinumab. Among patients on continuous 90 mg every 8 weeks treatment, the rate of antibodies to ustekinumab was 2.4%, similar to the rate seen in this group at 44 weeks.27 Of note, rates of antibodies to ustekinumab were higher among patients who were randomised to placebo (8.2%; 5/61).
4. DISCUSSION
IM‐UNITI is the 5‐year maintenance study in the Phase 3 pivotal programme for treatment of patients with Crohn's disease with ustekinumab. As previously shown, through 44 weeks of treatment, statistically significant evidence was generated demonstrating that treatment with subcutaneous ustekinumab 90 mg every 12 weeks or every 8 weeks was effective in maintaining clinical response and remission in patients with moderate‐to‐severe Crohn's disease.27
Our findings confirm that in ustekinumab induction responders, remission, and response rates were maintained through Week 92 of the study in both TNF antagonist‐refractory patients from UNITI‐1 and UNITI‐2 conventional therapy failure patients (predominantly TNF antagonist‐naïve). However, maintenance of remission from Week 44 to Week 92 of subcutaneous ustekinumab was slightly lower in the UNITI‐1 subset. Efficacy from Week 44 to Week 92 of subcutaneous ustekinumab maintenance in patients entering the long‐term extension was similar for both the every 8 weeks and every 12 weeks subcutaneous ustekinumab dose regimens, in both populations. In a combined analysis that included all randomised treatment groups, patients who had previously met loss of response criteria for dose adjustment before Week 44 had somewhat lower remission rates over the second year of treatment. Similar and consistent efficacy was observed for every 12 weeks and every 8 weeks dosing as measured by proportions of patients with normalised CRP and in those experiencing improvements in IBDQ and SF‐36 health‐related quality of life. Discontinuation rates were low, with over 90% of all patients completing Week 96 of the extension trial.
Many controlled maintenance studies of biological agents have been limited to 1 year or less in duration,7, 8, 11, 12 with the exceptions of adalimumab and vedolizumab.9, 10, 13 Recognising the challenges of cross‐study comparisons, due to differences in study populations, efficacy measures, statistical analysis, and study designs, it is difficult to compare these results with previous adalimumab or vedolizumab long‐term extension data. However, the maintenance of efficacy with ustekinumab in patients with Crohn's disease was stable over 92 weeks of treatment. This experience of ustekinumab in patients with Crohn's disease is consistent with that seen with maintenance of efficacy and excellent retention through 5 years reported from the ustekinumab psoriasis Phase 3 studies28, 29 and superior ustekinumab drug survival rates compared to TNF antagonists in the large psoriasis PSOLAR registry.26
Variability in efficacy from Weeks 44 to 92 of the long‐term extension was seen, depending on the analysis and the treatment group. Not unexpectedly, remission rates were higher with an as‐observed analysis. Remission rates were lower if all responders who entered IM‐UNITI were included in the analysis as opposed to only those who entered the long‐term extension. However, even with this most stringent analysis, remission rates remained stable in the study, with the clinically important observation that, even when analysed in this conservative fashion, approximately one‐half of induction responders were in remission at Week 92. These results are consistent with a recently published retrospective multicentre cohort study of 122 refractory Crohn's disease patients who were treated with ustekinumab with a median follow‐up of 26.6 months.30
In general, patients who had not failed an anti‐TNF therapy (UNITI‐2) demonstrated higher rates of remission at Week 92, consistent with the Week 44 results.27 While results were generally consistent between every 8 weeks and every 12 weeks treatment groups from Week 44 to Week 92, differences seen at Week 44 were generally sustained through Week 92. Clinical efficacy was consistently lower across endpoints for those patients who dose‐adjusted before entering the long‐term extension. Perhaps not surprisingly, upon entry into the long‐term extension, these patients (who had already previously met loss of response criteria prior to Week 44) had notably higher median CDAI scores, higher median CRP concentrations, lower median IBDQ scores, and lower rates of clinical remission, indicating that they are a subset with more severe disease. Among all treated patients (incorporating these patients as well as the nonrandomised and randomised groups), rates of response and remission were also slightly lower than the randomised every 8 weeks and every 12 weeks groups, but were similarly maintained. While some data have been published on predictors of response to ustekinumab in Crohn's disease patients, further analyses to identify patients who are most likely to achieve maximum benefit from ustekinumab would be helpful.31
Rates of patients who developed anti‐drug antibodies were low and formation of antibodies did not impact response and remission rates. In randomised patients who received continuous treatment, the number of patients with anti‐drug antibodies remained low and was consistent from Week 44 through Week 96.27 Randomised patients who received an induction dose of ustekinumab followed by placebo had higher rates of antibodies, suggesting that drug exposure followed by a significant gap in treatment may increase the risk of antibody formation. However, even this proportion with antibodies (8%) is low compared to anti‐drug antibody rates seen with other approved Crohn's disease biologics,32 particularly when those drugs are assessed using similar high sensitivity, drug tolerant assays.
While patients in this long‐term extension were not subject to dose escalation based on drug concentrations, a previous analysis of ustekinumab serum concentrations of UNITI‐1, UNITI‐2 and through Week 44 of IM‐UNITI demonstrated a relationship between serum concentrations and efficacy.33 This supports the use of the every 8 weeks dose regimen or the option to dose escalate if patients who start every 12 weeks dosage fail to achieve response or lose response, as was allowed before Week 44 of IM‐UNITI. However, future studies are needed to evaluate whether proactive therapeutic drug monitoring of ustekinumab to target levels improves long‐term Crohn's disease outcomes. Of note, in contrast to TNF antagonists, in the previous study,33 there was no significant impact of co‐treatment with immunomodulators on serum concentrations, possibly related to the low immunogenicity of ustekinumab.
In the study extension through Week 96, ustekinumab was well‐tolerated with a favourable safety profile that did not differentiate from placebo. Among all treated patients, events per hundred patient‐years of adverse events, serious adverse events, and serious infections were similar among the every 12 weeks and every 8 weeks groups, individually, with no evidence of a dose effect. Between the two dose groups and among combined ustekinumab groups, there was no difference in the number of these events compared to placebo. No serious opportunistic infections occurred in ustekinumab‐treated patients from Week 44 to Week 96. A nonserious infection of pulmonary tuberculosis occurred in a patient living in a region endemic for tuberculosis and who received ustekinumab every 8 weeks during the study extension. Rates of injection‐site reactions remained low with no reports of serious reactions, anaphylaxis, or serum sickness‐like reactions. Rates of malignancy were not increased. Three deaths were reported during the study extension with an observed rate of death comparable to the rate of death in a large cohort study of patients with Crohn's disease.34
There are limitations in the study extension design. First, patients were selected by the investigator to participate in the study extension because in their opinion, they might benefit from continued treatment (although 97.5% [719/737] of patients that reached Week 44 did so). This criterion may limit the generalisability of the findings to only those patients who responded to and tolerated ustekinumab through approximately 1 year of treatment. Second, because patients who entered the study extension on placebo represent a group of patients who were long‐term responders to ustekinumab induction or were true placebo responders and were therefore discontinued from the study when unblinding occurred, direct efficacy and statistical comparisons between placebo and ustekinumab treatment groups could not be performed over the long‐term extension duration. Hence, the emphasis of this manuscript is on efficacy measures among ustekinumab‐treated patients.
In summary, patients with moderately to severely active Crohn's disease, who received ustekinumab in the study extension, maintained clinical response and clinical remission through Week 92 of IM‐UNITI. Clinical results were supported by maintained normalisation of CRP concentrations and improved health‐related quality of life outcome measures. The safety profile observed for ustekinumab through Week 96 of treatment was consistent with the safety through Week 96 and with the established safety profile within the ustekinumab prescribing information with no new safety signals identified. Additional efficacy and safety data from IM‐UNITI will continue to be generated through 5 years of maintenance.
