

Abstract
Background
Injectable daxibotulinumtoxinA (an investigational botulinum toxin, RT002) may offer a more prolonged duration of response—and therefore less frequent dosing—than onabotulinumtoxinA.
Objectives
To perform a phase 2, open‐label, dose‐escalation study to assess the efficacy and safety of daxibotulinumtoxinA in cervical dystonia.
Methods
Subjects with moderate‐to‐severe isolated cervical dystonia were enrolled in sequential cohorts to receive a single open‐label, intramuscular dose of injectable daxibotulinumtoxinA of up to 200 U (n = 12), 200–300 U (n = 12), or 300–450 U (n = 13; https://clinicaltrials.gov identifier NCT02706795).
Results
Overall, 33/37 enrollees completed the trial. DaxibotulinumtoxinA was associated with mean reductions in Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS)‐Total score of 16.8 (38%) at week 4, 21.3 (50%) at week 6, and 12.8 (30%) at week 24. The proportion of subjects who were responders (achieved ≥ 20% reduction in TWSTRS‐Total score) was 94% at week 6 and 68% at week 24. The median duration of response (time until > 20% of the improvement in TWSTRS‐Total score achieved at week 4 was no longer retained or re‐treatment was needed) was 25.3 weeks (95% CI, 20.14–26.14 weeks). There were no serious adverse events and there was no apparent dose‐related increase in the incidence of adverse events. The most common treatment‐related adverse events were dysphagia (14%) and injection site erythema (8%).
Conclusions
Preliminary assessments suggest that injectable daxibotulinumtoxinA at doses up to 450 U is well tolerated and may offer prolonged efficacy in the treatment of cervical dystonia. Further studies involving larger numbers of patients are now warranted.
Keywords: botulinum toxin, CDIP, cervical dystonia, daxibotulinumtoxinA, TWSTRS
Introduction
Botulinum toxin (BoNT) is considered first‐line treatment for cervical dystonia1, 2 and can reduce abnormal neck posture and pain, and lessen impairments in activities of daily living. With currently available BoNT type A treatments, injections are repeated approximately every 12 weeks,3 but many patients feel the need for earlier reinjection4 and would prefer a longer‐lasting treatment.3 The development of a BoNT with a longer duration of benefit could provide more sustained improvement, lengthen the interval between injections, and lower the number of visits for injections (potentially reducing health care costs).
Injectable daxibotulinumtoxinA (RT002) is an investigational BoNT that is in clinical development for the treatment of cervical dystonia, glabellar lines,5 and plantar fasciitis.6 Injectable daxibotulinumtoxinA is a purified 150 kDa BoNT type A (RTT150) that is devoid of accessory proteins and formulated with a proprietary stabilizing excipient peptide (RTP004) in a lyophilized powder. The peptide is composed of two protein transduction domains on a backbone of consecutive lysines.7 The lysines carry a positive charge and result in the peptide binding avidly to RTT150 through electrostatic bonds. DaxibotulinumtoxinA is stable at room temperature and is produced without serum albumin or any other animal or human blood products.
In preclinical studies, daxibotulinumtoxinA has been shown to exhibit less diffusion than onabotulinumtoxinA and data suggest that it could offer a relatively greater duration of effect after injection into a target muscle.8 In clinical studies, injectable daxibotulinumtoxinA has shown a prolonged duration of response in the treatment of glabellar lines5, 9—with a significantly longer median duration of response than onabotulinumtoxinA (24 weeks versus 19 weeks; p = 0.030) when daxibotulinumtoxinA was used at the dose being evaluated in phase 3 glabellar line trials (40 U) and onabotulinumtoxinA was used at the dose for which it is approved for glabellar lines (20 U).5
An open‐label dose‐escalation study was performed to provide an initial assessment of the magnitude and duration of efficacy of injectable daxibotulinumtoxinA in adults with isolated cervical dystonia.
Methods
Study Design
Subjects could be enrolled in this phase 2 dose‐escalation study if they were judged clinically likely to benefit from daxibotulinumtoxinA at a low dose (cohort 1), a medium dose (cohort 2), or a high dose (cohort 3). Enrollment in cohorts 2 and 3 could only proceed after 6‐week safety data from the preceding cohort had been reviewed by an independent data monitoring committee. Subjects from cohorts 1 or 2 were eligible for reenrollment into cohort 3 if all inclusion criteria continued to be met.
The study was approved by the appropriate institutional review boards in accordance with applicable laws and conducted according to the principles of the Declaration of Helsinki. All subjects signed informed consent.
Subjects
Subjects were eligible for inclusion in the study if they had a diagnosis of isolated cervical dystonia of at least moderate severity, defined as a total score on the Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS)10 of at least 20, and a score on the TWSTRS severity subscale of at least 15. Subjects were also required to be 30–75 years of age. Medications for dystonia were required to remain at stable doses for at least 3 months prior to study entry and for the duration of the study. Exclusion criteria included cervical dystonia with predominant retrocollis or anterocollis posture, cervical dystonia that was attributable to an underlying etiology (e.g., trauma or drug‐induced dystonia), and the use of any BoNT product within the 6 months preceding the screening visit.
