

Abstract
Variants in the neuronal sodium channel gene SCN8A have been implicated in several neurological disorders. Early infantile epileptic encephalopathy type 13 results from de novo gain‐of‐function mutations that alter the biophysical properties of the channel. Complete loss‐of‐function variants of SCN8A have been identified in cases of isolated intellectual disability. We now report a novel heterozygous SCN8A variant, p.Pro1719Arg, in a small pedigree with five family members affected with autosomal dominant upper limb isolated myoclonus without seizures or cognitive impairment. Functional analysis of the p.Pro1719Arg variant in transfected neuron‐derived cells demonstrated greatly reduced Nav1.6 channel activity without altered gating properties. Hypomorphic alleles of Scn8a in the mouse are known to result in similar movement disorders. This study expands the phenotypic and functional spectrum of SCN8A variants to include inherited nonepileptic isolated myoclonus. SCN8A can be considered as a candidate gene for isolated movement disorders without seizures.
Keywords: movement disorder, myoclonus, Nav1.6, sodium channel
The SCN8A gene (MIM# 600702) encodes the neuronal voltage‐gated sodium channel Nav1.6, which is localized at the axon initial segment and functions in initiation and propagation of action potentials. Nav1.6 is also concentrated at nodes of Ranvier in myelinated neurons, including sciatic nerve, and is expressed at a high level in cerebellar Purkinje cells. During the past 5 years, SCN8A has been found to have a major role in sporadic epilepsy. De novo missense mutations of SCN8A have been identified in more than 200 individuals with early‐onset epileptic encephalopathy (EIEE13; MIM# 614558). Approximately 25% of affected individuals carry recurrent mutations, many of which occur at CpG dinucleotides in arginine codons (Wagnon & Meisler, 2015). Functional analysis has demonstrated that the most common mechanism is gain‐of‐function alterations in biophysical properties of Nav1.6 that result in neuronal hyperexcitability (Meisler et al., 2016; Veeramah et al., 2012). A small number of inherited variants have been associated with less severe conditions, including benign familial infantile seizures accompanied by paroxysmal dyskinesia (BFIS5, MIM# 617080) (Anand et al., 2016; Butler et al., 2017; Gardella et al., 2016; Han, Jang, Lee, Shin, & Park, 2017). Heterozygous loss‐of‐function mutations of SCN8A have been associated with isolated cognitive impairment in two individuals with de novo mutations (Wagnon et al., 2017) and one family with the protein truncation variant p.Pro1719Argfs*6 (MIM# 614306). In the mouse, homozygosity for partial loss‐of‐function alleles of Scn8a results in movement disorders including ataxia, tremor, and dystonia, whereas homozygosity for null alleles results in paralysis and juvenile lethality (Kearney et al., 2002; O'Brien & Meisler, 2013).
We now describe a missense variant of SCN8A in a family with nonepileptic essential myoclonus. The proband is a 35‐year‐old female with onset of isolated myoclonus at age 5 (Figure 1A, II‐2). She was diagnosed with upper limb action‐induced nonepileptic myoclonus, part of the myoclonus‐dystonia spectrum (MIM# 159900). The myoclonus is alcohol‐responsive in this individual. The proband has normal cognition and no other neurological abnormalities including dystonic posturing. Her affected nephew (Figure 1A, III‐2) is a 7‐year‐old male with onset of action‐induced myoclonus in the upper limbs at age 4, and evidence of subcortical origin from electrophysiological studies. EMG recordings in the upper limbs showed irregular, multifocal, myoclonic bursts, with duration ranging from 50 to 200 msec. Myoclonic jerk frequency was increased with maintenance of a posture or with action. EEG back‐averaging did not reveal a cortical correlate. His motor milestones were delayed, for example, independent walking at age three, but he does not have intellectual disability and does not exhibit dystonia or other neurological abnormalities. The clinical features of these two family members are summarized in Supplemental Table 1. Three additional family members were reported as affected by a similar movement disorder (Figure 1A, I‐2, II‐3, and III‐1), but were not examined directly. The pedigree is consistent with autosomal dominant inheritance.
Figure 1.
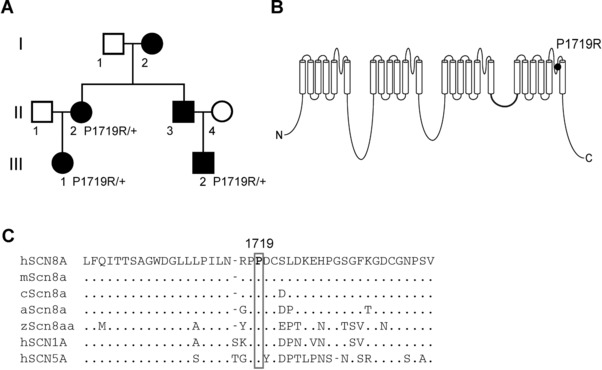
Inheritance of the SCN8A variant P1719R in a family with essential myoclonus. A: Pedigree demonstrating coinheritance of myoclonus and the SCN8A variant (c. 5156C > G, P1719R). Filled symbols, isolated myoclonus. B: Four‐domain structure of the voltage‐gated sodium channel alpha subunit. P1719R is located in the pore loop of domain IV. C: Evolutionary conservation of residue P1719R in multiple species. h, human; m, mouse; c, chicken; a, anole; z, zebrafish
In order to identify the genetic basis for the disorder, we performed whole‐exome sequencing (WES) in individuals II‐2 and III‐2. The study was approved by the University College London ethics committee (UCLH project 06/N076), and written informed consent was obtained from all participating subjects. DNA was extracted from peripheral lymphocytes following a standard protocol. Point mutations and copy‐number variants in SGCE, the most common genetic cause of autosomal dominant myoclonus‐dystonia (MIM# 159900), were previously excluded (Carecchio et al., 2013).
WES was carried out at the Institute of Neurology, University College London, using Illumina, (Santa Clara, CA, USA) Nextera Rapid Capture according to the manufacturer's recommendations. Indexed and pooled libraries were sequenced on Illumina's HiSeq3000. Bioinformatic analysis was performed as previously described (Mencacci et al., 2015).
Synonymous and noncoding variants were excluded from further examination. Variants were filtered to include only those that were shared by the two affected individuals and absent from public genomic databases (dbSNP 137, 1000 Genomes project, NHLBI Exome Variant Server, Complete Genomics 69 and Exome Aggregation Consortium [ExAC]). Finally, variants were required to have a CADD score greater than 20, indicative of greater potential for pathogenicity (https://cadd.gs.washington.edu/info) (Kircher et al., 2014).
After filtering, 11 variants remained (Supplemental Table 2). Among these, SCN8A is the only gene with previous association with movement disorders and a high intolerance score for missense and loss‐of‐function variants (z = 7.7 and pLI = 1.0) (exac.broadinstitute.org). Sanger sequencing analysis showed that the SCN8A mutation was also present in individual III‐1 (Figure 1A).
Affected individuals are heterozygous for the single nucleotide variant c.5156C > G in the SCN8A gene (RefSeq NM_014191.3), resulting in the amino acid substitution p.Pro1719Arg (P1719R). This variant is absent in ∼130,000 individuals listed in the gnomAD database (https://gnomad.broadinstitute.org). Proline residue 1719 is located in the pore loop of domain IV of the protein that confers sodium selectivity to the channel (Figure 1B). The pore loops are highly conserved through evolution and SCN8A residue 1719 is invariant in vertebrate orthologs and the human paralogs SCN1A and SCN5A, which diverged from SCN8A more than 400 million years ago (Figure 1C). Substitution of a charged arginine for the rigid proline residue is a chemically nonconservative change likely to alter secondary structure of the channel protein. This variant has been submitted as entry # 00163785 to the locus specific public database https://www.lovd.nl/SCN8A.
To determine experimentally whether P1719R is a deleterious variant, we carried out functional comparison of mutant and wildtype Nav1.6. The substitution P1719R was introduced into the tetrodotoxin (TTX)‐resistant derivative of the full‐length Nav1.6 cDNA clone, as previously described (Wagnon et al., 2016). Site‐directed mutagenesis was carried out with the QuikChange II XL kit (Agilent Technologies, Santa Clara, CA, USA). The 6‐kb open reading frame was re‐sequenced to confirm the absence of other mutations.
Channel activity was characterized in DRG‐neuron derived ND7/23 cells (Sigma–Aldrich, St. Louis, MO, USA). Cells were cultured and transfected as previously described (Wagnon et al., 2016). Forty‐eight hours after transfection with 5 μg of Nav1.6 alpha subunit cDNA and 0.5 μg of the fluorescent m‐Venus bioreporter, electrophysiological recordings of fluorescent cells were made. Recording was carried out in the presence of 500 nM TTX to block endogenous sodium currents. Currents were recorded using the whole‐cell configuration of the patch clamp recording technique as previously described (Wagnon et al., 2016).
Cells expressing the Nav1.6‐P1719R variant demonstrated inward sodium current that was significantly reduced compared with cells expressing wild‐type channels: −27.3 ± 5.3 pA/pF (n = 9) for mutant channel compared with −108 ± 21 pA/pF (n = 9) for wild‐type channel (P < 0.01) (Figure 2A and B). The voltage dependence of mutant channel activation and inactivation did not differ from wildtype (Figure 2C–F). The finding of partially reduced channel activity for P1719R is novel, since previously characterized deleterious variants resulted either in shifted voltage dependence of activation (Estacion et al., 2014), delayed channel inactivation (Wagnon et al., 2016), or complete loss of channel activity (Wagnon et al., 2017).
Figure 2.
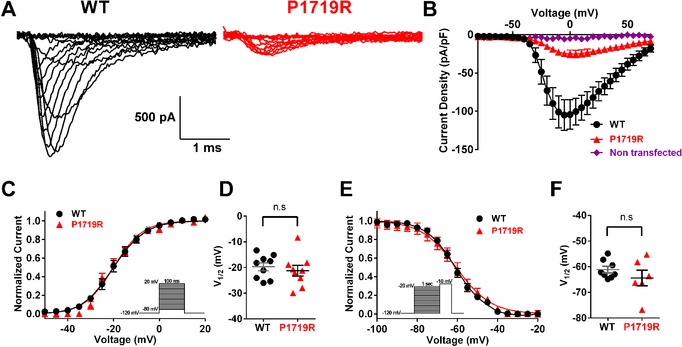
The SCN8A variant P1719R results in partial loss of Nav1.6 function. A: Representative traces of sodium currents recorded from ND7/23 cells that were transfected with Nav1.6 cDNA. Black, wild‐type (WT); red, P1719R. B: Averaged current–voltage (I–V) relation for WT (black, n = 9) and P1719R (red, n = 9) channels, and nontransfected cells (purple, n = 12). C: Voltage dependence of channel activation. Smooth lines correspond to the least squares fit when average data were fit to a single Boltzmann equation. D: Scatter plot of voltage at half‐maximal activation (V 1/2) for cells expressing WT and P1719R channels. E: Voltage dependence of steady‐state inactivation. Smooth lines correspond to the least squares fit when average data were fit to a single Boltzmann equation. F: Scatter plot of voltage at half‐maximal inactivation (V 1/2) for cells expressing WT and P1719R channels
Individuals with epileptic encephalopathy due to gain‐of‐function mutations of SCN8A often exhibit movement disorders and other neurological signs such as hypotonia, ataxia, dystonia, hyperreflexia, and choreoathetosis (Meisler et al., 2016). Severe hypotonia in these individuals is responsible for loss of intentional movement in 50% of cases (Meisler et al., 2016). SCN8A variants have not been previously reported in individuals with isolated movement disorders. This absence may reflect the lack of sequencing data in this population or the failure to recognize the relevance of missense variants. The identification of SCN8A‐P1719R cosegregating with isolated nonepileptic myoclonus demonstrates that mutation of SCN8A can result in isolated movement disorders in patients.
Variants of SCN8A with gain‐of‐function and elevated channel activity, either due to premature channel opening or delayed channel closing, result in epileptic encephalopathy (Meisler et al., 2016). At the other extreme, heterozygosity for complete loss‐of‐function alleles of SCN8A can result in isolated intellectual disability (Trudeau, Dalton, Day, Ranum, & Meisler, 2006; Wagnon et al., 2017). In rare cases, seizures have been seen in individuals with loss‐of‐function alleles of SCN8A lacking residual activity (Blanchard et al., 2015; Moller et al., 2016). Partial loss‐of‐function with residual activity, as demonstrated here for P1719R, has not previously been described for a pathogenic variant of human SCN8A. The dominant phenotype of this pore loop mutation could result from impaired trafficking of the coexpressed wild‐type Nav1.6, possible because of the recently described dimerization of the α subunit (Clatot et al, 2017). As another possibility, this mutation could impair glycosylation or trafficking of the channel protein (e.g., Jones et al., 2016). The data presented here demonstrate that heterozygosity for partial loss‐of‐function of SCN8A can be associated with movement disorders without seizures.
In the mouse, homozygosity for the hypomorphic Scn8amedJ allele, a splice site variant with 90% reduction in Nav1.6 protein, results in severe progressive dystonia (Sprunger, Escayg, Tallaksen‐Greene, Albin, & Meisler, 1999). A second hypomorphic allele, Scn8a9J, results in early onset tremor and adult onset dystonia (Jones et al., 2016). Complete loss of mouse Scn8a associated with homozygosity for a null allele results in hind limb paralysis and death (Burgess et al., 1995). Conditional inactivation of Scn8a restricted to cerebellar Purkinje and granule layer neurons also leads to pronounced ataxia (Levin et al., 2006), and dysfunction of cerebellar neurons could contribute to myoclonus in the family described here. Cerebellar dysfunction has been implicated in the pathogenesis of other forms of nonepileptic myoclonus such as SGCE‐related myoclonus‐dystonia (Carbon et al., 2013; Hubsch et al., 2011). Furthermore, we observed improvement of myoclonus with alcohol intake in the adult subject with SCN8A‐P1719R, an effect likely to occur through modulation of the activity of cerebellar neurons.
Three affected family members were demonstrated to carry the SCN8A variant. A fourth affected family member, II‐3, is an obligate carrier since his sister and son carry the variant. The fifth affected individual, III‐1, who was not available for testing, is likely to carry this variant since her clinical symptoms correspond to the other four affected family members. SCN8A‐P1719R thus appears to be a fully penetrant pathogenic variant responsible for autosomal dominant isolated myoclonus in this family, with no evidence of maternal imprinting as observed in myoclonus dystonia due to mutations of SGCE. In distinction from SGCF‐related myoclonus dystonia, the disease course in affected individuals from our kindred seems to be nonprogressive and no relevant psychiatric disturbances were observed. However other features (i.e., childhood‐onset, distribution of symptoms in the upper part of the body, response to alcohol, and a subcortical origin of myoclonus), partially overlap with the clinical phenotype associated with mutations of SGCE, which is generally dominated by myoclonus accompanied by mild dystonia. These findings extend the phenotypic spectrum associated with SCN8A to include a movement disorder without seizures or intellectual disability.
Supporting information
Supp. Table S1. Clinical features of two probands with the Scn8a † variant p.Pro1719Arg
Supporting information
ACKNOWLEDGMENT
The genetic study was undertaken at University College London Hospitals and University College London (UCL), who receive support from the Department of Health's National Institute for Health Research (NIHR) Biomedical Research Centers funding streams.
DISCLOSURE STATEMENT
The authors declare no conflict of interest.
Wagnon JL, Mencacci NE, Barker BS, et al. Partial loss‐of‐function of sodium channel SCN8A in familial isolated myoclonus. Human Mutation. 2018;39:965–969. https://doi.org/10.1002/humu.23547
Contract grant sponsor: National Institutes of Health (R01 NS34509, R01 NS103090); University of Virginia; EAN; Robert Bosch Foundation.
Communicated by Garry R. Cutting
REFERENCES
- Anand, G. , Collett‐White, F. , Orsini, A. , Thomas, S. , Jayapal, S. , Trump, N. , … Jayawant, S. (2016). Autosomal dominant SCN8A mutation with an unusually mild phenotype. European Journal of Paediatric Neurology, 20(5)761–765. https://doi.org/10.1016/j.ejpn.2016.04.015 [DOI] [PubMed] [Google Scholar]
- Blanchard, M. G. , Willemsen, M. H. , Walker, J. B. , Dib‐Hajj, S. D. , Waxman, S. G. , Jongmans, M. C. , … Kamsteeg, E. J. (2015). De novo gain‐of‐function and loss‐of‐function mutations of SCN8A in patients with intellectual disabilities and epilepsy. Journal of Medical Genetics, 52(5), 330–337. https://doi.org/10.1136/jmedgenet-2014-102813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess, D. L. , Kohrman, D. C. , Galt, J. , Plummer, N. W. , Jones, J. M. , Spear, B. , & Meisler, M. H. (1995). Mutation of a new sodium channel gene, Scn8a, in the mouse mutant ‘motor endplate disease’. Nature Genetics, 10(4), 461–465. https://doi.org/10.1038/ng0895-461 [DOI] [PubMed] [Google Scholar]
- Butler, K. M. , da Silva, C. , Shafir, Y. , Weisfeld‐Adams, J. D. , Alexander, J. J. , Hegde, M. , & Escayg, A. (2017). De novo and inherited SCN8A epilepsy mutations detected by gene panel analysis. Epilepsy Research, 129, 17–25. https://doi.org/10.1016/j.eplepsyres.2016.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon, M. , Raymond, D. , Ozelius, L. , Saunders‐Pullman, R. , Frucht, S. , Dhawan, V. , … Eidelberg, D. (2013). Metabolic changes in DYT11 myoclonus‐dystonia. Neurology, 80(4), 385–391. https://doi.org/10.1212/WNL.0b013e31827f0798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carecchio, M. , Magliozzi, M. , Copetti, M. , Ferraris, A. , Bernardini, L. , Bonetti, M. , … Valente, E. M. (2013). Defining the epsilon‐sarcoglycan (SGCE) gene phenotypic signature in myoclonus‐dystonia: A reappraisal of genetic testing criteria. Movement Disorders, 28(6), 787–794. https://doi.org/10.1002/mds.25506 [DOI] [PubMed] [Google Scholar]
- Clatot, J. , Hoshi, M. , Wan, X. , Liu, H. , Jain, A. , Shinlapawittayatorn, K. , … Deschenes, I. (2017). Voltage‐gated sodium channels assemble and gate as dimers. Nature Communications, 8(1), 2077 https://doi.org/10.1038/s41467-017-02262-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estacion, M. , O'Brien, J. E. , Conravey, A. , Hammer, M. F. , Waxman, S. G. , Dib‐Hajj, S. D. , & Meisler, M. H. (2014). A novel de novo mutation of SCN8A (Nav1.6) with enhanced channel activation in a child with epileptic encephalopathy. Neurobiology of Disease, 69, 117–123. https://doi.org/10.1016/j.nbd.2014.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardella, E. , Becker, F. , Moller, R. S. , Schubert, J. , Lemke, J. R. , Larsen, L. H. , … Weber, Y. G. (2016). Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Annals of Neurology, 79(3), 428–436. https://doi.org/10.1002/ana.24580 [DOI] [PubMed] [Google Scholar]
- Han, J. Y. , Jang, J. H. , Lee, I. G. , Shin, S. , & Park, J. (2017). A novel inherited mutation of SCN8A in a Korean family with benign familial infantile epilepsy using diagnostic exome sequencing. Annals of Clinical and Laboratory Science, 47(6), 747–753. [PubMed] [Google Scholar]
- Hubsch, C. , Vidailhet, M. , Rivaud‐Pechoux, S. , Pouget, P. , Brochard, V. , Degos, B. , … Roze, E. (2011). Impaired saccadic adaptation in DYT11 dystonia. Journal of Neurology, Neurosurgery, and Psychiatry, 82(10), 1103–1106. https://doi.org/10.1136/jnnp.2010.232793 [DOI] [PubMed] [Google Scholar]
- Jones, J. M. , Dionne, L. , Dell'Orco, J. , Parent, R. , Krueger, J. N. , Cheng, X. , … Meisler, M. H. (2016). Single amino acid deletion in transmembrane segment D4S6 of sodium channel Scn8a (Nav1.6) in a mouse mutant with a chronic movement disorder. Neurobiology of Disease, 89, 36–45. https://doi.org/10.1016/j.nbd.2016.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney, J. A. , Buchner, D. A. , De Haan, G. , Adamska, M. , Levin, S. I. , Furay, A. R. , … Meisler, M. H. (2002). Molecular and pathological effects of a modifier gene on deficiency of the sodium channel Scn8a (Na(v)1.6). Human Molecular Genetics, 11(22), 2765–2775. [DOI] [PubMed] [Google Scholar]
- Kircher, M. , Witten, D. M. , Jain, P. , O'Roak, B. J. , Cooper, G. M. , & Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46(3), 310–315. https://doi.org/10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin, S. I. , Khaliq, Z. M. , Aman, T. K. , Grieco, T. M. , Kearney, J. A. , Raman, I. M. , & Meisler, M. H. (2006). Impaired motor function in mice with cell‐specific knockout of sodium channel Scn8a (NaV1.6) in cerebellar purkinje neurons and granule cells. Journal of Neurophysiology, 96(2), 785–793. https://doi.org/10.1152/jn.01193.2005 [DOI] [PubMed] [Google Scholar]
- Meisler, M. H. , Helman, G. , Hammer, M. F. , Fureman, B. E. , Gaillard, W. D. , Goldin, A. L. , … Scheffer, I. E. (2016). SCN8A encephalopathy: Research progress and prospects. Epilepsia, 57(7), 1027–1035. https://doi.org/10.1111/epi.13422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mencacci, N. E. , Rubio‐Agusti, I. , Zdebik, A. , Asmus, F. , Ludtmann, M. H. , Ryten, M. , … Wood, N. W. (2015). A missense mutation in KCTD17 causes autosomal dominant myoclonus‐dystonia. American Journal of Human Genetics, 96(6), 938–947. https://doi.org/10.1016/j.ajhg.2015.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller, R. S. , Larsen, L. H. , Johannesen, K. M. , Talvik, I. , Talvik, T. , Vaher, U. , … Dahl, H. A. (2016). Gene panel testing in epileptic encephalopathies and familial epilepsies. Molecular Syndromology, 7(4), 210–219. https://doi.org/10.1159/000448369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien, J. E. , & Meisler, M. H. (2013). Sodium channel SCN8A (Nav1.6): Properties and de novo mutations in epileptic encephalopathy and intellectual disability. Frontiers in Genetics, 4, 213 https://doi.org/10.3389/fgene.2013.00213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprunger, L. K. , Escayg, A. , Tallaksen‐Greene, S. , Albin, R. L. , & Meisler, M. H. (1999). Dystonia associated with mutation of the neuronal sodium channel Scn8a and identification of the modifier locus Scnm1 on mouse chromosome 3. Human Molecular Genetics, 8(3), 471–479. [DOI] [PubMed] [Google Scholar]
- Trudeau, M. M. , Dalton, J. C. , Day, J. W. , Ranum, L. P. , & Meisler, M. H. (2006). Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation. Journal of Medical Genetics, 43(6), 527–530. https://doi.org/10.1136/jmg.2005.035667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeramah, K. R. , O'Brien, J. E. , Meisler, M. H. , Cheng, X. , Dib‐Hajj, S. D. , Waxman, S. G. , … Hammer, M. F. (2012). De novo pathogenic SCN8A mutation identified by whole‐genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. American Journal of Human Genetics, 90(3), 502–510. https://doi.org/10.1016/j.ajhg.2012.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon, J. L. , Barker, B. S. , Hounshell, J. A. , Haaxma, C. A. , Shealy, A. , Moss, T. , … Meisler, M. H. (2016). Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Annals of Clinical and Translational Neurology, 3(2), 114–123. https://doi.org/10.1002/acn3.276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon, J. L. , Barker, B. S. , Ottolini, M. , Park, Y. , Volkheimer, A. , Valdez, P. , … Meisler, M. H. (2017). Loss‐of‐function variants of SCN8A in intellectual disability without seizures. Neurology Genetics, 3(4), e170 https://doi.org/10.1212/NXG.0000000000000170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon, J. L. , & Meisler, M. H. (2015). Recurrent and non‐recurrent mutations of SCN8A in epileptic encephalopathy. Frontiers in Neurology, 6, 104 https://doi.org/10.3389/fneur.2015.00104 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supp. Table S1. Clinical features of two probands with the Scn8a † variant p.Pro1719Arg
Supporting information