AUTHORSHIP
Guarantor of the article: Dr. Brian G Feagan.
Author contributions: C Gasink, D Jacobstein, and J Johanns participated in the conception and design of the study, participated in acquisition/collection of data, analysis and interpretation of data, and drafted/revised the manuscript for important intellectual content. W Sandborn, P Rutgeerts, BE Sands, SB Hanauer, S Targan, S Ghosh, WJS de Villiers, J‐F Colombel and BG Feagan participated in conception and design of the study, analysis and interpretation on the data, and drafted/revised the manuscript for important intellectual content. B Zou participated in the analysis and interpretation of the data and drafted/revised the manuscript critically for important intellectual content.
All authors approved the final version of the manuscript for submission, including the authorship list.
Supporting information
ACKNOWLEDGEMENTS
The authors thank Ye Miao, employed by Janssen Research & Development for statistical support and Benjamin Yeager for his assistance with data review and preparation. Editorial support was provided by Kirsten Schuck Gross, BS, James P. Barrett, BS (Janssen Scientific Affairs, LLC), and Cynthia Guzzo, MD (HireGenics), Duluth, GA and funded by Janssen Scientific Affairs, LLC.
Declaration of personal interests: WJ Sandborn reports grants, personal fees and nonfinancial support from Abbvie; grants and personal fees from Prometheus Laboratories, grants and personal fees from Boehringer Ingelheim, grants and personal fees from Takeda, grants and personal fees from Atlantic Pharmaceuticals, grants and personal fees from Janssen, grants and personal fees from Bristol‐Myers Squibb, grants and personal fees from Genentech, grants and personal fees from Nutrition Science Partners, personal fees from Kyowa Hakko Kirin, personal fees from Millennium Pharmaceuticals, personal fees from Celgene Cellular Therapeutics, personal fees from Santarus, personal fees from Salix Pharmaceuticals, personal fees from Catabasis Pharmaceuticals, personal fees from Vertex Pharmaceuticals, personal fees from Warner Chilcott, personal fees from Gilead Sciences, personal fees from Cosmo Pharmaceuticals, personal fees from Ferring Pharmaceuticals, personal fees from Sigmoid Biotechnologies, personal fees from Tillotts Pharma, personal fees from Am Pharma BV, personal fees from Dr. August Wolff, personal fees from Avaxia Biologics, personal fees from Zyngenia, personal fees from Ironwood Pharmaceuticals, personal fees from Index Pharmaceuticals, personal fees from Nestle, personal fees from Lexicon Pharmaceuticals, personal fees from UCB Pharma, personal fees from Orexigen, personal fees from Luitpold Pharmaceuticals, personal fees from Baxter Healthcare, personal fees from Ferring Research Institute, personal fees from Amgen, personal fees from Novo Nordisk, personal fees from Mesoblast Inc., personal fees from Shire, personal fees from Ardelyx Inc., personal fees from Actavis, personal fees from Seattle Genetics, personal fees from MedImmune (AstraZeneca), personal fees from Actogenix NV, personal fees from Lipid Therapeutics Gmbh, personal fees from Eisai, personal fees from Qu Biologics, personal fees from Toray Industries Inc., personal fees from Teva Pharmaceuticals, personal fees from Eli Lilly, personal fees from Chiasma, personal fees from TiGenix, personal fees from Adherion Therapeutics, personal fees from Immune Pharmaceuticals, personal fees from Celgene, personal fees from Arena Pharmaceuticals, personal fees from Ambrx Inc., personal fees from Akros Pharma, personal fees from Vascular Biogenics, personal fees from Theradiag, personal fees from Forward Pharma, personal fees from Regeneron, personal fees from Galapagos, personal fees from Seres Health, personal fees from Ritter Pharmaceuticals, personal fees from Theravance, personal fees from Palatin, personal fees from Biogen, personal fees from Western University (owner of Robarts Clinical Trials). P Rutgeerts has received consulting fees from Johnson & Johnson, Merck Sharp and Dohme, AbbVie, UCB Pharma, Bristol‐Myers Squibb, Amgen–MedImmune–AstraZeneca, Pfizer, Tillotts Pharma, Falk Pharma, and Takeda, lecture fees from Johnson & Johnson, Merck Sharp and Dohme, AbbVie, and UCB Pharma, and personal fees from Robarts Clinical Trials. BE Sands has received consulting fees and research grants from AbbVie, Pfizer, Amgen, Bristol‐Myers Squibb, Celgene, Janssen, and Takeda, and has received consulting fees from Boehringer Ingelheim, Akros Pharma, Arena Pharmaceuticals, Forward Pharma, Immune Pharmaceuticals, Lilly, Synergy Pharmaceuticals, Theravance, Receptos, TiGenix, TopiVert Pharma, MedImmune, Vedanta Biosciences, Allergan, UCB Pharma, EnGene, Target PharmaSolutions, Lycer, Lyndra, Vivelix Pharmaceuticals, Oppilan Pharma, and Gilead; SB Hanauer has received grant support and fees for consulting, lectures, and serving on an advisory board from Janssen, Abbvie, and Takeda; consulting and or advisory board fees from Pfizer, Receptos, Novartis, Gilead, Boehringer‐Ingelheim, Seres Therapeutics, Ferring, Bristol‐Myers Squibb, Amgen, Genentech, Merck, Samsung Bioepis, Protagonist, Salix, TiGenix, Allergan, Celgene, Vhsquared; and grant support from Abbvie, Janssen, Pfizer, Celgene, Boehringer‐Ingelheim, Takeda, Gilead. S Targan has received consulting fees from Janssen. S Ghosh has received fees for serving on steering committees for Janssen, AbbVie, Pfizer, Receptos, Aerpio, Takeda, Gilead, Boehringer Ingelheim and Novartis, fees for serving on a drug‐monitoring committee for Janssen, and serving on advisory boards for AbbVie, Janssen, and Takeda. WJS de Villiers has received consulting fees from Janssen. J‐F Colombel has received personal fees from AbbVie, Amgen, Boehringer Ingelheim, Celgene, Celltrion, Enterome, Ferring, Genentech, Janssen–Johnson & Johnson, MedImmune, Merck, Pfizer, Protagonist, Second Genome, Seres, Shire, Takeda, Theradiag, and PPM Services, grant support from AbbVie, Janssen–Johnson & Johnson, and Takeda, and holds stock options in Genfit and Intestinal Biotech Development. BG Feagan has received fees for serving on advisory boards from AbbVie, Allergan, Amgen, AstraZeneca, Avaxia Biologics, Boehringer Ingelheim, Bristol‐Myers Squibb, Celgene, Elan–Biogen, Ferring, Genentech–Roche, Janssen–Johnson &Johnson, Merck, Millennium, Nestlé, Novo Nordisk, Novartis, Pfizer, Prometheus, Protagonist, Receptos, Salix, Sigmoid Pharma, Takeda, Teva, TiGenix, Tillotts Pharma, and UCB Pharma, consulting fees from AbbVie, Actogenix, Akros, Albireo Pharma, Allergan, Amgen, AstraZeneca, Avaxia Biologics, Avir Pharma, Axcan, Baxter Healthcare, Biogen Idec, Boehringer Ingelheim, Bristol‐Myers Squibb, Calypso Biotech, Celgene, Elan–Biogen, EnGene, Ferring, Genentech–Roche, GiCare Pharma, Gilead Sciences, Given Imaging, GlaxoSmithKline, Ironwood, Janssen Biotech–Centocor, Janssen–Johnson & Johnson, Kyowa Hakko Kirin, Eli Lilly, Merck, Mesoblast Pharma, Millennium, Nestlé, Novo Nordisk, Novartis, Pfizer, Prometheus, Protagonist, Receptos Salix, Sanofi, Shire, Sigmoid Pharma, Synergy Pharma, Takeda, Teva, TiGenix, Tillotts Pharma, UCB Pharma, Vertex, VHsquared, Wyeth, Zealand, and Zygenia, lecture fees from AbbVie, Janssen–Johnson & Johnson, Takeda, and UCB Pharma, and grant support from AbbVie, Amgen, AstraZeneca, Bristol‐ Meyers Squibb, Janssen Biotech–Centocor, Janssen–Johnson & Johnson, Pfizer, Receptos, Sanofi, and Takeda, and being a consultant to Robarts Clinical Trials. C Gasink, was an employee at Janssen Research & Development at the time of the study and is a current employee of Janssen Scientific Affairs, LLC. He owns stock or stock options. D Jacobstein, B Zou, and J Johanns were employees at Janssen Research & Development at the time of the study and own stock or stock options.
APPENDIX 1.
AUTHORS’ COMPLETE AFFILIATIONS
William J. Sandborn, University of California San Diego, La Jolla, CA, USA; Paul Rutgeerts, University Hospital, Gasthuisberg, Leuven, Belgium; Christopher Gasink, Janssen Scientific Affairs, LLC, Horsham, PA, USA; Douglas Jacobstein, Janssen Research & Development, LLC, Spring House, PA, USA; Bin Zou, Janssen Research & Development, LLC, Spring House, PA, USA; Jewel Johanns, Janssen Research & Development, LLC, Spring House, PA, USA; Bruce E. Sands, Icahn School of Medicine at Mt. Sinai Hospital, New York, NY, USA; Stephen B. Hanauer, Northwestern University Feinberg School of Medicine, Chicago, IL, USA; Stephan Targan, Cedars‐Sinai Medical Center, Los Angeles, CA, USA; Subrata Ghosh, NIHR Biomedical Research Centre and University of Birmingham, Birmingham UK; Willem J.S. de Villiers, Stellenbosch University, Stellenbosch, South Africa; Jean‐Frédéric Colombel, Icahn School of Medicine at Mt. Sinai Hospital, New York, NY, USA; Brian G. Feagan, Robarts Clinical Trials, Western University, London, Ontario, Canada.
Sandborn WJ, Rutgeerts P, Gasink C, et al. Long‐term efficacy and safety of ustekinumab for Crohn's disease through the second year of therapy. Aliment Pharmacol Ther. 2018;48:65–77. https://doi.org/10.1111/apt.14794
The Handling Editor for this article was Professor Jonathan Rhodes, and it was accepted for publication after full peer‐review.
Authors’ complete affiliations are listed in Appendix section.
Funding information
This study was funded by Janssen Research & Development, LLC. Writing and editorial support for this paper was funded in part by Janssen Research & Development, LLC.
Some of the data displayed in this article were presented at ECCO 2017 (oral presentation; Barcelona, Spain) and Digestive Disease Week 2017 (poster presentation; Chicago, IL).
