

SUMMARY
Excessive consumption of sweets is a risk factor for metabolic syndrome. A major chemical feature of sweets is fructose. Despite strong ties between fructose and disease, the metabolic fate of fructose in mammals remains incompletely understood. Here we use isotope tracing and mass spectrometry to track the fate of glucose and fructose carbons in vivo, finding that dietary fructose is cleared by the small intestine. Clearance requires the fructose-phosphorylating enzyme ketohexokinase. Low doses of fructose are ~90% cleared by the intestine, with only trace fructose but extensive fructose-derived glucose, lactate, and glycerate found in the portal blood. High doses of fructose (≥1 g/kg) overwhelm intestinal fructose absorption and clearance, resulting in fructose reaching both the liver and colonic microbiota. Intestinal fructose clearance is augmented both by prior exposure to fructose and by feeding. We propose that the small intestine shields the liver from otherwise toxic fructose exposure.
In Brief
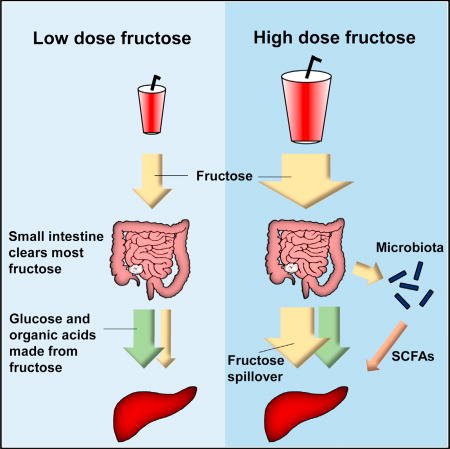
While it is commonly believed that the liver is the main site of fructose metabolism, Jang et al. show that it is actually the small intestine that clears most dietary fructose, and this is enhanced by feeding. High fructose doses spill over to the liver and to the colonic microbiota.
INTRODUCTION
Glucose and fructose are both hexose sugars with the same molecular formula (C6H12O6). Glucose is the predominant constituent of carbohydrates, which are not sweet. In contrast, monomeric and dimeric sugars (e.g., sucrose, which is glucose-fructose) taste sweet. Driven by the pleasurable experience of sweet taste, over the past two centuries, per capita consumption of dietary fructose has increased 100-fold. Fructose now accounts for ~10% of caloric intake in the United States (Bray et al., 2004; Marriott et al., 2009).
High fructose consumption is appreciated as a culprit in metabolic disease. Epidemiological studies indicate a strong correlation between high fructose intake and obesity, non-alcoholic fatty liver disease, type 2 diabetes, kidney dysfunction, and cardiovascular disease (Caliceti et al., 2017; Jegatheesan and De Bandt, 2017). However, the biological mechanisms underlying this link are still controversial (Niewoehner, 1986; Chong et al., 2007; Bravo et al., 2013; Johnson et al., 2013; Macdonald, 2016). Sweet taste suppresses satiety, and thereby enhances overall food consumption, which may lead to obesity and associated diseases (Bray et al., 2004; Tappy and Lê, 2010). In addition, differences in the metabolism of fructose versus glucose may contribute to metabolic disease.
The canonical pathway of glucose metabolism is glycolysis, which begins with phosphorylation of glucose on its 6-position, followed by reversible isomerization to make fructose 6-phosphate (F6P). In many microbes, fructose is phosphorylated on its 6-position and thereby follows nearly the same metabolic pathway as glucose. In mammals, however, fructose phosphorylation occurs on the 1-position, not 6-position, catalyzed by the enzyme ketohexokinase (Khk) (Heinz et al., 1968). The location of this initial phosphorylation is a pivotal difference, as fructose 1-phosphate (F1P) can be directly cleaved into three-carbon units, whereas F6P must be phosphorylated on its 1-position by phosphofructokinase, the most heavily regulated enzyme of glycolysis, before such cleavage. Thus, fructose bypasses the gating step of glycolysis. Moreover, its metabolism generates, in addition to the standard glycolytic intermediate dihydroxyacetone phosphate (DHAP), a non-phosphorylated three-carbon unit in the form of glyceraldehyde (Heinz et al., 1968).
While the biochemical pathway of fructose metabolism has been elucidated, how this pathway fits into the whole-body metabolism remains only partially understood. By bypassing phosphofructokinase, it is possible that fructose assimilation consumes ATP in a poorly controlled manner, leading to decreased energy charge, AMP accumulation, purine consumption by AMP deaminase, and eventual uric acid production (Johnson et al., 2013). Indeed, fructose feeding is a classical means of activating AMP deaminase flux in hepatocytes and at the whole-body level increases uric acid (Caliceti et al., 2017). The molecular links between these events and development of metabolic syndrome remain, however, unclear. Moreover, efforts to understand the fate of glyceraldehyde have been limited.
In terms of the roles of different organs, fructose is taken in from the lumen of the small intestine via Glut5 (Douard and Ferraris, 2008). Khk has two splicing isoforms, Khk-A and Khk-C, with Khk-C the major phosphorylating enzyme due to its lower Km (Diggle et al., 2010). Khk-C is expressed in intestine, liver, kidney, and pancreas (Ishimoto et al., 2012). Knockout mice for Khk show defective fructose metabolism with very high fructose levels in blood and urine, demonstrating that Khk is essential for fructose metabolism (Ishimoto et al., 2012; Patel et al., 2015a). Because liver expresses the highest level of Khk-C and high fructose intake causes liver-related pathologies (Ishimoto et al., 2012, 2013; Lanaspa et al., 2013; Zhang et al., 2017), it has been assumed that liver is the primary site of dietary fructose metabolism, with few studies examining the role of fructose in other organs (Miller et al., 1956; Herman et al., 1972; Froesch, 1972; Mirtschink et al., 2015).
Despite its name, the small intestine is a large organ, with a total mass similar to that of the liver. It requires a substantial amount of energy to drive ion pumps for nutrient transport and to support epithelial cell turnover. While the role of intestine in nutrient absorption and hormone secretion is well established, as is the importance of the large intestine (colon) microbiome, intestinal metabolism per se and its effect on systemic metabolism are poorly understood. Although liver and kidney are major gluconeogenic organs, intestine also significantly contributes to whole-body gluconeogenesis (Croset et al., 2001; Mithieux et al., 2004). Intestine-specific knockout mice for glucose 6-phosphatase (G6pc) indicate that intestine contributes ~25% of systemic gluconeogenesis both after prolonged fasting and in diabetes (Soty et al., 2014).
Here, we examine quantitatively the contribution of different organs to fructose metabolism, using isotope tracers and metabolomics. We find that small intestine plays a major role in dietary fructose metabolism, converting fructose to glucose and other circulating metabolites. In this manner, the small intestine shields the liver from fructose exposure. High doses of fructose overwhelm this shielding capacity. We hypothesize that the balance between fructose consumption and intestinal fructose clearance capacity determines liver exposure to dietary fructose and thereby fructose toxicity.
RESULTS
Dietary Fructose Is Rapidly Converted to Circulating Metabolites
To study systemic fructose metabolism quantitatively, we gavaged mice with a 1:1 mixture of fructose:glucose (the ratio naturally found in sucrose and similar to the 55:45 ratio found in high-fructose corn syrup). Either the fructose or the glucose was 13C labeled to enable us to follow its metabolism by liquid chromatography-mass spectrometry (LC-MS) (Figure 1A). Except for dose-response studies, all experiments were done at a dose of 0.5 g/kg of each hexose (1 g/kg total), which for a 60 kg person is equivalent on a g/kg basis to a total of 60 g sugar (~500 mL soda) and on a body surface area basis to 5 g sugar (~40 mL soda) (Sun and Empie, 2012; Kolderup and Svihus, 2015). After gavage, 13C-labeled glucose rose to ~2 mM circulating levels over 20 min and then gradually declined (Figure 1B). In contrast, oral administration of labeled fructose resulted in only low levels (peak ~0.1 mM) of circulating labeled fructose (Figure 1B).
Figure 1. Isotope Tracing Reveals Differential Formation of Circulating Metabolites from Oral Glucose versus Fructose.

(A) Experimental scheme. Mice received an oral gavage of 1:1 glucose:fructose (one unlabeled and the other U-13C labeled, 0.5 g/kg each) and metabolite labeling was measured by LC-MS. Glc, glucose; Fruc, fructose.
(B) Oral fructose does not circulate primarily as fructose. Data show blood concentrations of the administered labeled hexose (either U-13C-glucose or U-13C-fructose, with the other administered in unlabeled form) and associated area under the curve (AUC0–120 min) for the labeled hexose normalized such that glucose AUC = 100% (N = 4).
(C) Oral fructose forms circulating glucose and organic acids. Heatmap shows the percentage of labeled carbon atoms in the indicated circulating metabolites from the administered U-13C-hexose (N = 4).
(D and E) Graphs show corresponding blood concentrations of labeled glucose (D) and glycerate (E) and associated normalized AUC0–120 min.
(F) Normalized labeled AUC0–120 min for other organic acids (N = 4).
(G) Mice received sucrose (1 g/kg) instead of the 1:1 mixture of glucose:fructose, with either the glucose or the fructose moiety of sucrose labeled (N = 3). The fructose moiety of oral sucrose forms circulating glucose.
(H) Mice received a 1:1 mixture of unlabeled glucose and partially labeled fructose (carbons 1, 2, 3 or 4, 5, 6 labeled). Circulating glycerate, and to a lesser extent several other organic acids, is preferentially formed from carbons 4, 5, 6 of fructose. Red line indicates equal labeling from carbons 1, 2, 3 versus 4, 5, 6 (N = 4 for 1, 2, 3 and N = 3 for 4, 5, 6).
Data are means and error bars are ±SE. *p < 0.05 by a two-tailed unpaired Student’s t test. Labeled concentrations refer to the sum of all labeled forms, in which each form is weighted by fraction carbon atoms labeled. AUCs are calculated by the trapezoidal rule. See also Figure S1.
To find where fructose carbons go, we performed time course metabolomics analysis for labeled metabolites in the systemic circulation. This revealed more extensive conversion of fructose than of glucose into a variety of circulating metabolites (Figure 1C). Notably, a major portion of orally delivered fructose carbons were detected as glucose in circulation (Figure 1D). In addition, glycerate was produced from fructose 11-fold more than from glucose (Figure 1E). Fructose carbons also contributed more significantly to circulating TCA intermediates, amino acids (glutamate, glutamine, and alanine), and urea cycle metabolites (ornithine and citrulline) (Figure 1F). No difference was observed in lactate, gluconate, or serine (Figure 1F). These results indicate that dietary fructose is actively converted into glucose, glycerate, and a variety of other organic acids.
Sucrose is the primary source of fructose in most non-processed foods. Whether free fructose, as is found in high-fructose corn syrup, produces distinct physiological effects compared with sucrose remains unclear (Bray et al., 2004; Yu et al., 2013; Rosas-Villegas et al., 2017). To investigate sucrose metabolism, we gavaged mice with sucrose, which was 13C labeled on either its fructose or its glucose subunit. Labeling of glucose, glycerate, and other circulating metabolites from the labeled sucrose mirrored the results with the 1:1 fructose:glucose mixtures: 13C-glucose appeared directly in the circulation, whereas 13C-fructose was converted into glucose and organic acids (Figures 1G, S1A, and S1B). These results suggest that sucrose and free fructose are metabolically equivalent and are consistent with literature indicating that consumption of sugar and of high-fructose corn syrup has similar pathological effects (Tappy and Lê, 2010).
To further understand the metabolic fate of fructose, we gavaged mice with fructose labeled on its first three carbons, which become DHAP, versus the last three carbons, which become glyceraldehyde (13C-1,2,3 versus 13C-4,5,6). Synthesis of glucose from fructose drew equally from both halves of the molecule, as did lactate production. In contrast, circulating TCA intermediates were made preferentially from the back end of fructose. Most strikingly, circulating glycerate was produced almost solely from the back end of fructose (Figure 1H). This is chemically logical, as the back end of fructose is known to be converted into glyceraldehyde, but nevertheless strikingly high-lights the potential for fructose to produce non-canonical circulating metabolites that are not similarly made from ingestion of other carbohydrates.
Small Intestine Is a Primary Organ for Dietary Fructose Clearance
Transcriptional data show the highest expression of Khk-C in the liver, suggesting that liver is the key site of fructose catabolism (Ishimoto et al., 2012, 2013; Lanaspa et al., 2013). As an alternative perspective on the importance of different organs to fructose catabolism, we investigated the abundance of the fructose-specific metabolite F1P from 13C-fructose across different tissues (Figure 2A). In agreement with Khk-C expression data (Ishimoto et al., 2012), intravenous infusion of 13C-fructose with unlabeled glucose revealed that liver, kidney, pancreas, and small intestine accumulate substantial levels of labeled F1P (Figure 2B). In contrast, oral gavage of 13C-fructose with unlabeled glucose showed predominant F1P accumulation in the small intestine (jejunum > duodenum > ileum) with only a small amount of labeled F1P appearing in the liver (Figures 2C and S2A). The abundance of labeled glycerate, another fructose-specific metabolite, was also much higher (80-fold) in small intestine than any other organ (Figure S2B). Other metabolites that were made preferentially from fructose in the small intestine include glycerol-3-phosphate and several organic acids (Figure S2A). For comparison, the abundance of labeled glucose 6-phosphate (G6P) from 13C-glucose was greatest in skeletal muscle and liver and minimal in small intestine (Figure 2D). These data suggest that the small intestine largely passively transmits glucose from the intestine into the body, whereas it actively metabolizes fructose.
Figure 2. Dietary Fructose Is Metabolized by the Small Intestine.

(A) Schematic of glucose and fructose metabolism.
(B) Circulating fructose is metabolized in the small intestine, liver, kidney, and pancreas. Mice received a 1:1 mixture of unlabeled glucose and U-13C-fructose via a 2 hr continuous intravenous infusion (0.02 µmol/g/min each) and labeled F1P, the direct product of fructose phosphorylation, was measured in tissues (N = 5).
(C) Oral fructose labels F1P in the small intestine. Mice received an oral gavage of 1:1 unlabeled glucose:U-13C-fructose (0.5 g/kg each), and labeled F1P was measured in tissues (N = 5). Bar graph to the right shows AUC0–45 min for tissue F1P.
(D) Oral glucose labels G6P in muscle. Mice received an oral gavage of 1:1 U-13C-glucose:unlabeled fructose (0.5 g/kg each), and labeled G6P, the direct product of glucose phosphorylation, was measured in tissues (N = 4). Bar graph to the right shows AUC0–45 min for tissue G6P.
Data are means and error bars are ±SE. F1,6BP, fructose 1,6-bisphosphate; 3-PG, 3-phosphoglycerate; GA, glyceraldehyde; Quad, quadriceps; TA, tibialis anterior; iWAT and gWAT, inguinal and gonadal white adipose tissue; BAT, brown adipose tissue. See also Figure S2.
Accumulation of a metabolic intermediate, however, does not always correlate with pathway flux. To more directly and quantitatively assess the contribution of the small intestine to fructose clearance, we examined metabolites in the portal vein, which connects the small intestine to the liver. Notably, absorbed nutrients pass from the intestine to the liver via the portal vein before reaching the systemic circulation (Figure 3A). Measurements of bile acid and butyrate confirmed effective portal vein sampling (Figure S3A). Upon oral gavage of 13C-glucose with unlabeled fructose, labeled glucose rapidly appears in the portal vein. Similarly, when unlabeled glucose is given with 13C-fructose, labeled fructose quickly appears in the portal blood. However, the resulting portal vein concentrations of labeled fructose are more than 10-fold lower than those of labeled glucose (Figure 3B). Indeed, when labeled fructose is administered, labeled glucose quickly exceeds labeled fructose in the portal vein (Figure 3C). The extent of glucose labeling in the portal circulation exceeded that in the systemic circulation, indicating glucose production from fructose specifically in the intestine (Figure S3B). In the animals administered 13C-fructose, examination of the isotope labeling pattern of portal vein glucose revealed a predominance of M+3 labeling (three among six carbons are labeled), indicating that the six carbons of fructose are cleaved into three-carbon units and combine with three-carbon units from other nutrients before being converted into glucose by intestinal cells (Figure 3D). Thus, the small intestine imports, phosphorylates, and cleaves fructose, converting the resulting three-carbon units into glucose and other metabolites.
Figure 3. Most Fructose Is Cleared in the Small Intestine.

(A) Schematic of digestive system and location of portal vein.
(B) Oral fructose is processed prior to entering the portal vein. Data show portal vein concentrations of the administered labeled hexose (either U-13C-glucose or U-13C-fructose, with the other administered in unlabeled form) and associated normalized portal vein AUC0–30 min (N = 4 for labeled glucose and N = 5 for labeled fructose).
(C) Oral fructose forms portal vein glucose. Mice received a mixture of unlabeled glucose and U-13C-fructose and the concentration of labeled glucose and fructose in the portal vein was measured.
(D) Intestinal gluconeogenesis from oral fructose scrambles the hexose carbon atoms. Labeling pattern of portal vein glucose in mice receiving 1:1 glucose:fructose with either the glucose or the fructose U-13C labeled.
(E and F) In Khk knockout (KO) mice, oral labeled fructose appears in the portal vein as labeled fructose (E) and not labeled glucose (F). Bars indicate labeled portal vein AUC0–30 min of the indicated hexose, normalized to wild-type (WT) mice (N = 5).
(G) Normalized AUC0–30 min for labeled F1P in jejunum and liver from WT and Khk KO mice (N = 5).
Data are means and error bars are ±SE. See also Figure S3.
We also performed similar analysis in portal blood of Khk knockout mice (Diggle et al., 2010). Compared with control mice, Khk knockout mice displayed ~13-fold higher fructose in portal blood (Figure 3E). On the other hand, labeled glucose made from 13C-fructose was barely detected in portal blood (Figure 3F). No F1P production was detected in small intestine or liver in Khk knockout mice (Figure 3G). Thus, the small intestine clearance of fructose depends on Khk.
The small intestine takes in arterial blood and releases portal venous blood, with the portal blood enriched in metabolites absorbed from the intestinal lumen or made by intestinal metabolism (Figure 4A). Following 13C-fructose feeding, intestinal production of a metabolite from 13C-fructose results in a higher labeled metabolite concentration in the portal relative to systemic blood. To quantitate the small intestinal flux from fructose into glucose and other metabolic products, we measured the concentrations of these labeled products in the portal vein and the systemic circulation, at multiple time points after gavage of unlabeled glucose with 13C-fructose. At each time point, the portal- systemic concentration difference reflects the intestinal production flux of the metabolite at that moment. Taking the area under the curve (AUC) provides a quantitative measure of the total intestinal conversion of dietary fructose into other metabolites (see Supplemental Note).
Figure 4. Quantitative Analysis of Intestinal Fructose Metabolism.

(A) Illustration of small intestine and associated circulation and mathematical equation for quantitation of intestinal fructose metabolism based on systemic-portal vein labeled metabolite concentration differences.
(B) Mice received a mixture of U-13C-glucose and unlabeled fructose and the concentration of labeled glucose in the portal vein and tail vein was measured (N = 3).
(C–F) Mice received a mixture of unlabeled glucose and U-13C-fructose and the concentration of labeled glucose (C), lactate (D), alanine (E), and fructose (F) in the portal vein and tail vein was measured (N = 4).
(G) Fate of fructose in the intestine. Stacked bars show the fraction of fructose arriving to the intestine that is converted into each of the indicated metabolic products.
Data are means and error bars are ±SE.
Data were normalized based on the corresponding AUC for 13C-glucose after gavage of 13C-glucose with unlabeled fructose (Figure 4B). The portal-systemic concentration difference AUC (ΔAUC) of labeled glucose, upon administration of 13C-fructose with unlabeled glucose (ΔAUCglc←fruc), was 42% of that observed upon administration of 13C-glucose with unlabeled fructose (ΔAUCglc←glc). Assuming that most oral glucose passes intact into the portal vein, this implies that ~42% of the oral fructose dose is converted to glucose by the small intestine (Figure 4C). If a substantial fraction of glucose is consumed by the intestine rather than passively absorbed into the portal blood, our assumption of complete glucose absorption will lead to our overestimating the fraction of oral fructose that is converted into portal glucose.
Analogous to the measurement of intestinal fructose to glucose conversion, calculations can be made for other metabolic products, comparing their labeled portal-systemic concentration difference AUC when labeled fructose is administered with that of 13C-glucose when labeled glucose is administered, and correcting for the different numbers of carbon atoms in the different metabolic products. By this approach, we find that the small intestine converts ~20% and ~10% of fructose to lactate and alanine, respectively (Figures 4D and 4E). Only ~14% of fructose is released by the small intestine intact (Figure 4F). Additional smaller contributions (~3%) come from other organic acids such as glycerate, TCA intermediates, and amino acids (Figure 4G). Thus, the small intestine converts dietary fructose into glucose and organic acids.
High-Dose Fructose Saturates Intestinal Capacity and the Extra Fructose Is Digested by Liver and Microbiota
While extensive data now link fructose to metabolic diseases, whether fructose is toxic per se, or toxic only in excessive amounts, remains unclear (Niewoehner et al., 1984; Stanhope et al., 2015). Resolving this question is of paramount importance for dietary recommendations. To gain insights into the metabolic consequences of fructose dose, we gavaged unlabeled glucose and 13C-fructose, in a 1:1 ratio, at doses from 0.25 g/kg to 2 g/kg each (Marriott et al., 2009; Macdonald, 2016). Intestinal glucose production from fructose increases linearly up to 0.5 g/kg and then begins to saturate (Figure 5A). In tandem with the saturation of gluconeogenesis, direct passage of fructose into the portal circulation steeply increases (Figure 5A). Accordingly, the ratio of labeled fructose to labeled glucose in portal blood dramatically increases with higher doses of fructose (Figure 5B, note that the y axis is in a log scale). In tandem, while labeled F1P in the jejunum is nearly maximal at 0.5 g/kg fructose, labeled F1P in the liver more than doubles between 0.5 g/kg and 1 g/kg (Figure 5C). Thus, the small intestine nearly completely clears low doses of fructose, but passes higher doses of fructose to the liver.
Figure 5. Intestinal Fructose Metabolism Is Saturable, with Excess Fructose Cleared by the Liver and the Colonic Microbiome.

(A) Intestinal conversion of fructose to glucose saturates at high fructose doses. Mice received increasing doses of a 1:1 mixture of unlabeled glucose and U-13C-fructose, and the portal vein blood AUC0–30 min for labeled glucose and labeled fructose was measured (N = 3–5).
(B) Ratio of labeled oral fructose appearing in the portal vein as fructose versus glucose, derived from data in (A). Note that the y axis is in a log scale.
(C) High-dose fructose increases F1P in the liver. Mice received increasing doses of a 1:1 mixture of unlabeled glucose and U-13C-fructose, and intestinal and liver AUC0–45 min for labeled F1P was measured (N = 4–6).
(D) High doses of fructose overflow into the colon. Mice received increasing doses of a 1:1 mixture of unlabeled glucose and U-13C-fructose by oral gavage and feces were sampled after 60 min (N = 5).
(E) The colonic microbiome converts labeled oral fructose into F6P, but not F1P; sampling as in (D) (N = 4).
(F) The colonic microbiome further metabolizes oral fructose into TCA intermediates and essential amino acids. Mice received a high-dose oral gavage of 1:1 glucose:fructose (one unlabeled and the other U-13C-labeled, 2 g/kg each) and feces were sampled after 120 min (N = 3).
TIC, total ion counts. Data are means and error bars are ±SE. See also Figure S4.
In addition to the saturation of intestinal gluconeogenesis around 1 g/kg fructose, we also observed a flattening of the total amount of fructose-derived carbon in the portal vein (Figure 5A). In contrast, for dietary glucose, no such flattening was observed (Figure S4A). A simple explanation is saturation of fructose, but not glucose absorption from the gut lumen (Kiyasu and Chaikoff, 1957). Consistent with this, high fructose doses result in undigested fructose in feces (Figure 5D). This fructose is then utilized by intestinal microorganisms via hexokinase, as demonstrated by time- and dose-dependent increases in the small intestinal and to a much greater extent cecal contents and feces of labeled F6P, but not F1P (Figures 5E and S4B). Furthermore, intestinal bacteria use fructose carbons to generate TCA intermediates, essential amino acids, and short-chain fatty acids (Figures 5F, S4C, and S4D). The production of these species from labeled fructose was not observed in antibiotic-treated mice (Figures S4C–S4F). We did not observe detectable levels of bacteria-produced metabolic products containing fructose carbons in the systemic circulation (Figure S4G). In the case of amino acids, this likely reflects that the bacterial contribution, at least from this single-dose gavage, is negligible compared with the flux from protein in food. In the case of the short-chain fatty acids, it likely reflects nearly complete hepatic clearance, with butyrate readily detected in the portal, but not systemic, circulation (Figure S3A). Collectively, these data show that dietary fructose in excess of intestinal metabolic capacity spills over to liver and the microbiome, where it may cause disease by impacting hepatic function or microbial composition (Di Luccia et al., 2015; Zhang et al., 2017).
Intestinal Capacity for Fructose Metabolism Is Improved by Previous Fructose Consumption and by Feeding
The above results indicate that intestinal fructose absorption is incomplete at high doses. Studies have shown that in pups, previous fructose exposure enhances fructose absorption and clearance by inducing genes related to fructose metabolism in the small intestine (David et al., 1995; Cui et al., 2004; Patel et al., 2015b). To test if this adaptation occurs also in adults, we fed 10- to 12-week-old mice high doses of glucose and fructose (2 g/kg each) once daily for 5 days and quantified systemic fructose metabolism on days 1, 3, and 5. On day 3, we observed increased direct fructose absorption into the systemic circulation (Figure S5A), enhanced gluconeogenesis from fructose (Figure 6A), and elevated circulating and small intestinal glycerate (Figures S5B and S5C). No significant differences were observed between day 3 and day 5. Thus, a few days of prior exposure are sufficient to enhance fructose absorption and catabolism.
Figure 6. Intestinal Fructose Metabolism Is Enhanced by Previous Fructose Consumption and by Feeding.

(A) Mice received a 1:1 mixture of glucose and fructose (2 g/kg each) by daily gavage, with the fructose U-13C labeled on the measurement day. Data show systemic venous blood concentrations of labeled glucose derived from the oral labeled fructose (N = 5).
(B and C) Relative transcript levels of Glut5 (B) and G6pc (C) in liver and jejunum normalized such that liver from the saline-treated group = 1. Tissues were harvested 2 hr after oral gavage of saline or glucose + fructose (2 g/kg each) (N = 4).
(D) Mice received fructose in their drinking water (15% w/v) for 1 week, followed by gavage of a 1:1 mixture of unlabeled glucose and U-13C-labeled fructose (2 g/kg each) on day 7. Fructose was then removed from the drinking water for 1 week and the 2 g/kg gavage repeated. Data show systemic venous blood concentrations of labeled glucose derived from the oral labeled fructose (N = 5).
(E) Mice received increasing doses of a 1:1 mixture of unlabeled glucose and U-13C-fructose and the portal vein blood AUC0–30 min for labeled glucose was measured, after fasting for 6 hr or re-feeding for 2 hr (N = 3–5).
(F) Ratio of labeled oral fructose appearing in the portal vein as fructose versus glucose. Note that the y axis is in a log scale.
(G) Overflow of fructose into the feces. Feces were sampled 60 min after oral gavage (N = 4).
Data are means and error bars are ±SE. See also Figures S5 and S6.
To investigate the mechanism of the adaptation, we measured gene expression levels in the liver and the small intestine. Following fructose gavage, we observed a remarkably rapid and strong induction of Glut5 and the key gluconeogenic gene G6pc in small intestine, both of which increased more than 20-fold over 2 hr (Figures 6B and 6C). Induction of these genes did not occur in response to glucose alone (Figure S8A). The extent of their induction by fructose was much smaller in liver (Figures 6B, 6C, and S5D). Other genes related to fructose absorption and metabolism such as triokinase (Tkfc), aldolase B, and fructose 1,6-bisphosphatase (Fbp1) also increased by >3-fold in small intestine in response to fructose (Figure S5E).
We next tested if the adaptive increase of fructose absorption and metabolism is reversible. Mice were given fructose ad libitum in their drinking water for 1 week followed by no fructose exposure for an additional week. Systemic fructose metabolism was measured at days 7 and 14. Circulating labeled glucose from 13C-fructose rose at day 7 and returned to baseline by day 14 (Figure 6D). These results indicate that fructose absorption and metabolism is adaptive and reversible.
To maintain whole-body glucose homeostasis, gluconeogenesis is tightly regulated by the fed/fasted cycle. We were curious if fructose metabolism differs between the fed and the fasted states. We found a striking increase in the efficiency of intestinal fructose conversion into glucose in fed mice (Figures 6E and 6F). Fructose levels in feces were much lower in the re-fed group (Figure 6G), indicating enhanced intestinal fructose absorption as well. No significant difference was observed in dietary glucose absorption between the fed and the fasted states (Figure S6A). Surprisingly, insulin, a potent suppressor of hepatic gluconeogenesis, did not impair intestinal conversion of fructose to glucose (Figures S6B–S6D). Together, these data indicate that, in contrast to conventional gluconeogenesis in the liver and kidney, fructose-fueled intestinal gluconeogenesis is not suppressed but rather enhanced in the fed state.
DISCUSSION
The liver plays a major role in carbohydrate homeostasis, controlling glucose levels by synthesizing and degrading glycogen and making glucose via gluconeogenesis. In addition, the liver has generally been assumed to be the main site of fructose metabolism (Caliceti et al., 2017; Jegatheesan and De Bandt, 2017). This assumption is consistent with the liver’s general metabolic importance, high levels of expression of fructose catabolic enzymes, and sensitivity to fructose, which causes fatty liver disease (Ishimoto et al., 2012, 2013; Lanaspa et al., 2013; Zhang et al., 2017).
Here we re-assessed fructose metabolism at the whole-body level in mice using isotope tracers and mass spectrometry. By selectively labeling either glucose or fructose, we were able to administer the two sugars in a physiological 1:1 ratio, while tracking each individual sugar’s metabolic fate. We were surprised to find that, upon oral administration of labeled fructose, F1P accumulates much more in the small intestine than in the liver. This motivated us to sample blood from the portal vein, which connects the small intestine to the liver. In the portal vein, we observed that most dietary fructose has already been converted into glucose and various organic acids (lactate, glycerate, TCA intermediates, and amino acids).
The extent of passage of unmetabolized fructose through the small intestine to the liver depended on dose. Conversion of doses between mice and humans is not straightforward. Across mammals, total metabolic activity more closely mirrors body surface area than mass. For a typical adult mouse, daily intake is ~12 kcal, versus ~2,400 kcal for an adult human. One sensible way of converting doses of macronutrients is based on caloric intake: a dose of 0.5 g/kg fructose in mouse is ~0.5% of daily calorie intake, or the same as 3 g of fructose in a person (one orange or about 2 ounces of soda). Thus, the doses that we study here are in the range of typical human fructose consumption (Marriott et al., 2009; Macdonald, 2016).
Another concern is the extent of conservation of the small intestine’s role across species. The gross anatomy of the gastrointestinal system is similar across mammals: the small intestine drains to the portal circulation, which feeds the liver before connecting to the systemic circulation. But the relative sizes, metabolic activities, and functional roles of the organs may have diverged across evolution. Our observation of fructose conversion to glucose by the small intestine echo results of Ockerman and Lundborg (1965), who obtained evidence for similar conversion in humans based on detecting increased glucose levels in a mesenteric vein after intraduodenal injection of fructose. Thus, intestinal clearance of dietary fructose by the small intestine may be a general feature of mammalian metabolism (Ockerman and Lundborg, 1965; Bismut et al., 1993).
How, then, does fructose cause fatty liver? One possibility is that the small intestine (or intestinal microbiota) converts fructose into a hepatotoxic metabolite. For example, the microbiota makes butyrate, and the intestine makes copious glycerate from fructose, but not from glucose. Using positional isotope-labeled fructose, we show that glycerate is made selectively from fructose carbons 4, 5, 6. This makes biochemical sense, as the canonical fructose catabolic pathway converts these carbons into glyceraldehyde, whose oxidation can produce glycerate (Heinz et al., 1968). The effects of glycerate on liver and other organs merit further research. Beyond glycerate, we provide a thorough catalog of metabolites that are made preferentially from fructose relative to glucose (Figures S2A and S3B). For example, glycerol-3-phosphate could provide the backbone for phospholipid or triglyceride synthesis in the intestine (Figure S2A) (Macdonald, 2016). In addition to these direct fructose metabolic products, it is possible that fructose may trigger release from the intestine of other metabolites or signaling molecules that impact the liver (Crescenzo et al., 2017).
Another possible mechanism by which fructose may induce liver toxicity is via itself reaching the liver (Kim et al., 2016; Zhang et al., 2017). Fructose may cause liver ATP depletion (as Khk consumes ATP) or lipogenesis (as fructose catabolism bypasses the key regulated step of glycolysis, phosphofructokinase, and thereby provides an uncontrolled source of trioses). While we find that low doses of fructose are ~90% cleared in the small intestine, higher doses pass substantially (>30%) to the liver. This reflects saturation of small intestine fructose clearance. Based on these findings, we propose that the small intestine shields the liver from fructose and that excessive doses of fructose overwhelm the small intestine, spilling over to the liver where they cause toxicity (Figure 7). Liver- or intestine-specific Khk knockout mice will be valuable tools to test this idea (Diggle et al., 2010).
Figure 7. Illustration of the Roles of Intestine and Liver in Fructose Metabolism.

(A) It is commonly assumed that the small intestine passively transports fructose to portal circulation and the liver is a major organ for fructose metabolism.
(B) We show that low-dose dietary fructose is cleared by the small intestine, which converts it into glucose and organic acids. High fructose doses overwhelm the intestinal capacity for fructose metabolism and extra fructose spills over to the liver.
Intestinal fructose clearance is enhanced both by prior fructose exposure and by feeding. In the small intestine, fructose strongly and quickly induces genes involved in its uptake and catabolism, including the fructose transporter Glut5 and G6pc, which is required for eventual production of glucose from fructose. While intestinal levels of G6pc are typically about 30-fold lower than those in the liver, they are induced nearly 30-fold by fructose (Figure 6C). The increase in fructose clearance capacity in the fed state occurs rapidly and independent of prior fructose exposure and may involve metabolic or hormonal signals that alter enzyme activities or localization. Notably, while insulin suppresses classical gluconeogenesis, gluconeogenic genes induced by fructose feeding in the liver are not suppressed by insulin (Niewoehner et al., 1984; Kim et al., 2016), and production of glucose from fructose in the intestine is not suppressed but rather enhanced by feeding.
In addition to potential biochemical adaptations in the intestine that facilitate fructose-driven gluconeogenesis in the fed state, improved intestinal fructose clearance in the fed state may reflect slower fructose passage into the intestine. A key difference between the health effects of fiber-rich fruits (and perhaps even solid sweets like cake) and juices/sodas is their rate of intestinal fructose release. Based on our findings, although we did not directly modulate fructose delivery rate, it is likely that the appearance rate of free fructose in the small intestine plays a critical role in dictating its metabolic fate: like the lower doses in our experiments, a slower rate of fructose appearance will result in more complete intestinal fructose clearance, whereas higher doses and faster rates result in fructose overflow to the liver.
While much work remains to identify the sites and mechanisms of fructose toxicity, our fundamental findings that physiological fructose doses are cleared in small intestine, toxic doses spill over to the liver, and such spillover is decreased in the fed state are consistent with old-fashioned wisdom about sweets: eat sweets in moderation after meals. They provide a scientific rationale for the emerging epidemiological consensus that sodas and juices, while once viewed as benign because they are fat free, are actually hepatotoxic. And they argue for particular care in avoiding sweetened beverages between meals. By providing a new perspective on these basic dietary choices, this work also highlights the potential for isotope-tracer studies that measure mammalian metabolic activity to more broadly inform the connections between metabolism, diet, and disease.
Study Limitations
There is substantial epidemiological and experimental evidence linking fructose consumption to metabolic disease, especially fatty liver. The link between fructose and metabolic disease, however, remains controversial (van Buul et al., 2014; Caliceti et al., 2017; Jegatheesan and De Bandt, 2017). Nothing in the present manuscript addresses whether fructose is more toxic than other sugars or carbohydrates.
We do, however, definitively determine the main site of dietary fructose clearance in mice: the small intestine. Because higher doses of fructose overwhelm the small intestine and spill over to the liver, it is tempting to speculate that fructose metabolism in the small intestine is safe (physiologic), whereas fructose metabolism in the liver drives metabolic disease (pathologic, at least for individuals with consistent access to abundant high-calorie foods). We do not, however, test this hypothesis. Indeed, it is possible that intestinal metabolism of fructose drives metabolic disease.
Another important limitation regards the dose response to fructose. In fasted mice, there is a shift toward greater hepatic fructose metabolism between 0.25 g/kg and 1 g/kg fructose gavage. We consider it likely that this basic trend is conserved across many mammals—lower doses of fructose are cleared by the intestine and higher doses spill over to liver—but our current data are limited to C57BL/6 mice. Moreover, even if the basic trend is conserved, the dose response may vary. Understanding the associated dose-response pattern in humans is of critical importance, but not addressed experimentally here.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
EXPERIMENTAL MODEL AND SUBJECT DETAILS
- METHOD DETAILS
-
○Sample Collection
-
○Metabolite Extraction
-
○LC-MS
-
○Acetate measurement by GC-MS
-
○RNA Extraction and Quantitative RT-PCR
-
○
QUANTIFICATION AND STATISTICAL ANALYSIS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| D-FRUCTOSE (U-13C6, 99%) | Cambridge Isotope Laboratories | Cat#CLM-1553-PK |
| D-GLUCOSE (U-13C6, 99%) | Cambridge Isotope Laboratories | Cat#CLM-1396-PK |
| D-SUCROSE (FRUCTOSE-13C6, 98%) | Cambridge Isotope Laboratories | Cat#CLM-9811-PK |
| D-SUCROSE (GLUCOSE-13C6, 98%) | Cambridge Isotope Laboratories | Cat#CLM-8091-PK |
| D-[1,2,3-13C3]fructose | Omicron Biochemicals | Cat#FRU-027 |
| D-[4,5,6-13C3]fructose | Omicron Biochemicals | Cat#FRU-023 |
| XBridge BEH Amide XP column | Waters | Cat#176002889 |
| Atlantis T3 column | Waters | Cat#186003719 |
| 2,3,4,5,6-pentafluorobenzyl bromide | Sigma | Cat#101052 |
| HP-5MS 5% phenyl methyl silox column | Agilent | Cat#19091S-433 |
| Critical Commercial Assays | ||
| RNeasy Mini kit | Qiagen | Cat#74106 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368814 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | Charles River Laboratories | Cat#027 |
| Mouse: KHK whole-body knockout mice | the University of Leeds | N/A |
| Mouse: Vascular-catheterized C57BL/6 | Charles River Laboratories | Custom-ordered |
| Oligonucleotides | ||
| Primers for quantitative RT-PCR, see Table S1 | This paper | N/A |
| Software and Algorithms | ||
| MAVEN software | http://genomics-pubs.princeton.edu/mzroll/index.php | N/A |
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joshua Rabinowitz (joshr@princeton.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal studies followed protocols approved by the Princeton University Institutional Animal Care and Use Committee. Khk knockout mice were kindly provided from the University of Leeds to Pfizer. All experiments with the Khk knockout mice were performed at Pfizer and were carried out in strict accordance with federal, state, local, and institutional guidelines governing the use of laboratory animals in research and were reviewed and approved by Pfizer Institutional Animal Care and Use Committee. The mice were on normal light cycle (8 AM – 8 PM). Animals were randomized to treatment groups. In vivo infusion was performed on 9-week old male C57BL/6 mice pre-catheterized on the right jugular vein. Infusion was performed for 2 h to achieve isotopic steady state. The mouse infusion setup (Instech Laboratories, Plymouth Meeting, PA) included a tether and swivel system so that the animal has free movement in the cage. 1:1 mixture of [U-13C]-fructose and unlabeled glucose was prepared as solutions in normal saline (0.5 Meach) and infused via the catheter at a constant rate (0.02 µmol/g/min for each hexose). This dose was used to minimize perturbation of glucose homeostasis by infusion. (Circulating glucose levels were measured and did not change detectably.) For oral gavage experiment, 9–14 week old male C57BL/6 mice were used. On the day of the experiment, mice were transferred to new cages without food around 10 AM for fasting. At 3 PM, mice were fed (10 µl/g body weight) 1:1 mixture of glucose and fructose (either labeled or unlabeled) or sucrose in normal saline (0.9% NaCl in water) via a plastic feeding tube (Instech Laboratories, Plymouth Meeting, PA). To probe the fed state, mice were fasted from 10 AM to 6 PM (with lights on), lights were turned off at 6 PM, and chow was placed back in the cages to allow mice to eat for 2 h in the dark. At 8 PM, oral gavage was performed. For daily fructose feeding experiment, mice were fed 1:1 mixture of glucose and fructose (2 g/kg each) at 6 PM for 5 days. For ad lib fructose feeding experiment, mice were fed fructose in the drinking water (15% w/v).
METHOD DETAILS
Sample Collection Blood samples (~20 µl) were collected by tail bleeding and placed on ice in the absence of anticoagulant for 20 min, and centrifuged at 16,000 × g for 10 min at 4°C. For tissue harvest, mice were anesthetized with 1–3% isoflurane via a nose cone and tissues were quickly dissected and snap frozen (<5 sec) in liquid nitrogen with a pre-cooled Wollenberger clamp (Wollenberger et al., 1960). To eliminate the potential effect of heterogeneity in metabolism across different lobes of the liver, the whole liver was harvested and grounded to homogenous power before metabolite extraction. For microbiota analysis, luminal contents of the intestine were collected by squeezing them onto aluminum foil. The remaining empty intestine was snap frozen without additional rinsing because the rinsing and resulting delay in quenching of metabolism affects metabolite levels. For portal vein blood isolation, mice were anesthetized with 1–3% isoflurane via a nose cone. Then, the abdominal cavity was opened and adipose tissues and intestines were displaced to identify the portal vein. The portal vein was cut with sharp scissors and leaking blood (~50 µl) was immediately collected by a pipette with a blunted 200 µl tip. Successful isolation of portal vein blood was verified by much higher (>20×) concentrations of microbiota- derived bile acid (deoxycholic acid) and butyrate than blood from tail vein or carotid artery. Serum and tissue samples were kept at −80°C until LC-MS analysis.
Metabolite Extraction
To extract metabolites from serum samples, 100 µl −20°C 40:40:20 methanol:acetonitrile:water (extraction solvent) was added to 5 µl of serum sample and incubated on ice for 10 min, followed by vortexing and centrifugation at 16,000 × g for 10 min at 4°C. The supernatant (first extract) was transferred to a new tube. Then, 50 µl extraction solution was added to resuspend the pellet, followed by vortexing and centrifugation at 16,000 × g for 10 min at 4°C. The supernatant (second extract) was combined with the first extract. Then, 3 µl of the 150 µl extract was loaded to LC-MS. To extract metabolites from tissue samples, frozen tissue samples were ground at liquid nitrogen temperature with a Cryomill (Retsch, Newtown, PA). The resulting tissue powder was weighed (~20 mg). The extraction was then done by adding −20°C extraction solvent to the powder and incubating in −20°C overnight, followed by vortexing and centrifugation at 16,000 × g for 10 min at 4°C. The volume of the extraction solution (µl) was 40 × the weight of tissue (mg) to make an extract of 25 mg tissue per ml solvent.
LC-MS
Serum and tissue extracts were analyzed by LC-MS, using two different LC-MS methods chosen for optimal separation of glucose and fructose (in serum) and of hexose phosphate species (from tissues). Serum extracts were analyzed (without drying) using a quad-rupole-orbitrap mass spectrometer (Q Exactive, Thermo Fisher Scientific, San Jose, CA) operating in negative ion mode, coupled to hydrophilic interaction chromatography via electrospray ionization and used to scan from m/z 70 to 1000 at 1 Hz and 75,000 resolution. LC separation was on a XBridge BEH Amide column (2.1 mm × 150 mm, 2.5 µm particle size, 130A ° pore size) using a gradient of solvent A (20 mM ammonium acetate, 20 mM ammonium hydroxide in 95:5 water: acetonitrile, pH 9.45) and solvent B (acetonitrile). Flow rate was 150 µl/min. The LC gradient was: 0 min, 85% B; 2 min, 85% B; 3 min, 80% B; 5 min, 80% B; 6 min, 75% B; 7 min, 75% B; 8 min, 70% B; 9 min, 70% B; 10 min, 50% B; 12 min, 50% B; 13 min, 25% B; 16 min, 25% B; 18 min, 0% B; 23 min, 0% B; 24 min, 85% B; 30 min, 85% B. Autosampler temperature was 5°C, and injection volume was 3 µL. Tissue extracts were dried under nitrogen gas flow and re-dissolved in LC-MS grade water. Metabolites were analyzed via reverse-phase ion-pairing chromatography coupled to an Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific, San Jose, CA). The mass spectrometer was operated in negative ion mode with resolving power of 100,000 at m/z 200 and scan range of m/z 75–1000. The LC method was modified from an earlier method (Lu et al., 2010), using an Atlantis T3 column (150 mm 3 2.1 mm, 3 µm particle size, 100 Å pore size), with a gradient of solvent A (97:3 water:methanol with 10 mM tributylamine and 15 mM acetic acid) and solvent B (methanol). The LC gradient was 0 min, 0% B, 200 µl/min; 2 min, 0% B, 200 µl/min; 4 min, 20% B, 200 µl/min; 13 min, 80% B, 200 µl/min; 17 min, 100% B, 200 µl/min; 17.5 min, 100% B, 300 µl/min; 20 min, 100% B, 300 µl/min; 20.5 min, 0% B, 300 µl/min; 24 min, 0% B, 300 µl/min; 25 min, 0% B, 200 µl/min. Other LC parameters, common to both methods, were column temperature 25°C, autosampler temperature 5°C, and injection volume 10 µL. Data were analyzed using the MAVEN software (Melamud et al., 2010). Isotope labeling was corrected for natural 13C abundance (Su et al., 2017).
Acetate measurement by GC-MS
For acetate measurement by GC-MS, 15 µL of blood samples were mixed with 100 µL methanol, dried, and incubated at 65°C for 1 hour with 75 µL of 100 mM 2,3,4,5,6-pentafluorobenzyl bromide for derivatization. Subsequently, derivatized samples were dried and resuspended in 100 µL hexane, 70 µL of which were loaded in sample vials prepared with glass inserts. Analysis used a 7890A GC coupled to a 7200 QTOF mass spectrometer (Agilent, Santa Clara, CA). Chromatographic separation was achieved using an HP-5MS 5% phenyl methyl silox column. Helium was used as the mobile phase and flowed at a rate of 1.2 mL/min. 1 µL of sample was injected in splitless mode with inlet temperature 250°C. The GC temperature program was as follows: hold at 35°C for 6 min, increase 30°C per minute up to 220°C, and finally hold at 220°C for 5 min. Data were collected in full scan mode, and filtered using the exact masses for unlabeled, 1-labeled, 2-labeled acetate.
RNA Extraction and Quantitative RT-PCR
mRNA was isolated using an RNeasy Mini kit and reverse-transcribed with a High-Capacity cDNA Reverse Transcription Kit according to the manufacturers’ protocols. See Table S1 for primers used for quantitative RT-PCR.
QUANTIFICATION AND STATISTICAL ANALYSIS
P-values were calculated using a two-tailed unpaired Student’s t-test. P < 0.05 was considered statistically significant. Data are displayed as mean ± SE. All of the statistical details of experiments can be found in the figure legends. N represent the number of animals used.
Supplementary Material
Highlights.
Isotope tracing reveals that the small intestine metabolizes most dietary fructose
High-dose fructose saturates intestinal fructose clearance capacity
Excess fructose spills over to the liver and colonic microbiota
Intestinal fructose clearance is enhanced by feeding
Acknowledgments
This research was supported by NIH Pioneer Award 1DP1DK113643 and Diabetes Research Center Grant P30 DK019525. C.J. is a postdoctoral fellow of the American Diabetes Association (#1-17-PDF-076). S.H. is a Merck Fellow of the Life Sciences Research Foundation. G.L. is a postdoctoral fellow of the LAM Foundation (LAM00100F01-14) and Tuberous Sclerosis Alliance (TSA-01-14). Khk knockout mice were kindly provided from the University of Leeds. We thank members of the Rabinowitz lab, Lewis Cantley, Miguel Lanaspa, Richard Johnson, Michael Neinast, Shogo Wada, and Zoltan Arany for scientific discussions.
DECLARATION OF INTERESTS
W. Liu, G.J.T., and M.J.B. are employees of Pfizer. J.D.R. receives research funding from Pfizer and collaborates with Colorado Research Partners.
Footnotes
Supplemental Information includes a Supplemental Note, six figures, and one table and can be found with this article online at https://doi.org/10.1016/j.cmet.2017.12.016.
AUTHOR CONTRIBUTIONS
C.J. and J.D.R. designed the study. C.J. performed most experiments. S.H. developed the equations and wrote the Supplemental Note. S.H., A.J.C., and R.J.M. helped with animal experiments and sample preparation. W. Lu contributed to mass spectrometry method development and analyses. G.L. performed the gene expression studies. W. Liu, G.J.T., and M.J.B. designed and performed the Khk knockout mouse studies. C.J. and J.D.R. wrote the manuscript. All authors discussed the results and commented on the manuscript.
References
- Bismut H, Hers HG, Van Schaftingen E. Conversion of fructose to glucose in the rabbit small intestine. A reappraisal of the direct pathway. Eur. J. Biochem. 1993;213:721–726. doi: 10.1111/j.1432-1033.1993.tb17812.x. [DOI] [PubMed] [Google Scholar]
- Bravo S, Lowndes J, Sinnett S, Yu Z, Rippe J. Consumption of sucrose and high-fructose corn syrup does not increase liver fat or ectopic fat deposition in muscles. Appl. Physiol. Nutr. Metab. 2013;38:681–688. doi: 10.1139/apnm-2012-0322. [DOI] [PubMed] [Google Scholar]
- Bray GA, Nielsen SJ, Popkin BM. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr. 2004;79:537–543. doi: 10.1093/ajcn/79.4.537. [DOI] [PubMed] [Google Scholar]
- Caliceti C, Calabria D, Roda A, Cicero AFG. Fructose intake, serum uric acid, and cardiometabolic disorders: a critical review. Nutrients. 2017;9:E395. doi: 10.3390/nu9040395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong MF, Fielding BA, Frayn KN. Mechanisms for the acute effect of fructose on postprandial lipemia. Am. J. Clin. Nutr. 2007;85:1511–1520. doi: 10.1093/ajcn/85.6.1511. [DOI] [PubMed] [Google Scholar]
- Crescenzo R, Mazzoli A, Di Luccia B, Bianco F, Cancelliere R, Cigliano L, Liverini G, Baccigalupi L, Iossa S. Dietary fructose causes defective insulin signalling and ceramide accumulation in the liver that can be reversed by gut microbiota modulation. Food Nutr. Res. 2017;61:1331657. doi: 10.1080/16546628.2017.1331657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croset M, Rajas F, Zitoun C, Hurot JM, Montano S, Mithieux G. Rat small intestine is an insulin-sensitive gluconeogenic organ. Diabetes. 2001;50:740–746. doi: 10.2337/diabetes.50.4.740. [DOI] [PubMed] [Google Scholar]
- Cui XL, Soteropoulos P, Tolias P, Ferraris RP. Fructose-responsive genes in the small intestine of neonatal rats. Physiol. Genomics. 2004;18:206–217. doi: 10.1152/physiolgenomics.00056.2004. [DOI] [PubMed] [Google Scholar]
- Di Luccia B, Crescenzo R, Mazzoli A, Cigliano L, Venditti P, Walser JC, Widmer A, Baccigalupi L, Ricca E, Iossa S. Rescue of fructose-induced metabolic syndrome by antibiotics or faecal transplantation in a rat model of obesity. PLoS One. 2015;10:e0134893. doi: 10.1371/journal.pone.0134893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggle CP, Shires M, McRae C, Crellin D, Fisher J, Carr IM, Markham AF, Hayward BE, Asipu A, Bonthron DT. Both isoforms of ketohexokinase are dispensable for normal growth and development. Physiol. Genomics. 2010;42:235–243. doi: 10.1152/physiolgenomics.00128.2010. [DOI] [PubMed] [Google Scholar]
- David ES, Cingari DS, Ferraris RP. Dietary induction of intestinal fructose absorption in weaning rats. Pediatr. Res. 1995;37:777–782. doi: 10.1203/00006450-199506000-00017. [DOI] [PubMed] [Google Scholar]
- Douard V, Ferraris RP. Regulation of the fructose transporter GLUT5 in health and disease. Am. J. Physiol. Endocrinol. Metab. 2008;295:E227–E237. doi: 10.1152/ajpendo.90245.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froesch ER. Fructose metabolism in adipose tissue. Acta Med. Scand. Suppl. 1972;542:37–46. doi: 10.1111/j.0954-6820.1972.tb05317.x. [DOI] [PubMed] [Google Scholar]
- Heinz F, Lamprecht W, Kirsch J. Enzymes of fructose metabolism in human liver. J. Clin. Invest. 1968;47:1826–1832. doi: 10.1172/JCI105872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman RH, Stifel FB, Greene HL, Herman YF. Intestinal metabolism of fructose. Acta Med. Scand. Suppl. 1972;542:19–25. doi: 10.1111/j.0954-6820.1972.tb05315.x. [DOI] [PubMed] [Google Scholar]
- Ishimoto T, Lanaspa MA, Le MT, Garcia GE, Diggle CP, Maclean PS, Jackman MR, Asipu A, Roncal-Jimenez CA, Kosugi T, et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc. Natl. Acad. Sci. USA. 2012;109:4320–4325. doi: 10.1073/pnas.1119908109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimoto T, Lanaspa MA, Rivard CJ, Roncal-Jimenez CA, Orlicky DJ, Cicerchi C, McMahan RH, Abdelmalek MF, Rosen HR, Jackman MR, et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology. 2013;58:1632–1643. doi: 10.1002/hep.26594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jegatheesan P, De Bandt JP. Fructose and NAFLD: the multifaceted aspects of fructose metabolism. Nutrients. 2017;9:230. [Google Scholar]
- Johnson RJ, Nakagawa T, Sanchez-Lozada LG, Shafiu M, Sundaram S, Le M, Ishimoto T, Sautin YY, Lanaspa MA. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes. 2013;62:3307–3315. doi: 10.2337/db12-1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Krawczyk SA, Doridot L, Fowler AJ, Wang JX, Trauger SA, Noh HL, Kang HJ, Meissen JK, Blatnik M, et al. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J. Clin. Invest. 2016;126:4372–4386. doi: 10.1172/JCI81993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyasu JY, Chaikoff IL. On the manner of transport of absorbed fructose. J. Biol. Chem. 1957;224:935–939. [PubMed] [Google Scholar]
- Kolderup A, Svihus B. Fructose metabolism and relation to atherosclerosis, type 2 diabetes, and obesity. J. Nutr. Metab. 2015;2015:823081. doi: 10.1155/2015/823081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanaspa MA, Ishimoto T, Li N, Cicerchi C, Orlicky DJ, Ruzycki P, Rivard C, Inaba S, Roncal-Jimenez CA, Bales ES, et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat. Commun. 2013;4:2434. doi: 10.1038/ncomms3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Clasquin MF, Melamud E, Amador-Noguez D, Caudy AA, Rabinowitz JD. Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Anal. Chem. 2010;82:3212–3221. doi: 10.1021/ac902837x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marriott BP, Cole N, Lee E. National estimates of dietary fructose intake increased from 1977 to 2004 in the United States. J. Nutr. 2009;139:1228S–1235S. doi: 10.3945/jn.108.098277. [DOI] [PubMed] [Google Scholar]
- Macdonald IA. A review of recent evidence relating to sugars, insulin resistance and diabetes. Eur. J. Nutr. 2016;55:17–23. doi: 10.1007/s00394-016-1340-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamud E, Vastag L, Rabinowitz JD. Metabolomic analysis and visualization engine for LC-MS data. Anal. Chem. 2010;82:9818–9826. doi: 10.1021/ac1021166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M, Craig JW, Drucker WR, Woodward H., Jr The metabolism of fructose in man. Yale J. Biol. Med. 1956;29:335–360. [PMC free article] [PubMed] [Google Scholar]
- Mirtschink P, Krishnan J, Grimm F, Sarre A, Hörl M, Kayikci M, Fankhauser N, Christinat Y, Cortijo C, Feehan O, et al. HIF-driven SF3B1 induces KHK-C to enforce fructolysis and heart disease. Nature. 2015;522:444–449. doi: 10.1038/nature14508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mithieux G, Bady I, Gautier A, Croset M, Rajas F, Zitoun C. Induction of control genes in intestinal gluconeogenesis is sequential during fasting and maximal in diabetes. Am. J. Physiol. Endocrinol. Metab. 2004;286:E370–E375. doi: 10.1152/ajpendo.00299.2003. [DOI] [PubMed] [Google Scholar]
- Niewoehner CB, Gilboe DP, Nuttall GA, Nuttall FQ. Metabolic effects of oral fructose in the liver of fasted rats. Am. J. Physiol. 1984;247:E505–E512. doi: 10.1152/ajpendo.1984.247.4.E505. [DOI] [PubMed] [Google Scholar]
- Niewoehner CB. Metabolic effects of dietary versus parenteral fructose. J. Am. Coll. Nutr. 1986;5:443–450. doi: 10.1080/07315724.1986.10720147. [DOI] [PubMed] [Google Scholar]
- Ockerman PA, Lundborg H. Conversion of fructose to glucose by human jejunum absence of galactose-to-glucose conversion. Biochim. Biophys. Acta. 1965;105:34–42. doi: 10.1016/s0926-6593(65)80173-4. [DOI] [PubMed] [Google Scholar]
- Patel C, Sugimoto K, Douard V, Shah A, Inui H, Yamanouchi T, Ferraris RP. Effect of dietary fructose on portal and systemic serum fructose levels in rats and in KHK−/− and GLUT5−/− mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015a;309:G779–G790. doi: 10.1152/ajpgi.00188.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel C, Douard V, Yu S, Tharabenjasin P, Gao N, Ferraris RP. Fructose-induced increases in expression of intestinal fructolytic and gluconeogenic genes are regulated by GLUT5 and KHK. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015b;309:R499–R509. doi: 10.1152/ajpregu.00128.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Villegas A, Sánchez-Tapia M, Avila-Nava A, Ramírez V, Tovar AR, Torres N. Differential effect of sucrose and fructose in combination with a high fat diet on intestinal microbiota and kidney oxidative stress. Nutrients. 2017;9:E393. doi: 10.3390/nu9040393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soty M, Penhoat A, Amigo-Correig M, Vinera J, Sardella A, Vullin-Bouilloux F, Zitoun C, Houberdon I, Mithieux G. A gut-brain neural circuit controlled by intestinal gluconeogenesis is crucial in metabolic health. Mol. Metab. 2014;4:106–117. doi: 10.1016/j.molmet.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanhope KL, Medici V, Bremer AA, Lee V, Lam HD, Nunez MV, Chen GX, Keim NL, Havel PJ. A dose-response study of consuming high-fructose corn syrup-sweetened beverages on lipid/lipoprotein risk factors for cardiovascular disease in young adults. Am. J. Clin. Nutr. 2015;101:1144–1154. doi: 10.3945/ajcn.114.100461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Lu W, Rabinowitz JD. Metabolite spectral accuracy on orbitraps. Anal. Chem. 2017;89:5940–5948. doi: 10.1021/acs.analchem.7b00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SZ, Empie MW. Fructose metabolism in humans - what isotopic tracer studies tell us. Nutr. Metab. (Lond) 2012;9:89. doi: 10.1186/1743-7075-9-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tappy L, Lê KA. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010;90:23–46. doi: 10.1152/physrev.00019.2009. [DOI] [PubMed] [Google Scholar]
- van Buul VJ, Tappy L, Brouns FJ. Misconceptions about fructose-containing sugars and their role in the obesity epidemic. Nutr. Res. Rev. 2014;27:119–130. doi: 10.1017/S0954422414000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollenberger A, Ristau O, Schoffa G. Eine einfache Technik der extrem schnellen Abkühlung größerer Gewebestücke. Pflügers Arch. Gesamte Physiol. Menschen Tiere. 1960;270:399–412. [PubMed] [Google Scholar]
- Yu Z, Lowndes J, Rippe J. High-fructose corn syrup and sucrose have equivalent effects on energy-regulating hormones at normal human consumption levels. Nutr. Res. 2013;33:1043–1052. doi: 10.1016/j.nutres.2013.07.020. [DOI] [PubMed] [Google Scholar]
- Zhang DQ, Tong X, VanDommelen K, Gupta N, Stamper K, Brady GF, Meng Z, Lin JD, Rui LY, Omary MB, et al. Lipogenic transcription factor ChREBP mediates fructose-induced metabolic adaptations to prevent hepatotoxicity. J. Clin. Invest. 2017;137:2855–2867. doi: 10.1172/JCI89934. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.