

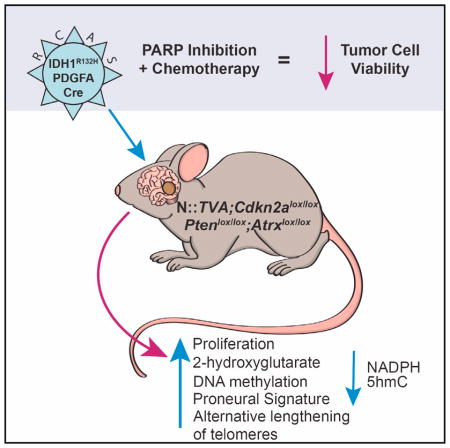
SUMMARY
Isocitrate dehydrogenase 1 (IDH1) is the most commonly mutated gene in grade II–III glioma and secondary glioblastoma (GBM). A causal role for IDH1R132H in gliomagenesis has been proposed, but functional validation in vivo has not been demonstrated. In this study, we assessed the role of IDH1R132H in glioma development in the context of clinically relevant cooperating genetic alterations in vitro and in vivo. Immortal astrocytes expressing IDH1R132H exhibited elevated (R)-2-hydroxyglutarate levels, reduced NADPH, increased proliferation, and anchorage-independent growth. Although not sufficient on its own, IDH1R132H cooperated with PDGFA and loss of Cdkn2a, Atrx, and Pten to promote glioma development in vivo. These tumors resembled pro-neural human mutant IDH1 GBM genetically, histologically, and functionally. Our findings support the hypothesis that IDH1R132H promotes glioma development. This model enhances our understanding of the biology of IDH1R132H-driven gliomas and facilitates testing of therapeutic strategies designed to combat this deadly disease.
In Brief
Philip et al. show that mutant IDH1 cooperates with PDGFA and loss of Cdkn2a, Atrx, and Pten to promote gliomagenesis in vivo in a mouse model of glioma. These tumors resemble proneural human mutant IDH1 glioblastoma and exhibit enhanced sensitivity to PARP inhibition in combination with chemotherapy.
INTRODUCTION
Gliomas are the most common primary CNS malignancy, but the molecular mechanisms responsible for their development and progression are not fully understood. In 2008, high-throughput sequencing of World Health Organization (WHO) grade IV glioblastoma multiforme (GBM) tumors identified a novel mutation at codon 132 (R132) in isocitrate dehydrogenase 1 (IDH1) in 12% of the samples analyzed (Parsons et al., 2008). Further studies have found this mutation, or an analogous mutation at codon 172 (R172) in isocitrate dehydrogenase 2 (IDH2), to be present in ~80% of WHO grade II–III gliomas and secondary GBM (Yan et al., 2009b). This mutation had never before been linked to cancer and the mechanism(s) by which it promotes tumor development is under intense investigation. Subsequent studies have identified IDH mutations in acute myelogenous leukemia (AML), cholangiocarcinoma, cartilaginous tumors, prostate cancer, papillary breast carcinoma, melanoma, acute lymphoblastic leukemia, angioimmunoblastic T cell lymphoma, and primary myelofibrosis indicating that these genes may be important players in multiple tumor types (reviewed in Cohen et al., 2013).
IDH1 and IDH2 form homodimers in the cytosol and mitochondria, respectively. Dimeric IDH contains two active sites, each composed of amino acid residues from both subunits. Thus, dimerization is essential for its enzymatic activity (Xu et al., 2004). IDH proteins catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG) in a two-step reaction that generates reduced nicotinamide adenine dinucleotide phosphate (NADPH) from NADP+. The IDH1 R132 and IDH2 R172 residues are located in the active sites of the enzyme and are critical for isocitrate binding (Parsons et al., 2008). Mutated IDH proteins utilize NADPH to reduce α-KG to R(−)-2-hydroxyglutarate (2-HG), which is supported by findings that 2-HG levels are elevated in mutant IDH1 gliomas (Dang et al., 2009). Thus, mutant IDH lowers the bioavailability of α-KG, while increased 2-HG competitively inhibits α-KG-dependent dioxygenases, including histone demethylases and the TET family of 5-methylcytosine (5mC) hydroxylases, which mediate DNA demethylation (Xu et al., 2011). As a result, gliomas harboring mutant IDH manifest a glioma-CpG island methylator phenotype (G-CIMP), which epigenetically alters the expression of numerous genes through DNA hypermethylation (Noushmehr et al., 2010).
Recent genomic analysis of diffuse, low-grade gliomas (LGGs) (WHO grades II or III) identified three subclasses consisting of wild-type IDH and mutant IDH with, or without, 1p/19q co-deletion (Brat et al., 2015). IDH mutation appears to be an early event in glioma development due to its presence in lower grade tumors. Nearly 90% of LGGs with an IDH mutation but no 1p/19q co-deletion also contained TP53 mutations and inactivating alterations of Alpha Thalassemia/Mental Retardation Syndrome X-Linked (ATRX). Amplification of loci encoding Platelet-Derived Growth Factor Receptor A (PDGFRA), Cyclin Dependent Kinase 4 (CDK4), and MYC was also reported (Brat et al., 2015). These alterations are similar to those observed in the proneural GBM subclass (Verhaak et al., 2010). We and others have further defined these subclasses based on copy number alterations. The IDH mutation subclass without 1p/19q co-deletion includes loss of 9p and 10q as well as gain of chromosomes 7 and 12q. Genes at these loci include Cyclin Dependent Kinase Inhibitor 2A (CDKN2A; 9p21.3), Phosphatase and Tensin Homolog Deleted on Chromosome Ten (PTEN; 10q23), Platelet-Derived Growth Factor Alpha (PDGFA; 7q11.23), CDK4 (12q13), and Mouse Double Minute 2 Homolog (MDM2; 12q15) (Cimino et al., 2017). Furthermore, we observed that progression of lower grade mutant IDH gliomas to GBM was associated with loss of chromosomal regions surrounding PTEN (Cohen et al., 2015).
Functional validation of these alterations in gliomagenesis has been hampered by difficulties establishing glioma mouse models (reviewed in Lenting et al., 2017). Expression of a conditional knockin allele of mutant IDH1 using Nestin-Cre was perinatal lethal in all mice, while expression of mutant IDH1 using GFAP-Cre was perinatal lethal in 92% of mice; no gliomas were observed in surviving mice (Sasaki et al., 2012). Restricted expression of IDH1R132H to the subventricular zone (SVZ) in adult mice using tamoxifen-inducible Nestin-CreER(T2) resulted in reduced α-KG, increased 2-HG and DNA methylation, enhanced proliferation of SVZ cells, and infiltration of neuronal and glial progenitor cells into neighboring regions. However, no gliomas were observed in these mice, which suggests that expression of mutant IDH1 alone is insufficient for glioma development (Bardella et al., 2016).
In this study, we delivered mutant IDH1 postnatally to nestin-expressing cells using the established RCAS/TVA glioma model. IDH1R132H cooperated with PDGFA and loss of Cdkn2a, Atrx, and Pten to transform immortal astrocytes in vitro and promote glioma development in vivo. Our findings functionally validate the role of IDH1R132H in driving glioma formation.
RESULTS
IDH1R132H Promotes Astrocyte Growth
To evaluate the role(s) of specific genes in glioma development, we and others have used an established mouse model system based on the RCAS/TVA retroviral vector system to induce malignant gliomas in vivo (Holland, 2000). Using this somatic-cell gene delivery method, we initially assessed the effect of IDH1R132H expression on the growth of primary astrocytes derived from Nestin::TVA (N::TVA);Cdkn2alox/lox mice. No differences in astrocyte proliferation were observed between cells infected with RCAS-Cre alone or in combination with either RCAS-IDH1 or RCAS-IDH1R132H, despite Cre-mediated loss of Cdkn2a and expression of wild-type (WT) or mutant IDH1 (data not shown). Genomic analysis of LGG subgroups shows that 86% of mutant IDH1 without 1p/19q loss have inactivation of ATRX. ATRX is a core component of the chromatin remodeling complex active in the maintenance of telomeres. Loss of ATRX function appears to promote alternative lengthening of telomeres (ALT), which occurs in the absence of telomerase activity and is a potential precursor to genomic instability (Hu et al., 2016). Therefore, we crossed conditional Atrx (Atrxlox) mice (Garrick et al., 2006) with N::TVA;Cdkn2alox/lox mice to generate N::TVA;Cdkn2alox/lox;Atrxlox/lox mice. Primary astrocytes isolated from these mice were infected with RCAS-Cre alone or in combination with either RCAS-IDH1 or RCAS-IDH1R132H, which are both hemagglutinin (HA) tagged to distinguish virally delivered IDH1 expression from endogenous IDH1 (Figure S1A, left panel). While no significant difference in proliferation was observed between uninfected N::TVA; Cdkn2alox/lox;Atrxlox/lox astrocytes compared to astrocytes infected with RCAS-Cre alone, a 1.5-fold increase in proliferation was observed when these astrocytes were infected with both RCAS-Cre and RCAS-IDH1R132H relative to RCAS-Cre alone (p = 0.004; Figure S1B).
Flavahan et al. previously demonstrated aberrant PDGFRA gene activation in cells harboring mutant IDH as a result of hyper-methylation at cohesin and CCCTC-binding factor (CTCF)-binding sites, which disrupted binding of CTCF, a methylation-sensitive insulator protein (Flavahan et al., 2016). In addition, IDH mutation clusters from human gliomas frequently include gain of chromosome 7 (Cimino et al., 2017), which contains platelet-derived growth factor alpha (PDGFA;7q11.23), and we have observed specific upregulation of Pdgfra mRNA in the presence of PDGFA expression in vivo (Shin et al., 2017). Therefore, we also evaluated the effect of PDGFRA signaling, via PDGFA expression, on the proliferation rate of N::TVA;Cdkn2alox/lox; Atrxlox/lox astrocytes following infection with RCAS-PDGFA and RCAS-Cre alone or in combination with either RCAS-IDH1 or RCAS-IDH1R132H (Figures S1A and S1B). Significant, but modest, differences in proliferation were observed between uninfected N::TVA;Cdkn2alox/lox;Atrxlox/lox astrocytes, cells infected with each virus alone, or cells infected with the combination of RCAS-PDGFA, RCAS-Cre, and RCAS-IDH1R132H (p < 0.05; Figure S1B). Because expression of PDGFA only modestly increased cellular proliferation in this context, we crossed N::TVA;Cdkn2alox/lox;Atrxlox/lox mice with conditional Pten (Ptenlox) mice (Zheng et al., 2008) to generate N::TVA; Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox mice and model loss of chromosome 10q, which we observed associated with progression of lower grade mutant IDH1 gliomas to GBM (Cohen et al., 2015). Primary astrocytes from these mice were infected with RCAS-PDGFA and RCAS-Cre alone or in combination with either RCAS-IDH1 or RCAS-IDH1R132H and expression was confirmed by western blot (Figure S1A, right panel). Significant differences in proliferation were observed between uninfected N::TVA; Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox astrocytes and astrocytes infected with each virus individually (p < 0.04), but a nearly 10-fold increase in proliferation was observed when these cells were infected with the combination of RCAS-PDGFA, RCAS-Cre, and RCAS-IDH1R132H as compared with uninfected astrocytes or astrocytes infected with RCAS-PDGFA, RCAS-Cre, and RCAS-IDH1 (p < 0.0001; Figure 1A). These data demonstrate that IDH1R132H promotes the growth of immortal Cdkn2a-deficient mouse astrocytes in vitro, and this effect is enhanced by expression of PDGFA in combination with loss of Atrx and Pten.
Figure 1. IDH1R132H Promotes Growth of Immortal Astrocytes, Consumes NADPH, and Increases 2-HG Levels.

(A) Cell proliferation was determined by MTT assay of astrocytes derived from N::TVA;Cdkn2alox/lox; Atrxlox/lox;Ptenlox/lox mice and infected with viruses containing Cre, IDH1R132H, IDH1, and/or PDGFA as indicated.
(B) Anchorage-independent growth of N::TVA; Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox astrocytes infected with viruses containing the genes indicated was analyzed by soft agar colony formation assay.
(C) NADPH production was measured from N::TVA;Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox astrocytes infected with viruses containing the genes indicated.
(D) 2-HG production from N::TVA; Cdkn2alox/lox; Atrxlox/lox;Ptenlox/lox astrocytes infected with viruses containing the genes indicated was detected by LC/MS.
(E) Anchorage-independent growth of N::TVA; Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox astrocytes infected with viruses containing the genes indicated and/or treated with 2-HG (checked bars) was analyzed by soft agar colony formation assay.
Data are expressed as the mean ± SEM with three replicates. Statistical significance is denoted: *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S1.
IDH1R132H Promotes Growth in Soft Agar
To evaluate the ability of IDH1R132H to promote anchorage-independent growth, colony formation of N::TVA;Cdkn2alox/lox; Atrxlox/lox;Ptenlox/lox astrocytes was measured in soft agar. We demonstrated previously that astrocytes lacking Cdkn2a are immortal in culture but are not transformed as evidenced by their inability to form colonies in the semi-solid soft agar matrix (Robinson et al., 2010). Similarly, very few colonies were detected in astrocytes lacking Cdkn2a, Atrx, and Pten (Figure 1B); however, IDH1R132H expression significantly increased anchorage-independent growth in this context (p = 0.008; Figure 1B). Furthermore, PDGFA combined with IDH1R132H expression significantly increased colony number and size, compared with PDGFA expression alone (p < 0.01; Figures 1B and S1C). Interestingly, anchorage-independent growth was significantly reduced when WT IDH1 was co-expressed with PDGFA in this context (p= 0.01; Figure1B). Expression of WT IDH1in immortalastrocytes likely increases cellular levels of α-KG, which has been shown to have anti-tumor activity both in vitro and in vivo (reviewed in Zdzisińska et al., 2017). These findings demonstrate that mutant IDH1 promotes anchorage-independent growth of immortal Cdkn2a-deficient mouse astrocytes in vitro in PDGFA-activated cells with loss of Atrx and Pten.
IDH1R132H Consumes NADPH and Produces 2-HG
The physiological role of IDH1 is to convert isocitrate to α-KG and produce NADPH (Xu et al., 2004). R132H substitution in IDH1 alters the structure of the enzyme’s active site and its enzymatic activity (Parsons et al., 2008). Thus, rather than generating NADPH, IDH1R132H consumes NADPH to generate 2-HG (Dang et al., 2009). To determine the ability of WT IDH1 and IDH1R132H to generate NADPH in N::TVA;Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox astrocytes, we compared the levels of NADPH produced by these cells following infection with RCAS-Cre and RCAS-IDH1 or RCAS-IDH1R132H in the presence or absence of RCAS-PDGFA. In the presence of isocitrate, cells expressing WT IDH1 generated significantly higher levels of NADPH compared with cells expressing IDH1R132H (p < 0.001; Figure 1C). To assess the neomorphic enzymatic activity of IDH1R132H, the level of 2-HG produced in each astrocyte culture expressing IDH1R132H alone, PDGFA alone, PDGFA and IDH1R132H, or PDGFA and WT IDH1 was quantitated using liquid chromatography/mass spectrophotometry (LC/MS). As expected, significantly higher levels of 2-HG were detected in cells expressing IDH1R132H compared with the parental cells regardless of the presence or absence of PDGFA (p < 0.03). Astrocytes with IDH1R132H expression produced more than 100-fold greater levels of 2-HG than the parental cells (Figure 1D). In contrast, there was no significant difference in 2-HG levels between cells expressing PDGFA alone or cells expressing PDGFA in combination with WT IDH1 when compared with the parental cells (Figure 1D). These data demonstrate that mouse astrocytes expressing IDH1R132H behave similarly to human glioma cells harboring IDH1R132H with respect to decreased NADPH production and increased levels of 2-HG.
Effects of IDH1R132H Are Mediated by 2-HG
To investigate whether the effects of IDH1R132H in this context are mediated by 2-HG, N::TVA;Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox astrocytes expressing Cre alone or Cre and PDGFA were suspended in soft agar and treated with vehicle (DMSO) or a cell membrane-permeable [trifluoromethyl benzyl (TFMB)-esterified] version of (R)-2HG (Losman et al., 2013). Treatment of these cells with TFMB-(R)-2HG significantly enhanced anchorage-independent growth in all cells analyzed relative to their vehicle-treated controls. In cells expressing PDGFA and Cre, addition of TFMB-(R)-2HG mimicked the anchorage-independent growth observed in cells expressing IDH1R132H. Interestingly, TFMB-(R)-2HG further enhanced colony formation in cells expressing IDH1R132H (Figure 1E). These data suggest that the effects of IDH1R132H on anchorage-independent growth are mediated by 2-HG.
IDH1R132H Promotes Glioma Formation In Vivo
Previous studies have demonstrated that IDH1R132H expression alone is insufficient to initiate glioma development in mice (Amankulor et al., 2017; Bardella et al., 2016), and our data support these findings. Informed by our in vitro studies, we evaluated the effect of IDH1R132H expression in cooperation with additional genetic alterations in vivo. While intracranial delivery of RCAS-Cre and RCAS-IDH1R132H in combination did not result in glioma development or a decrease in survival within the experimental time frame of 150 days in N::TVA;Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox mice (Table S1), the addition of RCAS-PDGFA led to glioma development in 88% (14/16) of injected mice in this context (Figure 2; Tables S1 and S2). Median survival was 43.5 ± 11 days in this cohort. In contrast, intracranial delivery of RCAS-Cre and RCAS-PDGFA with or without RCAS-IDH1 into N::TVA;Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox mice resulted in tumor development in only 20% (3/15) of injected mice for both conditions. The difference between either of these cohorts and the mutant IDH1 cohort is highly significant (p = 3.8 × 10−5; and 4.0 × 10−4, respectively; Figure 2A). A significant difference in tumor incidence and median survival was also observed between N::TVA;Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox mice and all other strains of mice evaluated (N::TVA;Atrxlox/lox, N::TVA;Atrxlox/lox;Ptenlox/lox, N::TVA;Cdkn2alox/lox;Atrxlox/lox, N::TVACdkn2alox/lox;Ptenlox/lox and N::TVA; Ptenox/lox) injected with RCAS-PDGFA, RCAS-Cre, and RCAS-IDH1R132H (p < 0.0001) (Figures 2B and S2; Tables S1 and S2). This demonstrates the significant cooperativity and necessity of these combined genetic alterations in promoting tumor development and demonstrates that IDH1R132H promotes glioma formation in this context.
Figure 2. IDH1R132H Cooperates with PDGFA and Loss of Cdkn2a, Atrx, and Pten to Promote Glioma Formation In Vivo.

(A) Kaplan-Meier tumor-free survival analysis of N::TVA;Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox mice infected with viruses containing PDGFA and Cre (n = 15, dotted line), PDGFA, Cre, and IDH1 (n = 15, dashed line), or PDGFA, Cre, and IDH1R132H (n = 16, solid line).
(B) Kaplan-Meier tumor-free survival analysis of the mouse strains indicated infected with viruses containing PDGFA, Cre, and IDH1R132H.
Statistical significance is denoted by ***p < 0.001. ns, not significant. See also Figure S2 and Tables S1 and S2.
Brain sections from all mice were examined following necropsy and were scored by a board-certified neuropathologist (J.A.S.) as “no tumor,” “low-grade tumor,” or “high-grade tumor” based on histological features apparent in H&E-stained sections. Low-grade tumors demonstrated single cell infiltration with nuclear atypia including hyperchromasia and pleomorphism, while high-grade tumors also exhibited mitotic figures, vascular proliferation, and/or areas of pseudopalisading necrosis (Figures 3A and S3). All of the tumors that developed in the N::TVA;Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox mice were high grade except one tumor in the RCAS-PDGFA and RCAS-Cre cohort. Cre-mediated recombination of Atrx, Cdkn2a, and Pten was assessed by immunohistochemistry (IHC) for ATRX, p19ARF, and PTEN, respectively (Figure S4). The majority of cells in the low-grade tumor in the RCAS-PDGFA and RCAS-Cre cohort continued to express ATRX (data not shown). High-grade tumors were positive for endomucin, an endothelial cell marker, indicating aberrant proliferative vasculature (Figure S4). Virally delivered RCAS-IDH1R132H was detected by IHC with an antibody that specifically detects IDH1R132H and both wild-type and mutant IDH1 were also detected using an antibody that recognizes the HA epitope tag (Figure 3A). All tumors highly expressed PDGFA (Figures 3A and S4) and the oligodendrocyte marker, Olig2, but IHC for the astrocyte marker, glial fibrillary acidic protein (GFAP), appeared to be limited to non-tumorigenic reactive astrocytes (Figure 3A). The high-grade tumors in each cohort were assessed for proliferation by IHC for Ki67 (Figure 3B). The mean percentage of Ki67 positive cells was quantified from three high-powered fields using ImageJ (Almeida et al., 2012). The average percentage of Ki67 positive cells was 21.2% ± 1.5% in the PDGFA and Cre tumors, 27.1% ± 2.3% in the PDGFA, Cre and IDH1 tumors, and 26.2% ± 4.6% in the PDGFA, Cre, and IDH1R132H tumors. No significant difference was observed between any of the pairwise comparisons.
Figure 3. Histological Examination of Gliomas from N::TVA;Cdkn2alox/lox;Atrxlox/lox; Ptenlox/lox-Injected Mice.

(A) Representative H&E and IHC images for indicated antibodies IDH1R132H, HA, Oligo2, GFAP, and PDGFA for gliomas from each cohort of mice injected with viruses containing PDGFA and Cre alone (right column) or in combination with IDH1R132H (left column) or WT IDH1 (middle column).
(B) Representative high-power H&E and Ki67 images of tumors generated in mice injected with viruses containing PDGFA and Cre alone (right column) or in combination with IDH1R132H (left column) or WT IDH1 (middle column) as indicated. Scale bar represents 100 μm.
See also Figures S3 and S4.
IDH1R132H Gliomas with ATRX Loss Display an ALT Phenotype
ALT is an adaptive mechanism utilized by tumor cells that lack telomerase activity and hence rely on a telomerase-independent mechanism for maintenance of their telomere length (Hu et al., 2016). The ALT phenotype is characterized by the co-localization of the telomeres with ALT-associated promyelocytic leukemia (PML)-like bodies (APBs), which are a subset of PML bodies that are present only in ALT positive and telomerase negative tumors (Draskovic et al., 2009; Jiang et al., 2009). To detect the presence of APBs and their co-existence with telomeres, we performed immuno-FISH with telomere probes and an anti-PML antibody. As expected, Atrx-deficient tumors exhibited co-localization of APBs with telomeres, whereas WT Atrx tumors did not (Figure 4).
Figure 4. IDH1R132H Gliomas with Atrx Loss Display an ALT Phenotype.
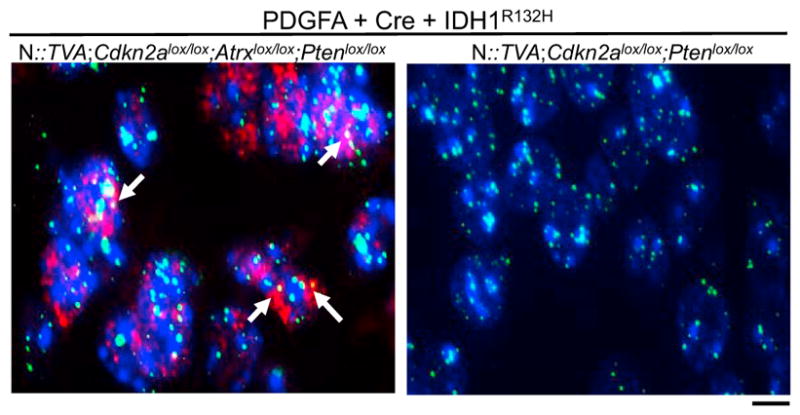
Representative images of immuno-FISH on brain sections from N::TVA; Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox and N::TVA;Cdkn2alox/lox;Ptenlox/lox gliomas from mice injected with viruses containing PDGFA, Cre, and IDH1R132H. White arrows indicate the co-localization of ALT-associated promyelocytic leukemia-like bodies (red) with telomeres (green). Background nuclear DNA is stained with Hoechst 33342 (blue). Scale bar represents 10 μm.
IDH1R132H-Driven Tumors Most Closely Resemble the Proneural Subtype
Human GBM has been classified into four different subtypes consisting of proneural, classical, mesenchymal, and neural (Verhaak et al., 2010). High-grade IDH1R132H-driven tumors in mice resemble human GBM histologically, but it is unclear which subtype, if any, they model. To determine whether the IDH1R132H-driven mouse gliomas resembled a particular subtype, we performed gene expression analysis on RNA extracted from formalin-fixed paraffin-embedded (FFPE) high-grade gliomas generated in N::TVA;Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox mice by delivery of RCAS-PDGFA, RCAS-Cre, and RCAS-IDH1R132H as well as high-grade gliomas generated with RCAS-PDGFA and RCAS-Cre with RCAS-IDH1 in the same mouse strain. Three tumors from each cohort were analyzed by RNA sequencing. We have previously shown that the transcript coverage and exonic mapping rate we observed are typical for FFPE tissue (Shin et al., 2017). We performed gene set enrichment analysis (GSEA) to compare the expression pattern of these tumors with the datasets from the four GBM subtypes as described previously (Lu et al., 2016). Based on GSEA, the signature gene sets associated with human proneural GBM subtype were significantly enriched in the gene expression profile of IDH1R132H-driven mouse tumors with a significant normalized enrichment score (NES) of 2.11. None of the signature gene sets from any of the other subtypes were significantly associated with the IDH1R132H-driven tumors (Figure 5).
Figure 5. GSEA Comparison between Mouse and Human GBM.

GSEA enrichment plots showing the comparison of gene expression profiles in high-grade tumors derived from N::TVA;Cdkn2alox/lox;Atrxlox/lox; Ptenlox/lox mice infected with viruses containing PDGFA, Cre, and IDH1R132H with the TCGA signature gene sets in neural, proneural, classical, and mesenchymal GBM as indicated. NES, normalized enrichment score; p value represents the statistical significance of the enrichment score; FDR, false discovery rate.
IDH1R132H Alters the Methylation Landscape in Mouse Gliomas
2-HG is a competitive inhibitor of α-KG-dependent dioxygenases including the TET family of 5mC hydroxylases. In human cells, DNA methylation primarily occurs as a 5mC modification of the cytosine bases in CpG dinucleotides. 5mC is an important epigenetic mark that is involved in the control of gene transcription. However, recent studies have identified 5-hydroxymethylcyto-sine (5hmC), which is formed by oxidation of 5mC by TET enzymes (Tahiliani et al., 2009). Xu et al. demonstrated that mutant IDH1 and IDH2 inhibit 5hmC production by TET (Xu et al., 2011). To assess this phenotype in the tumors that developed in our mouse model, we performed IHC for 5hmC on all tumors that developed in injected N::TVA;Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox mice. Consistent with decreased activity of TET in cells expressing IDH1R132H, we observed significantly decreased 5hmC staining in IDH1R132H tumors compared with tumors expressing WT IDH1 or PDGFA and Cre alone (Figures 6A and 6B). A modest, but significant, decrease in 5hmC staining was also observed in the WT IDH1 tumors compared to the PDGFA and Cre-only tumors. This may be explained by the finding that wild-type IDH1 can also catalyze the conversion of α-KG to 2-HG, albeit less efficiently than mutant IDH (Pietrak et al., 2011).
Figure 6. IDH1R132H Reduces 5hmC Levels and Alters the Methylation Landscape in Mouse Gliomas.

(A) Representative IHC images of 5hmC expression in gliomas generated in N::TVA;Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox mice injected with viruses containing PDGFA and Cre alone (right column) or in combination with IDH1R132H (left column) or WT IDH1 (middle column) as indicated. Scale bar represents 100 μm.
(B) Quantification of 5hmC IHC in all three tumor types.
(C) Heatmap comparing the relative difference of the regional fraction methylation of differentially methylated CpG regions between IDH1R132H and IDH1 samples.
Columns correspond to samples and rows correspond to regions organized into 6 groups by k-means algorithm. See also Figure S5.
Decreased 5hmC levels in IDH1R132H tumors suggested that the mouse tumors exhibit a hypermethylation phenotype similar to what is observed in the human disease. To further validate this finding, the DNA methylomes in the mouse gliomas were profiled using methyl-seq. As an initial analysis to demonstrate the similarity between samples, a clustered heatmap was generated and the sample to sample distance was measured using Euclidean distance (Figure S5A). Differentially methylated CpG regions in both IDH1R132H and IDH1 expressing cells were identified and clustered using K-means algorithm. Expression of mutant IDH1 caused a marked increase in hypermethylation at a large number of CpG regions compared to WT IDH1 (Figures 6 and S5B). Two robust differential DNA methylation clustering patterns, which correlated with IDH1R132H samples with high methylation levels and WT IDH1 with relatively lower methylation levels, were observed (Figure 6). Further analysis of the methylation targets between IDH1R132H and WT IDH1 tumors revealed 16,179 regions that were differentially methylated, including 3,433 differentially methylated CpG regions that intersect within 3 kb of a promoter. There were 13,570 regions that were hypermethylated and 2,609 that were hypomethylated in IDH1R132H tumors compared with WT IDH1 tumors.
IDH1R132H-Expressing Cells Demonstrate Enhanced Sensitivity to PARP Inhibitors
Previous studies revealed that IDH1 mutant cells are hypersensitive to poly-(adenosine 5′-diphosphateribose) polymerase (PARP) inhibitors (Lu et al., 2017; Sulkowski et al., 2017). This phenotype was further validated by others in glioma cell lines ectopically expressing IDH1R132H (Lu et al., 2017), in primary patient-derived IDH1R132H glioma cells, and in subcutaneous xenograft tumor models (Sulkowski et al., 2017). We assessed the cytotoxicity of the PARP inhibitor, olaparib, alone and in combination with temozolomide (TMZ), a DNA alkylating agent that is currently the standard of care for patients with IDH mutant gliomas, using primary tumor cells derived from IDH1R132H and WT IDH1 gliomas as well as astrocytes from N::TVA; Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox mice infected with viruses containing PDGFA, Cre, and IDH1R132H or IDH1. We observed enhanced sensitivity to olaparib in cells harboring IDH1R132H compared with WT IDH1. IC50 values from dose-response curves of olaparib were 26.6 and 85.3 μM for IDH1R132H astrocytes and tumor cells, respectively (Figure 7A), while the IC50 values of their WT counterparts were >100 μM (Figure 7B). Furthermore, TMZ significantly enhanced the cytotoxic effects of olaparib in IDH1R132H cells. Addition of TMZ at 10, 25, and 50 μM potentiated the cytotoxicity of olaparib selectively in IDH1R132H-expressing cells, reducing the olaparib IC50 from 26.6 to 23.4 μM, 9.51 μM, and 3.76 μM in astrocytes and from 85.3 to 32.4 μM, and 18.9 to 13.9 μM in tumor cells, respectively (Figure 7A). No significant potentiation was observed in cells expressing WT IDH1 (Figure 7B). In agreement with a previous study that reported enhanced vulnerability of glioma cells ectopically expressing IDHR132H to TMZ (Lu et al., 2017), we also observed sensitivity in astrocytes expressing IDH1R132H (Figure 7C, inset). However, the combination of TMZ and olaparib resulted in a significant reduction in cell viability compared with either agent alone for both astrocytes and tumor cells expressing IDH1R132H, suggesting that this is an effective therapeutic strategy for the treatment of IDH1R132H tumors (Figures 7C and 7D).
Figure 7. Astrocytes and Glioma Cells Harboring IDH1R132H Demonstrate Enhanced Sensitivity to Inhibition of PARP-Mediated DNA Repair and Potentiation by Alkylating Chemotherapy.

(A) Dose-response curves for N::TVA;Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox mouse astrocytes (left panel) and tumor cells (right panel) expressing PDGFA, Cre, and IDH1R132H treated with olaparib and TMZ.
(B) Dose-response curves for N::TVA;Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox mouse astrocytes and tumor cells expressing PDGFA, Cre, and WT IDH1 treated with olaparib and TMZ.
(C) Quantification of cell viability in mouse astrocytes expressing wild-type or mutant IDH1 treated with olaparib alone (25 μM), TMZ alone (10 μM, 50 μM; inset), and olaparib in combination with TMZ.
(D) Quantification of cell viability in wild-type and mutant IDH1 glioma-derived cells treated with olaparib alone (25 μM), TMZ alone (10 μM, 50 μM; inset), and in combination with TMZ.
Data are expressed as the mean ± SEM with three replicates. Significance is denoted: *p < 0.05, **p < 0.01, ***p < 0.001. lengthening of telomeres-associated PML bodies by p53/p21 requires HP1 proteins. J. Cell Biol. 185, 797–810.
DISCUSSION
In this study, we provide in vivo evidence that IDH1R132H promotes gliomagenesis. We observed that IDH1R132H cooperates with PDGFA and loss of the tumor suppressors Cdkn2a, Atrx, and Pten to induce tumor development but was not sufficient to promote tumor development on its own. Our findings are consistent with a previous study in which specific expression of IDH1R132H in nestin-expressing cells within the SVZ in adult mice did not produce tumors but led to increased 2-HG, enhanced proliferation, and infiltration of neuronal and glial progenitor cells into neighboring regions (Bardella et al., 2016). Modrek et al. reported similar findings using a human neural stem cell model in the context of mutant IDH1 with loss of TP53 and ATRX (Modrek et al., 2017). Likewise, Amankulor et al. (2017) did not observe glioma development using the RCAS/TVA glioma model when mutant IDH1 was expressed alone or in combination with TP53 loss and/or Cdkn2a loss. Gliomas were induced when combined with PDGFA, but there was no significant difference in survival between mice harboring tumors expressing either wild-type or mutant IDH1 in the context of homozygous Cdkn2a loss. Similarly, we did not observe a significant difference in survival between mice harboring tumors expressing either PDGFA alone or in combination with wild-type or mutant IDH1 in the context of homozygous Cdkn2a loss or in combination with Pten loss. Our data suggest that loss of all three tumor suppressors Cdkn2a, Atrx, and Pten in combination is necessary for IDH1R132H and PDGFA to significantly enhance tumor penetrance and decrease tumor latency (Figures 2 and S2).
Loss of Cdkn2a eliminates expression of INK4A and alternative reading frame (ARF), which function to regulate the retinoblastoma (RB) and TP53 pathways, respectively. In the absence of these cell-cycle checkpoints, cells are able to bypass senescence until their telomeres become critically short and they enter crisis. Loss of Atrx enables these cells to use recombination as a mechanism to lengthen their telomeres and escape crisis in the absence of telomerase activation. However, it was recently reported that dysregulation of both the TP53 and RB pathways as well as ATRX loss was not sufficient to drive this process in human astrocytes. Interestingly, combined loss of ATRX and expression of mutant IDH1 was required in T53/RB-deficient human astrocytes to produce the ALT phenotype, bypass telomere crisis, and escape cell death (Mukherjee et al., 2018). Nonetheless, this combination was still unable to promote tumor development in vivo within the experimental time frame of 150 days (Table S1), which suggests that further cooperating events are required. The PI3K/AKT pathway is one of the most commonly altered signaling pathways in cancer. It is activated by a number of mechanisms including but not limited to signaling from receptor tyrosine kinases (RTK) and loss of the tumor suppressor PTEN. Activation of the PI3K/AKT signaling pathway not only promotes cell survival, progression through the cell cycle, and cell migration but also has profound effects on cellular metabolism including up-regulation of glutamine transporters (Boehmer et al., 2003). The ability of mutant IDH1 to produce 2-HG is limited by the availability of its substrate α-KG. To generate 2-HG from α-KG, mutant IDH1 relies on wild-type IDH1 to convert isocitrate to α-KG. This may explain why the wild-type allele is retained and mutant IDH1 is almost always heterozygous. 2-HG has also been shown to be derived from glutamine (Dang et al., 2009). Glutamine enters the cell via transporters and is converted to glutamate by mitochondrial glutaminase. Glutamate can then be converted to α-KG by either glutamate dehydrogenase or by amino-transferases. Activation of PI3K/AKT signaling by PDGFA and loss of PTEN likely leads to increased glutamine uptake, which further cooperates with mutant IDH1 to promote tumorigenesis by providing a source of α-KG for further production of 2-HG. We are currently evaluating this hypothesis.
The mouse tumors resemble high-grade human mutant IDH1 gliomas genetically, histologically, and functionally. The majority of human IDH1 mutant gliomas contain inactivating alterations of ATRX. Loss of 9p (CDKN2A; 9p21.3) and 10q (PTEN; 10q23) as well as gain of chromosomes 7 (PDGFA; 7q11.23) and 12q (CDK4; 12q13 and MDM2; 12q15) have also been observed (Cimino et al., 2017). The majority of mutant IDH1 gliomas also contain TP53 mutations (94%) (Brat et al., 2015), which was not directly evaluated in this study but is currently under investigation. Deletion of the Cdkn2a locus results in loss of the tumor suppressor ARF, which normally regulates TP53 function via MDM2, and loss of the tumor suppressor p16, which normally regulates Rb function via CDK4/6. Therefore, disruption of both the TP53 and Rb pathways was evaluated through deletion of Cdkn2a.
Histologically, the mutant IDH1 mouse tumors were highly proliferative, contained abnormal vasculature, and exhibited areas of pseudopalisading necrosis (Figures 3, S3, and S4). Functionally, mouse astrocytes containing IDH1R132H produced over 100-fold greater levels of 2-HG than the parental cells or cells containing WT IDH1 (Figure 1D). This increase is similar to the fold changes observed in patient tumor samples relative to normal brain tissue (Dang et al., 2009). We also observed the presence of APBs and their co-existence with telomeres in Atrx-deficient tumors, which suggests that the mechanism by which these tumor cells bypass crisis as a result of shortened telomeres is through ALT (Figure 4). We and others have previously shown that mouse gliomas driven by PDGFA in the context of Cdkn2a and Pten loss resemble the proneural subtype of human GBM (Ozawa et al., 2014; Shin et al., 2017). Comparison of the IDH1R132H tumors to the human subtype signatures demonstrated that they positively correlated with the pronerual signature (Figure 5), which is characterized by IDH mutation and alterations in PDGFRA signaling in the human disease (Verhaak et al., 2010). Bardella et al. (2016) performed a similar GSEA of conditional knockin mice induced to express IDH1R132H in SVZ cells at 5 weeks of age and also observed a significant association with the proneural subtype.
The TET family of 5mC hydroxylases, which mediate DNA demethylation (Xu et al., 2011), are inhibited by high 2-HG levels. As a result, gliomas harboring mutant IDH manifest a G-CIMP, which epigenetically alters the expression of numerous genes through DNA hypermethylation (Noushmehr et al., 2010). We observed significantly decreased 5hmC staining in IDH1R132H tumors compared with tumors expressing WT IDH1, which is consistent with decreased activity of TET in cells expressing IDH1R132H (Figures 6A and 6B). Global assessment of DNA methylation in the mouse tumors revealed a large number of differentially methylated CpG regions between IDH1R132H tumors and WT IDH1 tumors (Figures 6C and S5). These observations suggest that IDH1R132H induces a global hypermethylation phenotype in the mouse gliomas similar to the human disease.
In a prospective analysis, grade II–IV glioma patients with mutated IDH1 or IDH2 had significantly longer overall survival than patients without IDH mutation (Sanson et al., 2009). This finding has been confirmed independently by others (Weller et al., 2009; Yan et al., 2009b). Interestingly, the presence of an IDH mutation was found to be an independent marker for better prognosis (Sanson et al., 2009). GBM patients with mutated IDH1 or IDH2 have an average survival of 31 versus 15 months for patients without the mutation (Sanson et al., 2009; Yan et al., 2009a). Patients with anaplastic astrocytomas containing IDH mutations also had a statistically significant increase in average overall survival when compared with patients without IDH mutations (Yan et al., 2009b). These clinical findings suggested that tumors harboring mutations in IDH are more sensitive to conventional chemotherapy and radio-therapy, and this was confirmed by two independent clinical studies, but the mechanistic basis for these observation was unclear (Cairncross et al., 2014; Tran et al., 2014). Sulkowski et al. (2017) recently shed light on this mechanism by demonstrating that 2-HG produced by mutant IDH induces a defect in homologous recombination (HR), via inhibition of histone lysine demethylases, and reduced ability to repair DNA double-strand breaks. Cancers with defects in DNA repair are often highly sensitive to PARP inhibitors. Therapeutics developed based on this approach have been effective in treating hereditary breast and ovarian cancers, which have defects in BRCA1 and BRCA2, and the Food and Drug Administration (FDA) has approved two PARP inhibitors, rucaparib and olaparib, to treat certain BRCA mutant ovarian cancers. Sulkowski et al. further demonstrated that mutant IDH tumor cells are sensitive to olaparib. We confirmed these findings and demonstrated potentiation of olaparib with the alkylating agent TMZ in both astrocytes and tumor-derived cells lines (Figure 7).
The discovery of mutations in IDH has cast a new light on the molecular landscape in glioma and is changing the paradigm for how the disease is defined and treated. Several small molecule inhibitors targeting mutant IDH have been generated and are in various stages of development (Dang et al., 2016). A selective inhibitor of mutant IDH1 inhibited the production of 2-HG and the growth of cells expressing IDH1R132H, but this was independent of its epigenetic effects (Rohle et al., 2013). Demonstration of in vivo efficacy of these and other therapies in a relevant glioma model would further support translation to the clinic. The model we described in this study will aid in furthering our understanding of the biology of mutant IDH1 gliomas and is ideally suited for assessing rational therapeutic strategies designed to combat this deadly disease.
EXPERIMENTAL PROCEDURES
Further details and an outline of resources used in this work can be found in Supplemental Experimental Procedures.
Statistical Methods
Survival data analysis was performed using a log-rank test of the Kaplan-Meier estimate of survival. Cell proliferation and NADPH assay p values were determined by two-way ANOVA (*p < 0. 05, **p < 0. 01, ***p < 0. 001). To compare means, two-tailed Student’s t test was used. Ki67-stained cells were quantitated using ImageJ software as described (Almeida et al., 2012). p values below 0.05 were considered significant.
Mice
N::TVA;Cdkn2alox/lox;Atrxlox/lox;Ptenlox/lox mice were produced by crossing pre-existing strains. All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Utah. Both male and female newborn through adult mice were used in this study.
Supplementary Material
Highlights.
IDH1R132H and PDGFA cooperate with loss of Cdkn2a, Atrx, and Pten in gliomagenesis
2-HG mediates the oncogenic effects of IDH1R132H
IDH1R132H-driven tumors mimic the human disease and resemble the proneural subtype
IDH1R132H tumor cells have enhanced sensitivity to PARP inhibitors
Acknowledgments
We thank the members of the Koh, VanBrocklin, and Holmen labs as well as E. Holland, R. DePinho, and M. Bosenberg for providing mouse strains, reagents, and advice. We thank the Huntsman Cancer Institute (HCI) Vivarium staff for assistance with mouse husbandry. We thank Tim Parnell for bioinformatics expertise and Rowan Arave for assistance with the graphical abstract. We acknowledge the use of the Mass Spectrometry Core, the DNA Synthesis Core, the DNA Sequencing Core, the Small Animal Imaging Core, and the Drug Discovery Core at the University of Utah. We also acknowledge use of the HCI Shared Resources for High-Throughput Genomics and Bioinformatics analysis and the Biorepository Molecular Pathology (BMP) Research Histology Section supported by P30CA042014 awarded to HCI from the National Cancer Institute (NCI). This work was supported by the National Institute of Neurological Disorders and Stroke (R01NS075155) and NCI (F30CA203096).
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession number for RNA sequencing (RNA-seq) and methylation array data reported in this paper is GEO: GSE107616.
Supplemental Information includes Supplemental Experimental Procedures, five figures, and two tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.03.133.
AUTHOR CONTRIBUTIONS
B.P., D.X.Y., M.R.S., C.H.S., J.P.R., H.C., G.L.R., M.W.V., and S.L.H. contributed to the experimental design. B.P. performed the majority of the experiments. S.N.A. was responsible for breeding and genotyping. A.E.W. and S.R.T. performed immunohistochemistry. J.A.S. provided pathological analysis. R.J.G. created the Atrx-floxed mice. R.E.L. prepared the TFMB-(R)-2HG. B.P., D.X.Y., M.R.S., and S.L.H. prepared the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Almeida JS, Iriabho EE, Gorrepati VL, Wilkinson SR, Grüneberg A, Robbins DE, Hackney JR. ImageJS: Personalized, participated, pervasive, and reproducible image bioinformatics in the web browser. J Pathol Inform. 2012;3:25. doi: 10.4103/2153-3539.98813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amankulor NM, Kim Y, Arora S, Kargl J, Szulzewsky F, Hanke M, Margineantu DH, Rao A, Bolouri H, Delrow J, et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017;31:774–786. doi: 10.1101/gad.294991.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardella C, Al-Dalahmah O, Krell D, Brazauskas P, Al-Qahtani K, Tomkova M, Adam J, Serres S, Lockstone H, Freeman-Mills L, et al. Expression of Idh1R132Hin the murine subventricular zone stem cell niche recapitulates features of early gliomagenesis. Cancer Cell. 2016;30:578–594. doi: 10.1016/j.ccell.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehmer C, Okur F, Setiawan I, Bröer S, Lang F. Properties and regulation of glutamine transporter SN1 by protein kinases SGK and PKB. Biochem Biophys Res Commun. 2003;306:156–162. doi: 10.1016/s0006-291x(03)00921-5. [DOI] [PubMed] [Google Scholar]
- Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, Morozova O, et al. Cancer Genome Atlas Research Network. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372:2481–2498. doi: 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairncross JG, Wang M, Jenkins RB, Shaw EG, Giannini C, Brachman DG, Buckner JC, Fink KL, Souhami L, Laperriere NJ, et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J Clin Oncol. 2014;32:783–790. doi: 10.1200/JCO.2013.49.3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimino PJ, Zager M, McFerrin L, Wirsching HG, Bolouri H, Hentschel B, von Deimling A, Jones D, Reifenberger G, Weller M, Holland EC. Multidimensional scaling of diffuse gliomas: Application to the 2016 World Health Organization classification system with prognostically relevant molecular subtype discovery. Acta Neuropathol Commun. 2017;5:39. doi: 10.1186/s40478-017-0443-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AL, Holmen SL, Colman H. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep. 2013;13:345. doi: 10.1007/s11910-013-0345-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A, Sato M, Aldape K, Mason CC, Alfaro-Munoz K, Heathcock L, South ST, Abegglen LM, Schiffman JD, Colman H. DNA copy number analysis of Grade II–III and Grade IV gliomas reveals differences in molecular ontogeny including chromothripsis associated with IDH mutation status. Acta Neuropathol Commun. 2015;3:34. doi: 10.1186/s40478-015-0213-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, Yen K, Attar EC. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann Oncol. 2016;27:599–608. doi: 10.1093/annonc/mdw013. [DOI] [PubMed] [Google Scholar]
- Draskovic I, Arnoult N, Steiner V, Bacchetti S, Lomonte P, Londoño-Vallejo A. Probing PML body function in ALT cells reveals spatiotemporal requirements for telomere recombination. Proc Natl Acad Sci USA. 2009;106:15726–15731. doi: 10.1073/pnas.0907689106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suvà ML, Bernstein BE. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529:110–114. doi: 10.1038/nature16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrick D, Sharpe JA, Arkell R, Dobbie L, Smith AJ, Wood WG, Higgs DR, Gibbons RJ. Loss of Atrx affects trophoblast development and the pattern of X-inactivation in extraembryonic tissues. PLoS Genet. 2006;2:e58. doi: 10.1371/journal.pgen.0020058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland EC. A mouse model for glioma: Biology, pathology, and therapeutic opportunities. Toxicol Pathol. 2000;28:171–177. doi: 10.1177/019262330002800122. [DOI] [PubMed] [Google Scholar]
- Hu Y, Shi G, Zhang L, Li F, Jiang Y, Jiang S, Ma W, Zhao Y, Songyang Z, Huang J. Switch telomerase to ALT mechanism by inducing telomeric DNA damages and dysfunction of ATRX and DAXX. Sci Rep. 2016;6:32280. doi: 10.1038/srep32280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W-Q, Zhong Z-H, Nguyen A, Henson JD, Toouli CD, Braithwaite AW, Reddel RR. Induction of alternative. 2009 doi: 10.1083/jcb.200810084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenting K, Verhaak R, Ter Laan M, Wesseling P, Leenders W. Glioma: Experimental models and reality. Acta Neuropathol. 2017;133:263–282. doi: 10.1007/s00401-017-1671-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, Kaelin WG., Jr (R)-2-hy-droxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Chen Y, Zhao C, Wang H, He D, Xu L, Wang J, He X, Deng Y, Lu EE, et al. Olig2-dependent reciprocal shift in PDGF and EGF receptor signaling regulates tumor phenotype and mitotic growth in malignant glioma. Cancer Cell. 2016;29:669–683. doi: 10.1016/j.ccell.2016.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Kwintkiewicz J, Liu Y, Tech K, Frady LN, Su YT, Bautista W, Moon SI, MacDonald J, Ewend MG, et al. Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res. 2017;77:1709–1718. doi: 10.1158/0008-5472.CAN-16-2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrek AS, Golub D, Khan T, Bready D, Prado J, Bowman C, Deng J, Zhang G, Rocha PP, Raviram R, et al. Low-grade astrocytoma mutations in IDH1, P53, and ATRX cooperate to block differentiation of human neural stem cells via repression of SOX2. Cell Rep. 2017;21:1267–1280. doi: 10.1016/j.celrep.2017.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee J, Johannessen TA, Ohba S, Chow TT, Jones LE, Pandita A, Pieper RO. Mutant IDH1 co-operates with ATRX loss to drive the alternative lengthening of telomere (ALT) phenotype in glioma. Cancer Res. 2018 doi: 10.1158/0008-5472.CAN-17-2269. Published online March 15, 2018. https://doi.org/10.1158/0008-5472.CAN-17-2269. [DOI] [PMC free article] [PubMed]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. Cancer Genome Atlas Research Network. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T, Riester M, Cheng YK, Huse JT, Squatrito M, Helmy K, Charles N, Michor F, Holland EC. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014;26:288–300. doi: 10.1016/j.ccr.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrak B, Zhao H, Qi H, Quinn C, Gao E, Boyer JG, Concha N, Brown K, Duraiswami C, Wooster R, et al. A tale of two subunits: How the neomorphic R132H IDH1 mutation enhances production of αHG. Biochemistry. 2011;50:4804–4812. doi: 10.1021/bi200499m. [DOI] [PubMed] [Google Scholar]
- Robinson JP, VanBrocklin MW, Guilbeault AR, Signorelli DL, Brandner S, Holmen SL. Activated BRAF induces gliomas in mice when combined with Ink4a/Arf loss or Akt activation. Oncogene. 2010;29:335–344. doi: 10.1038/onc.2009.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F, El Hallani S, Boisselier B, Mokhtari K, Hoang-Xuan K, Delattre JY. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. 2009;27:4150–4154. doi: 10.1200/JCO.2009.21.9832. [DOI] [PubMed] [Google Scholar]
- Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio II, Cairns RA, McCracken S, Wakeham A, Haight J, et al. D-2-hydroxygluta-rate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012;26:2038–2049. doi: 10.1101/gad.198200.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin CH, Robinson JP, Sonnen JA, Welker AE, Yu DX, VanBrocklin MW, Holmen SL. HBEGF promotes gliomagenesis in the context of Ink4a/Arf and Pten loss. Oncogene. 2017;36:4610–4618. doi: 10.1038/onc.2017.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, Liu Y, Sundaram RK, Hegan DC, Fons NR, et al. 2-Hy-droxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med. 2017:9. doi: 10.1126/scitranslmed.aal2463. Published online February 1, 2017 https://doi.org/10.1126/scitranslmed.aal2463. [DOI] [PMC free article] [PubMed]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran AN, Lai A, Li S, Pope WB, Teixeira S, Harris RJ, Woodworth DC, Nghiemphu PL, Cloughesy TF, Ellingson BM. Increased sensitivity to radiochemotherapy in IDH1 mutant glioblastoma as demonstrated by serial quantitative MR volumetry. Neurooncol. 2014;16:414–420. doi: 10.1093/neuonc/not198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. Cancer Genome Atlas Research Network. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller M, Felsberg J, Hartmann C, Berger H, Steinbach JP, Schramm J, Westphal M, Schackert G, Simon M, Tonn JC, et al. Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: A prospective translational study of the German Glioma Network. J Clin Oncol. 2009;27:5743–5750. doi: 10.1200/JCO.2009.23.0805. [DOI] [PubMed] [Google Scholar]
- Xu X, Zhao J, Xu Z, Peng B, Huang Q, Arnold E, Ding J. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J Biol Chem. 2004;279:33946–33957. doi: 10.1074/jbc.M404298200. [DOI] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Bigner DD, Velculescu V, Parsons DW. Mutant metabolic enzymes are at the origin of gliomas. Cancer Res. 2009a;69:9157–9159. doi: 10.1158/0008-5472.CAN-09-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009b;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdzisińska B, Żurek A, Kandefer-Szerszeń M. Alpha-ketoglutarate as a molecule with pleiotropic activity: Well-known and novel possibilities of therapeutic use. Arch Immunol Ther Exp (Warsz) 2017;65:21–36. doi: 10.1007/s00005-016-0406-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, Perry SR, Tonon G, Chu GC, Ding Z, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455:1129–1133. doi: 10.1038/nature07443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.