REFERENCES
- 1. Torres J, Mehandru S, Colombel JF, et al. Crohn's disease. Lancet. 2017;389:1741‐1755. [DOI] [PubMed] [Google Scholar]
- 2. Gomollon F, Dignass A, Annese V, et al. 3rd European evidence‐based consensus on the diagnosis and management of Crohn's disease 2016: part 1: diagnosis and medical management. J Crohns Colitis. 2017;11:3‐25. [DOI] [PubMed] [Google Scholar]
- 3. Lichtenstein GR, Yan S, Bala M, et al. Infliximab maintenance treatment reduces hospitalizations, surgeries, and procedures in fistulizing Crohn's disease. Gastroenterology. 2005;128:862‐869. [DOI] [PubMed] [Google Scholar]
- 4. Katz JA, Antoni C, Keenan GF, et al. Outcome of pregnancy in women receiving infliximab for the treatment of Crohn's disease and rheumatoid arthritis. Am J Gastroenterol. 2004;99:2385‐2392. [DOI] [PubMed] [Google Scholar]
- 5. Pearson DC, May GR, Fick G, et al. Azathioprine for maintaining remission of Crohn's disease. Cochrane Database Syst Rev. 2000;(2):CD000067. [DOI] [PubMed] [Google Scholar]
- 6. Feagan BG, Fedorak RN, Irvine EJ, et al. A comparison of methotrexate with placebo for the maintenance of remission in Crohn's disease. North American Crohn's Study Group Investigators. N Engl J Med. 2000;342:1627‐1632. [DOI] [PubMed] [Google Scholar]
- 7. Hanauer SB, Feagan BG, Lichtenstein GR, et al. Maintenance infliximab for Crohn's disease: the ACCENT I randomised trial. Lancet. 2002;359:1541‐1549. [DOI] [PubMed] [Google Scholar]
- 8. Sands BE, Anderson FH, Bernstein CN, et al. Infliximab maintenance therapy for fistulizing Crohn's disease. N Engl J Med. 2004;350:876‐885. [DOI] [PubMed] [Google Scholar]
- 9. Panaccione R, Colombel JF, Sandborn WJ, et al. Adalimumab maintains remission of Crohn's disease after up to 4 years of treatment: data from CHARM and ADHERE. Aliment Pharmacol Ther. 2013;38:1236‐1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Panaccione R, Colombel JF, Sandborn WJ, et al. Adalimumab sustains clinical remission and overall clinical benefit after 2 years of therapy for Crohn's disease. Aliment Pharmacol Ther. 2010;31:1296‐1309. [DOI] [PubMed] [Google Scholar]
- 11. Schreiber S, Khaliq‐Kareemi M, Lawrance IC, et al. Maintenance therapy with certolizumab pegol for Crohn's disease. N Engl J Med. 2007;357:239‐250. [DOI] [PubMed] [Google Scholar]
- 12. Sandborn WJ, Colombel JF, Enns R, et al. Natalizumab induction and maintenance therapy for Crohn's disease. N Engl J Med. 2005;353:1912‐1925. [DOI] [PubMed] [Google Scholar]
- 13. Vermeire S, Loftus EV Jr, Colombel JF, et al. Long‐term efficacy of vedolizumab for Crohn's disease. J Crohns Colitis. 2017;11:412‐424. [DOI] [PubMed] [Google Scholar]
- 14. Sands BE, Feagan BG, Rutgeerts P, et al. Effects of vedolizumab induction therapy for patients with Crohn's disease in whom tumor necrosis factor antagonist treatment failed. Gastroenterology. 2014;147:618‐627, e3. [DOI] [PubMed] [Google Scholar]
- 15. Anderson PJ. Tumor necrosis factor inhibitors: clinical implications of their different immunogenicity profiles. Semin Arthritis Rheum. 2005;34:19‐22. [DOI] [PubMed] [Google Scholar]
- 16. Strand V, Balsa A, Al‐Saleh J, et al. Immunogenicity of biologics in chronic inflammatory diseases: a systematic review. BioDrugs. 2017;31:299‐316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nielsen OH, Bjerrum JT, Seidelin JB, et al. Biological treatment of Crohn's disease. Dig Dis. 2012;30(Suppl 3):121‐133. [DOI] [PubMed] [Google Scholar]
- 18. Connell WR, Kamm MA, Ritchie JK, et al. Bone marrow toxicity caused by azathioprine in inflammatory bowel disease: 27 years of experience. Gut. 1993;34:1081‐1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lemaitre M, Kirchgesner J, Rudnichi A, et al. Association between use of thiopurines or tumor necrosis factor antagonists alone or in combination and risk of lymphoma in patients with inflammatory bowel disease. JAMA. 2017;318:1679‐1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Connell WR, Kamm MA, Dickson M, et al. Long‐term neoplasia risk after azathioprine treatment in inflammatory bowel disease. Lancet. 1994;343:1249‐1252. [DOI] [PubMed] [Google Scholar]
- 21. Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha‐neutralizing agent. N Engl J Med. 2001;345:1098‐1104. [DOI] [PubMed] [Google Scholar]
- 22. Khanna D, McMahon M, Furst DE. Safety of tumour necrosis factor‐alpha antagonists. Drug Saf. 2004;27:307‐324. [DOI] [PubMed] [Google Scholar]
- 23. STELARA [package insert]. Horsham, PA: Janssen Biotech, Inc, 2017. [Google Scholar]
- 24. Papp KA, Griffiths CE, Gordon K, et al. Long‐term safety of ustekinumab in patients with moderate‐to‐severe psoriasis: final results from 5 years of follow‐up. Br J Dermatol. 2013;168:844‐854. [DOI] [PubMed] [Google Scholar]
- 25. Papp K, Gottlieb AB, Naldi L, et al. Safety surveillance for ustekinumab and other psoriasis treatments from the Psoriasis Longitudinal Assessment and Registry (PSOLAR). J Drugs Dermatol. 2015;14:706‐714. [PubMed] [Google Scholar]
- 26. Menter A, Papp KA, Gooderham M, et al. Drug survival of biologic therapy in a large, disease‐based registry of patients with psoriasis: results from the Psoriasis Longitudinal Assessment and Registry (PSOLAR). J Eur Acad Dermatol Venereol. 2016;30:1148‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feagan BG, Sandborn WJ, Gasink C, et al. Ustekinumab as induction and maintenance therapy for Crohn's disease. N Engl J Med. 2016;375:1946‐1960. [DOI] [PubMed] [Google Scholar]
- 28. Kimball AB, Papp KA, Wasfi Y, et al. Long‐term efficacy of ustekinumab in patients with moderate‐to‐severe psoriasis treated for up to 5 years in the PHOENIX 1 study. J Eur Acad Dermatol Venereol. 2013;27:1535‐1545. [DOI] [PubMed] [Google Scholar]
- 29. Kimball AB, Gordon KB, Fakharzadeh S, et al. Long‐term efficacy of ustekinumab in patients with moderate‐to‐severe psoriasis: results from the PHOENIX 1 trial through up to 3 years. Br J Dermatol. 2012;166:861‐872. [DOI] [PubMed] [Google Scholar]
- 30. Wils P, Bouhnik Y, Michetti P, et al. Long‐term efficacy and safety of ustekinumab in 122 refractory Crohn's disease patients: a multicentre experience. Aliment Pharmacol Ther. 2018;47:588‐595. [DOI] [PubMed] [Google Scholar]
- 31. Barre A, Colombel JF, Ungaro R. Review article: predictors of response to vedolizumab and ustekinumab in inflammatory bowel disease. Aliment Pharmacol Ther. 2018;47:896‐905. [DOI] [PubMed] [Google Scholar]
- 32. Moss AC, Brinks V, Carpenter JF. Review article: immunogenicity of anti‐TNF biologics in IBD ‐ the role of patient, product and prescriber factors. Aliment Pharmacol Ther. 2013;38:1188‐1197. [DOI] [PubMed] [Google Scholar]
- 33. Adedokun OJ, Xu Z, Gasink C, et al. Pharmacokinetics and exposure response relationships of ustekinumab in patients with Crohn's disease. Gastroenterology. 2018;154:1660‐1671. [DOI] [PubMed] [Google Scholar]
- 34. Jess T, Loftus EV Jr, Harmsen WS, et al. Survival and cause specific mortality in patients with inflammatory bowel disease: a long term outcome study in Olmsted County, Minnesota, 1940‐2004. Gut. 2006;55:1248‐1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials