

ABSTRACT
Biomaterial vaccines offer new capabilities that can be exploited for both infectious disease and cancer. We recently developed a novel vaccine platform based on self‐assembly of immune signals into immune polyelectrolyte multilayers (iPEMs). These iPEM vaccines are electrostatically assembled from peptide antigens and nucleic acid‐based toll‐like receptor agonists (TLRas) that serve as molecular adjuvants. Gold nanoparticles (AuNPs) coated with iPEMs stimulate effector cytokine secretion in vitro and expand antigen‐specific T cells in mice. Here we investigated how the dose, injection route, and choice of molecular adjuvant impacts the ability of iPEMs to generate T cell immunity and anti‐tumor response in mice. Three injection routes—intradermal, subcutaneous, and intramuscular—and three iPEM dosing levels were employed. Intradermal injection induced the most potent antigen‐specific T cell responses and, for all routes, the level of response was dose‐dependent. We further discovered that these vaccines generate durable memory, indicated by potent, antigen‐specific CD8+ T cell recall responses in mice challenged with vaccine 49 days after a prime‐boost immunization regimen. In a common exogenous antigen melanoma model, iPEM vaccines slowed or stopped tumor growth more effectively than equivalent ad‐mixed formulations. Further, iPEMs containing CpG—a TLR9a—were more potent compared with iPEMs containing polyIC, a TLR3a. These findings demonstrate the ability of iPEMs to enhance response to several different classes of vaccine cargos, supporting iPEMs as a simple vaccine platform that mimics attractive features of other nanoparticles using immune signals that can be self‐assembled or coated on substrates. Biotechnol. Bioeng. 2017;114: 423–431. © 2016 The Authors. Biotechnology and Bioengineering Published by Wiley Periodicals, Inc.
Keywords: vaccine, adjuvant, polyelectrolyte multilayer, immunology, nanotechnology, cancer
Introduction
An ongoing translational challenge facing vaccines and immunotherapies is the need for better control over the characteristics of the immune responses these vaccines generate. Generation of immunity is influenced by the combination of signals, dose, and injection route with which vaccines are administered, including the efficiency with which each component drains to lymph nodes (LNs) and spleen (Mueller and Germain, 2009; Zinkernagel et al., 1997). Biomaterials offer features—co‐delivery and cargo protection, for example—that can help control these characteristics (Andorko et al., 2015; Swartz et al., 2012). However, nanoparticle vaccines and next generation adjuvants are becoming increasingly complex relative to traditional vaccines from the perspective of synthesis, purification, characterization, and mechanism of action (Wu et al., 2014). These manufacturing and consistency hurdles are major challenges for next generation vaccine technologies (Gammon et al., 2016; Josefsberg and Buckland, 2012). Thus, materials that simplify vaccine composition and delivery could support more effective translation (Melchels et al., 2015).
We recently reported a new nanomaterial vaccine platform based on polyelectrolyte multilayer (PEMs) films assembled entirely from immune signals (Chiu et al., 2015a; Zhang et al., 2015b). These immune PEMs (iPEMs) are electrostatically assembled on AuNPs using alternating deposition of nucleic acid adjuvants that stimulate the toll‐like receptor (TLR) 3 pathogen‐sensing pathway, and a model peptide antigen modified to display cationic amino acids to support assembly (Zhang et al., 2015b). In our previous study we showed that AuNPs without iPEM coatings did not activate dendritic cells, drive T cell proliferation, or cause secretion of inflammatory (e.g., IL‐1β) or effector (e.g., IFN‐γ, TNF) cytokines. Similarly, other groups have shown that this immunologically inert characteristic makes AuNPs a useful tool for studying immune function, for example, by functionalizing AuNPs with specific chemistries, different shapes, or other properties. (Moyano et al., 2012; Niikura et al., 2013). Thus we used AuNPs as templates for building iPEMs, which allow direct control over the loading of each vaccine signal (e.g., TLR agonists, peptide antigen) and coating onto substrates over a wide length scale. In cell culture, iPEMs activate dendritic cells and stimulate pro‐immune cytokines, while in mice, iPEMs drain to LNs and drive potent expansion of antigen‐specific CD8+ T cells (Chiu et al., 2015a; Zhang et al., 2015b).
In assessing the efficacy of iPEMs, or any vaccine system, injection route, dose, and vaccine composition play an important role in determining the immunogenicity. Administration route, for example, is critical in determining not only the potency, but also in polarizing the type of response (Eggert et al., 1999; Goetsch et al., 2000). This influence exists because the anatomical and physiological environments at the injection site and during trafficking of antigen imprint unique cues on both the innate and adaptive immune cells involved in antigen presentation and in effector response (Pavot et al., 2012; Zhang et al., 2015a). The impact of injection route on efficacy has been studied using a range of vaccine classes (Bremer et al., 2014; Jelley‐Gibbs et al., 2012; Luhrs et al., 2001; Pittman, 2002). In one example, virus‐like particles (VLPs) have been employed to investigate the impact of injection route on trafficking and immunization in response to simian‐human immunodeficiency VLPs (Cubas et al., 2009). This study revealed that intradermal (i.d.) injection resulted in the highest level of antibody‐mediated and cellular immunity, and these effects resulted from better lymphatic drainage and entry to the subcapsular sinuses. This improved drainage resulted in the greatest formation of germinal centers—structures integral in the generation or high affinity antibodies.
Along similar lines, newer selective adjuvants—such as TLRas—can often drive efficient responses even if drainage to LNs or spleen is more limited (Wilson et al., 2007). This is an advantage over more traditional adjuvants such as alum, which do not offer selective triggering of specific immune pathways (Andorko et al., 2015). In particular, toll‐like receptors have been under intense study as vaccine adjuvants because these ligands trigger the TLR pathways that have evolved to detect molecular patterns common in pathogens (e.g., viruses, bacteria), but that are uncommon in humans (Steinhagen et al., 2011). Further, many recent studies indicate that delivery of multi‐functional adjuvants—for example, combinations of TLR agonists (TLRas)—that activate several distinct TLRs or inflammatory pathways can lead to significantly more robust immunity (Bagchi et al., 2007; Duthie et al., 2011; Kasturi et al., 2011; Pavot et al., 2014; Steinhagen et al., 2011; Tom et al., 2013; Wu et al., 2014; Zhu et al., 2010). Thus, technologies that provide modularity to deliver TLRas or defined adjuvants alone or in combination, without extensive reformulation, could improve the robustness of vaccines.
Toward the goals above, here we studied the immunogenicity of iPEMs as a function of vaccine dose, adjuvant choice, immunization route, and injection schedule. These vaccines are self‐assembled from a model antigen, SIINFEKL, and TLRas for either the TLR3 (polyIC) or TLR9 (CpG) pathways. We show these iPEMs can be assembled and that in mice, intra‐dermal injection leads to the most potent and durable expansion of antigen‐specific T cells. These effects are dose‐dependent and, importantly, drive long‐lasting memory that generates a strong recall response during vaccine challenge 49 days after a prime‐boost regimen. In mice, iPEMs provide protection against a melanoma model expressing an exogenous antigen in a manner that is a function of both the formulation (i.e., ad‐mixed vs. iPEMs) and the choice of TLRa.
Materials and Methods
Materials
Peptides from ovalbumin (SIINFEKL, “SIIN”; SIINFEKL‐R9; “SIIN*”) were synthesized by GenScript. The peptides had a purity >98% and were synthesized with or without a fluorescein (FITC) label on the N‐terminus. PolyIC was purchased from Invivogen. CpG (5′ T‐C‐C‐A‐T‐G‐A‐C‐G‐T‐T‐C‐C‐T‐G‐A‐C‐G‐T‐T 3′) was synthesized by IDT. Gold(III) chloride trihydrate (99.9%) and chitosan (MW = 2000) were from Sigma. TE buffer was purchased from Amresco (Solon, OH). RPMI‐1640 media was purchased from Lonza (Allendale, NJ) and fetal bovine serum (FBS) was supplied by Corning (Tewksbury, MA). 2‐Mercaptoethanol was purchased from Sigma–Aldrich (St. Louis, MO). HEPES and non‐essential amino acids were purchased from VWR (Radnor, PA). L‐Glutamine, Penicillin‐Streptomycin, and DAPI were purchased from Thermo Fisher Scientific (Grand Island, NY). Spleen Dissociation Medium was from STEMCELL Technologies (Vancouver, British Columbia, Canada). CD11c microbeads were purchased from Miltenyi Biotec (Cambridge, MA). Fluorescent antibody conjugates were purchased from BD (San Jose, CA).
Cells and Animals
All animals used for in vivo and in vitro studies were female C57BL/6J mice (4–12 weeks, stock #000664) purchased from Jackson Laboratories (Bar Harbor, ME). All animal studies were carried out in compliance with Federal, State, and local guidelines, and using protocols reviewed and approved by the University of Maryland's Institutional Animal Care and Use Committee (IACUC).
iPEM Preparation and Characterization
AuNPs were prepared and coated with iPEMs using an alternating deposition process similar to that which we recently reported (Zhang et al., 2015b). Briefly, 50 mL of chitosan solution (0.3%, w/v) in 1% acetic acid was heated to 100°C and mixed with 40 µL aqueous chloroauric acid (HAuCl4, 0.01 M). The solution was maintained at 100°C for 25 min to obtain a red colored dispersion. 1.9 mg of AuNP was collected by centrifugation (15,000 rcf, 15 min) and re‐suspended 100 µL of DI water. AuNPs were then added to 900 µL of polyIC or CpG solution (for each cargo, 500 µg/mL in DI water adjusted to pH 4.0), mixed by pipetting, and placed in a sonic water bath for 45 s at room temperature. The suspension was maintained for 5 min, collected by centrifugation at 4°C (12,500 rcf, 15 min), and then washed with DI water to obtain AuNP‐polyIC or AuNP‐CpG. Following centrifugation and resuspension in a fresh aliquot of 100 µL of DI water, polyIC or CpG‐coated AuNPs were incubated with 900 µL of peptide SIIN* (pH = 5.0, 500 µg/mL) and washed as above to obtain AuNP‐(polyIC/SIIN*) or AuNP‐(CpG/SIIN*). These steps were repeated until the desired numbers of layers of each component were deposited. The sizes of iPEM‐modified AuNPs were measured by dynamic light scattering (DLS) using a Zetasizer Nano Z Analyzer. Loading of TLRas and peptides on AuNP were characterized by UV‐Vis absorbance of deposition solutions using the Beer–Lambert law at a wavelength of 260 nm for polyIC or CpG, and standard curves prepared at 488 nm for FITC‐labeled peptides.
In Vivo Immunization Studies
Mice, in groups of 4–5 as indicated, were unimmunized or immunized via intramuscular (i.m.), intradermal (i.d.), or subcutaneous (s.c.) injection using a volume of 25 µL at each flank. In some studies, groups received equivalent doses of the indicated vaccines in either soluble form (“Soluble”) prepared by mixing just prior to injection, or formulated as iPEMs formulated by coating four layers on AuNPs [i.e., AuNP‐(polyIC/SIIN*)2 or AuNP‐(CpG/SIIN*)2]. Mice were primed on day 0 and, in some studies, received a booster injection on either day 14 or 21. For long‐term recall studies, mice received a vaccine challenge on day 70 using the same route and form as the priming and booster injections.
In Vivo Analysis of Antigen‐Specific CD8+ T Cell Expansion
Peripheral blood was collected from mice at day 0, 7, 14, 21, 28, and 35. For recall studies, blood samples were drawn from mice on days 70 and 77. The blood samples were treated twice with ACK lysing buffer (1 mL, Life Technologies) for 5 min, collected by centrifugation (800g, 5 min), then washed twice in PBS. Cells were blocked with Fc block (BD Biosciences), followed by SIINFEKL MHC‐I tetramer (PE conjugate, BioLegend) for 30 min (25× dilution) and for CD8a (100× dilution, APC conjugate; BD Biosciences) for 20 min. The stained cells were washed and re‐suspended in DAPI (1000× dilution), analyzed by flow cytometry on a BD FACS CantoII, then data was processed using FlowJo.
Tumor Studies
Mice in groups of 4–5 were vaccinated on day 0, followed with a booster injection on day 14. On day 21, mice were implanted with 3 × 105 B16 melanoma cells expressing ovalbumin (OVA) protein. The B16‐OVA tumor cells were a generous gift from Dr. Kenneth Rock. Mice were monitored daily for body weight and tumor burden. Tumor sizes were measured as the product of two orthogonal diameters. Mice were euthanized when aggregate tumor burden reached IACUC‐approved endpoints of 150 mm2.
Statistical Analysis
One‐way ANOVA with a Tukey post‐test run with Graphpad Prism (version 6.02) was employed for statistical analysis. P values of *, <0.05; **, <0.01; and ***, <0.001 were used to indicate statistical significance. Data are reported as mean values ± standard error of the mean (SEM). All experiments were conducted using replicates of four samples (e.g., cell culture wells) or animal group sizes of 4–5 mice per group. Data shown in all figures are representative examples of 2–4 experiments with similar results.
Results
We first assembled iPEMs on AuNPs using CpG (polyanionic) and SIINFEKL peptide modified with nonaarginine (SIIN*) to confer cationic charge. (CpG/SIIN*)2 iPEM particles exhibited sizes of 103 ± 32 nm (Fig. S1), and loading levels of 96.2 ± 9.3 μg CpG per 1 mg of AuNPs and 65.6 ± 4.7 μg SIIN* per 1 mg of AuNPs. To confirm iPEMs remain intact—rather than dissembling and releasing components in dissociated or free forms—we next built AuNP‐(CpG/SIIN*)2 iPEMs from fluorescent SIIN* and fluorescent CpG, then incubated these particles in PBS. At defined times, the iPEM‐coated AuNPs were collected and the amount of release of each component (i.e., SIIN*, CpG) in the supernatant were measured by fluorimetry. After 48 h, less than 4% of SIIN* was released, while no CpG was detected, suggesting iPEMs are stable after assembly (Fig. S2). To assess the impact of injection route on iPEM‐induced T cell response, we immunized mice with (CpG/SIIN*)2 using either i.m., s.c., or i.d. injection routes. Mice were primed on day 0 and boosted on day 21 using a homologous prime‐boost regimen (Fig. 1A). For each injection route, vaccination resulted in detectable levels of SIINFEKL‐specific CD8+ T cells 1 week after the priming injection; however, only the i.d. route drove a statistically significant increase (Fig. 1B and C). These T cell populations then contracted by day 21, at which time mice were boosted. One week later (day 28), all groups exhibited increased T cell responses that were larger than the primary responses for each group at day 7, as well as the baseline value of 0.30 ± 0.04% measured in the naïve group at day 28 (Fig. 1D). We again observed that the magnitude of SIIN‐specific CD8+ T cell response was a function of injection route, with frequencies of 4.54 ± 1.11%, 2.62 ± 0.29%, and 1.32 ± 0.33%, for i.d., s.c., and i.m. injection, respectively (Fig. 1D). There were statistical significances between the i.d. versus naïve (***P < 0.001), s.c. versus naïve (*P < 0.05) and i.d. versus i.m (**P < 0.01) groups. These increases in antigen‐specific cell populations were clearly observable in representative flow cytometry scatter plots from each group, with values of 0.27%, 1.26%, 2.99%, and 3.84% for the naïve, i.m., s.c., and i.d. groups, respectively (Fig. 1E). Thus, iPEMs generate primary responses and more potent recall responses that are both antigen‐specific and a function of the injection route used for immunization.
Figure 1.
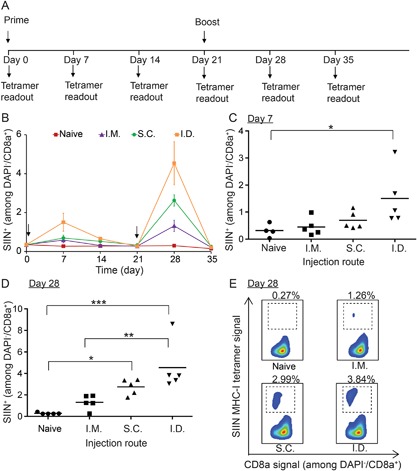
Impact of injection route on expansion of SIIN‐specific CD8+ T cells. Three injection routes were employed: i.m., s.c., and i.d. (A) Schematic representation depicting experiment design and vaccination regimen. Mice were primed on day 0 and boosted on day 21. (B) Frequency of SIIN‐specific CD8+ T cells over 35 days. Statistical analysis of SIIN‐specific CD8+ T cell level in each group on (C) day 7 and (D) day 28. (E) Representative scatter plots showing distributions of SIIN‐specific and CD8+ T cells on day 28. In each group, the mice were injected with iPEMs composed of 65.6 µg SIIN* and 96.2 µg CpG. *P < 0.05; **P < 0.01; ***P < 0.001.
To test the durability and memory capacity of T cell responses generated by iPEM vaccines, the groups in Figure 1 were monitored until T cells contracted (day 35), then a vaccine challenge was administered 35 days later (day 70) using the same formulations and routes employed during priming and boosting (Fig. 2A). At day 35, no significance differences were observed between groups, with SIIN‐specific T cell frequencies in the naïve, i.m., s.c., and i.d. groups of 0.16 ± 0.04%, 0.22 ± 0.03%, 0.24 ± 0.06%, and 0.21 ± 0.05%, respectively. As expected, just prior to the vaccine challenge on day 70, baseline levels were observed in these same groups: 0.09 ± 0.02%, 0.09 ± 0.01%, 0.03 ± 0.01%, and 0.16 ± 0.05% (Fig. 2B and C). Seven days after the vaccine challenge (day 77), iPEMs drove a potent recall response that was dependent on injection route (Fig. 2D and E). For each route, these values were greater than both the naïve group, and the corresponding value observed with each route after the booster injection. In particular, the frequency of antigen‐specific T cells in naïve, i.m., s.c., and i.d. groups were 0.26 ± 0.02%, 2.55 ± 0.40%, 4.85 ± 0.45%, and 7.15 ± 1.61% on day 77, respectively, (Fig. 2D and E) compared with 0.29 ± 0.04%, 1.32 ± 0.33%, 2.62 ± 0.29%, and 4.54 ± 1.11% on day 28 in each group (Fig. 1D and E). Together, the results in Figures 1 and 2 demonstrate iPEMs generate responses that are strong and durable, providing synergistic recall responses 49 days after a booster injection.
Figure 2.
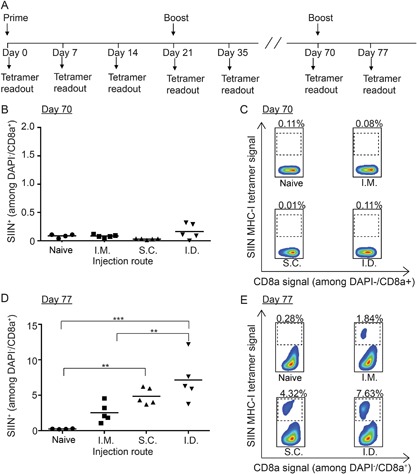
Durability of SIIN‐specific CD8+ T cells in mice immunized with iPEMs. (A) Schematic representation depicting experiment design and vaccination regimen. Mice were primed on day 0 and boosted on day 21 as in Figure 1, then boosted on day 70. Frequency (B, D) and representative flow cytometry scatter plots (C, E) of SIIN‐specific CD8+ T cells in peripheral blood of mice on day 70 (B, C) and day 77 (D, E). **P < 0.01; ***P < 0.001.
After identifying i.d. injection as the most efficient route for inducing antigen‐specific CD8+ T cell responses, we used this route to study the impact of iPEM dose on expansion of this cell population. Three dosing levels were tested, with a base dose (1×) corresponding to 16 μg SIIN* and 24 μg CpG, 4× (corresponding to the dose in Figs. 1 and 2), or a 16× dose. For each group, the prime (day 0) and booster injections (day 14) were identical (Fig. 3A). Vaccination with iPEMs resulted in an increase in SIIN‐specific CD8+ T cells by day 7, with the magnitude of response correlated to dose (Fig. 3B). The frequencies of SIIN‐specific CD8+ T cells one week after the prime injection were 0.41 ± 0.07%, 0.80 ± 0.14%, 1.13 ± 0.19%, and 1.81 ± 0.11% for mice in the naive, 1×, 4×, and 16× groups, respectively (Fig. 3C). There were statistical difference among the 16× versus naïve (P < 0.0001), 4× versus naïve (P < 0.05), 16× versus 1× (P < 0.001), and 16× versus 4× (P < 0.05) groups. Similar trends, but much larger in magnitude, were measured one week after the booster injection (day 21), yielding SIIN‐specific CD8+ T cell frequencies of 0.30 ± 0.13%, 1.43 ± 0.44%, 4.20 ± 1.62%, and 14.38 ± 1.88% for mice in the naive, 1×, 4×, and 16× groups, respectively. (Fig. 3D and E). These large frequencies were also reflected in the development of distinct populations of antigen‐specific CD8+ T cells in the flow cytometry scatter plots (Fig. 3E). Together, the data in this figure indicate combination of the optimal route and dose of iPEMs generates SIIN‐specific CD8+ T cell frequencies in peripheral blood as high as 15%.
Figure 3.
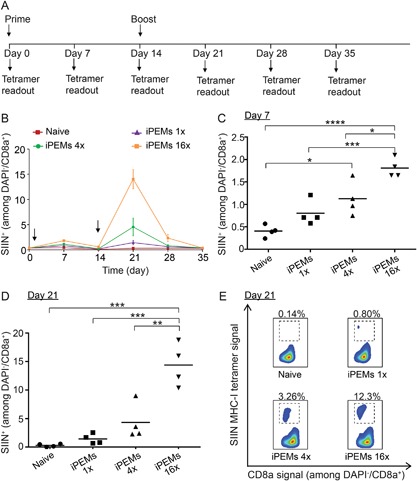
(A) Schematic representation depicting experiment design and vaccination regimen. Mice were primed on day 0 and boosted on day 14. (B) Expansion of SIIN‐specific CD8+ T cells mice as a function of iPEM dose over 35 days. Frequency of SIIN‐specific CD8+ T cells in each group on (C) day 7 and (D) day 21. (E) Representative flow cytometry scatter plots showing distributions of SIIN‐specific CD8+ T cells on day 21. *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001.
We next tested the functional capacity of the iPEM‐coated AuNPs using a melanoma model expressing ovalbumin (B16‐OVA). Since iPEMs allow modular assembly of essentially any polyion, in these studies we compared vaccines containing SIIN* and one of two TLRas, either polyIC (TLR3) or CpG (TLR9), which has recently been the focus of human cancer vaccines (Duthie et al., 2011; Steinhagen et al., 2011). Mice received vaccines formulated as (polyIC/SIIN*)2 iPEMs, (CpG/SIIN*)2 iPEMs, or as equivalent doses of each soluble component prepared by simple mixing just prior to injection. iPEMs coated on AuNPs exhibited a size of 90.5 ± 29.7 nm, with polyIC loading of 104.0 ± 5.5 μg per 1 mg of AuNPs and SIIN* loading of 64.9 ± 1.5 μg per 1 mg of AuNPs. Mice were primed on day 0, boosted on day 14, and then tumor challenge was performed on day 22 (Fig. 4A). On day 28, MHC‐I tetramer staining revealed a large population of circulating SIIN‐specific CD8+ T cells in peripheral blood collected from several of the treatment groups (Fig. 4B and C). Frequency measurements correlated to values of 0.35 ± 0.06%, 1.03 ± 0.31%, 8.29 ± 0.62%, 9.72 ± 1.90%, and 14.9 ± 1.80%, respectively for naïve, soluble polyIC/SIIN, soluble CpG/SIIN, (polyIC/SIIN*)2 iPEMs, and (CpG/SIIN*)2 iPEMs. In particular, iPEMs drove greater expansion compared to the corresponding soluble formulations, and, further, CpG generally induced larger responses compared to polyIC. Correlating with T cell expansion levels observed during tumor challenge, (CpG/SIIN*)2 iPEMs were most effective in slowing tumor growth compared to all other formulations (Fig. 4D–F, Fig. S3). (CpG/SIIN*)2 iPEMs conferred survival to 50% of mice for the duration of the 84 day study, compared to 25% in the soluble groups, and 0% in the other groups (Fig. 4D). iPEM‐mediated enhancements in anti‐tumor immunity were also reflected in the median survival for each group, which were 42, 42, 47.5, 53.5, and 73.5 days for naïve, soluble (polyC/SIIN), soluble (CpG/SIIN), (polyIC/SIIN*)2 iPEM, and (CpG/SIIN*)2 iPEM groups, respectively (Fig. 4D). Of note, on the day the first animal from the (SIIN*/CpG)2 iPEM group developed a tumor (Day 40), the mean burden in this group was 4.0 ± 3.7 mm2 compared with 107.2 ± 27.0, 101.9 ± 37.2, 29.1 ± 17.3, and 6.18 ± 4.72 mm2 in naive, soluble (polyC/SIIN), soluble (CpG/SIIN), and (polyIC/SIIN*)2 iPEM groups, respectively (Fig. 4E and F). Thus CpG‐containing iPEMs increase survival relative to soluble formulations, while iPEMs containing either CpG or polyIC slowed tumor growth relative to soluble formulations.
Figure 4.
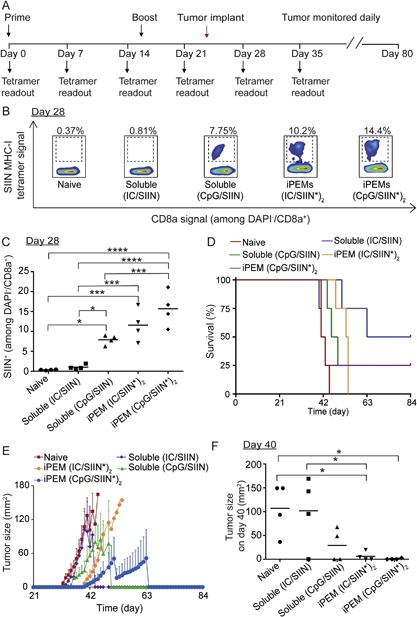
Enhancement of anti‐tumor immunity by iPEMs. (A) Schematic representation depicting experiment design and tumor challenge regimen. Mice were primed on day 0 and boosted on day 14 with iPEMs or soluble mixture of antigen and adjuvant containing the equivalent doses. On day 21, mice were challenged with 3 × 105 B16‐OVA cells. (B) Representative flow cytometry scatter plots and (C) Frequency of antigen‐specific CD8+ T cell expansion on day 28 (i.e., 7 days after tumor implantation). (D) Survival and (E) aggregate tumor burden for each group over 84 days. (F) Tumor burden for each group on day 40, the onset of the first tumor in the iPEM (CpG/SIIN*)2 group. For all formulations, doses—in soluble or iPEM form—were 65.6 μg SIIN, and either 105.2 μg polyIC or 96.2 μg CpG. *P < 0.05; ***P < 0.001; ****P < 0.0001.
Discussion
In our previous study with iPEMs we showed these structures elicit strong T cell responses in mice (Zhang et al., 2015b). To build our understanding of the iPEM platform, here we investigated the impact of injection route, doses, and TLRa choice on T cell expansion and anti‐tumor immunity. We found that all three types of injection routes (i.e., i.m., s.c., i.d.) expand antigen‐specific T cells, but that i.d. injections are most effective. One factor likely contributing to this finding is the presence of skin‐resident immune cells (e.g., Langerhans cells) able to efficiently capture antigens directly from the skin (Heath and Carbone, 2013). Similar results have been observed in route comparison studies for other types of vaccines (Heath and Carbone, 2013; Zinkernagel et al., 1997). Further, many lymphatic structures are present in the intradermal region of skin, where two plexuses compose lymphatic vessels: one of the plexuses drains into larger lymphatic vessels in the lower dermis, and the other drains to the subcutaneous zone in the superficial part of skin (Skobe and Detmar, 2000). These networks can facilitate trafficking of antigens—including iPEMs—directly from the skin and through lymphatic vessels to draining lymph nodes. In comparison to i.d. injection, i.m. and s.c. injections resulted in lower levels of antigen‐specific T cells. This was likely due to less efficient delivery of iPEMs to draining lymph nodes. During i.m. injection, for example, lymphatic capillaries do not exist in most muscular bundles; this route typically relies on exceeding the void volume of the muscle with the injection fluid to cause inflammation and recruit immune antigen presenting cells able to internalize antigens or vaccines (Lu and Hogenesch, 2013).
As with injection route, most vaccines also exhibit a dose effect, a characteristic that underscores the importance of potency for vaccine efficacy and with respect to manufacturing and feasibility. As alluded to in the introduction, synthetic adjuvants also provide new opportunity to gain more rational control over immunogenicity and immune polarization (Wu et al., 2014). At the same time, recent studies are demonstrating the potential of adjuvants that activate more diverse sets of stimulatory immune pathways—TLRs, for example—that can drive inflammation to enhance both innate and adaptive immune responses (Steinhagen et al., 2011). In our studies, iPEMs allowed facile incorporation of TLR3 or TLR9 agonists using identical protocols. The simplicity of this approach—and the elimination of all other components, along with heating, cooling, solvents, and other complicating manufacturing steps—illustrates some of the attractive features of iPEMs for vaccine delivery. Additionally, while TLRas are being exploited as mono‐adjuvants in numerous preclinical and clinical studies (Duthie et al., 2011), an exciting area of discovery centers on recent reports showing mixtures of TLRas can generate adjuvant functions broader than those associated with the individual agonists; these effects can be synergistic, or even suppressive (Bagchi et al., 2007; Tom et al., 2013). For example, preclinical reports demonstrate co‐administration of TLR2a (macrophage‐activating lipoprotein 2; MALP‐2), TLR3a (polyinosine‐polycytidylic acid, polyIC), and TLR9a (CpG) drives synergistic T cell‐mediated antiviral responses (Zhu et al., 2010). Recent work also demonstrates that co‐activating TLR4 and TLR7 can be exploited to enhance antibody‐mediated immunity (Kasturi et al., 2011). Additional new studies indicate that activating several distinct classes of pathogen‐associated molecular pattern (PAMPS) receptors—TLRs and NOD‐like receptors, for example—can enhance immunity by generating multiple types of antibody isotypes (i.e., IgA, IgG) (Pavot et al., 2014). The modularity of iPEMs is well‐suited for such strategies, and could be exploited in future studies to formulate vaccines with tunable control over the absolute and relative loading of several TLRas used to build iPEMs.
Along these same lines, the form of how signals in iPEMs are delivered and trafficked in immune cells could provide additional opportunities to bias response. In our past work, for example, we observed that iPEMs efficiently co‐deliver antigen and adjuvant to antigen presenting cells in vitro and to draining lymph nodes in mice (Chiu et al., 2015b, 2016). In our current study, we found that iPEM components do not dissociate from AuNP templates, while others have shown generally that PEMs layers are interpenetrating within the films (Pavoor et al., 2004). Together, these data suggest that immune cells might be able to simultaneously process both components, irrespective of the specific film architectures, though the internalization and trafficking mechanisms remain to be elucidated.
In mice, our dose study revealed expansion of SIIN‐specific T cells induced by iPEMs was strong and dose dependent. Since antigen‐specific T cells play an important role in anti‐tumor immunity, we employed an exogenous antigen melanoma model as an initial functional test bed to assess iPEM‐coated AuNP vaccines. During tumor studies, the highest frequency of antigen‐specific CD8+ T cell response [i.e., (SIIN*/CpG)2 iPEMs] correlated with the efficacy. This result is not unexpected, as many recent clinical cancer vaccine trials in humans have shown promise using TLR9 agonists (e.g., CpG) as mono‐adjuvants or in combination with other adjuvants or immunotherapies (Shirota and Klinman, 2014; Shirota et al., 2015). Interestingly, tetramer frequencies associated with other iPEM configurations [e.g., (SIIN*/polyIC)2 iPEMs] only loosely correlated to efficacy; this finding is likely due to the limited impact of TLR3 agonists for driving anti‐tumor efficacy, along with the more general range of hurdles facing existing treatments. As one example, tumors establish a suppressive microenvironment that inactivates infiltrating immune cells targeting the tumor. Thus, expansion of a large set of tumor specific T cells is only part of the goal, as these cells must exhibit the ability to maintain functionality in the tumor environment. This latter idea might suggest additional opportunities to incorporate cues into iPEMs that help establish more potent or higher affinity T cells. Notwithstanding this challenge and the need for testing in more clinically relevant cancer models; overall the findings above indicate that antigen‐specific T cells can be rationally expanded by juxtaposing different TLRas with the same antigen in iPEMs, and that the responses generated by these structures are functional in slowing or preventing tumor growth in mice.
To test the durability of iPEM vaccines, we performed a recall study 70 days after priming mice (49 days after boosting). This vaccine challenge caused a dramatic increase in SIIN‐specific CD8+ T cells. This result is of general interest for vaccine design because the data indicate iPEMs induce immune cells that exhibit memory capacity (i.e., successive encounters with antigen resulted in increasingly strong antigen‐specific responses). In the context of cancer, this idea is particularly important because one of the major problems with existing treatments is relapse. Thus, one likely strategy for deploying cancer vaccines is as an adjunctive or combination therapy (e.g., with chemotherapy or checkpoint blockade) (Gammon et al., 2016; Hu and Zhang, 2012). For example, during resection, incompletely removed tumors—or remaining peripheral tumor cells—can cause relapse; so, therapies that provide enduring immune cell populations able to destroy residual cells could be instrumental in helping to overcome this hurdle. Follow‐on studies with iPEMs will be needed to directly confirm the presence and phenotype of memory T cells, how effective these populations may be in anti‐tumor immunity (e.g., through adoptive‐transfer studies), as well as whether design of iPEMs with multiple TLRas—or designed ratios of TLRas—correlate with improvements to effector or memory responses.
Conclusion
Building on our initial report, here we studied the roles of injection route, dose, and TLRa adjuvant selection on expansion and efficacy of anti‐tumor T cells. Our study reveals the potency of iPEMs is a function of dose, injection route, and composition. Importantly, the responses generated by iPEMs are durable, enabling strong recall responses 49 days after a prime‐boost immunization regimen. In the tumor model, iPEM vaccines can inhibit tumor growth more effectively than a mixture of soluble antigen and adjuvant containing an equivalent dose of cargos. Together, these results demonstrate the simple, modular nature of iPEMs, with features that could enable more rational design and easier translation owing to more defined vaccine compositions.
This work was supported in part by NSF CAREER Award #1351688, Alliance for Cancer Gene Therapy Young Investigator Award #15051543, and the University of Maryland Division of Research (Tier 1). C.M.J is a Damon Runyon‐Rachleff Innovator supported by the Damon Runyon Foundation (#DRR3415) and a Young Investigator supported by the Melanoma Research Alliance (#348963). J.I.A. is a trainee on NIH Grant #T32 AI089621 and a Graduate Fellow supported by the American Association of Pharmaceutical Scientists Foundation.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Figure S1. Dynamic light scattering size distribution of iPEMs in PBS after assembly using a configuration of AuNPs‐(CpG/SIIN*)2.
Figure S2. Stability of iPEMs as indicated by measurement of the concentration of each iPEM component in the supernatant of PBS solutions used to incubate iPEMs after assembly.
Figure S3. Tumor growth curves for each animal in the tumor challenge study depicted in Figure 4 of the main text.
Conflict of interest: The authors declare no conflicts of interest.
References
- Andorko JI, Hess KL, Jewell CM. 2015. Harnessing biomaterials to engineer the lymph node microenvironment for immunity or tolerance. AAPS J 17(2):323–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagchi A, Herrup EA, Warren HS, Trigilio J, Shin HS, Valentine C, Hellman J. 2007. MyD88‐dependent and MyD88‐independent pathways in synergy, priming, and tolerance between TLR agonists. J Immunol 178(2):1164–1171. [DOI] [PubMed] [Google Scholar]
- Bremer PT, Schlosburg JE, Lively JM, Janda KD. 2014. Injection route and TLR9 agonist addition significantly impact heroin vaccine efficacy. Mol Pharm 11(3):1075–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YC, Gammon JM, Andorko JI, Tostanoski LH, Jewell CM. 2015a. Modular vaccine design using carrier‐Free capsules assembled from polyionic immune signals. ACS Biomater Sci Eng 1(12):1200–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YC, Gammon JM, Andorko JI, Tostanoski LH, Jewell CM. 2015b. Modular vaccine design using carrier‐Free capsules assembled from polyionic immune signals. ACS Biomater Sci Eng 1(12):1200–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YC, Gammon JM, Andorko JI, Tostanoski LH, Jewell CM. 2016. Assembly and immunological processing of polyelectrolyte multilayers composed of antigens and adjuvants. ACS Appl Mater Interfaces 8(29):18722–18731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubas R, Zhang S, Kwon SK, Sevick‐Muraca EM, Li M, Chen CY, Yao QZ. 2009. Virus‐like particle (VLP) lymphatic trafficking and immune response generation after immunization by different routes. J Immunother 32(2):118–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duthie MS, Windish HP, Fox CB, Reed SG. 2011. Use of defined TLR ligands as adjuvants within human vaccines. Immunol Rev 239:178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggert AAO, Schreurs MWJ, Boerman OC, Oyen WJC, de Boer AJ, Punt CJA, Figdor CG, Adema GJ. 1999. Biodistribution and vaccine efficiency of murine dendritic cells are dependent on the route of administration. Cancer Res 59(14):3340–3345. [PubMed] [Google Scholar]
- Gammon JM, Dold NM, Jewell CM. 2016. Improving the clinical impact of biomaterials in cancer immunotherapy. Oncotarget 7:15421–15443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetsch L, Plotnicky‐Gilquin H, Champion T, Beck A, Corvaia N, Stahl S, Bonnefoy JY, Nguyen TN, Power UF. 2000. Influence of administration dose and route on the immunogenicity and protective efficacy of BBG2Na, a recombinant respiratory syncytial virus subunit vaccine candidate. Vaccine 18(24):2735–2742. [DOI] [PubMed] [Google Scholar]
- Heath WR, Carbone FR. 2013. The skin‐resident and migratory immune system in steady state and memory: Innate lymphocytes, dendritic cells and T cells. Nat Immunol 14(10):978–985. [DOI] [PubMed] [Google Scholar]
- Hu CM, Zhang L. 2012. Nanoparticle‐based combination therapy toward overcoming drug resistance in cancer. Biochem Pharmacol 83(8):1104–1111. [DOI] [PubMed] [Google Scholar]
- Jelley‐Gibbs D, Winfrey C, Eaton S, Haynes L. 2012. Protective efficacy of CD4 memory T cells depends on route of administration of inactivated influenza vaccine. J Immunol 188:113.31. [Google Scholar]
- Josefsberg JO, Buckland B. 2012. Vaccine process technology. Biotechnol Bioeng 109(6):1443–1460. [DOI] [PubMed] [Google Scholar]
- Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, Ravindran R, Stewart S, Alam M, Kwissa M, Villinger F, Murthy N, Steel J, Jacob J, Hogan RJ, García‐Sastre A, Compans R, Pulendran B. 2011. Programming the magnitude and persistence of antibody responses with innate immunity. Nature 470(7335):543–U136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Hogenesch H. 2013. Kinetics of the inflammatory response following intramuscular injection of aluminum adjuvant. Vaccine 31(37):3979–3986. [DOI] [PubMed] [Google Scholar]
- Luhrs P, Kutil R, Buschle M, Schmidt W, Stingl G, Schneeberger A. 2001. Modulating specific immune responses by varying the route of vaccine administration. J Invest Dermatol 117(2):451–451. [Google Scholar]
- Melchels FP, Fehr I, Reitz AS, Dunker U, Beagley KW, Dargaville TR, Hutmacher DW. 2015. Initial design and physical characterization of a polymeric device for osmosis‐driven delayed burst delivery of vaccines. Biotechnol Bioeng 112(9):1927–1935. [DOI] [PubMed] [Google Scholar]
- Moyano DF, Goldsmith M, Solfiell DJ, Landesman‐Milo D, Miranda OR, Peer D, Rotello VM. 2012. Nanoparticle hydrophobicity dictates immune response. J Am Chem Soc 134(9):3965–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SN, Germain RN. 2009. Stromal cell contributions to the homeostasis and functionality of the immune system. Nat Rev Immunol 9(9):618–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niikura K, Matsunaga T, Suzuki T, Kobayashi S, Yamaguchi H, Orba Y, Kawaguchi A, Hasegawa H, Kajino K, Ninomiya T, Ijiro K, Sawa H. 2013. Gold nanoparticles as a vaccine platform: Influence of size and shape on immunological responses in vitro and in vivo. ACS Nano 7(5):3926–3938. [DOI] [PubMed] [Google Scholar]
- Pavoor PV, Bellare A, Strom A, Yang DH, Cohen RE. 2004. Mechanical characterization of polyelectrolyte multilayers using quasi‐static nanoindentation. Macromolecules 37(13):4865–4871. [Google Scholar]
- Pavot V, Rochereau N, Genin C, Verrier B, Paul S. 2012. New insights in mucosal vaccine development. Vaccine 30(2):142–154. [DOI] [PubMed] [Google Scholar]
- Pavot V, Rochereau N, Rességuier J, Gutjahr A, Genin C, Tiraby G, Perouzel E, Lioux T, Vernejoul F, Verrier B, Paul S. 2014. Cutting edge: New chimeric NOD2/TLR2 adjuvant drastically increases vaccine immunogenicity. J Immunol 193(12):5781–5785. [DOI] [PubMed] [Google Scholar]
- Pittman PR. 2002. Aluminum‐containing vaccine associated adverse events: Role of route of administration and gender. Vaccine 20:S48–S50. [DOI] [PubMed] [Google Scholar]
- Shirota H, Klinman DM. 2014. Recent progress concerning CpG DNA and its use as a vaccine adjuvant. Expert Rev Vaccines 13(2):299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirota H, Tross D, Klinman DM. 2015. CpG oligonucleotides as cancer vaccine adjuvants. Vaccines (Basel) 3(2):390–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skobe M, Detmar M. 2000. Structure, function, and molecular control of the skin lymphatic system. J Investig Dermatol Symp Proc 5(1):14–19. [DOI] [PubMed] [Google Scholar]
- Steinhagen F, Kinjo T, Bode C, Klinman DM. 2011. TLR‐based immune adjuvants. Vaccine 29(17):3341–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz MA, Hirosue S, Hubbell JA. 2012. Engineering approaches to immunotherapy. Sci Transl Med 4(148). [DOI] [PubMed] [Google Scholar]
- Tom JK, Mancini RJ, Esser‐Kahn AP. 2013. Covalent modification of cell surfaces with TLR agonists improves & directs immune stimulation. Chem Commun (Camb) 49(83):9618–9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson KD, Raney SG, Sekirov L, Chikh G, deJong SD, Cullis PR, Tam YK. 2007. Effects of intravenous and subcutaneous administration on the pharmacokinetics, biodistribution, cellular uptake and immunostimulatory activity of CpG ODN encapsulated in liposomal nanoparticles. Int Immunopharmacol 7(8):1064–1075. [DOI] [PubMed] [Google Scholar]
- Wu TYH, Singh M, Miller AT, De Gregorio E, Doro F, D'Oro U, Skibinski DAG, Mbow ML, Bufali S, Herman AE, others. 2014. Rational design of small molecules as vaccine adjuvants. Sci Transl Med 6(263). [DOI] [PubMed] [Google Scholar]
- Zhang L, Wang W, Wang S. 2015a. Effect of vaccine administration modality on immunogenicity and efficacy. Expert Rev Vaccines 14(11):1509–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang PP, Chiu YC, Tostanoski LH, Jewell CM. 2015b. Polyelectrolyte multilayers assembled entirely from immune signals on gold nanoparticle templates promote antigen‐Specific t cell response. ACS Nano 9(6):6465–6477. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Egelston C, Gagnon S, Sui Y, Belyakov IM, Klinman DM, Berzofsky JA. 2010. Using 3 TLR ligands as a combination adjuvant induces qualitative changes in T cell responses needed for antiviral protection in mice. J Clin Invest 120(2):607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkernagel RM, Ehl S, Aichele P, Oehen S, Kundig T, Hengartner H. 1997. Antigen localisation regulates immune responses in a dose‐ and time‐dependent fashion: A geographical view of immune reactivity. Immunol Rev 156:199–209. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Figure S1. Dynamic light scattering size distribution of iPEMs in PBS after assembly using a configuration of AuNPs‐(CpG/SIIN*)2.
Figure S2. Stability of iPEMs as indicated by measurement of the concentration of each iPEM component in the supernatant of PBS solutions used to incubate iPEMs after assembly.
Figure S3. Tumor growth curves for each animal in the tumor challenge study depicted in Figure 4 of the main text.