

Abstract
Voltage‐gated Ca2+ (CaV) channels mediate Ca2+ entry into excitable cells to regulate a myriad of cellular events following membrane depolarization. We report the engineering of RGK GTPases, a class of genetically encoded CaV channel modulators, to enable photo‐tunable modulation of CaV channel activity in excitable mammalian cells. This optogenetic tool (designated optoRGK) tailored for CaV channels could find broad applications in interrogating a wide range of CaV‐mediated physiological processes.
Keywords: calcium signaling, ion channels, optogenetics, protein engineering, synthetic biology
Voltage‐gated Ca2+ (CaV) channels constitute the major route of Ca2+ entry into cells of the nervous and cardiovascular systems, as well as other electrically excitable cells.1 CaV channels respond to membrane depolarization to permit Ca2+ influx, thereby playing instrumental roles in Ca2+‐dependent physiological processes, including neurotransmitter/hormone release, gene expression, and muscle contraction.1b, 1c Deregulated CaV channels can give rise to pathophysiological conditions ranging from cardiovascular disorders to psychiatric conditions.2 Consequently, CaV channels are important targets for therapeutic intervention and physiological regulation.3 Currently widely used CaV channel blockers (e.g., dihydropyridines) have prominent drawbacks, including off‐target toxicity, lack of spatial control, and non‐reversibility.3b, 4 New interventional approaches to control CaV channels are thus needed.
Optogenetics, which involves the incorporation of synthetic photosensitive modules into cells of living tissues to control cellular activities with high spatiotemporal precision, provides an ideal solution to overcome the drawbacks associated with conventional CaV channel blockers.5 The Ras‐like GTPases Rad/Rem/Gem/Kir (RGK), which function as negative regulators of CaV channels, are considered to be the prime candidates for generating optogenetic tools to modulate CaV channels.6 Given that membrane anchoring is necessary for RGKs to exert their suppressive effects on CaV channels,6b, 7 we reasoned that CaV channels could be remotely modulated by harnessing light to control the translocation of RGK to the plasma membrane (PM). We therefore engineered a set of optogenetic constructs by using a light‐sensitive heterodimerization system to control the subcellular localization of engineered Rem (Scheme 1 and Figure S1 in the Supporting Information). We chose the optical dimerizer pair iLID (LOV2‐ssrA) and sspB because of their small size, fast photoresponsive kinetics, low background interaction, and minimized perturbation to endogenous signaling pathways.8
Scheme 1.
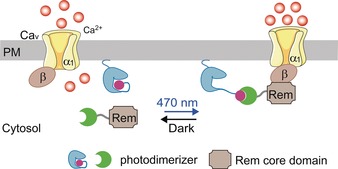
Design of optoRGK to photo‐tune CaV channel activity. Spatiotemporal control of the Rem1 core domain is achieved by utilizing the LOV2‐ssrA/sspB optical dimerizer pair. The light‐inducible cytosol‐to‐PM translocation of Rem enables inducible suppression of CaV channel activity. Green crescent=1st photodimerizer component (sspB), blue structure=membrane tethered LOV2, pink sphere=2nd photodimerizer component (ssrA), LOV2+ssrA=improved light‐induced dimer (iLID) protein.
To enable light‐inducible cytosol‐to‐PM translocation of engineered Rem, we set out to install sspB into different positions of Rem1‐266 tagged with the red fluorescent protein mCherry (mCherry‐Rem1‐266) via flexible linkers with varying lengths (Figures S1–3). In parallel, we tethered iLID tagged with the yellow fluorescent protein Venus (Venus‐iLID) to the PM with a C‐terminal PM‐targeting sequence (CAAX) derived from KRas4B8 or the PM‐tethering motif (Lyn11) from the tyrosine‐protein kinase Lyn.9 We then co‐transfected these two constructs into HeLa cells and examined the reversible recruitment of cytosolic Rem1‐266 to the PM (Figure 1 a, Figures S1–4). In the dark, mCherry‐Rem1‐266–sspB was evenly distributed in the cytosol (Figure 1 a). Upon blue light illumination, photoexcitation produced a covalent adduct between residue C450 of LOV2 and the cofactor FMN, thereby exposing the caged ssrA component of iLID to restore its interaction with sspB. Consequently, mCherry‐Rem1‐266–sspB was recruited toward the PM within seconds (t1/2, on=3.2±1.0 sec). Upon withdrawal of light, mCherry‐Rem1‐266–sspB dissociated from the PM‐resident ssrA and diffused back to the cytosol (t1/2, off=23.0±2.4 sec; Figure 1, Figure S3, and Movie S1). After screening over 20 constructs with different combinations of key elements (various Rem fragments, linkers, PM‐targeting motifs, and insertion sites), we identified mCherry‐Rem11‐266–sspB/iLID‐CAAX as an ideal candidate because it exhibited an optimal performance with high sensitivity and relatively fast kinetics (Figures S1–S3). To enable a near 1:1 co‐expression of the two components using a single construct within the same cells, we used a multicistronic expression system by utilizing the self‐cleaving 2A peptide (P2A)to generate fluorescent protein (FP)‐tagged (mCherry or CFP) Rem11‐266–sspB‐P2A‐iLID‐CAAX.10 We named the system optoRGK and used the FP‐tagged constructs for further characterization and applications in excitable cells.
Figure 1.
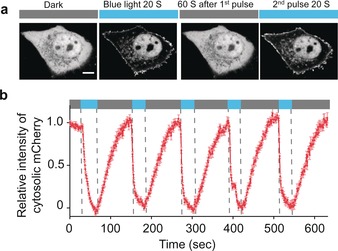
Visualization of the reversible recruitment of the Rem core domain (Rem1‐266, without the C‐terminus) to the PM in response to blue‐light illumination. a) Representative confocal images showing light‐inducible cytosol‐to‐PM translocation of mCherry‐Rem11‐266–sspB in HeLa cells co‐expressing Venus‐iLID‐CAAX. The images represent the same cell in the dark (black bar) or exposed to blue light at 470 nm (40 μW mm−2; blue bar). Scale bar: 5 μm. b) Quantification of cytosolic mCherry signals of optoRGK over five repeated light–dark cycles. n=34 cells from three independent experiments. Error bars denote the SEM.
To determine whether optoRGK could photo‐modulate CaV channels, we expressed optoRGK in HEK293 cells stably expressing the human CaV1.2 channel components (HEK‐CaV1.2)11 and evaluated membrane‐depolarization‐induced Ca2+ entry with a red genetically encoded Ca2+ sensor jRGCaMP1b12 (Figure 2 a,b) or the green Ca2+ dye Fluo‐4 (Figure S3). 50 mm potassium chloride (KCl) was employed to induce membrane depolarization. In the dark, addition of KCl elicited a pulse of CaV‐mediated Ca2+ influx in both control and optoRGK‐expressing cells (Figure 2, 1st cycle). Upon blue‐light illumination, cells overexpressing optoRGK showed a significant reduction in KCl‐induced Ca2+ entry compared to control cells (Figure 2 a, 2nd cycle). Furthermore, Ca2+ influx could be restored in the absence of blue light (Figure 2 a, 3rd cycle), thereby attesting to the reversibility of optoRGK in the regulation of CaV channels. In parallel, we performed electrophysiological studies to independently confirm optoRGK‐mediated, light‐switchable modulation of CaV channels in HEK293‐CaV1.2 cells. In the dark, cells expressing optoRGK showed robust whole‐cell currents with a typical CaV I‐V relationship and amplitudes similar to those of control cells (maximal peak current density≈21.2 pA/pF, n=12; Figure 2 c,d). However, upon receiving blue‐light stimulation, the amplitudes of depolarization‐induced whole‐cell currents were significantly diminished (maximal peak current density≈6.3 pA/pF, n=15; Figure 2 c,d). These results clearly established optoRGK as a genetically encoded light‐switchable channel modulator that allows optical inhibition of CaV channels in excitable cells.
Figure 2.
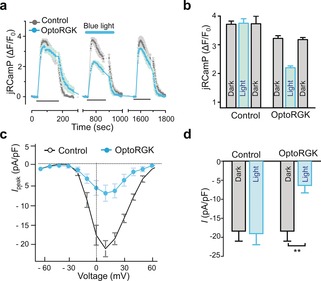
OptoRGK‐mediated photoswitchable inhibition of Ca2+ entry through CaV1.2 channels. a) Ca2+ influx in HEK‐CaV1.2 cells transiently expressing optoRGK and the red Ca2+ sensor jRCaMP1b with and without blue‐light stimulation. Cells transfected with the empty vector are used as control. Membrane‐depolarization‐induced Ca2+ entry was elicited by adding 50 mm KCl (black line below the curves; three repeated cycles) to transfected cells. Blue line represents light stimulation under 470 nm with a power density of 40 μW mm−2. b) Bar graphs showing the statistical results of mean Ca2+ entry for each cycle. c) The current–voltage relationships of CaV channels in HEK‐Cav1.2 cells transfected with optoRGK. Cells were either shielded from light or exposed to blue light prior to electrophysiological recording. d) Bar graphs showing the statistical results of peak whole‐cell currents induced by pulses of +10 mV depolarization (pA/pF) in HEK‐CaV1.2 cells before and after photo‐stimulation. All data were presented as mean±SEM. **P<0.01 (paired Student's t‐test).
We next moved on to test optoRGK in C2C12 cells, a mouse myoblast cell line13 with functional CaV channels.6b Again, we observed light‐dependent inhibition of Ca2+ influx in this excitable cell line (Figure S5). To further test optoRGK in more physiologically relevant systems, we introduced it into HL‐1 cardiomyocytes, a well‐characterized atrial myocyte culture line derived from the adult mouse that retains many of the differentiated properties of cardiac cells,14 including rhythmic oscillations of cytosolic Ca2+ (Movie S2). By using Fluo‐4 as the Ca2+ indicator, we first evaluated rhythmic Ca2+ oscillations in HL‐1 cells with and without overexpression of full length Rem or its truncated version Rem1‐266 (Figure S6, Movies S3 and S4). Both control cells and HL‐1 cells transfected with mCherry‐Rem1‐266–sspB (Figure S6b) exhibited rhythmic Ca2+ oscillations, while cardiomyocytes expressing the full‐length Rem failed to evoke Ca2+ transients (Figure S6a). These findings are consistent with the results obtained from C2C12 myoblast cells (Figure S5) and other types of excitable cells.15
Having validated the use of HL‐1 cardiomyocytes to test our tool, we next examined the rhythmic oscillations of cytosolic Ca2+ (Fluo‐4 signals as readout) in HL‐1 cells expressing optoRGK. Upon blue‐light illumination, mCherry‐Rem11‐266–sspB translocated from the cytosol to close to the PM within several seconds (Figure S7), accompanied by the attenuation of oscillatory Ca2+ signals (Figure 3 a, bottom, and Movie S5). By contrast, the control cells displayed regular Ca2+ oscillations under blue light. To further validate whether such action was reversible in HL‐1 cells, we used the red Ca2+ indicator Cal‐590 (excitation at 562 nm without pre‐activating optoRGK) rather than Fluo‐4 to monitor Ca2+ oscillations (Figure 3 b). HL‐1 cells expressing optoRGK showed regular Ca2+ spikes in the dark. However, upon blue‐light illumination, the rhythmic oscillations were substantially attenuated. Notably, regular Ca2+ oscillations were restored in the same HL‐1 cardiomyocyte after removal of the light source (Figure 3 b).
Figure 3.
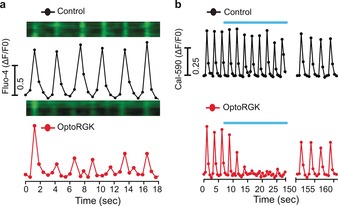
OptoRGK‐mediated light‐inducible inhibition of the rhythmic oscillations of cytosolic Ca2+ in cardiac cells. a) Ca2+ oscillations reported by Fluo‐4 in HL‐1 cells with (bottom) and without (top) expression of optoRGK. Kymographs of Fluo‐4 signals in a representative HL‐1 cell are shown above the traces. Excitation was set at 488 nm to record Fluo‐4 signals while simultaneously photoactivating optoRGK. b) Ca2+ oscillations in HL‐1 cells monitored by Cal‐590. Blue bar=blue light illumination at 470 nm (40 μW mm−2).
