

Abstract
This study evaluated the pharmacokinetics (PK), efficacy, safety, and tolerability of bleselumab – a fully‐human anti‐CD40 monoclonal recombinant IgG4. Patients with moderate‐to‐severe psoriasis were randomized on day 1 to receive bleselumab or placebo on days 1, 15 and 29 in a dose‐escalation of bleselumab at 0.1, 0.3, 1.0 or 3.0 mg/kg. The safety‐analysis set (SAF) and full‐analysis set (FAS) included all patients who received bleselumab or placebo, and the PK‐analysis set (PKAS) included patients in the SAF with ≥1 quantifiable serum bleselumab concentration. Serial blood samples were collected after each dose, and the bleselumab serum concentration was measured. After each dose, the area‐under‐the‐concentration–time curve over 336 hours (AUC336) and the maximum serum concentration (Cmax), and dose proportionality of AUC336 and Cmax were determined. The psoriasis area and severity index (PASI) score, the physician static global assessment (PSGA) score, the percentage body surface area (%BSA) affected with psoriasis, adverse events and laboratory parameters were assessed. Sixty patients were randomized and included in the SAF/FAS (bleselumab, n = 49; placebo, n = 11); 48 formed the PKAS. Bleselumab Cmax and AUC336 were more than dose proportional in the range 0.1–3.0 mg/kg, suggesting nonlinear PK after single/multiple doses. No clinically significant infusion reactions, cytokine‐release syndrome, or thromboembolic events were reported. Bleselumab did not improve the PASI scores, PSGA scores, or %BSA versus placebo. Transient elevation of alanine aminotransferase and aspartate aminotransferase levels by >3 × upper limit of normal were observed in four (8.2%) and two (4.1%) patients, respectively, in the 1.0 or 3.0 mg/kg groups. Patients with liver function test increases had no concurrent changes in bilirubin. Bleselumab demonstrated nonlinear PK after single and multiple doses, with few adverse reactions.
Keywords: bleselumab, moderate‐to‐severe plaque psoriasis, pharmacokinetics, randomized controlled trial
1. INTRODUCTION
Psoriasis is a common, chronic skin disease, with a prevalence of 0.7–2.9% across Europe and North America (Papp, Gulliver, Lynde, Poulin, Ashkenas, Canadian Psoriasis Guidelines Committee, 2011; Parisi, Symmons, Griffiths, Ashcroft, Identification and Management of Psoriasis and Associated ComorbidiTy (IMPACT) project team, 2013). Clinical moderate‐to‐severe psoriasis is characterized by scaling and erythema, with symptoms of itching, physical irritation, pain and soreness (Helmick, Lee‐Han, Hirsch, Baird, & Bartlett, 2014; Stern, Nijsten, Feldman, Margolis, & Rolstad, 2004; World Health Organization, 2016). Symptoms of psoriasis limit the patient's ability to perform daily activities, and reduce their quality of life (Sampogna, Tabolli, Abeni, IDI Multipurpose Psoriasis Research on Vital Experiences (IMPROVE) investigators, 2012; van de Kerkhof et al., 2015).
Psoriasis lesions are associated with over production of keratinocytes, and dermal angiogenesis with T cell infiltration (Cai, Fleming, & Yan, 2012; Karczewski, Dobrowolska, Rychlewska‐Hańczewska, & Adamski, 2016; World Health Organization, 2016). The underlying pathophysiology involves CD40 signalling, induced by its ligand, CD154 (Pasch et al., 2004). CD40 is a cell surface tumor necrosis factor (TNF) receptor expressed by a variety of cells, including B lymphocytes, dendritic cells, keratinocytes, endothelial cells and epithelial cells, while CD154 is expressed on activated CD4+ T cells (Peters, Stunz, & Bishop, 2009). CD40 and CD154 interactions are involved in immune regulation and play a role in autoimmune disorders and transplant rejection (Jacobson et al., 2007; Larsen et al., 1996; Peters et al., 2009). It is hypothesized that blocking the interaction between CD40 and CD154 may control psoriasis.
Bleselumab (ASKP1240) is a fully‐human anti‐CD40 monoclonal antibody (immunoglobulin G4) that inhibits immune responses by blocking CD40–CD154 interaction between T cells, B cells, and antigen‐presenting cells (Okimura et al., 2014). This variant of anti‐CD40 is expressed in Chinese hamster ovary cells, and is recovered from culture medium, and subsequently purified (Ishida et al., 2002; Okimura et al., 2014).
The results from in vitro and in vivo studies suggest a potential therapeutic role for bleselumab as an immunosuppressive agent for patients with an autoimmune disease, such as psoriasis (Okimura et al., 2014). In vitro, bleselumab was found to be a concentration‐dependent inhibitor of CD154‐induced peripheral blood mononuclear cell proliferation (Okimura et al., 2014). Furthermore, in a cynomolgus monkey model (Okimura et al., 2014), 3 weeks of weekly bleselumab infusions attenuated delayed hypersensitivity and tetanus toxoid‐mediated specific antibody formation (Okimura et al., 2014). These immunosuppressive effects correlated with CD40 receptor saturation, suggesting a potential therapeutic role for bleselumab as an immunosuppressive agent for patients with diseases such as psoriasis. Bleselumab may also prevent rejection in solid organ transplantation (Oura et al., 2012; Song et al., 2014).
In a Phase I study in healthy volunteers, bleselumab demonstrated good tolerability: adverse events (AEs) collected over 1 week following exposure to bleselumab were generally mild in intensity, and there were no discontinuations due to AEs (Goldwater et al., 2013). There were also no treatment‐related adverse effects on the cardiovascular, respiratory, renal, or central nervous system in cynomolgus monkeys receiving weekly bleselumab doses up to 100 mg/kg for 4 weeks (Okimura et al., 2014). Of note, in healthy volunteers, bleselumab demonstrated nonlinear pharmacokinetics (PK) in the dose range of 0.1–10 mg/kg, which was evidenced by disproportionate increases in systemic exposure with increasing dose (Goldwater et al., 2013). However, no PK, safety or tolerability data are currently available for patients with psoriasis. This study evaluated the PK, efficacy, safety, and tolerability of intravenous doses of bleselumab in patients with moderate‐to‐severe plaque psoriasis.
2. MATERIAL AND METHODS
2.1. Study design and patients
This was a multicenter, randomized, double‐blind, placebo‐controlled, sequential‐dose group, multiple‐dose escalation study of bleselumab (Kyowa Hakko Kirin Co. Ltd, in partnership with Astellas Pharma, Inc., Japan) conducted at five sites in Canada, three sites in Australia, and four sites in New Zealand (http://clinicaltrials.gov NCT01585233). The study was started on 30 April 2012, after approval by the Independent Ethics Committee or Institutional Review Board, and was carried out in accordance with Good Clinical Practice, International Committee on Harmonisation guidelines, and the Declaration of Helsinki. All patients provided written confirmation of informed consent prior to screening.
Patients were included if they were aged 18–75 years, had moderate‐to‐severe plaque psoriasis for ≥6 months, had ≥5% body surface area (BSA) with plaque psoriasis, and were candidates for phototherapy and/or systemic therapy. In order to enhance enrolment, the inclusion criteria were amended to recruit patients with plaque psoriasis %BSA ≥ 5%, compared with ≥10% before the amendment. This change reduced the baseline severity and extent of psoriasis in the 1.0 and 3.0 mg/kg groups. Patients were excluded if they had been treated with either systemic biologic (including efalizumab), or nonbiologic immunosuppressants within the last 30 days or 5 half‐lives (56 days, or 5 half‐lives for biologics), whichever was longer, prior to the first dose of the study drug. A description of exclusion criteria can be found at http://clinicaltrials.gov (https://clinicaltrials.gov/ct2/show/NCT01585233).
After a 28‐day screening period, the patients were randomized on day 1 (baseline) to bleselumab or placebo, using a 4:1 ratio (bleselumab: placebo) within each dose cohort. The bleselumab dose groups were 0.1, 0.3, 1.0 or 3.0 mg/kg (Figure 1), with no change in study drug dosing permitted once treatment was initiated. The study drug was administered by intravenous infusion over 30 minutes on days 1, 15 and 29, and patients were followed up for an additional 12 weeks (up to day 113) (Figure 1). Patients could continue to receive permitted concomitant medications and therapies.
Figure 1.
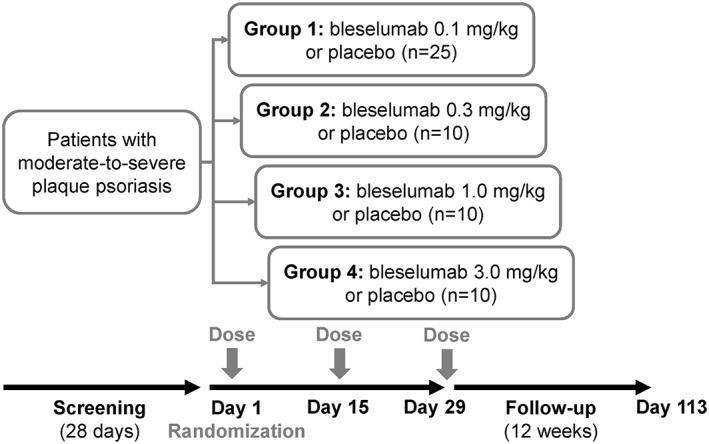
Study design
As this was a sequential dose‐escalation‐by‐cohort study, the safety data collected were reviewed prior to the completion of each active‐treatment cohort, in order to make dose escalation decisions for the subsequent study cohort. However, no statistical analyses or by‐cohort unblinding of treatment assignments were planned or performed at the time of these data reviews.
2.2. Pharmacokinetics
Blood samples for the PK assessment of serum bleselumab concentrations were collected on days 1, 15 and 29, pre‐infusion (0 hours), and at 30 minutes, 2.5 hours and 4.5 hours after infusion.
Concentrations of serum bleselumab were measured using a sandwich immunoassay with electrochemiluminescence detection (Goldwater et al., 2013). The lower limit of quantification (LLOQ) for the assay was 81.92 ng/ml, and the assay was linear over the range of 81.92–50,000 ng/ml. A standard curve was constructed by using four quality control samples. The precision and accuracy of the quality controls were ≤ 9.7% and within ±6.7%, respectively. The titer of anti‐bleselumab antibodies in serum was assessed using an electrochemiluminescence bridging immunogenicity assay (Kernstock, 2010), and the occurrence of neutralizing antibodies was recorded.
2.3. Endpoints
The primary PK endpoints were the bleselumab area under the serum concentration–time curve over 336 hours (AUC336) and the maximum concentration (Cmax) after dosing on days 1, 15 and 29. Secondary PK endpoints were the trough concentration (Ctrough) on days 15, 29 and day 43, and the elimination half‐life (T½) of bleselumab on day 29.
Secondary efficacy endpoints were the mean change from baseline to 8 weeks in psoriasis area and severity index (PASI) score (classic and linear), the Physician Static Global Assessment (PSGA) score, and the percentage BSA affected with psoriasis (%BSA). The proportion of patients achieving treatment success at 8 weeks, defined as a score of 1 (almost clear) or 0 (clear) using the PSGA, was also evaluated.
The safety and tolerability were assessed throughout the study. Treatment‐emergent AEs (TEAEs) were recorded, and included AEs that started/worsened after the initial dose of study drug, up to and including day 113. Change from baseline (pre‐dose) over the follow‐up period was assessed for clinical laboratory parameters, including hematology, blood chemistry, fasting lipid profile, cytokines [interleukin (IL)‐2, IL‐6, interferon (IFN)‐γ, and TNFα], coagulation and thrombotic pathways, and urinalysis. Other measures were electrocardiograms (ECGs), and vital signs (systolic and diastolic blood pressure). Tolerability parameters also included the occurrence of anti‐bleselumab antibody formation (and whether they were neutralizing).
2.4. Sample size calculation
The primary objective of the study was originally to assess bleselumab efficacy, and the primary efficacy endpoint was the change of PASI score from baseline to 8 weeks. Based on the original protocol, a sample size of 20 patients per bleselumab treatment group and 20 pooled placebo patients, assuming a difference of 30% and equal standard deviation of 30% with α = 0.05, resulted in 86% power for a two‐sided test. Based on the first protocol amendment, the sample size was reduced from 20 to 16 patients per group, the minimum number of patients exposed to study drug that were required for statistically adequate power (80% for detecting a clinically meaningful difference of 30% between any bleselumab treatment group and pooled placebo). However, in order to focus on the PK of bleselumab, which is essential to support PK and pharmacodynamics modeling for bleselumab dose selection in future clinical studies, efficacy became a secondary objective. A further amendment therefore resulted in a reduced target sample size (from 16 to 10 patients) per active treatment arm, after the 0.1 mg/kg bleselumab group, based on previous experience with studies of a comparable design and at a similar stage in drug development. As such, the planned sample size was 55 patients (25 patients in Group 1, and 10 patients in each of Groups 2, 3 and 4, allocated within each group in a 4:1 ratio of bleselumab to placebo) (Figure 1).
2.5. Randomization and statistical analyses
The Sponsor's Global Data Science department, or designee, generated the randomization schedule using a fixed block size. Eligible patients were assigned a number and centrally randomized in a double‐blind manner, using an interactive voice or web response system. In order to maintain blinding of the individual administering the study drug, only the pharmacist or investigator's designee at the investigational site was unblinded, and could dispense the study drug.
The safety‐analysis set (SAF) and full‐analysis set (FAS) comprised all randomized patients who received bleselumab or placebo. The FAS and SAF were used to analyse efficacy and safety endpoints, respectively, and the SAF was also used to assess demographic and baseline characteristics. The PK‐analysis set (PKAS) included patients in the SAF with at least one quantifiable serum concentration of bleselumab, and was used to analyse the PK endpoints.
Descriptive statistics were used to summarize parameters for individual bleselumab treatment groups, the pooled bleselumab group, and the pooled placebo group. Serum bleselumab concentrations below the assay LLOQ were set to 0. When half or more of the individual data in a group were below the LLOQ, the standard deviation (SD) and coefficient of variation were not calculated. The PK parameters were computed using WinNonlin software (Pharsight Corp, Mountain View, California, USA) Version 6.2. AUC336, Cmax, Ctrough and T½ were determined using non‐compartmental methodology, and analyses were conducted by dosing day. The dose proportionality of AUC336 and Cmax was evaluated using separate power models by dosing day. The power model was analysed using linear regression with the natural logarithm of the PK parameter against the natural logarithm of dose. Dose proportionality was concluded if the 95% confidence interval for the slope included 1.
The original primary clinical criteria for efficacy were based on the mean change in PASI score from baseline to 8 weeks. It was assumed that the SD of the change in PASI score was equal across all treatment groups. In order to test the null hypothesis of equal changes from baseline to week 8 in PASI score across the treatment groups, analysis of covariance was used with baseline PASI score as a covariate, treatment group as a fixed effect, and site as a random effect. If the covariate was not statistically significant, then the analysis was repeated, without baseline PASI score. Pairwise comparisons of the change from baseline to week 8 for each active treatment group versus pooled placebo were performed. Similar analyses were also performed for PSGA score and %BSA. Missing efficacy data at week 8 was imputed using the last observation carried forward.
All statistical tests were two‐sided with α = 0.05 and no adjustments for multiplicity were made. Data were analysed using SAS® Version 9.3 on Unix.
3. RESULTS
3.1. Patient characteristics
Overall, 115 patients provided informed consent (first patient enrolled 30 April 2012), 55 were screening failures, and 60 were randomized and included in the FAS and SAF, of whom 48 comprised the PKAS (Figure 2). Of the 60 randomized patients, 31/60 (51.7%) were randomized after the primary objective of the study changed from efficacy to safety, and the %BSA was reduced from 10% to 5% for study entry. The date of the last study evaluation was 7 January 2015.
Figure 2.
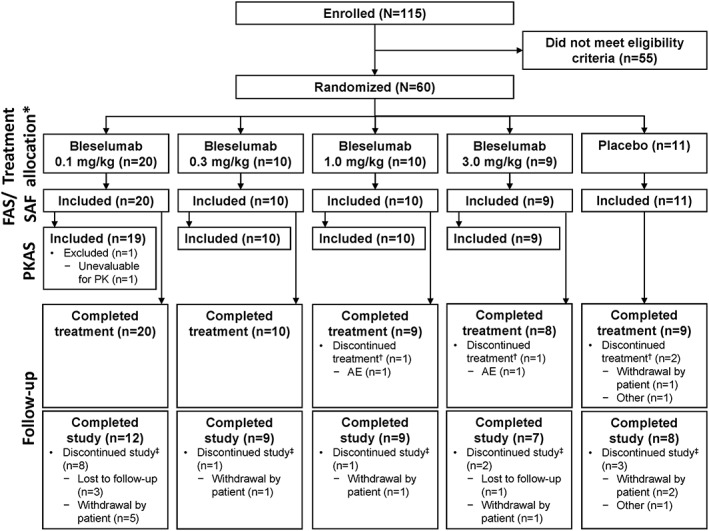
Flow of patients through the study. *All randomized patients received their allocated intervention. †Patients who did not complete the last treatment period evaluation visit (day 29). ‡Patients who did not complete the last study evaluation visit (day 113). AE, adverse event; FAS, full‐analysis set; PK, pharmacokinetics; PKAS, pharmacokinetics‐analysis set; SAF, safety‐analysis set
Patient demographics and baseline characteristics are presented in Table 1. The mean PASI score and %BSA affected by psoriasis was higher in the placebo and bleselumab 0.1 mg/kg groups compared with the other study arms (Table 1). The mean ± SD treatment durations were 28.8 ± 3.2 (range 15–37) days with bleselumab and 26.8 ± 5.6 (range 15–30) days with placebo.
Table 1.
Patient demographics and baseline characteristics (SAF)
| Parameter | Placebo (n = 11) | Bleselumab dose (mg/kg) | ||||
|---|---|---|---|---|---|---|
| 0.1 (n = 20) | 0.3 (n = 10) | 1.0 (n = 10) | 3.0 (n = 9) | Total (n = 49) | ||
| Male, n (%) | 6 (54.5) | 11 (55.0) | 8 (80.0) | 6 (60.0) | 7 (77.8) | 32 (65.3) |
| Race, n (%) | ||||||
| Caucasian | 10 (90.9) | 18 (90.0) | 10 (100.0) | 8 (80.0) | 8 (88.9) | 44 (89.8) |
| Asian | 0 | 1 (5.0) | 0 | 2 (20.0) | 0 | 3 (6.1) |
| Other | 1 (9.1) | 1 (5.0) | 0 | 0 | 1 (11.1) | 2 (4.1) |
| Age, years | ||||||
| Mean ± SD | 40.7 ± 10.9 | 44.4 ± 13.4 | 49.2 ± 16.0 | 49.2 ± 11.8 | 47.8 ± 8.8 | 47.0 ± 12.8 |
| Median | 41.0 | 45.5 | 49.0 | 51.0 | 47.0 | 47.0 |
| Minimum; maximum | 27; 57 | 21; 68 | 25; 70 | 28; 72 | 31; 63 | 21; 72 |
| Weight, kg | ||||||
| Mean ± SD | 95.3 ± 15.3 | 93.9 ± 19.0 | 89.4 ± 22.2 | 93.2 ± 18.3 | 98.8 ± 31.2 | 93.7 ± 21.7 |
| Median | 100 | 94.5 | 82.5 | 86.7 | 95.4 | 87.8 |
| Minimum; maximum | 68.9; 114 | 69.5; 134 | 69.5; 142 | 68.3; 118 | 62.0; 157 | 62.0; 157 |
| Height, cm | ||||||
| Mean ± SD | 172 ± 7.5 | 172 ± 9.6 | 176 ± 8.9 | 165 ± 7.0 | 173 ± 9.1 | 172 ± 9.3 |
| Median | 171 | 174 | 176 | 166 | 175 | 172 |
| Minimum; maximum | 162; 186 | 152; 186 | 165; 190 | 153; 173 | 160; 188 | 152; 190 |
| BMI, kg/m2 | ||||||
| Mean ± SD | 32.3 ± 5.7 | 31.5 ± 4.9 | 28.7 ± 4.7 | 34.4 ± 8.0 | 32.6 ± 7.6 | 31.7 ± 6.2 |
| Median | 33.0 | 32.2 | 27.6 | 33.5 | 29.1 | 29.9 |
| Minimum; maximum | 24.7; 40.5 | 22.2; 39.5 | 23.4; 39.3 | 23.2; 44.8 | 22.9; 44.5 | 22.2; 44.8 |
| PASI score | ||||||
| Classic | ||||||
| Mean ± SD | 15.6 ± 7.0 | 17.5 ± 7.4 | 12.0 ± 3.6 | 11.2 ± 3.3 | 14.0 ± 13.9 | 14.4 ± 8.1 |
| Median | 15.5 | 16.2 | 12.5 | 11.4 | 7.8 | 12.2 |
| Minimum; maximum | 7; 34 | 8; 43 | 7; 18 | 6; 17 | 5; 47 | 5; 47 |
| Linear | ||||||
| Mean ± SD | 10.4 ± 7.8 | 10.5 ± 7.7 | 6.4 ± 2.9 | 4.1 ± 1.6 | 9.1 ± 13.0 | 8.1 ± 7.7 |
| Median | 7.7 | 9.2 | 5.8 | 4.2 | 4.3 | 5.5 |
| Minimum; maximum | 3; 31 | 4; 37 | 3; 12 | 2; 6 | 2; 42 | 2; 42 |
| PSGA score | ||||||
| Mean ± SD | 2.5 ± 0.5 | 3.1 ± 0.7 | 2.6 ± 0.7 | 2.6 ± 0.7 | 2.8 ± 0.7 | 2.8 ± 0.7 |
| Median | 3.0 | 3.0 | 2.5 | 2.5 | 3.0 | 3.0 |
| Minimum; maximum | 2; 3 | 2; 4 | 2; 4 | 2; 4 | 2; 4 | 2; 4 |
| BSA, % | ||||||
| Mean ± SD | 26.2 ± 21.9 | 21.4 ± 12.0 | 15.5 ± 6.3 | 9.9 ± 3.8 | 17.4 ± 19.3 | 17.1 ± 12.2 |
| Median | 20.0 | 18.3 | 12.5 | 9.3 | 10.7 | 12.5 |
| Minimum; maximum | 9; 86 | 10; 58 | 10; 26 | 5; 17 | 6; 67 | 5; 67 |
BMI, body mass index; BSA, body surface area; PASI, Psoriasis Area and Severity Index; PSGA, Physician Static Global Assessment; SAF, safety‐analysis set; SD, standard deviation.
Overall, 15 (25.0%) randomized patients discontinued the study (Figure 2). Four (6.7%) patients receiving bleselumab were lost to follow‐up. Eight (13.3%) patients receiving bleselumab and three (5.0%) patients receiving placebo withdrew from the study, and the reasons for withdrawal are presented in Supplementary Table 1.
3.2. Pharmacokinetics
The mean serum concentration–time profiles for bleselumab are presented in Figure 3. The mean serum AUC336 and Cmax appeared to increase after multiple doses in the bleselumab 1.0 and 3.0 mg/kg groups, suggesting accumulation, whereas there were no apparent increases in AUC336 or Cmax in the 0.1 and 0.3 mg/kg groups (Table 2). The increases in AUC336 and Cmax were more than dose proportional at dose ranges of 0.1 to 3.0 mg/kg on days 1, 15 and 29, indicating nonlinear PK after single and multiple doses of bleselumab (Table 3).
Figure 3.
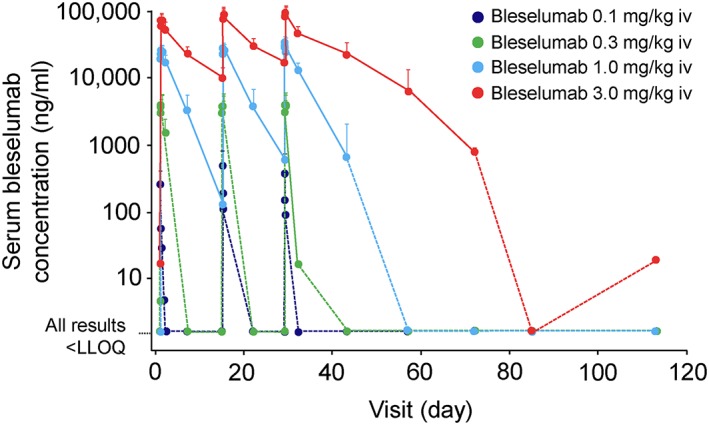
Mean + SD serum bleselumab concentrations (semi‐log scale) (PKAS). Mean serum concentrations of 0 were assigned a value of 0.02 × LLOQ to enable these points to appear on the plot, since the log value of 0 is undefined. A dotted line was used to connect these converted points to prior and subsequent points on the graph. Where at least half of individual data in a group at a given time point were below the LLOQ, SD was not calculated. IV, intravenous; LLOQ, lower limit of quantification; PKAS, pharmacokinetics‐analysis set; SD, standard deviation
Table 2.
Serum PK parameters for bleselumab 0.1, 0.3, 1.0 and 3.0 mg/kg doses on days 1, 15 and 29 (PKAS)
| Parameter | Bleselumab dose (mg/kg) | |||
|---|---|---|---|---|
| 0.1 | 0.3 | 1.0 | 3.0 | |
| Baseline (day 1) | ||||
| AUC336, μg·h/ml | ||||
| n | 18 | 10 | 10 | 9 |
| Mean ± SD | 0.727 ± 1.22 | 174 ± 77.5 | 1865 ± 672 | 9398 ± 1990 |
| Median | 0.290 | 198 | 1631 | 8775 |
| Minimum; maximum | 0; 4.50 | 0; 260 | 1211; 3292 | 6241; 12,310 |
| %CV | 167.2 | 44.6 | 36.0 | 21.2 |
| Cmax, μg/ml | ||||
| n | 18 | 10 | 10 | 9 |
| Mean ± SD | 0.264 ± 0.140 | 4.24 ± 1.66 | 27.0 ± 6.00 | 77.3 ± 17.4 |
| Median | 0.232 | 4.56 | 25.1 | 77.1 |
| Minimum; maximum | 0; 0.498 | 0; 5.85 | 20.3; 38.2 | 51.7; 98.9 |
| %CV | 53.0 | 39.0 | 22.2 | 22.5 |
| Day 15 | ||||
| AUC336, μg·h/ml | ||||
| n | 17 | 10 | 10 | 8 |
| Mean ± SD | 10.1 ± 28.1 | 333 ± 135 | 2373 ± 998 | 12,311 ± 3612 |
| Median | 0.741 | 362 | 2276 | 11,838 |
| Minimum; maximum | 0.138; 116 | 0.161; 475 | 930; 4487 | 7266; 17,731 |
| %CV | 278.5 | 40.4 | 42.1 | 29.3 |
| Cmax, μg/ml | ||||
| n | 18 | 10 | 10 | 9 |
| Mean ± SD | 0.522 ± 0.371 | 4.26 ± 1.58 | 28.7 ± 5.56 | 90.7 ± 20.8 |
| Median | 0.385 | 4.63 | 29.1 | 81.1 |
| Minimum; maximum | 0.110; 1.63 | 0.128; 5.93 | 18.2; 35.0 | 56.8; 116 |
| %CV | 71.0 | 37.1 | 19.4 | 22.9 |
| Ctrough, μg/ml | ||||
| n | 18 | 10 | 10 | 9 |
| Mean ± SD | 0.131 (NA) | 10.3 ± 4.09 | ||
| Median | 0* | 0* | 0 | 9.89 |
| Minimum; maximum | 0; 1.20 | 4.92; 17.3 | ||
| %CV | NA | 39.5 | ||
| Day 29 | ||||
| AUC336, μg·h/ml | ||||
| n | 18 | 10 | 7 | 7 |
| Mean ± SD | 2.27 ± 3.35 | 161 ± 66.6 | 3046 ± 1098 | 14,543 ± 4495 |
| Median | 0.457 | 176 | 2580 | 15806 |
| Minimum; maximum | 0.140; 11.6 | 0.201; 241 | 2060; 4887 | 6524; 20,838 |
| %CV | 147.5 | 41.5 | 36.0 | 30.9 |
| Cmax, μg/ml | ||||
| n | 18 | 10 | 9 | 8 |
| Mean ± SD | 0.402 ± 0.291 | 4.36 ± 1.62 | 32.9 ± 7.04 | 101 ± 28.8 |
| Median | 0.326 | 4.61 | 33.1 | 99.9 |
| Minimum; maximum | 0.112; 1.19 | 0.159; 6.29 | 22.9; 45.2 | 60.6; 141 |
| %CV | 72.3 | 37.1 | 21.4 | 28.4 |
| Ctrough, μg/ml | ||||
| n | 18 | 9 | 10 | 8 |
| Mean ± SD | 0.585 (NA) | 17.3 ± 6.50 | ||
| Median | 0* | 0* | 0 | 17.5 |
| Minimum; maximum | 0; 4.27 | 6.95; 26.4 | ||
| %CV | NA | 37.6 | ||
| T½, hours | ||||
| n | 0† | 0† | 7 | 6 |
| Mean ± SD | 55.1 ± 24.9 | 189 ± 41.3 | ||
| Median | NA | NA | 47.0 | 194 |
| Minimum; maximum | 37.4; 108 | 126; 230 | ||
| %CV | 45.1 | 21.9 | ||
| Day 43 | ||||
| Ctrough, μg/ml | ||||
| n | 16 | 9 | 7 | 7 |
| Mean ± SD | 0.699 ± 1.36 | 22.8 ± 11.3 | ||
| Median | 0* | 0* | 0.099 | 25.0 |
| Minimum; maximum | 0; 3.73 | 0; 36.0 | ||
| %CV | 194.6 | 49.5 | ||
Mean bleselumab Ctrough levels were below the assay LLOQ (81.92 ng/ml) with doses of 0.1 and 0.3 mg/kg. Where at least half of the individual data in a group were below the LLOQ, SD and %CV were not calculated.
T½ was not available for bleselumab doses of 0.1 and 0.3 mg/kg, due to too few quantifiable concentrations to calculate T½. AUC336, area under the concentration–time curve over 336 hours; Cmax, maximum concentration; Ctrough, trough concentration; CV, coefficient of variation; LLOQ, lower limit of quantification; NA, not available; PKAS, pharmacokinetics‐analysis set; SD, standard deviation; T½, elimination half‐life.
Table 3.
Statistical assessment of dose proportionality for bleselumab (PKAS)
| Dose range (mg/kg) | Visit | Parameter | Slope estimate (95% CI) |
|---|---|---|---|
| 0.1–3.0 | Baseline (day 1) | AUC336, μg·h/ml | 2.92 (2.59, 3.24) |
| Cmax, μg/ml | 1.67 (1.53, 1.81) | ||
| Day 15 | AUC336, μg·h/ml | 2.67 (2.24, 3.11) | |
| Cmax, μg/ml | 1.58 (1.41, 1.74) | ||
| Day 29 | AUC336, μg·h/ml | 2.88 (2.47, 3.29) | |
| Cmax, μg/ml | 1.68 (1.50, 1.87) |
Dose proportionality was concluded if the 95% confidence interval for the slope included 1. AUC336, area under the concentration–time curve over 336 hours; CI, confidence interval; Cmax, maximum concentration; PKAS, pharmacokinetics‐analysis set.
The mean Ctrough increased with higher doses of bleselumab (Table 2). On day 29, the mean ± SD T½ was 55.1 ± 24.9 and 189 ± 41.3 hours with bleselumab 1.0 and 3.0 mg/kg, respectively (Table 2). There were too few quantifiable serum bleselumab concentrations to calculate the T½ for doses of 0.1 and 0.3 mg/kg.
3.3. Efficacy
There was no difference in PASI scores, PSGA scores, and %BSA at week 8 between bleselumab and placebo groups. Furthermore, the least‐squares (LS) mean change in these parameters from baseline to week 8 was not significantly different between each active treatment group versus the pooled placebo. While not reaching statistical significance, bleselumab 3.0 mg/kg demonstrated some pharmacologic activity at week 8, based on the numeric decrease in LS mean PASI score, PSGA score, and %BSA versus placebo (Supplementary Table 2).
At week 8, three (27.3%) patients receiving placebo, and six (12.2%) patients receiving bleselumab [two (10.0%), one (10.0%), one (10.0%) and two (22.2%) patients in the bleselumab 0.1, 0.3, 1.0 and 3.0 mg/kg groups, respectively] achieved treatment success, as measured by a PSGA assessment of clear or almost clear.
3.4. Treatment‐emergent adverse events
Overall, 43 (87.8%) and 9 (81.8%) patients in the total bleselumab and placebo groups experienced TEAEs (Table 4), of which most were mild or moderate in severity. Twenty‐three (46.9%) patients treated with bleselumab and eight (72.7%) patients receiving placebo experienced TEAEs considered possibly or probably drug related; most occurred in the bleselumab 3.0 mg/kg group. Of the TEAEs occurring in ≥3 patients in the SAF, the most common in bleselumab‐treated patients were upper respiratory tract infection (22.4% of patients), headache (16.3%), gastroenteritis (14.3%), and nasopharyngitis (12.2%) (Supplementary Table 3).
Table 4.
Overview of TEAEs (SAF)
| Category | Placebo (n = 11) | Bleselumab dose (mg/kg) | ||||
|---|---|---|---|---|---|---|
| 0.1 (n = 20) | 0.3 (n = 10) | 1.0 (n = 10) | 3.0 (n = 9) | Total (n = 49) | ||
| TEAE | 9 (81.8) | 16 (80.0) | 9 (90.0) | 9 (90.0) | 9 (100) | 43 (87.8) |
| Drug‐related* TEAE | 8 (72.7) | 6 (30.0) | 6 (60.0) | 3 (30.0) | 8 (88.9) | 23 (46.9) |
| Deaths | 0 | 0 | 0 | 0 | 0 | 0 |
| Serious TEAE | 1 (9.1) | 2 (10.0) | 0 | 1 (10.0) | 1 (11.1) | 4 (8.2) |
| Drug‐related* serious TEAE | 0 | 0 | 0 | 1 (10.0) | 1 (11.1) | 2 (4.1) |
| TEAE leading to permanent discontinuation of study drug | 0 | 0 | 0 | 1 (10.0) | 1 (11.1) | 2 (4.1) |
| Drug‐related* TEAE leading to permanent discontinuation of study drug | 0 | 0 | 0 | 1 (10.0) | 1 (11.1) | 2 (4.1) |
Data are reported as n (%).
Possibly or probably drug related, as assessed by the investigator, or records where the relationship was missing. SAF, safety‐analysis set; TEAE, treatment‐emergent adverse event.
Serious TEAEs were experienced by four (8.2%) patients taking bleselumab (Table 4). In the bleselumab 0.1 mg/kg group, one patient had malignant melanoma, and one experienced an overdose, due to errors in the study drug dosing procedures at the study site. The dose of bleselumab was not changed for the patient with overdose, and the TEAE resolved, with minimal or no impact on the patient's safety. One patient in the bleselumab 1.0 mg/kg group experienced a grade 3 liver function test (LFT) abnormality, considered possibly related to the study drug. In the bleselumab 3.0 mg/kg group, one patient experienced a grade 2 event of angina pectoris considered possibly related to the study drug; the event started on day 62 and resolved on day 82.
The TEAEs led to treatment discontinuation in two (4.1%) patients receiving bleselumab (one each in the 1.0 and 3.0 mg/kg groups; abnormal LFT, as described above, and study‐drug induced hepatitis that did not resolve, respectively). No patients died during the study.
3.5. Clinical laboratory evaluations, vital signs and electrocardiograms
Supplementary Table 4 presents the clinical laboratory evaluations at days 1 and 113. The mean hematology values remained within reference ranges throughout the study, and individual values outside the reference ranges were not considered potentially clinically relevant.
Patients in the bleselumab 1.0 and 3.0 mg/kg dose groups had a greater incidence of elevated ALT and AST levels compared with the placebo and bleselumab 0.1 mg/kg groups. Four patients (8.2%) had ALT >3×ULN (two each in the 1.0 and 3.0 mg/kg groups), and two patients (4.1%) had AST levels >3×ULN (one each in the 1.0 and 3.0 mg/kg groups). One patient receiving bleselumab 1.0 mg/kg developed ALT >3×ULN and AST > 3×ULN on day 15; bleselumab therapy was stopped after dosing on day 15, and the elevated ALT and AST resolved by day 57. Another patient receiving bleselumab 1.0 mg/kg experienced ALT >3×ULN, beginning on day 7 and resolving on day 15, without bleselumab dose adjustment. In a patient receiving bleselumab 3.0 mg/kg, ALT >3×ULN and AST > 3×ULN were noted on day 9, but resolved by day 58 and 44, respectively; this patient stopped receiving bleselumab on day 15, due to the development of hepatitis. One patient receiving bleselumab 3.0 mg/kg had ALT >3×ULN on day 29; the dose of bleselumab was not changed, and all LFTs were within normal limits by day 71. However, no patients experienced bilirubin >2×ULN. Elevated LFTs were not accompanied by clinical symptoms and resolved whether the study drug was discontinued or not. There were no Hy's law cases observed during the study.
The mean cytokine measurements remained low across treatment groups, and there was no evidence of cytokine release syndrome. Furthermore, there were no obvious differences across groups, or trends over time, in mean changes from baseline to day 29 or day 113 in mean urinalysis and coagulation/thrombotic values, and there was no evidence of thromboembolic events (Supplementary Table 4).
Mean vital signs remained within the normal range throughout the study, although one patient (bleselumab 3.0 mg/kg group) experienced a TEAE of increased blood pressure. The increase in blood pressure occurred on day 85 (162/98 mmHg), and was 136/88 mmHg by day 112. Abnormal ECGs were observed across treatment groups; however, only one patient (bleselumab 1.0 mg/kg group) had a clinically significant abnormality in their ECG (atrial fibrillation on day 30); this was considered unrelated to study drug, and the patient completed the study. The same patient also experienced two other atrial fibrillation events (days 57 and 113), which were not considered clinically significant.
3.6. Antibody development
In the bleselumab groups, 27/49 (55.1%) patients tested positive for anti‐bleselumab antibodies at least once during the study: 9/49 (18.4%) patients tested positive pre‐dose at baseline, and 26/49 (53.1%) patients tested positive after baseline. Excluding the patients who had positive results for anti‐bleselumab antibodies pre‐dose at baseline, 18/40 (45.0%) patients tested positive at some time point during the study [6/18 (33.3%), 5/7 (71.4%), 4/7 (57.1%) and 3/8 (37.5%) in the 0.1, 0.3, 1.0, and 3.0 mg/kg groups, respectively]. In these patients, the earliest detection of anti‐bleselumab antibody was day 15 in the 0.1 and 0.3 mg/kg groups, day 29 in the 1.0 mg/kg group, and day 113 in the 3.0 mg/kg group.
Neutralizing antibodies were recorded in four, four, seven, and three patients receiving bleselumab 0.1, 0.3, 1.0 and 3.0 mg/kg, respectively, and were sustained through to at least day 85. One patient (2.0%) in the 0.3 mg/kg dose group who tested positive for anti‐bleselumab neutralizing antibodies tested positive for anti‐bleselumab antibodies at baseline (and several other time points).
4. DISCUSSION
In this study of escalating doses of intravenous bleselumab between cohorts of patients with moderate‐to‐severe plaque psoriasis, bleselumab accumulated in the serum over time with higher doses (1.0 and 3.0 mg/kg). Furthermore, bleselumab AUC336 and Cmax demonstrated nonlinear PK at dose ranges of 0.1–3.0 mg/kg. Compared with the placebo, bleselumab did not improve PASI scores, PSGA scores and %BSA, irrespective of the dose administered. However, intravenous bleselumab was generally well tolerated.
In a Phase I, randomized, double‐blind, placebo‐controlled, dose‐escalation study in 109 healthy volunteers, bleselumab displayed disproportionate increases in systemic exposure with dose (Goldwater et al., 2013). The mean bleselumab Cmax and AUCinf (AUC from time 0 extrapolated to infinity) ranged from 0.7 to 251.6 μg/ml and 6.5 to 55,409.6 μg·h/ml, respectively, following bleselumab doses of 0.1–10 mg/kg (Goldwater et al., 2013). The AUC336 and Cmax values that we observed in psoriasis patients in the present study were generally comparable with those in the healthy volunteers over the same dose range (0.1–3.0 mg/kg). Additionally, as in the healthy volunteer study (Goldwater et al., 2013), bleselumab exhibited nonlinear PK in psoriasis patients in the dose range of 0.1–3.0 mg/kg, which was evidenced by disproportionate increases in systemic exposure with dose. Indeed, nonlinear PK has also been reported with other anti‐CD40 antibodies (Albach et al., 2018; Bensinger et al., 2012). Bleselumab is a monoclonal antibody, designed with a high affinity for a specific target. This high affinity binding to target is saturable, due to finite binding sites on cells, and subsequent turnover of the drug–target complex may contribute to the PK disposition of biologics (Dua, Hawkins, & van der Graaf, 2015). In this regard, saturation of target binding sites may be one of the causes of nonlinear PK seen with bleselumab.
This study was not powered to test the efficacy of bleselumab treatment in patients with psoriasis versus placebo. There was some evidence of pharmacologic activity at the 3.0 mg/kg dose relative to placebo, which supports the existing evidence that CD40 signalling plays a role in psoriasis disease activity (Pasch et al., 2004). However, due to changes in the number of patients in the 0.3 to 3.0 mg/kg groups, and study inclusion criteria that reduced the baseline severity and extent of psoriasis (from ≥10% to ≥5% BSA) for patients in the 1.0 and 3.0 mg/kg groups, no conclusions can be made regarding dose response and the efficacy of bleselumab in the treatment of psoriasis.
In our study, most patients (87.8%) receiving bleselumab experienced TEAEs during follow‐up, as did most patients receiving placebo (81.8%). Consistent with a Phase I study in healthy volunteers (Goldwater et al., 2013), the most common TEAEs reported with bleselumab in this study were upper respiratory tract infection and headache. Elevations in ALT and AST to >3×ULN, indicative of hepatic injury, but without either concomitant elevations of bilirubin >2×ULN or a clinically significant decrease in mean serum albumin, were reported for a few patients in the 1.0 and 3.0 mg/kg bleselumab groups. In a preclinical primate study of cynomolgus monkeys, bleselumab did not impact liver enzyme levels (Aoyagi et al., 2009; Song et al., 2014). The effect of bleselumab on elevation of ALT and AST levels (albeit transient) in our study remains unclear at present, and a larger clinical trial is needed to fully characterize the association of bleselumab with hepatocyte function.
The underlying pathophysiology of plaque psoriasis involves CD40 signalling, induced by its ligand, CD154 (Pasch et al., 2004). Anti‐CD154 monoclonal antibodies have been associated with a high incidence of thromboembolic complications (Larsen, Knechtle, Adams, Pearson, & Kirk, 2006). By contrast, in our study, targeting CD40 with bleselumab was not associated with thromboembolic events, which is consistent with a study in healthy volunteers (Goldwater et al., 2013). Coagulation parameters (e.g., prothrombin time) remained stable. Furthermore, bleselumab did not upregulate cell activation markers, such as thromboplastin, in vitro (Okimura et al., 2014).
Consistent with studies of bleselumab in healthy volunteers and cynomolgus monkeys (Aoyagi et al., 2009; Goldwater et al., 2013; Okimura et al., 2014; Song et al., 2014), ECG changes were generally not clinically significant and mean chemical laboratory and hematology parameters, and vital signs remained within reference ranges throughout the study. Furthermore, there was no clinical evidence of cytokine release syndrome (Goldwater et al., 2013). As bleselumab is under development as an adjunct immunosuppressive agent in transplantation and other possible immune diseases (Aoyagi et al., 2009), the safety data generated from our study helps to inform future trials in other indications.
In this study, antibodies to bleselumab were detected in approximately half of patients at least once, and across all dosing arms. In patients receiving bleselumab 0.1, 0.3 or 1.0 mg/kg, anti‐bleselumab antibodies developed before week 8, after the first dose. By comparison, the first post‐baseline detection of anti‐bleselumab antibodies in patients receiving bleselumab 3.0 mg/kg was after week 8. Anti‐bleselumab antibodies observed in this study may be either neutralizing or non‐neutralizing. While neutralizing antibodies blunt efficacy of the drug, non‐neutralizing antibodies do not affect efficacy (Krishna & Nadler, 2016; Shankar, Pendley, & Stein, 2007). Neutralizing antibodies were detected in all active treatment groups, in line with results from a Phase I study of healthy volunteers (Goldwater et al., 2013); however, it was beyond the scope of this study to differentiate pre‐existing from de novo neutralizing antibodies. It is, therefore, not possible to verify the effect of anti‐bleselumab antibodies on clinical efficacy.
This study has several limitations. Particularly in the 0.1 and 0.3 mg/kg groups, some serum bleselumab concentrations were below the assay LLOQ, which could have affected dose proportionality calculations. The efficacy results should be interpreted with caution due to the small sample size, and disparity in some baseline patient characteristics; the minimum %BSA for inclusion was reduced from 10% to 5% for study entry, which was likely responsible for the higher %BSA in the placebo and 0.1 mg/kg bleselumab groups versus other treatment arms. Due to the small number of patients, there are insufficient data to discuss the impact of antibody development on the nonlinearity of PK. However, the sample size was reduced when the original objective to assess the efficacy of bleselumab was amended to meet the primary PK endpoints of AUC336 and Cmax with escalating doses of bleselumab in patients with plaque psoriasis. This enabled essential PK data to be collected to support PK and pharmacodynamics modeling for bleselumab dose selection in future clinical studies.
5. CONCLUSION
This is the first study to analyse the PK parameters of intravenous infusion of bleselumab in patients with moderate‐to‐severe plaque psoriasis. Bleselumab accumulated in serum after repeated infusions with doses of 1.0 and 3.0 mg/kg, but not with doses of 0.1 and 0.3 mg/kg. Furthermore, PK parameters had a nonlinear relationship over the dose range of bleselumab tested (0.1, 0.3, 1.0 or 3.0 mg/kg). Bleselumab infusions were associated with few minor local reactions. In a limited number of patients treated with 1.0 and 3.0 mg/kg doses, transient elevations of liver transaminase enzymes were observed, which spontaneously resolved without sequelae. In conclusion, bleselumab was generally well tolerated in patients with moderate‐to‐severe plaque psoriasis.
FUNDING INFORMATION
This study was sponsored by Astellas Pharma, Inc. under the co‐development partnership with Kyowa Hakko Kirin Co., Ltd. Editorial support in the development of this manuscript was funded by Astellas Pharma, Inc.
CONFLICTS OF INTEREST
Mysore S. Anil Kumar, Ryo Tainaka, Udaya Valluri, Xuegong Wang, and Tong Zhu, are employees of Astellas. Kim Papp reports grants and personal fees from Astellas, during the conduct of the study. Outside the submitted work, Kim Papp reports grants and personal fees from AbbVie, Amgen, Anacor, Baxalta, Boehringer Ingelheim, Bristol‐Meyers Squibb, Celgene, Dermira, Dow Pharma, Janssen, Kyowa Hakko Kirin, Leo, Merck (MSD), Merck‐Serono, Novartis, Pfizer, Takeda, UCB, and Valeant, and personal fees from Akros, AstraZeneca, Baxter, Cipher, Eli Lilly, Forward Pharma, Meiji Seika Pharma, Mitsubishi Pharma, and Mylan. Christian Schwabe reports grants from Astellas, during the conduct of the study.
AUTHOR CONTRIBUTIONS
All authors analysed and interpreted the data, revised the manuscript critically for important intellectual content, and approved the final version of the manuscript for submission. Mysore S. Anil Kumar additionally conceptualized the manuscript publication, assembled the team, developed initial abstract and manuscript drafts, and obtained budget.
Supporting information
TABLE S1. Reasons for patient withdrawal from the study (SAF)
TABLE S2. Change from baseline to Week 8 in PASI score (classic and linear), PSGA score, and %BSA affected by psoriasis (FAS)
TABLE S3. TEAEs occurring in ≥3 patients from placebo or bleselumab total group in the SAF
TABLE S4. Clinical laboratory parameters on Days 1, 29 and 113 (SAF)
ACKNOWLEDGEMENTS
This study was sponsored by Astellas Pharma, Inc. The authors thank all investigators for their effort and cooperation. Daniella T. Draper, PhD, CMPP from Cello Health MedErgy (Europe) assisted in drafting the initial version of the manuscript under the direction of the authors, and provided editorial support throughout its development. Editorial support was funded by Astellas Pharma, Inc.
Anil Kumar MS, Papp K, Tainaka R, et al. Randomized, controlled study of bleselumab (ASKP1240) pharmacokinetics and safety in patients with moderate‐to‐severe plaque psoriasis. Biopharm Drug Dispos. 2018;39:245–255. https://doi.org/10.1002/bdd.2130
Contributor Information
Mysore S. Anil Kumar, Email: anil.kumar@astellas.com
Kim Papp, Email: kapapp@probitymedical.com.
Ryo Tainaka, Email: ryo.tainaka@astellas.com.
Udaya Valluri, Email: udaya.valluri@astellas.com.
Xuegong Wang, Email: xuegong.wang@astellas.com.
Tong Zhu, Email: tong.zhu@astellas.com.
Christian Schwabe, Email: christian.schwabe@clinicalstudies.co.nz.
REFERENCES
- Albach, F. N. , Wagner, F. , Hüser, A. , Igel, J. , Joseph, D. , Hilbert, J. , … Steffgen, J. (2018). Safety, pharmacokinetics and pharmacodynamics of single rising doses of BI 655064, an antagonistic anti‐CD40 antibody in healthy subjects: a potential novel treatment for autoimmune diseases. European Journal of Clinical Pharmacology, 74, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyagi, T. , Yamashita, K. , Suzuki, T. , Uno, M. , Goto, R. , Taniguchi, M. , … Todo, S. (2009). A human anti‐CD40 monoclonal antibody, 4D11, for kidney transplantation in cynomolgus monkeys: induction and maintenance therapy. American Journal of Transplantation, 9, 1732–1741. [DOI] [PubMed] [Google Scholar]
- Bensinger, W. , Maziarz, R. T. , Jagannath, S. , Spencer, A. , Durrant, S. , Becker, P. S. , … Stadtmauer, E. A. (2012). A phase 1 study of lucatumumab, a fully human anti‐CD40 antagonist monoclonal antibody administered intravenously to patients with relapsed or refractory multiple myeloma. British Journal of Haematology, 159, 58–66. [DOI] [PubMed] [Google Scholar]
- Cai, Y. , Fleming, C. , & Yan, J. (2012). New insights of T cells in the pathogenesis of psoriasis. Cellular & Molecular Immunology, 9, 302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Kerkhof, P. C. M. , Reich, K. , Kavanaugh, A. , Bachelez, H. , Barker, J. , Girolomoni, G. , … Lebwohl, M. G. (2015). Physician perspectives in the management of psoriasis and psoriatic arthritis: results from the population‐based Multinational Assessment of Psoriasis and Psoriatic Arthritis survey. Journal of European Academy of Dermatology and Venereology, 29, 2002–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dua, P. , Hawkins, E. , & van der Graaf, P. H. (2015). A tutorial on target‐mediated drug disposition (TMDD) models. CPT Pharmacometrics & Systems Pharmacology, 4, 324–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldwater, R. , Keirns, J. , Blahunka, P. , First, R. , Sawamoto, T. , Zhang, W. , … Holman, J. (2013). A Phase 1, randomized ascending single‐dose study of antagonist anti‐human CD40 ASKP1240 in healthy subjects. American Journal of Transplantation, 13, 1040–1046. [DOI] [PubMed] [Google Scholar]
- Helmick, C. G. , Lee‐Han, H. , Hirsch, S. C. , Baird, T. L. , & Bartlett, C. L. (2014). Prevalence of psoriasis among adults in the U.S.: 2003‐2006 and 2009‐2010 National Health and Nutrition Examination Surveys. American Journal of Preventive Medicine, 47, 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida, I. , Tomizuka, K. , Yoshida, H. , Tahara, T. , Takahashi, N. , Ohguma, A. , … Lonberg, N. (2002). Production of human monoclonal and polyclonal antibodies in TransChromo animals. Cloning and Stem Cells, 4, 91–102. [DOI] [PubMed] [Google Scholar]
- Jacobson, E. M. , Huber, A. K. , Akeno, N. , Sivak, M. , Li, C. W. , Concepcion, E. , … Tomer, Y. (2007). A CD40 Kozak sequence polymorphism and susceptibility to antibody‐mediated autoimmune conditions: the role of CD40 tissue‐specific expression. Genes and Immunity, 8, 205–214. [DOI] [PubMed] [Google Scholar]
- Karczewski, J. , Dobrowolska, A. , Rychlewska‐Hańczewska, A. , & Adamski, Z. (2016). New insights into the role of T cells in pathogenesis of psoriasis and psoriatic arthritis. Autoimmunity, 49, 435–450. [DOI] [PubMed] [Google Scholar]
- Kernstock, R. (2010). Bioanalytical Validation Report IC‐2009–002. Validation of an analytical method for the screening, confirmation, and titer for the presence of anti‐ASKP1240 antibodies in human serum using electrochemiluminescence detection. Skokie, IL, USA.
- Krishna, M. , & Nadler, S. G. (2016). Immunogenicity to biotherapeutics – the role of anti‐drug immune complexes. Frontiers in Immunology, 7, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen, C. P. , Alexander, D. Z. , Hollenbaugh, D. , Elwood, E. T. , Ritchie, S. C. , Aruffo, A. , … Pearson, T. C. (1996). CD40‐gp39 interactions play a critical role during allograft rejection. Suppression of allograft rejection by blockade of the CD40‐gp39 pathway. Transplantation, 61, 4–9. [DOI] [PubMed] [Google Scholar]
- Larsen, C. P. , Knechtle, S. J. , Adams, A. , Pearson, T. , & Kirk, A. D. (2006). A new look at blockade of T‐cell costimulation: a therapeutic strategy for long‐term maintenance immunosuppression. American Journal of Transplantation, 6, 876–883. [DOI] [PubMed] [Google Scholar]
- Okimura, K. , Maeta, K. , Kobayashi, N. , Goto, M. , Kano, N. , Ishihara, T. , … Mura, T. (2014). Characterization of ASKP1240, a fully human antibody targeting human CD40 with potent immunosuppressive effects. American Journal of Transplantation, 14, 1290–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oura, T. , Yamashita, K. , Suzuki, T. , Fukumori, D. , Watanabe, M. , Hirokata, G. , … Todo, S. (2012). Long‐term hepatic allograft acceptance based on CD40 blockade by ASKP1240 in nonhuman primates. American Journal of Transplantation, 12, 1740–1754. [DOI] [PubMed] [Google Scholar]
- Papp, K. , Gulliver, W. , Lynde, C. , Poulin, Y. , Ashkenas, J. , & Canadian Psoriasis Guidelines Committee (2011). Canadian guidelines for the management of plaque psoriasis: overview. Journal of Cutaneous Medicine and Surgery, 15, 210–219. [DOI] [PubMed] [Google Scholar]
- Parisi, R. , Symmons, D. P. M. , Griffiths, C. E. M. , Ashcroft, D. M. , & Identification and Management of Psoriasis and Associated ComorbidiTy (IMPACT) project team (2013). Global epidemiology of psoriasis: a systematic review of incidence and prevalence. Journal of Investigative Dermatology, 133, 377–385. [DOI] [PubMed] [Google Scholar]
- Pasch, M. C. , Timár, K. K. , van Meurs, M. , Heydendael, V. M. , Bos, J. D. , Laman, J. D. , & Asghar, S. S. (2004). In situ demonstration of CD40‐ and CD154‐positive cells in psoriatic lesions and keratinocyte production of chemokines by CD40 ligation in vitro. Journal of Pathology, 203, 839–848. [DOI] [PubMed] [Google Scholar]
- Peters, A. L. , Stunz, L. L. , & Bishop, G. A. (2009). CD40 and autoimmunity: the dark side of a great activator. Seminars in Immunology, 21, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampogna, F. , Tabolli, S. , Abeni, D. , & IDI Multipurpose Psoriasis Research on Vital Experiences (IMPROVE) investigators (2012). Living with psoriasis: prevalence of shame, anger, worry, and problems in daily activities and social life. Acta Dermato‐ Venereologica, 92, 299–303. [DOI] [PubMed] [Google Scholar]
- Shankar, G. , Pendley, C. , & Stein, K. E. (2007). A risk‐based bioanalytical strategy for the assessment of antibody immune responses against biological drugs. Nature Biotechnology, 25, 555–561. [DOI] [PubMed] [Google Scholar]
- Song, L. , Ma, A. , Dun, H. , Hu, Y. , Zeng, L. , Bai, J. , … Chen, H. (2014). Effects of ASKP1240 combined with tacrolimus or mycophenolate mofetil on renal allograft survival in cynomolgus monkeys. Transplantation, 98, 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern, R. S. , Nijsten, T. , Feldman, S. R. , Margolis, D. J. , & Rolstad, T. (2004). Psoriasis is common, carries a substantial burden even when not extensive, and is associated with widespread treatment dissatisfaction. Journal of Investigative Dermatology Symposium Proceedings, 9, 136–139. [DOI] [PubMed] [Google Scholar]
- World Health Organization (2016). Global Report on Psoriasis. Geneva: WHO. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1. Reasons for patient withdrawal from the study (SAF)
TABLE S2. Change from baseline to Week 8 in PASI score (classic and linear), PSGA score, and %BSA affected by psoriasis (FAS)
TABLE S3. TEAEs occurring in ≥3 patients from placebo or bleselumab total group in the SAF
TABLE S4. Clinical laboratory parameters on Days 1, 29 and 113 (SAF)