Treatment
DaxibotulinumtoxinA (Revance Therapeutics, Inc.) was supplied in vials containing 160 U of lyophilized product and was reconstituted and diluted to a 100 U/mL solution using 1.6 mL of sterile non‐preserved 0.9% sodium chloride solution. Subjects received a single intramuscular treatment of daxibotulinumtoxinA, with cohort 1 receiving a maximal dose of 200 U, cohort 2 receiving 200–300 U, and cohort 3 receiving 300–450 U. The dose of daxibotulinumtoxinA used (within the range for each cohort), the number of muscles injected, the number of injection sites in each muscle, and the use of electromyography were at the discretion of the treating investigator.
Outcome Measures
Subjects were evaluated at baseline and weeks 2, 4, 6, 9, 12, 16, 20, and 24. The primary efficacy outcome was the change in TWSTRS‐Total score from baseline to week 4. Secondary efficacy outcomes included the following:
Change from baseline in TWSTRS‐Total score at other time points besides week 4
Change from baseline in score for each of the three component TWSTRS subscales (severity, disability, and pain)
Percentage of treatment responders (defined as ≥ 20% reduction in TWSTRS‐Total score post‐treatment)
Duration of effect (defined as the duration of retention of ≥ 20% of the improvement in TWSTRS‐Total score achieved at week 4 or the time since injection until a subject expressed a need for treatment and the investigator agreed that it was necessary [whichever was earlier])
Clinical Global Impression of Change (CGIC) and Patient Global Impression of Change (PGIC) (9‐point scales used by investigators and subjects, respectively, to rate the global response to treatment as: 4 [complete improvement/abolishment of dystonia symptoms], 3 [marked improvement], 2 [moderate improvement], 1 [slight improvement], 0 [no change], ‐1 [slight worsening], ‐2 [moderate worsening], ‐3 [marked worsening], or ‐4 [very marked worsening])
Change in total score on the Cervical Dystonia Impact Profile (CDIP‐58), a disease‐specific patient‐rated questionnaire that assesses the impact of cervical dystonia on health‐related quality of life using a 100‐point scale (0 = no impact, 100 = most impact).11 The CDIP‐58 questionnaire is composed of eight subscales (head and neck, pain and discomfort, sleep, upper limb activities, walking, annoyance, mood, and psychosocial functioning)
TWSTRS scores were evaluated at all visits, the CGIC and PGIC were evaluated at all visits except baseline, and the CDIP‐58 was evaluated at all visits except weeks 2 and 9.
Subjects without improvement in TWSTRS‐Total score at week 4 were followed until week 9 and then discontinued the study. Subjects with improvement continued beyond week 9 for up to 24 weeks—but completed the study before week 24 once they no longer retained ≥ 20% of the improvement in TWSTRS‐Total score achieved at week 4, or if they expressed a need for treatment and the investigator agreed that it was necessary (whichever was earlier).
Subjects reported adverse events spontaneously and in response to specific querying about dysphagia and neck weakness. Adverse events were obtained at every study visit and by telephone interview at weeks 14, 18, and 22. The investigator assessed adverse events in terms of their relationship to the study drug and classified the severity of these events as mild (not noticeable to subject), moderate (may be of sufficient severity to make subject uncomfortable and influence daily activities, intervention may be needed), or severe (may cause severe discomfort, usually interferes with daily activities, subject may not be able to continue in study, intervention usually needed).
Statistical Analyses
The sample size was based on clinical considerations only and no formal sample size calculation was performed. As a result, the study was not powered to detect statistical significance and statistical analyses were largely descriptive and limited to calculating percentages, means, medians, and standard deviations. The analyses presented in this paper are based on observed data except that, if data for the primary endpoint (change from baseline in TWSTRS‐Total score at week 4) were missing, values were imputed with data from the closest time point after week 4.
Results
The study was conducted at seven neurology clinics and one physical medicine and rehabilitation clinic in the US between September 2015 and April 2017. Three additional sites participated in screening, but did not have subjects who enrolled.
Subjects
A total of 34 subjects enrolled in the study, with three subjects enrolling into two different cohorts (one into cohorts 1 and 3, two into cohorts 2 and 3). This resulted in 37 sets of data being obtained—12 in cohort 1, 12 in cohort 2, and 13 in cohort 3 (Figure 1A). An additional 19 subjects failed screening requirements (17 did not meet the inclusion and exclusion criteria and two withdrew before enrollment).
Figure 1.
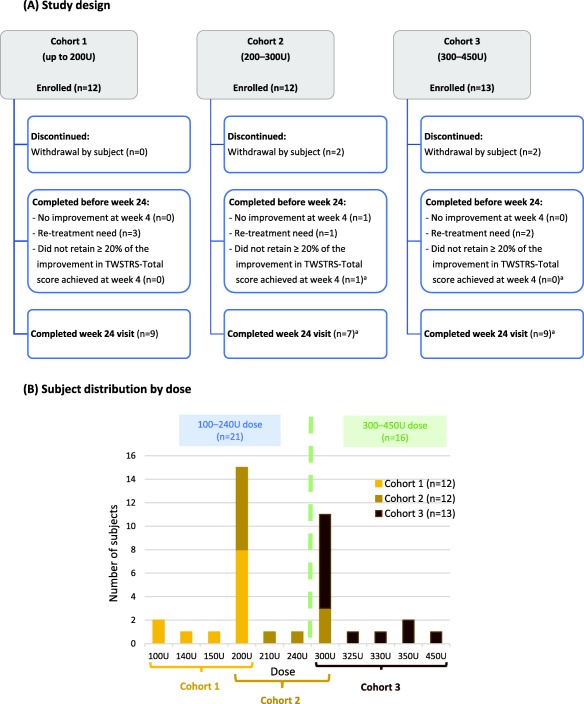
Study design and subject distribution by dose.
aThree subjects had a protocol deviation as they remained in the study until Week 24 even though they had ceased to be treatment responders at an earlier visit (at week 20 for 2 subjects in cohort 2 and at week 6 for 1 subject in cohort 3).
In total, 33/37 (89%) subjects completed the study and four subjects withdrew (three due to the time and/or travel commitment required and one due to loss to follow‐up). Among the 33 completing subjects, 25 were followed for 24 weeks (including three who should have completed at earlier visits but were protocol deviations as detailed in Figure 1A). Of the remaining eight subjects who completed before week 24, one had no improvement at week 4 (and therefore completed the study at week 9), six required re‐treatment between weeks 9 and 24, and one did not retain ≥ 20% of the improvement in TWSTRS‐Total score previously achieved at week 4.
There was overlap between cohorts in terms of the dose administered: eight subjects in cohort 1 and seven in cohort 2 received 200 U, and three subjects in cohort 2 and eight in cohort 3 received 300 U (Figure 1B). Therefore, for all evaluations, data were analyzed in two groups—a lower‐dose group (100–240 U, n = 21) and a higher‐dose group (300–450 U, n = 16; Figure 1B). All subjects were evaluated for efficacy (intention‐to‐treat analyses) and safety.
Demographics
Baseline demographics were generally similar between the two dose groups, except the higher‐dose group had a relatively lower proportion of females (63% vs. 86%) and a longer median duration of cervical dystonia (7.2 vs. 4.8 years; Table 1). Subjects had a mean TWSTRS‐Total score of 44.1 (Table 1).
Table 1.
Baseline Demographics
| DaxibotulinumtoxinA dose | |||
|---|---|---|---|
|
100–240 U (N = 21) |
300–450 U (N = 16) a |
100–450 U (N = 37) (Combined group) |
|
| Mean age ± SD, yrs | 54 ± 10.4 | 58 ± 10.6 | 56 ± 10.4 |
| [range] | [32–74] | [30–70] | [30–74] |
| Females, n (%) | 18 (86%) | 10 (63%) | 28 (76%) |
| Caucasians, n (%) | 18 (86%) | 14 (88%) | 32 (86%) |
| Duration of cervical dystonia, yrs | |||
| Mean ± SD | 7.3 ± 8.09 | 7.9 ± 7.13 | 7.6 ± 7.59 |
| Median [range] | 4.8 [0.0–24.1] | 7.2 [0.0–23.3] | 4.9 [0.0–24.1] |
| Prior treatment with botulinum toxin, n | 9 (43%) | 8 (50%) | 17 (46%) |
| Mean dose of daxibotulinumtoxinA ± SD, U | 188 ± 35.1 | 319 ± 39.3 | 244 ± 75.4 |
| Mean TWSTRS score ± SD | |||
| Total score | 44.4 ± 9.52 | 43.8 ± 10.20 | 44.1 ± 9.69 |
| Severity subscore | 20.5 ± 3.23 | 21.9 ± 3.89 | 21.1 ± 3.55 |
| Disability subscore | 12.6 ± 3.92 | 11.6 ± 4.51 | 12.2 ± 4.15 |
| Pain subscore | 11.3 ± 4.63 | 10.3 ± 4.55 | 10.8 ± 4.56 |
| CDIP‐58 total score | |||
| Mean ± SD | 53.9 ± 18.76 | 49.7 ± 14.90 | 52.1 ± 17.10 |
| Median [range] | 51.4 [22.8–90.7] | 55.9 [23.5–71.7] | 54.8 [22.8–90.7] |
Abbreviations: CDIP‐58, Cervical Dystonia Impact Profile; SD, standard deviation; TWSTRS, Toronto Western Spasmodic Torticollis Rating Scale
aThree were reenrolled from lower‐dose group.
Efficacy
The reductions in TWSTRS‐Total and subscale scores are shown in Figures 2A‐2D. For clarity, the graphs depict only the results from the lower‐ and higher‐dose groups; the overall group data (for both groups combined) are summarized below.
Figure 2.
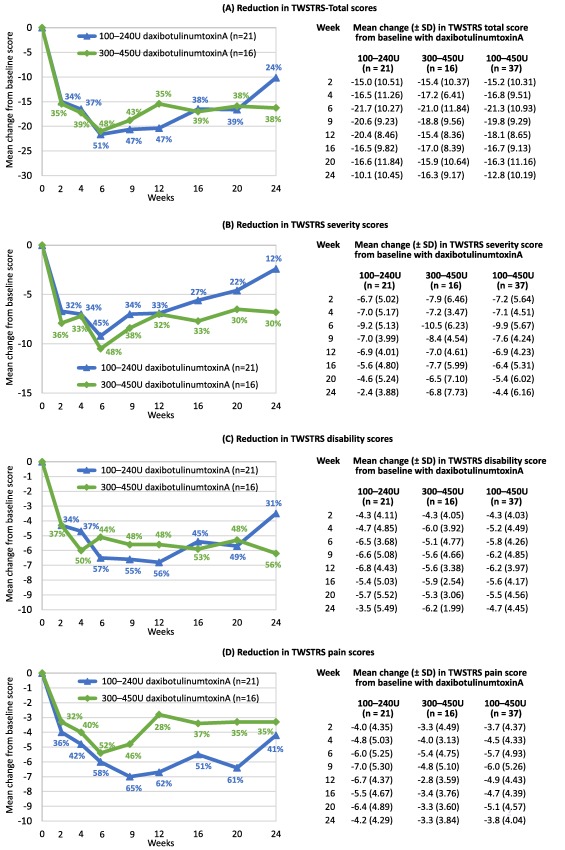
Reduction in TWSTRS‐Total score and subscores for severity, disability, and pain in the lower‐dose and higher‐dose groups.
The mean change in TWSTRS‐Total score at week 4 (the primary efficacy outcome) was ‐16.8 ± 9.51 in all subjects (Figure 2A). Converting the mean change in TWSTRS‐Total score to a percentage change reveals that, in both dose groups combined, daxibotulinumtoxinA resulted in a 38% reduction in TWSTRS‐Total score at week 4, a 50% reduction at week 6 (the peak reduction), a 42% reduction at week 12, and a 30% reduction at week 24 (Figure 2A).
At week 6, the overall reductions in mean TWSTRS severity, disability, and pain subscale scores were 47%, 50%, and 55%, respectively (Figures 2B, 2C, 2D). Peak reductions occurred at week 6 for severity, week 12 for disability (53%), and week 9 for pain (57%). Moreover, at week 24, the overall reductions in these subscale scores were 20%, 42%, and 39%, respectively.
The response rate (i.e., proportion of subjects achieving ≥ 20% reduction from baseline in TWSTRS‐Total score) across all subjects was 83% at week 4, peaked at 94% at week 6, and was still 68% at week 24 (Figure 3A). Among the 36 of 37 enrollees who had an improvement in TWSTRS‐Total score at week 4, the median duration of response (i.e., the duration over which subjects retained ≥ 20% of the improvement in TWSTRS‐Total score that each had achieved at week 4) was 25.3 weeks (95% CI, 20.14 to 26.14 weeks; Figure 3B).
Figure 3.
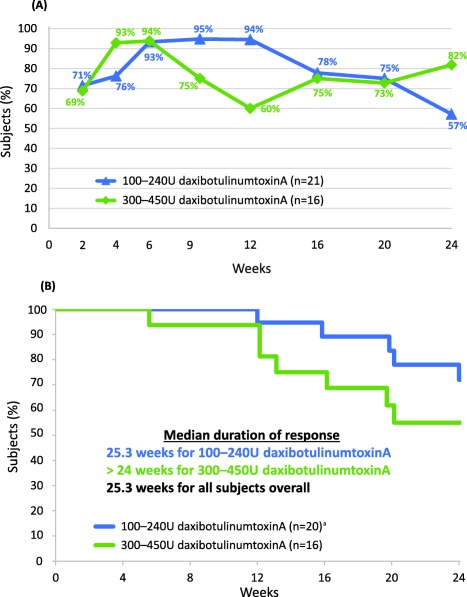
Response rate and duration of response. (A) Subjects achieving response (at least 20% improvement from baseline in TWSTRS‐Total score); (B) Subjects retaining at least 20% of the improvement in TWSTRS‐Total score achieved at week 4 (among subjects with improvement at week 4). Withdrawals due to need for re‐treatment are considered events.
aOne subject was excluded from analysis as their TWSTRS score was not improved at week 4
In one of the three reenrolled subjects, treatment response was last achieved at week 16 in the first enrollment and at week 9 in the second enrollment. In the other two reenrolled subjects, treatment response was last achieved at week 24 for both enrollments. During the second enrollment for one of these subjects, treatment response was transiently lost at week 12 (improvement from baseline in TWSTRS score being 19.2%, so failing to meet the 20% threshold for treatment response), but the subject remained in the study until week 24 (a protocol deviation) and met the criteria for treatment response at weeks 16, 20, and 24 without further treatment.
CGIC ratings of improvement were attained by 97% of subjects at week 2, 97% at week 4, 94% at week 6, 63% at week 16, and 40% at week 24 (Supporting Table S1). PGIC ratings of improvement were attained by 92% of subjects at week 2, 83% at week 4, 87% at week 6, 63% at week 16, and 60% at week 24 (Supporting Table S2). Subject ratings of the impact of cervical dystonia on health‐related quality of life were improved at all evaluations, with the median CDIP‐58 score reduced by at least 13 from week 4 to week 24 (Supporting Table S3).
Safety and Tolerability
DaxibotulinumtoxinA appeared to be generally safe and well tolerated. There were no serious adverse events and no discontinuations due to adverse events. Treatment‐related adverse events (i.e., adverse events that were possibly, probably, or definitely related to treatment) were generally transient and all were mild‐to‐moderate, except one that was considered severe (neck pain on days 10 and 11). The most commonly reported treatment‐related adverse events were dysphagia (14%, mean duration 35 days), injection site erythema (8%, mean duration 8 days), injection site pain (5%, mean duration 13 days), muscular weakness (5%, mean duration 51 days), injection site bruising (5%, mean duration 22 days), and muscle tightness (5%, mean duration 32 days; Table 2).
Table 2.
Incidence of Treatment‐Emergent Adverse Events Considered at Least Possibly Related to Treatment
| Adverse event | Subjects (%) | ||
|---|---|---|---|
|
100–240 U daxibotulinumtoxinA (N = 21) |
300–450 U daxibotulinumtoxinA (N = 16) |
100–450 U daxibotulinumtoxinA (N = 37) (Combined group) |
|
| Dysphagia |
3 (14%) [mild] |
2 (13%) [mild] |
5 (14%) a |
| Injection site erythema |
2 (10%) [mild] |
1 (6%) [mild] |
3 (8%) |
| Injection site pain |
1 (5%) [moderate] |
1 (6%) [mild] |
2 (5%) |
| Muscular weakness |
2 (10%) [1 mild, 1 moderate] |
_ | 2 (5%) |
| Injection site bruising |
2 (10%) [mild] |
_ | 2 (5%) |
| Muscle tightness |
1 (5%) [mild] |
1 (5%) [mild] |
2 (5%) b |
| Neck pain |
1 (5%) [severe] |
_ | 1 (3%) |
| Muscle spasms |
1 (5%) [moderate] |
_ | 1 (3%) |
| Trismus |
1 (5%) [moderate] |
_ | 1 (3%) |
| Fatigue | _ |
1 (5%) [mild] |
1 (3%) |
| Nausea | _ |
1 (5%) [mild] |
1 (3%) |
| Asthenia | _ |
1 (5%) [mild] |
1 (3%) |
aOne reenrolled subject reported treatment‐emergent dysphagia in both cohorts.
bOne reenrolled subject reported treatment‐emergent muscle tightness in both cohorts.
The subjects with treatment‐related dysphagia received 25–50 U daxibotulinumtoxinA unilaterally into the sternocleidomastoid muscle and 0–50 U into the levator scapulae muscle. The remaining subjects received up to 250 U in the sternocleidomastoid muscle and up to 80 U in the levator scapulae without developing treatment‐related dysphagia. Among the 31 subjects injected into the sternocleidomastoid muscle, one subject received 250 U in this muscle (150 U on one side split between 2 injections, plus 100 U on the other side in a single injection), one subject received 90 U in this muscle (50 U on one side split between 5 injections, plus 40 U on the other side split between 4 injections), and the other 29 subjects received a dose of 20–50 U (as a single injection in 21 subjects and split across 2‐to‐4 injections in eight subjects, five of whom had injections on a single side and three of whom had injections in both the right and left muscles).
Discussion
The results of this study demonstrate that daxibotulinumtoxinA appears to be well tolerated at doses up to 450 U, and can achieve clinically meaningful improvements in the signs and symptoms of cervical dystonia (as evaluated using TWSTRS scores)—not only at the primary end point of 4 weeks, but also from week 2 through week 24. In total, 36 of 37 subjects showed an improvement in TWSTRS‐Total score at week 4 and the median duration of their response was more than 25 weeks. Overall, the peak therapeutic benefit occurred at week 6 when the TWSTRS‐Total score was reduced by a mean of 21.3 points (a 50% reduction from the mean baseline score). In addition, much of the benefit was still maintained at week 24 when the mean reduction in TWSTRS‐Total score was 12.8 points (a 30% reduction from the mean baseline score) and 68% (25/37) of the enrollees were still treatment responders (i.e., had ≥ 20% reduction from baseline in TWSTRS‐Total score).
Investigator and subject assessments of the global impression of change indicated that more than 90% of subjects had an improvement in their cervical dystonia following treatment with daxibotulinumtoxinA. Furthermore, 60% of subject ratings considered improvement was still present at week 24 compared with 40% of investigator ratings. The CDIP‐58 data were consistent with the TWSTRS data in terms of both the magnitude and duration of treatment benefit and suggest that the overall impact of cervical dystonia was reduced.
The lower‐dose and higher‐dose groups both demonstrated efficacy and a prolonged duration of benefit. There was a greater reduction in mean TWSTRS pain score in the lower‐dose group than the higher‐dose group (Figure 2D), and this could be partly attributable to the lower‐dose group having relatively higher pain scores at baseline (11.3 vs. 10.3). The three subjects who reenrolled had lower baseline TWSTRS pain scores at their second enrollment (into the higher‐dose group) compared with their first enrollment (into the lower‐dose group) and it may be that some of the effect of daxibotulinumtoxinA on their pain persisted when they enrolled the second time. However, given the small sample size, the between‐group variability, and the open‐label design, it is not possible to draw any conclusions regarding dose‐response effects.
The mean 16.8‐point (38%) reduction in TWSTRS‐Total score at week 4 is greater than that reported in trials with other BoNTs. For example, a mean reduction in TWSTRS‐Total score of 9.9 has been reported for incobotulinumtoxinA 120 U,12 10.9 for incobotulinumtoxinA 240 U,12 9.3 for botulinum toxin type B 5,000 U,13 11.7 for botulinum toxin type B 10,000 U,13 9.9 for abobotulinumtoxinA 500 U,14 and a least squares mean reduction of approximately 14 for abobotulinumtoxinA 500 U.15 However, these previous studies were larger, double blind, and placebo controlled, so these differences may not be meaningful. Previous studies with other BoNTs have used a variety of different definitions for response rates14, 15, 16 and duration of response14, 17, 18 (or have not reported them at all) and so similar comparisons are not so feasible for these outcome measures. In terms of reductions in CDIP‐58 score, the mean reductions of 15.0 in this study at week 4 and 12.6 at week 24 are similar to those in previous trials with other BoNTs at week 4.15, 19
DaxibotulinumtoxinA appeared to be generally safe and well tolerated, with no dose‐related increase in the incidence of treatment‐related adverse events. In common with other BoNTs, dysphagia was the most common adverse event and its incidence in this trial (14% related to treatment, 16% regardless of relationship to treatment) appears in the same range as in studies with other BoNT type A products already approved to treat cervical dystonia (7–16% related to treatment, 7–19% regardless of relationship to treatment).12, 15, 20
A significant limitation of the study was its small size and open‐label design. In addition, reenrolling three subjects into a second cohort could have introduced a selection bias toward good efficacy and tolerability. However, the results of the study suggest that daxibotulinumtoxinA can reduce the clinical manifestations of cervical dystonia for potentially an extended period of time relative to other currently available BoNTs, and is generally well tolerated across a range of doses. The results presented here provide strong evidence to support a large randomized, double‐blind, comparative study to further evaluate the magnitude and duration of clinical response to daxibotulinumtoxinA in cervical dystonia.
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
J.J.: 1C, 3B
D.T.: 1C, 3B
A.P.: 1C, 3B
A.B.: 1C, 3B
M.E.: 1C, 3B
R.R.: 3B
C.O.: 3B
D.S.: 3B
G.S.: 3A
C.C.: 1A, 1C, 3B
Disclosures
Ethical Compliance Statement: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflict of Interest: Dr Jankovic has received research and/or training grants from Allergan, Dystonia Coalition, Dystonia Medical Research Foundation, and Revance Therapeutics, Inc. He has served as a consultant or as an advisory committee member for Allergan, Inc., Merz Pharmaceuticals, and Revance Therapeutics, Inc.
Dr Truong serves in the speaker bureau of US World Med and reports research support from Acadia Pharmaceutical Inc., Adamas Inc., Axovant Sciences Ltd., Bristol‐Myers Squibb, Civitas Therapeutics, Inc., Cynapsus Therapeutics Inc., Impax Laboratories, Inc., Intec Pharma, Ltd., Kyowa Kirin Pharmaceutical Development Inc., Lundbeck Inc., Neuroderm Inc., Parkinson Study Group, Sunovion Pharmaceuticals, Inc., and UCB Pharmaceutical, Inc.
Dr Patel has served as a consultant and received research grants from Revance Therapeutics, Inc., Allergan, Ipsen, and Merz. He is a speaker for Allergan, Ipsen, and Merz.
Dr Brashear has consulted with Ipsen and Revance Therapeutics, Inc. on protocol development for clinical trials. Dr Brashear's research is with Revance Therapeutics Inc., Ipsen, Merz, Allergan, and NINDS. All research payments are made directly to Wake Forest School of Medicine. Dr Brashear's conflict of interest is managed by Wake Forest School of Medicine. She has received book royalty payments from Demos and serves on the Board of Directors of the American Board of Psychiatry and Neurology.
Dr Evatt has served as a consultant and/or received research grants from Revance Therapeutics, Inc., Allergan, Inc., Ipsen, and Merz Pharmaceuticals, LLC.
Dr Rubio is an employee of, and holds stock or stock options in, Revance Therapeutics, Inc.
Dr Oh was an employee of, and held stock or stock options in, Revance Therapeutics, Inc.
Dr Snyder is an employee of, and holds stock or stock options in, Revance Therapeutics, Inc.
Dr Shears is an employee of Write on Target Ltd., which has received funding from Revance Therapeutics, Inc. for medical writing services.
Dr Comella serves on the editorial board of Clinical Neuropharmacology, Sleep Medicine, and Continuum. She receives research support from NIH R01NS074343, U54NS065701, Dystonia Medical Research Foundation, Merz Pharmaceuticals, Revance Therapeutics Inc., Retrophin, and Acorda Therapeutics. She receives compensation/honoraria for services as a consultant or advisory committee member for Acorda Therapeutics, Allergan, Inc., Lundbeck Ltd., Medtronic Inc., Merz Pharmaceuticals, Acadia Pharmaceuticals, Jazz Pharmaceuticals, Neurocrine Biosciences Inc., and Revance Therapeutics, Inc. She receives royalties from Cambridge University Press and Wolters Kluwer publishing, and research support from the Parkinson's Disease Foundation. Funding source for the study and for the development of the manuscript: Revance Therapeutics, Inc., Newark, CA.
Financial disclosures for the previous 12 months: Dr Jankovic has received research and/or training grants from: Adamas Pharmaceuticals, Inc; Allergan, Inc; Biotie Therapies; CHDI Foundation; Civitas/Acorda Therapeutics; Dystonia Coalition; Dystonia Medical Research Foundation; F. Hoffmann‐La Roche Ltd; Huntington Study Group; Kyowa Haako Kirin Pharma, Inc; Medtronic Neuromodulation; Merz Pharmaceuticals; The Michael J Fox Foundation for Parkinson Research; National Institutes of Health; Neurocrine Biosciences; NeuroDerm Ltd; Parkinson's Foundation; Parkinson Study Group; Pfizer Inc; Prothena Biosciences Inc; Psyadon Pharmaceuticals, Inc; Revance Therapeutics, Inc; Sangamo BioSciences, Inc.; St. Jude Medical; and Teva Pharmaceutical Industries Ltd. He has served as a consultant or as an advisory committee member for: Adamas Pharmaceuticals, Inc; Allergan, Inc; Merz Pharmaceuticals; Pfizer Inc; Prothena Biosciences; Revance Therapeutics, Inc; and Teva Pharmaceutical Industries Ltd. In addition, he has received royalties or other payments from: Cambridge; Elsevier; Future Science Group; Hodder Arnold; Medlink: Neurology; Lippincott Williams and Wilkins; and Wiley‐Blackwell. He has served on the editorial boards for: Expert Review of Neurotherapeutics; Medlink; Neurology in Clinical Practice; The Botulinum Journal; PeerJ; Therapeutic Advances in Neurological Disorders; Neurotherapeutics; Tremor and Other Hyperkinetic Movements; Journal of Parkinson's Disease; and UpToDate.
Dr Truong has received research support from Acadia Pharmaceutical Inc., Adamas Inc., Axovant Sciences Ltd., Bristol‐Myers Squibb, Civitas Therapeutics, Inc., Cynapsus Therapeutics Inc., Impax Laboratories, Inc., Intec Pharma, Ltd., Kyowa Kirin Pharmaceutical Development Inc., Lundbeck Inc., Neuroderm Inc., Parkinson Study Group, Revance Therapeutics, Inc., Sunovion Pharmaceuticals, Inc., and UCB Pharmaceutical, Inc. Dr Truong also serves in the speaker bureau of US World Med and receives royalties from Cambridge University Press, Wiley Blackwell Publishing company, and Demos Publishing company.
Dr Patel has been a consultant to and received funding for research from Allergan, Ipsen, Revance Therapeutics, Inc., and Merz. He has also received honoraria for speaking for Allergan, Ipsen, and Merz.
Dr Brashear has: owned stock in Care Directions (a start‐up company); been a consultant to Allergan, Ipsen, Revance Therapeutics Inc., and US WorldMeds; provided expert testimony; received honoraria from the American Academy of Neurology; received book royalties from Demos; received grants from NINDS, Allergan, Ipsen, Merz, and Revance Therapeutics, Inc.; and served on the board of the American Board of Psychiatry and Neurology.
Dr Evatt has received salary support as Acting Chief, Neurology, Atlanta VA Medical Center, Decatur, GA. The contents do not represent the views of the US Department of Veterans Affairs or the United States Government. Dr Evatt has received research grants from Sunovion Pharmaceuticals, Biotie Therapies, Sanofi US Services Inc., Civitas Therapeutics, BioHaven Pharmaceutical, and Revance Therapeutics, Inc.
Dr Rubio has been an employee of, and held stock/stock options in, Revance Therapeutics, Inc.
Dr Oh has been an employee of Glenmark Pharmaceuticals and Revance Therapeutics, Inc. and has held stock/stock options in Revance Therapeutics, Inc.
Dr Snyder has been an employee of Ipsen Biopharmaceutics, Inc. and Revance Therapeutics, Inc. and held stock/stock options in both companies.
Dr Shears has been an employee of Write on Target Ltd.
Dr Comella has received research support from the NIH R01NS074343, U54NS065701, Dystonia Medical Research Foundation, Merz Pharmaceuticals, Revance Therapeutics, Inc., Retrophin and Acorda Therapeutic. She receives compensation/honoraria for services as a consultant or an advisory committee member from Acorda Therapeutics, Allergan, Inc., Lundbeck Ltd., Medtronic Inc., Merz Pharmaceuticals, Acadia Pharmaceuticals, Jazz Pharmaceuticals, Neurocrine Biosciences Inc., and Revance Therapeutics, Inc. She receives royalties from Cambridge University Press and Wolters Kluwer publishing, and research support from the Parkinson's Disease Foundation.
Supporting information
Supporting information may be found in the online version of this article.
Table S1. Subjects showing an improvement rating on the Clinical Global Impression of Change
Table S2. Subjects showing an improvement rating on the Patient Global Impression of Change
Table S3. Change from baseline in Cervical Dystonia Impact Profile (CDIP‐58) total score, which assesses subject ratings of impact of cervical dystonia on health‐related quality of life on a score from 0 (no impact) to 100 (most impact)
Acknowledgments
We are grateful to our coinvestigators Norman Bettle, MD and Aparna Shukla, MD for their contributions to this study; and Yan Zheng, MS for her statistical expertise. Some of the data in this manuscript were presented in poster format at the International Congress of Parkinson's Disease and Movement Disorders, Vancouver, BC, June 4–8, 2017.
The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the United States government.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Contarino MF, Van Den Dool J, Balash Y, et al. Clinical practice: evidence‐based recommendations for the treatment of cervical dystonia with botulinum toxin. Front Neurol 2017;8:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jankovic J. Botulinum toxin: State of the art. Mov Disord 2017;32:1131–1138. [DOI] [PubMed] [Google Scholar]
- 3. Poliziani M, Koch M, Liu X. Striving for more good days: patient perspectives on botulinum toxin for the treatment of cervical dystonia. Patient Prefer Adherence 2016;10:1601–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Evidente VG, Pappert EJ. Botulinum toxin therapy for cervical dystonia: the science of dosing. Tremor Other Hyperkinet Mov (NY) 2014;4:273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carruthers J, Solish N, Humphrey S, et al. Injectable daxibotulinumtoxinA for the treatment of glabellar lines: a phase 2, randomized, dose‐ranging, double‐blind, multicenter comparison with onabotulinumtoxinA and placebo. Dermatol Surg 2017;43:1321–1331. [DOI] [PubMed] [Google Scholar]
- 6. https://Clinicaltrials.gov website. Available at: https://clinicaltrials.gov/ct2/results?cond=&term=Revance&cntry1=&state1=&recrs=#wrapper. Accessed July 20, 2017.
- 7. Glogau RG, Waugh JM. Preclinical transcutaneous flux experiments using a macromolecule transport system (MTS) peptide for delivery of botulinum toxin type A. Poster presented at the 66th annual meeting of the American Academy of Dermatology, San Antonio, Texas, February 1–5, 2008. Available at: http://www.revance.com/pdfs/Preclinical-transcutaneous-flux-experiment-type-a.pdf. Accessed August 14, 2017.
- 8. Stone HF, Zhu Z, Thach TQ, Ruegg CL. Characterization of diffusion and duration of action of a new botulinum toxin type A formulation. Toxicon 2011;58:159–167. [DOI] [PubMed] [Google Scholar]
- 9. Garcia‐Murray E, Velasco Villasenor ML, Acevedo B, et al. Safety and efficacy of RT002, an injectable botulinum toxin type A, for treating glabellar lines: results of a phase 1/2, open‐label, sequential dose‐escalation study. Dermatol Surg 2015;41(Suppl 1):S47–S55. [DOI] [PubMed] [Google Scholar]
- 10. Norwegian Health Informatics AS website . Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS). Available at: http://nevro.legehandboka.no/imagevault/publishedmedia/jipv0xim2ibb749yalju/6454-2-twstrs.pdf. Accessed July 19, 2017.
- 11. Cano SJ, Warner TT, Linacre JM, et al. Capturing the true burden of dystonia on patients: The Cervical Dystonia Impact Profile (CDIP‐58). Neurology 2004;63:1629–1633. [DOI] [PubMed] [Google Scholar]
- 12. Comella CL, Jankovic J, Truong DD, Hanschmann A, Grafe S; U.S. XEOMIN Cervical Dystonia Study Group . Efficacy and safety of incobotulinumtoxinA (NT 201, XEOMIN®, botulinum neurotoxin type A, without accessory proteins) in patients with cervical dystonia. J Neurol Sci 2011;308:103–109. [DOI] [PubMed] [Google Scholar]
- 13. Brashear A, Lew MF, Dykstra DD, et al. Safety and efficacy of NeuroBloc (botulinum toxin type B) in type A‐responsive cervical dystonia. Neurology 1999;53:1439–1446. [DOI] [PubMed] [Google Scholar]
- 14. Truong D, Duane DD, Jankovic J, et al. Efficacy and safety of botulinum type A toxin (Dysport) in cervical dystonia: results of the first US randomized, double‐blind, placebo‐controlled study. Mov Disord 2005;20:783–791. [DOI] [PubMed] [Google Scholar]
- 15. Poewe W, Burbaud P, Castelnovo G, et al. Efficacy and safety of abobotulinumtoxinA liquid formulation in cervical dystonia: A randomized‐controlled trial. Mov Disord 2016;31:1649–1657. [DOI] [PubMed] [Google Scholar]
- 16. Trosch RM, Espay AJ, Truong D, et al. Multicenter observational study of abobotulinumtoxinA neurotoxin in cervical dystonia: The ANCHOR‐CD registry. J Neurol Sci 2017;376:84–90. [DOI] [PubMed] [Google Scholar]
- 17. Marsh WA, Monroe DM, Brin MF, Gallagher CJ. Systematic review and meta‐analysis of the duration of clinical effect of onabotulinumtoxinA in cervical dystonia. BMC Neurol 2014;14:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Odergren T, Hjaltason H, Kaakkola S, et al. A double blind, randomised, parallel group study to investigate the dose equivalence of Dysport and Botox in the treatment of cervical dystonia. J Neurol Neurosurg Psychiatry 1998;64:6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fernandez HH, Pagan F, Danisi F, et al. on behalf of the XCiDaBLE Study Group . Prospective study evaluating incobotulinumtoxinA for cervical dystonia or blepharospasm: interim results from the first 145 subjects with cervical dystonia. Tremor Other Hyperkinet Mov (NY) 2013;3:tre‐03‐139‐2924‐1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Charles D, Brashear A, Hauser RA, et al. Efficacy, tolerability, and immunogenicity of onabotulinumtoxinA in a randomized, double‐blind, placebo‐controlled trial for cervical dystonia. Clin Neuropharmacol 2012;35:208–214. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information may be found in the online version of this article.
Table S1. Subjects showing an improvement rating on the Clinical Global Impression of Change
Table S2. Subjects showing an improvement rating on the Patient Global Impression of Change
Table S3. Change from baseline in Cervical Dystonia Impact Profile (CDIP‐58) total score, which assesses subject ratings of impact of cervical dystonia on health‐related quality of life on a score from 0 (no impact) to 100 (most impact)