Taken together, compared with traditional small‐molecule CaV channel blockers that often lack reversibility, selectivity, and tissue‐specificity, engineered RGK proteins could serve as promising candidates to enable spatiotemporal control of CaV channels with a simple pulse of light. This study complements the recent development of engineered stromal interaction molecule 1 (STIM1) to photo‐regulate endogenous Ca2+ channels in mammalian cells (e.g., optoSTIM116 and Opto‐CRAC17). We anticipate that the optoRGK tool developed in the current study will find broad applications in interrogating a wide range of Ca2+‐dependent physiological processes in mammals.
Proof‐of‐concept experiments have already demonstrated the potential of using RGK to treat heart disease.18 To test potential in vivo applications, we plan to express optoRGK in the atrioventricular node of rodent models with atrial fibrillation disease,19 and examine whether photostimulation could suppress aberrant atrioventricular nodal conduction to intervene atrial fibrillation.
Given that RGK proteins primarily exert suppressive effects on high‐voltage‐activated Ca2+ (CaV1/CaV2) channels20 and that LOV2‐based photoswitches have relatively slow kinetics, the efficacy of optoRGK will likely depend on the distribution and endogenous levels of CaV channel subtypes in different cell types and tissues. In its current configuration, optoRGK is well suited to modulate cardiomyocytes because of the abundant expression of l‐type CaV1.1/CaV1.2 channels and the relatively long duration of cardiac action potentials. Its compatibility with neurons and other types of electrically excitable cells (e.g., pancreatic beta cells) remains to be tested.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Acknowledgements
This work was supported by grants from NIH (R21GM126532 and R01GM112003), CPRIT (RP170660 and RR140053), the National Natural Science foundation of China (NSFC‐31471279 and NSFC‐81728019), the Recruitment Program for Young Professionals of China, the Fundamental Research Funds for the Central Universities (2017STUD20), the Welch Foundation (BE‐1913) and the American Cancer Society (RSG‐16–215‐01‐TBE).
G. Ma, J. Liu, Y. Ke, X. Liu, M. Li, F. Wang, G. Han, Y. Huang, Y. Wang, Y. Zhou, Angew. Chem. Int. Ed. 2018, 57, 7019.
Contributor Information
Prof. Youjun Wang, Email: wyoujun@bnu.edu.cn
Prof. Yubin Zhou, Email: yzhou@ibt.tamhsc.edu.
References
- 1.
- 1a. Yang S. N., Berggren P. O., Am. J. Physiol.: Endocrinol. Metab. 2005, 288, E16–28; [DOI] [PubMed] [Google Scholar]
- 1b. Atlas D., Annu. Rev. Biochem. 2013, 82, 607–635; [DOI] [PubMed] [Google Scholar]
- 1c. Catterall W. A., Annu. Rev. Cell Dev. Biol. 2000, 16, 521–555. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Splawski I., Timothy K. W., Sharpe L. M., Decher N., Kumar P., Bloise R., Napolitano C., Schwartz P. J., Joseph R. M., Condouris K., Tager-Flusberg H., Priori S. G., Sanguinetti M. C., Keating M. T., Cell 2004, 119, 19–31; [DOI] [PubMed] [Google Scholar]
- 2b. Simms B. A., Zamponi G. W., Neuron 2014, 82, 24–45. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Zamponi G. W., Striessnig J., Koschak A., Dolphin A. C., Pharmacol. Rev. 2015, 67, 821–870; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Zamponi G. W., Nat. Rev. Drug Discovery 2016, 15, 19–34. [DOI] [PubMed] [Google Scholar]
- 4. Sunkel C. E., Fau de Casa-Juana M., Cillero F. J., Priego J. G., Ortega M. P., J. Med. Chem. 1988, 31, 1886–1890. [DOI] [PubMed] [Google Scholar]
- 5. Wang M., Yu Y., Shao J., Heng B. C., Ye H., Quant. Biol. 2017, 1–13.29881643 [Google Scholar]
- 6.
- 6a. Béguin P., Nagashima K., Gonoi T., Shibasaki T., Takahashi K., Kashima Y., Ozaki N., Geering K., Iwanaga T., Seino S., Nature 2001, 411, 701–706; [DOI] [PubMed] [Google Scholar]
- 6b. Finlin B. S., Crump S. M., Satin J., Andres D. A., Proc. Natl. Acad. Sci. USA 2003, 100, 14469–14474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Correll R. N., Pang C., Finlin B. S., Dailey A. M., Satin J., Andres D. A., J. Biol. Chem. 2007, 282, 28431–28440; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Yang T., Suhail Y., Dalton S., Kernan T., Colecraft H. M., Nat. Chem. Biol. 2007, 3, 795–804. [DOI] [PubMed] [Google Scholar]
- 8. Guntas G., Hallett R. A., Zimmerman S. P., Williams T., Yumerefendi H., Bear J. E., Kuhlman B., Proc. Natl. Acad. Sci. USA 2015, 112, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hammond G. R., Fischer M. J., Anderson K. E., Holdich J., Koteci A., Balla T., Irvine R. F., Science 2012, 337, 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim J. H., Lee S. R., Li L. H., Park H. J., Park J. H., Lee K. Y., Kim M. K., Shin B. A., Choi S. Y., PloS One 2011, 6, e18556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Balasubramanian B., Imredy J. P., Kim D., Penniman J., Lagrutta A., Salata J. J., J. Pharmacol. Toxicol. Methods 2009, 59, 62–72. [DOI] [PubMed] [Google Scholar]
- 12. Dana H., Mohar B., Sun Y., Narayan S., Gordus A., Hasseman J. P., Tsegaye G., Holt G. T., Hu A., Walpita D., Patel R., Macklin J. J., Bargmann C. I., Ahrens M. B., Schreiter E. R., Jayaraman V., Looger L. L., Svoboda K., Kim D. S., eLife 2016, 5, e12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burattini S., Ferri P., Battistelli M., Curci R., Luchetti F., Falcieri E., Eur. J. Histochem. 2004, 48, 223–233. [PubMed] [Google Scholar]
- 14.
- 14a. Claycomb W. C., N. A. Lanson, Jr. , Stallworth B. S., Egeland D. B., Delcarpio J. B., Bahinski A., N. J. Izzo, Jr. , Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b. Sartiani L., Bochet P., Cerbai E., Mugelli A., Fischmeister R., J. Physiol. 2002, 545, 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Chang D. D., Colecraft H. M., J. Physiol. 2015, 593, 5075–5090; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Magyar J., Kiper C. E., Sievert G., Cai W., Shi G. X., Crump S. M., Li L., Niederer S., Smith N., Andres D. A., Satin J., Channels 2012, 6, 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Ma G., Wen S., He L., Huang Y., Wang Y., Zhou Y., Cell calcium 2017, 64, 36–46; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Kyung T., Lee S., Kim J. E., Cho T., Park H., Jeong Y. M., Kim D., Shin A., Kim S., Baek J., Kim J., Kim N. Y., Woo D., Chae S., Kim C. H., Shin H. S., Han Y. M., Kim D., Heo W. D., Nat. Biotechnol. 2015, 33, 1092–1096. [DOI] [PubMed] [Google Scholar]
- 17. He L., Zhang Y., Ma G., Tan P., Li Z., Zang S., Wu X., Jing J., Fang S., Zhou L., Wang Y., Huang Y., Hogan P. G., Han G., Zhou Y., eLife 2015, 4, e10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Makarewich C. A., Correll R. N., Gao H., Zhang H., Yang B., Berretta R. M., Rizzo V., Molkentin J. D., Houser S. R., Circ. Res. 2012, 110, 669–674; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Murata M., Cingolani E., McDonald A. D., Donahue J. K., Marban E., Circ. Res. 2004, 95, 398–405. [DOI] [PubMed] [Google Scholar]
- 19. Nishida K., Michael G., Dobrev D., Nattel S., EP Europace 2010, 12, 160–172. [DOI] [PubMed] [Google Scholar]
- 20. Yang T., Colecraft H. M., Biochim. Biophys. Acta Biomembr. 2013, 1828, 1644–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary