

Abstract
Tyrosine kinase receptors are one of the main targets in cancer therapy. They play an essential role in the modulation of growth factor signaling and thereby inducing cell proliferation and growth. Tyrosine kinase inhibitors such as neratinib bind to EGFR and HER2 receptors and exhibit antitumor activity. However, little is known about their detailed cellular uptake and metabolism. Here, we report for the first time the intracellular spatial distribution and metabolism of neratinib in different cancer cells using label‐free Raman imaging. Two new neratinib metabolites were detected and fluorescence imaging of the same cells indicate that neratinib accumulates in lysosomes. The results also suggest that both EGFR and HER2 follow the classical endosome lysosomal pathway for degradation. A combination of Raman microscopy, DFT calculations, and LC‐MS was used to identify the chemical structure of neratinib metabolites. These results show the potential of Raman microscopy to study drug pharmacokinetics.
Keywords: cancer, label-free imaging, metabolism, Raman spectroscopy, tyrosine kinase inhibitors
Tyrosine kinase receptors are essential in regulating important cellular processes such as migration, survival, proliferation, and differentiation.1 The ErbB family of tyrosine kinase receptors is over‐expressed in a variety of cancers and consist of four members: EGFR, HER2, HER3, and HER4.2 Controlling the EGFR pathway is one of the main approaches for targeted cancer therapy because of the overexpression of EGFR, which in turns stimulates tumorigenesis by modulating cellular proliferation, angiogenesis, and metastasis.
Non‐small cell lung cancer (NSCLC) patients harboring EGFR mutations (L858R) initially respond to first‐generation tyrosine kinase inhibitors (TKIs). Despite the impressive initial response to treatment, disease progression happens after an average of one year of the therapy. This is driven by a second point mutation in EGFR (T790M) in approximately 60 % of cases.3, 4, 5 The T790M EGFR mutation, which mediates resistance to first‐generation TKIs by acting as a “gatekeeper” mutation, induces steric hindrance in the adenosine triphosphate binding pocket, thus preventing inhibitor binding.4, 6
Neratinib (Nerlynx), a second‐generation TKI with dual specificity against EGFR and HER2, was found to be effective despite the T790M resistance mutation in EGFR and binds irreversibly to EGFR and HER2.7, 8 Neratinib was approved last year by the U.S. Food and Drug Administration for treatment of early‐stage HER2‐positive breast cancer.9 However, little is known about the intracellular distribution and metabolism of neratinib.10 The organelle distribution and the transportation of TKIs are also poorly understood. Drug distribution in cells is frequently performed using fluorescent‐labeled drug molecules, which are tested for their pharmacological efficacy. Despite the molecular specificity provided by fluorescence molecules, they are often much larger than the drug molecules of interest and can significantly alter the drug pharmaceutical activity. In contrast, confocal Raman microscopy is a label‐free imaging method that is suitable to visualize small molecules in cells.11, 12, 13, 14 However, the Raman sensitivity is limited and higher concentrations (2 mm–100 μm) of molecules were used in earlier studies to achieve suitable signal to noise ratios.11, 12, 13, 15, 16, 17
Here, we follow the uptake, intracellular spatial distribution, and metabolism of neratinib at significantly lower concentrations (0.5–10 μm) within different cancer cells using label‐free Raman microspectroscopy, therefore achieving a closer approximation of the clinical application levels. HER2‐positive breast cancer cells (SK‐BR‐3) and NSCLC cells with (NCI‐H1975) and without (Calu‐3) T790M EGFR mutations were used. Two neratinib metabolites were detected in cells. A combination of Raman microscopy, density functional theory (DFT) calculations, and liquid chromatography–mass spectrometry (LC‐MS) was used to identify the chemical structure of neratinib metabolites.
The Raman spectrum of neratinib and its chemical structure are shown in Figure S1. Neratinib has a strong C≡N stretching vibration band at 2208 cm−1. This band is located in the “Raman silent” region of cells (1800–2800 cm−1) and can be used as a label‐free marker to visualize the drug in cells. Figure 1 A depicts an integrated Raman intensity image of the band at 1425–1470 cm−1 (C−H deformation) of SK‐BR‐3 cells treated with neratinib, reflecting the different densities of various native cellular components as well as neratinib. Clearly noticeable is the nucleus surrounded by the cytoplasm. Panel B displays an integrated Raman intensity image in the 2200–2225 cm−1 region, representing neratinib. Panel C shows an overlay of the Panels A and B, indicating that neratinib is distributed in the cell. To confirm that neratinib is localized within the cell but not precipitated on the cell surface, we performed cross‐section Raman imaging along the x–z axis of the same cell as shown in Panels E–G. These images clearly demonstrate that neratinib is internalized within the cell. Similar results were obtained for NSCLC NCI‐H1975 and Calu‐3 cells (Figures S2 and S3).
Figure 1.
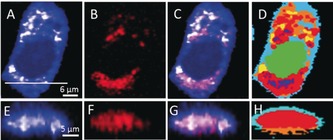
Raman imaging of SK‐BR‐3 cells treated with 5 μm neratinib for 8 h. Raman images reconstructed from the C−H deformation (A) and C≡N stretching (B) intensities. C) Overlay of Panels A and B. E–G) Cross‐section Raman images of the same cell measured along the x–z axis. Scanning positions are indicated by the white line in Panel A. D,H) HCA results based on the Raman data shown in Panels A and E.
A combination of Raman microspectroscopy and multivariate analyses was used to show different cellular components.11, 18, 19, 20 We have calculated the hierarchical cluster analysis (HCA) of Raman datasets of cells from several replicates (Figures S2–S6) and also for cells treated with different neratinib concentrations (Figures S7–S9). The HCA index‐color image (Figure 1 D) reproduces the position of the nucleus (Figure S10) as well as several regions within the cytoplasm. The red and blue clusters in the HCA image (Figure 1 D) also reproduce the localization of neratinib in the cell shown in Figure 1 B.
Figure 2 displays the Raman average spectra of the red (b) and blue (c) clusters of Figure 1 D in addition to the spectra of free neratinib (a) and SK‐BR‐3 cells (control, d). The average spectra of neratinib in cells (b,c) display features similar to those of the free neratinib spectrum (a), including the characteristic signal of the C≡N group. Thus, these results indicate that Raman microscopy coupled with HCA can be used to calculate the Raman spectra of neratinib in cells. Furthermore, the 2208 cm−1 band can be used as a label‐free marker band to monitor neratinib distribution in cells.
Figure 2.
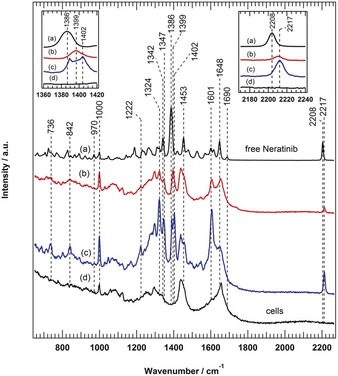
a) Raman spectrum of free neratinib. The average Raman spectra of drug regions within the cell of the red (b) and blue (c) clusters in Figure 1 D are shown. d) Raman spectrum of untreated SK‐BR‐3 cells (control). The insets show enlarged spectra around the characteristic 1386 and 2208 cm−1 bands.
The application of label‐free Raman microscopy to monitor the distribution of drugs in cells is not limited to molecules with a label‐free marker in the Raman silent region of cells (1800–2800 cm−1). Small‐molecule TKIs usually have strong and sharp Raman bands in the fingerprint region that can also be used to monitor drug distribution in cells.21 Neratinib has a strong Raman band at 1386 cm−1 (Figure 2 a) that can also be used and is even superior as a label‐free marker for neratinib in cells (Figure S11). Furthermore, similar Raman measurements were performed in which NCI‐H1975 cells were treated with 0.5 μm (250 ng mL−1) neratinib (Figure S12). This concentration is 200‐ and 4000‐fold lower than the concentrations used for the Raman imaging of erlotinib11 and another antitumor drug candidate,13 respectively, in colon cancer cells. It is not only one of the lowest concentrations of drugs in cancer cells detected by Raman microscopy, but it is also close to the detected neratinib concentration in the plasma of human patients (5.8–143.3 ng mL−1), after treatment with single oral doses.22, 23, 24
The Raman spectrum of cells (Figure 2 d) shows bands at ca. 1657 (amide I), ca. 1450 (C−H and CH2 bending deformation), ca. 1200–1400 (amide III of the peptide linkages), and ca. 1000 cm−1 (ring‐breathing mode of phenylalanine).25 These bands were also observed in the spectra of the neratinib‐containing clusters in cells (b,c), implying that cells contribute partially to these spectra. In addition, the Raman spectra (b,c) of neratinib localized in cells are similar to that of free neratinib (spectrum a). However, the C≡N band at 2208 cm−1 in the spectrum of free neratinib (a) is upshifted to 2217 cm−1 in both spectra (b,c) of neratinib in cells. The band at 1386 cm−1 in spectrum (a) is also upshifted to 1399 cm−1 in spectrum (b), while a doublet at 1402/1388 cm−1 appeared in spectrum (c). It should be noted that the 1402 cm−1 band does not exist in the spectra of free neratinib (a) and control (d). These changes in spectra (b) and (c) suggest that the structure of neratinib is altered due to metabolism but not as a result of the environmental effect of, for instance, lipids (Figures S13 and S14).
The Raman spectra of neratinib precursors and DFT calculations (Figure S15) suggest that the band at 1386 cm−1 of free neratinib originates mainly from its quinoline ring (C−C stretching and C−H bending). Since the 1386 cm−1 and 2208 cm−1 (C≡N, conjugated to the quinoline ring) bands of free neratinib experience significant wavenumber shifts in cells, the quinoline ring of neratinib is most likely altered in the metabolites of neratinib. Similar results were observed for NSCLC cells with and without T790M EGFR mutation (Figures S2 and S3). Furthermore, the Raman spectra of these metabolites contain a contribution from parent neratinib (Figure S16). The presence of parent neratinib alongside with metabolites in cells was confirmed by LC‐MS. Thus, we have observed the intracellular distribution of parent neratinib in addition to two metabolites in different cancer cells for the first time in a label‐free manner.
The Raman intensity of the C≡N band near 2208 cm−1 of neratinib displays a linear correlation with the drug concentration (Figure S17). Thus, the total concentration of neratinib and its metabolites localized within different cancer cells can be quantitatively estimated based on the calibration curve of neratinib solutions (2–15 mm). The calculated neratinib concentrations within cancer cells are 6.2–9.6(±1.0–1.2) mm, which are 1200–1900(±200–240) fold higher than the extracellular neratinib concentration (5 μm). These high intracellular neratinib concentrations account for the strong neratinib Raman signals in different cancer cells.
Neratinib targets EGFR and HER28 and it is important to monitor the distribution not only of neratinib in cells but also of these receptors as well as of lysosomes. Neratinib is a weak base that can be protonated in an acidic environment, and lysosomes are most likely sites for neratinib accumulation.26 The distribution of neratinib in three replicates of different cells was detected through Raman spectral imaging, while that of the lysosome, EGFR, and HER2 in the same cells was monitored through fluorescence microscopy (Figure 3 and Figures S18–S28). Figure 3 A displays only HCA clusters of neratinib metabolites taken from the index‐color image of Raman results (Figure 1 D). The red and pink (instead of blue) clusters represent M1 and M2 metabolites, respectively. Figure 3 B displays the distribution of lysosomes (green) surrounding the nucleus (blue) in the cell.
Figure 3.
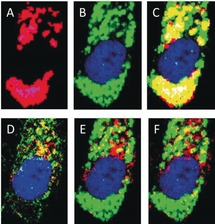
A) HCA of neratinib‐containing clusters in the cell, B) fluorescence image of the same cell that shows lysosomes (green) surrounding the nucleus (blue), and C) overlay (yellow and white) of Panels A and B. D) Fluorescence image of the same cell that shows EGFR (red) and HER2 (green) distribution around the nucleus. The overlay (yellow) of Panel B with EGFR (red) and HER2 (red) is shown in Panels E and F, respectively.
The overlay (Panel C) of Panels A and B indicates that most of the drug is accumulated in the lysosomes (yellow and white). It is suggested that a weakly basic drug induces a marked increase in lysosomal biogenesis, leading to a remarkable increase in lysosomes size and number, and finally drug accumulation within the lysosomes.26 Similar results were obtained here, where neratinib also induces an increase in the lysosomal biogenesis compared to the control (Figure S29). Figure 3 D shows the fluorescence imaging of nucleus (blue), EGFR (red), and HER2 (green). In the absence of neratinib, most of EGFR and HER2 is associated with the cell surface and in the cytoplasm (Figure S30), while neratinib treatment enhances the cytoplasmic accumulation of EGFR and HER2 (Figure 3 D and Figures S18–S28). These results suggest that neratinib induces an internalization of both EGFR and HER2. Panels E and F are the overlay images of lysosomes with EGFR and HER2, respectively. Some of these receptors are colocalized with lysosomes. It is reported that the internalized HER2 in SK‐BR‐3 cells treated with neratinib follows the classical endosome lysosomal pathway for degradation.27 Thus, the present results suggest that both EGFR and HER2 undergo neratinib‐induced endocytosis.
Neratinib possesses an α,β‐unsaturated carbonyl moiety (acrylamide group) which forms a covalent bond with Cys797 of EGFR and with Cys805 of HER27, 8 through a Michael addition reaction. Neratinib also retains electrophilic reactivity to the Cys residue of glutathione (GSH) by forming a GSH adduct.28 We investigated the chemical reactivity of neratinib with GSH (Figure S31). A new Raman band assigned to the C–S stretching29 vibration appeared at 673 cm−1, produced as a result of the formation of a covalent bond between the acrylamide moiety of neratinib and a Cys of GSH. This band was not detected in the Raman spectra of neratinib in cells, implying that the majority of the detected parent neratinib and its metabolites in cells are not covalently bound to EGFR or HER2.
The LC‐MS results of cells treated with neratinib indicated the presence of parent neratinib (M=556.2) and three major metabolites I (M=449.1), II (M=433.1), and III (M=461.1). Their possible structures are shown in Scheme 1. The identified metabolites do not carry the dimethylamino acrylamide moiety which would bind the Cys residue of EGFR or HER2. In addition, the intracellular concentration of neratinib is most likely much higher than that of EGFR and HER2, so that the fraction of covalently bound neratinib is at a very low concentration that may not be monitored within the detection limits of Raman microscopy. Thus, both Raman and LC‐MS results demonstrate that the detected neratinib metabolites in cells are not bound covalently to EGFR or HER2 proteins and are inactive. These results suggest that Raman microscopy has a potential for the measurement of the physiochemical properties of drug candidates at an early stage of drug discovery and development. These measurements are essential for reducing the present high attrition rates of novel drug candidates.30 The application of imaging modalities to drug discovery has a perspective to speed up preclinical medicinal chemistry optimisation cycles and enhance an in vitro to in vivo translation of drug candidates.31, 32
Scheme 1.
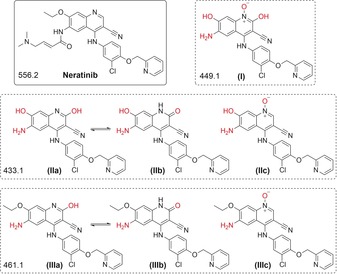
Neratinib and its metabolite candidates with the respective monoisotopic masses (g mol−1).
Furthermore, the neratinib metabolites identified by LC‐MS experienced hydroxylation, N‐oxidation, and dealkylation of the quinoline ring (Table S1 and Figures S32–S35). These structural changes may be induced by the cytochrome P450 enzyme (CYP450) superfamily, which is involved in most of the metabolic pathway.33, 34 It has been reported that neratinib is metabolized by the CYP450 isoform 3A4,24 which is expressed in SK‐BR‐3, NCI‐H1975, and Calu‐3 cells.35
In order to correlate the metabolites detected by Raman microspectroscopy (M1 and M2) with those identified by LC‐MS, we performed DFT calculations (Table 1 and Figure S36) for all metabolite candidates (Scheme 1). The data show good agreement between the calculated Raman bands (ca. 1386 and 2208 cm−1) of metabolites I and IIb/IIIb identified by LC‐MS with M1 and M2 detected by Raman microspectroscopy, respectively. Therefore, we suggest that the Raman spectrum of M1 (Figure 2 b) originates from metabolites I, while metabolites IIb and/or IIIb contribute to the Raman spectrum of M2 (Figure 2 c).
Table 1.
The experimental and DFT calculated Raman wavenumbers of characteristic bands of neratinib and its metabolites.
| Compound | Wavenumber [cm−1] | ||
|---|---|---|---|
| Raman: neratinib | 1386 | – | 2208 |
| M1 | 1399 (+13) | – | 2217 (+9) |
| M2 | 1388 (+2) | 1402 (+16) | 2217 (+9) |
| DFT: neratinib | 1381 | – | 2238 |
| metabolite I | 1395 (+14) | – | 2252 (+14) |
| metabolite IIa | 1364 (−17) | – | 2244 (+6) |
| metabolite IIb | 1383 (+2) | 1395 (+14) | 2244 (+6) |
| metabolite IIc | 1378 (−3) | – | 2251 (+13) |
| metabolite IIIa | 1369 (−12) | – | 2244 (+6) |
| metabolite IIIb | 1382 (+1) | 1401 (+20) | 2243 (+5) |
| metabolite IIIc | 1365 (−16) | 1373 (−8) | 2250 (+12) |
In conclusion, an intracellular spatial distribution of neratinib metabolites was monitored using label‐free Raman microspectroscopy. Most of the neratinib metabolites accumulated in lysosomes. Both EGFR and HER2 undergo neratinib‐induced endocytosis. New metabolites of neratinib were detected in breast cancer and NSCLC cells. A combination of Raman microspectroscopy, DFT calculations, and LC‐MS was used to identify the chemical structure of neratinib metabolites. These results show the potential of the Raman technique for pharmacokinetics assessment of drugs in cells.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dennis Petersen and Sascha D. Krauß for optimizing the Matlab scripts for data analysis, Jörn Güldenhaupt for preparing lipids, and Svenja Seger for assisting with the LC‐MS measurements. This research was supported by the Protein Research Unit Ruhr within Europe (PURE) and the Ministry of Innovation, Science and Research (MIWF) of North‐Rhine Westphalia, Germany, and the European Regional Development Fund, European Union and North‐Rhine Westphalia, Germany.
K. Aljakouch, T. Lechtonen, H. K. Yosef, M. K. Hammoud, W. Alsaidi, C. Kötting, C. Mügge, R. Kourist, S. F. El-Mashtoly, K. Gerwert, Angew. Chem. Int. Ed. 2018, 57, 7250.
Contributor Information
Karim Aljakouch, http://www.bph.rub.de/.
Dr. Samir F. El‐Mashtoly, Email: samir@bph.rub.de.
References
- 1. Lemmon M. A., Schlessinger J., Cell 2010, 141, 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yarden Y., Sliwkowski M. X., Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [DOI] [PubMed] [Google Scholar]
- 3. Sequist L. V., Waltman B. A., Dias-Santagata D., Digumarthy S., Turke A. B., Fidias P., Bergethon K., Shaw A. T., Gettinger S., Cosper A. K., et al., Sci. Transl. Med. 2011, 3, 75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pao W., Miller V. A., Politi K. A., Riely G. J., Somwar R., Zakowski M. F., Kris M. G., Varmus H., PLoS Med. 2005, 2, e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sharma S. V., Bell D. W., Settleman J., Haber D. A., Nat. Rev. Cancer 2007, 7, 169–181. [DOI] [PubMed] [Google Scholar]
- 6. Kwak E. L., Sordella R., Bell D. W., Godin-Heymann N., Okimoto R. A., Brannigan B. W., Harris P. L., Driscoll D. R., Fidias P., Lynch T. J., et al., Proc. Natl. Acad. Sci. USA 2005, 102, 7665–7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yun C.-H., Mengwasser K. E., Toms A. V., Woo M. S., Greulich H., Wong K.-K., Meyerson M., Eck M. J., Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feldinger K., Kong A., Breast Cancer Dove Med. Press 2015, 7, 147–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chan A., Delaloge S., Holmes F. A., Moy B., Iwata H., Harvey V. J., Robert N. J., Silovski T., Gokmen E., von Minckwitz G., et al., Lancet Oncol. 2016, 17, 367–377. [DOI] [PubMed] [Google Scholar]
- 10. Hug B., Abbas R., Leister C., Burns J., Sonnichsen D., Clin. Cancer Res. 2010, 16, 4016–4023. [DOI] [PubMed] [Google Scholar]
- 11. El-Mashtoly S. F., Petersen D., Yosef H. K., Mosig A., Reinacher-Schick A., Kötting C., Gerwert K., Analyst 2014, 139, 1155–1161. [DOI] [PubMed] [Google Scholar]
- 12. Tipping W. J., Lee M., Serrels A., Brunton V. G., Hulme A. N., Chem. Soc. Rev. 2016, 45, 2075–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meister K., Niesel J., Schatzschneider U., Metzler-Nolte N., Schmidt D. A., Havenith M., Angew. Chem. Int. Ed. 2010, 49, 3310–3312; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3382–3384. [Google Scholar]
- 14. Krafft C., Schmitt M., Schie I. W., Cialla-May D., Matthäus C., Bocklitz T., Popp J., Angew. Chem. Int. Ed. 2017, 56, 4392–4430; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4458–4500. [Google Scholar]
- 15. Wei L., Hu F., Shen Y., Chen Z., Yu Y., Lin C.-C., Wang M. C., Min W., Nat. Methods 2014, 11, 410–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smith G. P. S., McGoverin C. M., Fraser S. J., Gordon K. C., Adv. Drug Delivery Rev. 2015, 89, 21–41. [DOI] [PubMed] [Google Scholar]
- 17. Kann B., Offerhaus H. L., Windbergs M., Otto C., Adv. Drug Delivery Rev. 2015, 89, 71–90. [DOI] [PubMed] [Google Scholar]
- 18. Miljković M., Chernenko T., Romeo M. J., Bird B., Matthäus C., Diem M., Analyst 2010, 135, 2002–2013. [DOI] [PubMed] [Google Scholar]
- 19. El-Mashtoly S. F., Niedieker D., Petersen D., Krauss S. D., Freier E., Maghnouj A., Mosig A., Hahn S., Kötting C., Gerwert K., Biophys. J. 2014, 106, 1910–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. El-Mashtoly S. F., Yosef H. K., Petersen D., Mavarani L., Maghnouj A., Hahn S., Kötting C., Gerwert K., Anal. Chem. 2015, 87, 7297–7304. [DOI] [PubMed] [Google Scholar]
- 21. Fu D., Zhou J., Zhu W. S., Manley P. W., Wang Y. K., Hood T., Wylie A., Xie X. S., Nat. Chem. 2014, 6, 614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wong K.-K., Fracasso P. M., Bukowski R. M., Lynch T. J., Munster P. N., Shapiro G. I., Janne P. A., Eder J. P., Naughton M. J., Ellis M. J., et al., Clin. Cancer Res. 2009, 15, 2552–2558. [DOI] [PubMed] [Google Scholar]
- 23. Ito Y., Suenaga M., Hatake K., Takahashi S., Yokoyama M., Onozawa Y., Yamazaki K., Hironaka S., Hashigami K., Hasegawa H., et al., Jpn. J. Clin. Oncol. 2012, 42, 278–286. [DOI] [PubMed] [Google Scholar]
- 24. Abbas R., Hug B. A., Leister C., Burns J., Sonnichsen D., Br. J. Clin. Pharmacol. 2011, 71, 522–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matthäus C., Chernenko T., Newmark J. A., Warner C. M., Diem M., Biophys. J. 2007, 93, 668–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Keyvanjah K., DiPrimeo D., Li A., Obaidi M., Swearingen D., Wong A., Br. J. Clin. Pharmacol. 2017, 83, 554–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Y., Zhang J., Liu C., Du S., Feng L., Luan X., Zhang Y., Shi Y., Wang T., Wu Y., et al., Cancer Lett. 2016, 382, 176–185. [DOI] [PubMed] [Google Scholar]
- 28. Shibata Y., Chiba M., Drug Metab. Dispos. Biol. Fate Chem. 2015, 43, 375–384. [DOI] [PubMed] [Google Scholar]
- 29. Jess P. R. T., Smith D. D. W., Mazilu M., Dholakia K., Riches A. C., Herrington C. S., Int. J. Cancer 2007, 121, 2723–2728. [DOI] [PubMed] [Google Scholar]
- 30. Henchoz Y., Bard B., Guillarme D., Carrupt P.-A., Veuthey J.-L., Martel S., Anal. Bioanal. Chem. 2009, 394, 707–729. [DOI] [PubMed] [Google Scholar]
- 31. Conway J. R. W., Carragher N. O., Timpson P., Nat. Rev. Cancer 2014, 14, 314–328. [DOI] [PubMed] [Google Scholar]
- 32. Isherwood B., Timpson P., McGhee E. J., Anderson K. I., Canel M., Serrels A., Brunton V. G., Carragher N. O., Pharmaceutics 2011, 3, 141–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reactive Drug Metabolites (Ed.: A. S. Kalgutkar), Wiley-VCH, Weinheim, 2012. [Google Scholar]
- 34. ADME and Translational Pharmacokinetics/Pharmacodynamics of Therapeutic Proteins: Applications in Drug Discovery and Development (Eds.: H. Zhou, F.-P. Theil), Wiley, Hoboken, NJ, 2016. [Google Scholar]
- 35. Lee Y.-F., Lee C.-Y., Lai L.-C., Tsai M.-H., Lu T.-P., Chuang E. Y., Database 2018, 2018, bax101-bax101. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary