

ABSTRACT
Patients with anterior cruciate ligament (ACL) rupture are two times as likely to develop posttraumatic osteoarthritis (PTOA). Annually, there are ∼900,000 knee injuries in the United States, which account for ∼12% of all osteoarthritis (OA) cases. PTOA leads to reduced physical activity, deconditioning of the musculoskeletal system, and in severe cases requires joint replacement to restore function. Therefore, treatments that would prevent cartilage degradation post‐injury would provide attractive alternatives to surgery. Sclerostin (Sost), a Wnt antagonist and a potent negative regulator of bone formation, has recently been implicated in regulating chondrocyte function in OA. To determine whether elevated levels of Sost play a protective role in PTOA, we examined the progression of OA using a noninvasive tibial compression overload model in SOST transgenic (SOSTTG) and knockout (Sost‐/‐) mice. Here we report that SOSTTG mice develop moderate OA and display significantly less advanced PTOA phenotype at 16 weeks post‐injury compared with wild‐type (WT) controls and Sost‐/‐. In addition, SOSTTG built ∼50% and ∼65% less osteophyte volume than WT and Sost‐/‐, respectively. Quantification of metalloproteinase (MMP) activity showed that SOSTTG had ∼2‐fold less MMP activation than WT or Sost‐/‐, and this was supported by a significant reduction in MMP2/3 protein levels, suggesting that elevated levels of SOST inhibit the activity of proteolytic enzymes known to degrade articular cartilage matrix. Furthermore, intra‐articular administration of recombinant Sost protein, immediately post‐injury, also significantly decreased MMP activity levels relative to PBS‐treated controls, and Sost activation in response to injury was TNFα and NF‐κB dependent. These results provide in vivo evidence that sclerostin functions as a protective molecule immediately after joint injury to prevent cartilage degradation. © 2018 The Authors. Journal of Bone and Mineral Research Published by Wiley Periodicals Inc.
Keywords: OSTEOARTHRITIS, SCLEROSTIN, SOST, MMP, OSTEOPHYTE, WNT SIGNALING
Introduction
The increased risk of developing knee osteoarthritis (OA) after injury to the anterior cruciate ligament (ACL) has been well documented both clinically and in experimental models.1, 2 Clinical manifestation of posttraumatic osteoarthritis (PTOA) is characterized by narrowing of the joint space, emergence of osteophytes through osteoarthritic remodeling, cartilage erosion, and fibrillation.2 Biomechanical disturbances in the joint such as lateral subluxation of the tibia further the development of osteophytes in the lateral tibial‐femoral compartment and cause misalignment, rotation, and anterior subluxation of the joint; all these physical manifestations contribute to the emergence of intra‐articular lesions. Cartilage lesions become further exacerbated through molecular changes in the joint, including the increase in the production of matrix‐degrading enzymes, such as aggrecanases and metalloproteinases (MMPs). Elevated levels of catabolic enzymes enhance the loss of articular cartilage,3 increase the amount of pain experienced, and lead to impaired joint mobility in >50% of individuals who sustained an ACL tear.4, 5 In addition to changes in joint architecture and uneven biomechanical load distribution in the knee after ACL tear, the individual susceptibilities to inflammatory responses, enzymatic cartilage destruction, and osteophyte formation will also determine subsequent osteoarthritic outcomes.
Wnt/β‐catenin signaling was previously implicated in OA pathogenesis.6 Conditional activation of β‐catenin in the articular chondrocytes of adult mice resulted in reduced articular cartilage area, surface fibrillation, vertical clefting, and osteophyte formation, independent of trauma, suggesting that activation of Wnt signaling in the articular cartilage causes OA‐like phenotypes.7 Furthermore, β‐catenin has been shown to stimulate the activity of catabolic enzymes in the extracellular matrix (ECM) of the cartilage.8 These findings suggest that bone and cartilage are regulated by similar but functionally opposing mechanisms, where Wnt signaling is anabolic in bone but catabolic in the cartilage.9 Sclerostin (Sost) is a potent negative regulator of bone mass, where it normally inhibits Wnt signaling through interactions with low‐density lipoprotein receptor‐related protein (LRP) 5/6 co‐receptors.10, 11 In the absence of Sost protein, patients develop two types of hyperostosis, sclerosteosis and van Buchem disease.12, 13 Consistent with the human hyperosteosis, Sost‐deficient mice (Sost‐/‐) also acquire a generalized high bone mass phenotype.14, 15 Conversely, transgenic mice overexpressing SOST (SOSTTG or TG) are osteopenic.16
Until recently, Sost expression has been described as osteocyte‐specific, but several reports have now shown that Sost is also expressed in the articular cartilage. Elevated levels of Sost were observed in chondrocytes near damaged sites in the articular cartilage of mice and sheep subjected to surgical models of OA.6, 17 Similarly, transcriptional analysis found SOST to be upregulated ∼14‐fold in cartilage derived from biopsies of OA patients undergoing joint replacement surgery,18 suggesting that upregulation of SOST in cartilage may have a protective role. Although these observations have been correlative, in vivo evidence has been lacking in support of Sost as an anti‐catabolic agent in the joint. Here we investigated the role of Sost in the articular cartilage and found SOST to inhibit cartilage degradation subsequence to traumatic injury by downregulating catabolic enzymes whose expression is Wnt‐dependent. These findings suggest that elevated levels of Sost, immediately after injury, can aid the joint in maintaining its articular cartilage integrity in PTOA.
Materials and Methods
Mice strains and tibial compression OA injury
Sost‐/‐ and SOSTTG mice have been previously described and are on C57Bl/6 background.15, 16 Sost‐/‐ allele was generated by replacing the open reading frame with the LacZ reporter.19 SOSTTG express a ∼158‐kb human bacterial artificial chromosome (BAC; RM11‐209M4) that encompasses three genes (DUSP3, SOST, MEOX1) and the 90‐kb noncoding interval separating SOST from MEOX1; therefore, human SOST expression is physiological and regulated by endogenous human regulatory elements.16 Both Sost‐/‐ and SOSTTG were back‐crossed to C57Bl/6 >10 generations. Sost‐/‐ and SOSTTG were genotyped by PCR. Some SOSTTG animals display limb defects; only normal and animals missing one digit (thumb only) were used in these experiments. Mice were injured at 16 weeks of age using a previously described tibial compression OA injury model.1 In brief, a single dynamic compressive load was applied to the stationary (right) knee joint displacing the tibial condyle over the femoral condyle to induce an anterior cruciate ligament (ACL) rupture. In vivo experiments utilized a minimum of 5 animals (n ≥ 5) to reach statistical significance. All mice (C57Bl/6, Sost‐/‐ and SOSTTG) examined were bred in‐house and housed 2 to 3 animals per cage. Mice were allowed unlimited free range of food (Envigo, East Millstone, NJ, USA; cat. 2918, 18% protein) and water; mice were on a 12‐hour lights on/off cycle. All animal procedures were carried out in accordance with guidelines under the Institutional Animal Care and Use Committees at Lawrence Livermore National Laboratory and University of California, Davis.
Histology and OA evaluation
Injured and uninjured joints were dissected free of soft tissue, fixed, dehydrated, paraffin embedded, and sectioned as previously described.20 To visualize the cartilage, sagittal 6‐μm paraffin serial sections were stained with Safranin‐O (0.1%, Sigma, St. Louis, MO, USA; S8884) and Fast Green (0.05%, Sigma; F7252) using standard protocol from IHC world website. OA severity was evaluated at 1 day and 6‐, 12‐, and 16‐weeks after injury on sagittal sections using a modified osteoarthritis research society international (OARSI) scoring scale, as previously described.21 Starting from the synovium membrane to the articular cartilage, for each region, cartilage scoring began ∼0.4 mm out from the start of synovium. Blinded slides were evaluated by three scientists (two with and one without expertise in OA) utilizing a modified (sagittal) OARSI scoring parameter.21
Immunohistochemistry (IHC)
Sagittal serial sections were stained utilizing primary antibodies directed to mouse Sost (R&D Systems, Minneapolis, MN, USA; AF1589 [1 µg/mL]), human SOST (Abcam, Cambridge, MA, USA; ab75914 [100 µg/mL]), Collagen II (Abcam; ab21291 [1:50]), activated β‐catenin (Millipore, Billerica, MA, USA; 05‐665 [10 µg/mL]), MMP 2 (Abcam; ab110186 [5 µg/mL]), MMP 3 (Abcam; ab52915 [7.04 µg/mL]), MMP9 (Abcam; ab137867 [10.22 µg/mL]), MMP14 (Abcam; ab53712 [10 µg/mL]), and Furin (Abcam; ab3467 [20 µg/mL]). Trypsin/EDTA was used for antigen retrieval in 37°C for 30 minutes for all primary antibody except for activated β‐catenin and MMPs 2, 3, 9, and Furin, which required Uni‐trieve (Innovex Biosciences, Richmond, CA, USA) in 65°C for 30 minutes. After Uni‐trieve, activated β‐catenin and Furin require an additional retrieval with Proteinase K (Ambion, Austin, TX, USA; AM2546 [20 µg/mL]) for 20 minutes. This was followed by using Alexa‐Fluor 488 (green) or 594 (red) (Molecular Probes, Eugene, OR, USA) to determine protein expression.
Micro‐computed tomography (μCT)
The subchondral trabecular bone of the femoral epiphysis and the osteophyte volume quantifications were carried out as previously described.22
RNA sequencing (RNA‐seq)
Methodology was previously described.20 Briefly, whole knee joints were dissected, chopped, homogenized in Qiazol (79306, Qiagen, Valencia, CA, USA) lysis solution, and a fraction (1 mL) of the homogenate was used per RNA isolation. Isolated RNA (1–2 µg) was sequenced using Illumina (San Diego, CA, USA) HiSeq instrument, and a quality check was performed using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The reads were subsequently mapped to mouse genome (mm10) using TopHat.23, 24 Transcriptome assembly was performed using Cufflinks, and data was normalized using Cuffnorm.24, 25 A t test was performed to identify significantly (p < 0.05) differentially expressed (1.5‐fold) genes.
Quantitative real‐time PCR (qPCR)
Total RNA was purified using RNeasy Mini Kit (Qiagen), subsequent reverse transcription was done by Superscript III First‐Strand Synthesis System (Invitrogen, Carlsbad, CA, USA) with oligodT primers, and real‐time qPCR was performed with SYBR Select Master Mix (Applied Biosystems, Carlsbad, CA, USA); all were used according to the manufacturer's protocol. Applied Biosystems 7900HT Fast Real‐Time PCR System was utilized with the following cycling conditions: 50°C for 2 minutes for SYBR, then 95°C for 3 minutes (2 minutes for SYBR), followed by 40 cycles of 95°C for 3 seconds (10 seconds for SYBR) and 30 seconds at 60°C. Data were normalized to control genes (GAPDH), and fold changes were calculated using the comparative Ct method.26 Three independent replicates (each whole joint RNA) were analyzed for each genotype. The following primers were used: mouse Sost, 5′‐AGCCTTCAGGAATGATGCCAC‐3′ and 5′‐CTTTGGCGTCATAGGGATGGT‐3′; GAPDH, 5′‐CCAATGTGTCCGTCGTGGATCT‐3′ and 5′‐CCTCAGTGTAGCCCAAGATGC‐3′.
ChIP‐seq data analysis and transfections
NF‐κB binding sites were identified by mapping ENCODE NF‐κB (RELA) ChIP‐seq data sets to SOST locus (hg19) using UCSC genome browser. NF‐κB binding sites in hg19 coordinates were mapped to mm10 using ECR browser27 and conserved binding sites were identified. NF‐κB elements were PCRed from mouse genomic DNA using the primers listed in Table 1 and were cloned upstream of the TK promoter into the EcoRI site of GlucTK‐mini vector. ATDC5 cells were plated at 10% confluency in a 24‐well plate and cultured in differentiating media (DMEM/F12 + 5% FBS + insulin) for 7 days before transfection with media changes every 2 to 3 days. Plasmids were transfected using fugene 6 with a fugene/DNA ratio of 1:3. Twenty‐four hours after transfection, cells were treated with 100 ng/mL TNFα. Luciferase activity was measured 24 hours after TNFα treatment.
Table 1.
NF‐κB Putative Binding Sites Within Sost Locus
| NF‐κB binding sites (mm10) | Distance from TSS | Primers for PCR | Size (bp) | ECR name |
|---|---|---|---|---|
| chr11:101968161‐101969174 | –1146 a | ccagaatccacctgcctttcc/cctatctctctggaccctcct | 1111 | NF‐κ≡1 |
| chr11:101919485‐101920334 | +47530 | gggtatcgaatgcaggtcagc/gcttgcccaagtcacacaca | 927 | NF‐κ≡2 |
| chr11:101904120‐101905739 | +62895 | ggagggagccctttagttcag/caaggcatgggtctgaggac | 1672 | NF‐κ≡3 |
TSS = transcription start site; ECR = evolutionary conserved region.
ChipSeq hits map to promoter region, defined as 2 kb upstream of TSS. All binding sites map to ECRs (>100 bp; >70% human/mouse identity).
MMP activity (MMPSense) and intra‐articular (IA) injections
MMPSense (PerkinElmer, Waltham, MA, USA; NEV10168) was administered intravenously 5 hours after injury. Animals (PBS control [n = 5], IA experimental [n = 8]) were euthanized, skin was removed, and the joints were scanned using a Kodak (Rochester, NY, USA) image station 4000R digital imaging system at excitation and emission wavelengths of 750 ± 20 nm and 790 ± 20 nm, respectively. The fluorescent intensity of the uninjured knee joint was used as background and the fluorescent intensity of the injured knee reported here was background subtracted from the uninjured knee joint. Ten‐week‐old WT male mice were injured, received three doses of recombinant mouse Sost (rmSost, R&D Systems 1589‐ST‐025/CF; 4 μg/kg) intra‐articularly (10 μL volume) starting 4 hours after injury, and in vivo imaging was taken in the same manner as MMPSense. Refer Fig. 4 A for time line. IA administration of NF‐κB inhibitor (Bay‐11‐7082 [Sigma; 196870]; 4 μg/kg) and TNFα inhibitor (neutralizing monoclonal antibody [Abcam; ab185795]; 4 μg/kg) were introduced in the same manner as rmSost. Dimethyl sulfoxide (DMSO) and saline were used as controls for NF‐κB and TNFα inhibitor, respectively. Refer Fig. 3 A for time line.
Figure 4.
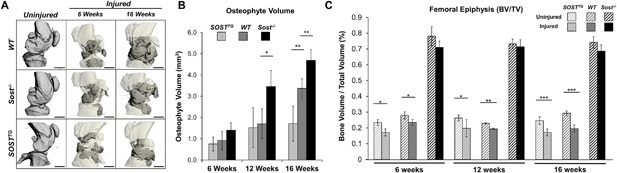
SOSTTG joints are protected from excessive osteophyte formation, while Sost‐/‐ joints are protected from subchondral bone loss in injured joints. μCT representation of mouse injured joints at 6 and 16 weeks post‐injury. Darker regions in the injured scans depict ectopic bone nodules (A). Osteophyte volume (gray area in A) was quantified at 6, 12, and 16 weeks post‐injury and compared between genotypes (B). Subchondral trabecular bone volume to total volume ratio was quantified and analyzed between injured and uninjured joints at 6, 12, and 16 weeks post‐injury. Scale bar = 1 mm. *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 3.
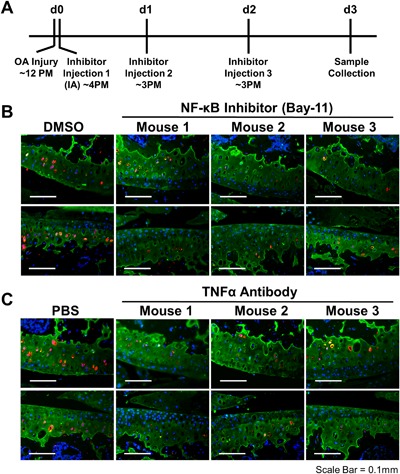
Sost activation in the injured joint is TNFα and NF‐κB dependent. Time line for IA administration of NF‐κB inhibitor (BAY‐11‐7082) and TNFα inhibitor (neutralizing antibody) (A). Immunohistochemical staining of cartilage (Col2a; green), Sost (red), and nucleus (DAPI; blue) between vehicle (DMSO or PBS) and BAY‐11‐7082 (B) or TNFα antibody (C) treated injured joints.
Statistical analysis
Two‐way ANOVA was performed using GraphPad (La Jolla, CA, USA) Prism 6 software when evaluating OA severity (aged‐matched uninjured and contralateral between genotypes). In addition, two‐way ANOVA of pairwise comparisons between injured and contralateral joints and among genotypes were also performed. A p value of less than 0.05 was considered statistically significant. The standard error of mean (SEM) were presented in the error bars.
Results
SOSTTG mice develop a less severe PTOA phenotype post‐ACL injury
Using a tibial compression PTOA mouse model,1 we examined whether chronic exposure to elevated levels of SOST would impact OA outcomes post‐injury. SOSTTG, Sost‐/‐, and C57Bl6 control (WT) mice were examined histologically and by micro‐computed tomography (μCT) at 1 day and 6‐, 12‐, and 16‐weeks after injury. C57Bl6 have been previously shown to have a mild PTOA phenotype at 8 weeks after injury,1 therefore carrying out the study to 16 weeks post‐injury would allow us to capture any beneficial effects overexpression of SOST would have upon PTOA development. Although the lateral compartments of the knees were relatively normal in all genotypes (Supplemental Fig. S1A), significant differences were observed in the medial compartments at every time point examined (Fig. 1). Injured and uninjured joints were indistinguishable, across all genotypes, at 1 day after injury (Supplemental Fig. S1B), indicating that the cartilage and bone were not damaged by the compressive overload.1 The biomechanical destabilization and lateral subluxation of the joint promoted significant erosion of both cartilage and bone on the femoral and tibial posterior side (Fig. 1 A, regions I and III) of the medial compartment of the joint in all genotypes.
Figure 1.
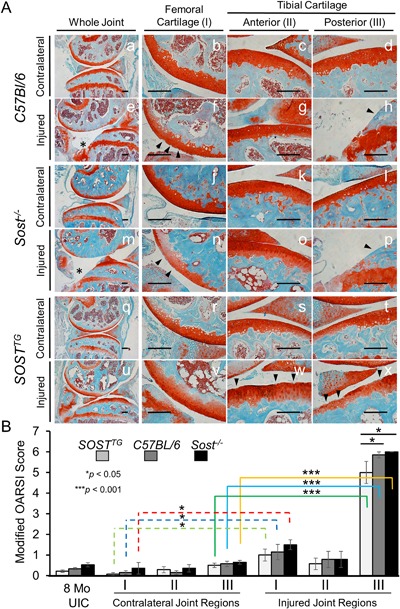
Moderate PTOA phenotype in SOSTTG compared with WT and Sost‐/‐. Histological staining of Safranin‐O and Fast Green on contralateral (left knee) at 16 weeks post‐injury (A). Cartilage integrity scoring using three distinct regions: femoral surface (I); anterior tibial surface (II); and the posterior tibial surface (III). Lower magnification (×5) of whole joint overview between C57Bl/6 (a, e), Sost‐/‐ (i, m), and SOSTTG (q, u); higher‐resolution (×20) images are provided for all other images. Injured and contralateral joints were examined and scored in three distinct regions using a modified OARSI scoring method (OA severity: 0∼2, mild; 3∼4, moderate; and 5∼6, severe). Uninjured control (UIC) and contralateral (left knee) were utilized as controls. Scale bar = 0.2 mm. *p < 0.05, ***p < 0.001. Erosion to the growth plate is marked by asterisks (e, m). Black arrows indicated regions of erosion.
However, at 16 weeks after injury, SOSTTG retained significantly more of the articular cartilage integrity throughout the joint, whereas WT and Sost‐/‐ joints displayed significant erosion below the growth plate of the posterior tibial plateaus (Fig. 1 A, region III). Interestingly, minor fibrillations and clefts down below the superficial zone of the articular cartilage were obvious in the femoral cartilage in all three genotypes (Fig. 1 A, region I), whereas the anterior tibial cartilage remains relatively unaffected (Fig. 1 A, region II). Examination of the sagittal views of the joints by a modified OARSI grading scale determined that SOSTTG had a significantly less severe cartilage loss phenotype than either Sost‐/‐ or WT joints (Fig. 1 B). Furthermore, although anterior‐tibial surface remained largely unchanged after injury, the femoral and tibial posterior compartment were significantly different between injured and contralateral joints. SOSTTG retained most cartilage integrity at 16 weeks after injury (Fig. 1 Av, w, x). The erosion on the posterior side of the tibia proceeded beyond the growth plate in both WT and Sost‐/‐ injured joints, whereas the growth plate was relatively intact in SOSTTG (Fig. 1 Ax) These results imply that chronic exposure to high SOST levels preserves cartilage thickness, suggesting that elevated levels of sclerostin in the joint improves subsequent OA outcomes in response to ACL rupture.
Sost/SOST is upregulated in the articular cartilage post‐injury by an NF‐κB dependent mechanism
Because Sost is not robustly expressed in the articular cartilage and Sost‐/‐ mice do not exhibit a dramatic PTOA‐like phenotype in uninjured controls,16, 28 we examined whether Sost expression is inducible post‐injury in the absence of cartilage erosion, as previously described.17 Low levels of Sost‐positive chondrocytes were observed in the deep zone of articular cartilage for both WT (Fig. 2 A) and SOSTTG (Fig. 2 E) uninjured (contralateral) joints. Consistent with previous reports by Chan and colleagues17 in a surgical model of OA, we found endogenous levels of Sost (Fig. 2 B, C, F, G) as well as transgenic levels of SOST (Fig. 2 J, K) to dramatically elevate in femoral and tibial cartilage, at 1 day after injury, primarily in the deep zone of the cartilage. Osteocyte expression in the femoral cortices was unaffected by injury (Fig. 2 D, H, L). Transcriptionally, we observed a significant increase in Sost at 3 and 4 days after injury (Fig. 2 M), and by RNA‐seq, injured SOSTTG had 1.5‐fold greater Sost/SOST levels (mouse + human) than injured controls (mouse only), at 1 day after injury. These findings suggest that sclerostin expression is inducible in the articular chondrocytes by traumatic joint injury and confirms the tissue‐specific overexpression of SOST in SOSTTG, which collectively express higher levels of Sost/SOST proteins in the articular chondrocytes than in WT injured joints. We examined all areas of the joint and found cells turning on Sost post‐injury only in the articular chondrocytes (Supplemental Fig. S2). The human transgene also contains two other transcripts, MEOX1 and DUSP3, therefore we also quantified human and mouse transcript levels in injured and uninjured joints, at 1 day after injury for these genes. Both MEOX1 and DUSP3 were expressed at very low levels (<6 FPKM), 8‐fold less than human SOST levels (50 FPKM) (Supplemental Fig. S3).
Figure 2.
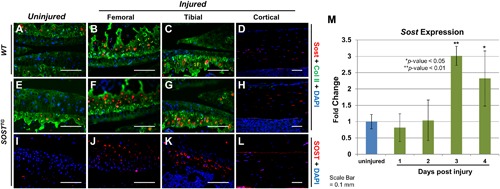
Sclerostin upregulates in the articular cartilage post‐injury. Sost immunostaining was conducted on contralateral WT (A) and SOSTTG (E and I) joints at 1 day after injury. Injured WT joints had elevated levels of Sost (B and C), while injured SOSTTG joints had elevated expression of both mouse (F and G) and human Sclerostin (J and K) 1 day after injury. No differences were observed in Sclerostin expression in the osteocytes of injured animals (D, H, and L). Real‐time quantitative PCR (qPCR) analysis of whole‐joint RNA of WTs at 1, 2, 3, and 4 days after injury (M). All images are at ×20 magnification. *p < 0.05, **p < 0.01.
Previous studies have shown that inflammatory cytokine TNFα activates Sost in an NF‐κB‐dependent manner in osteocyte‐like cells, and it was suggested that NF‐κB binds to the Sost promoter.29 To explore the possibility of direct transcriptional activation of Sost by inflammation, we searched the Sost genomic locus for NF‐κB confirmed DNA binding sites using chromatin immunoprecipitation data sets (ChIP‐seq). We identified three NF‐κB ChIP‐seq hits in the Sost locus that map to the promoter and two human/mouse evolutionary conserved regions (ECRs; >100 bp/>70% nucleotide identity) downstream of Sost and within the Van Buchem deletion region15, 16 (Table 1). Using ATDC5 cell line, we have confirmed that Sost expression is upregulated by TNFα treatment (Supplemental Fig. S4B) and that 2/3 Chip‐seq candidate regions (Supplemental Fig. S4A) exhibit enhancer activity in vitro (Supplemental Fig. S4C). To determine whether Sost activation in the injured joint is NF‐κB dependent, C57Bl/6 injured animals received three intra‐articular (IA) injections of an NF‐κB inhibitor (Bay‐11‐7082) or TNFα neutralizing antibody, starting 4 hours post‐injury (Fig. 3 A). Histological examination of injured joints showed that both the NF‐κB inhibitor and the TNFα antibody prevented the activation of Sost in the cartilage of the injured joints in contrast to IA injections with vehicle (DMSO or PBS) (Fig. 3 B, C). These data suggest that inflammatory cytokines such as TNFα in the injured joint activate Sost expression via its promoter and/or a distal ECR in chondrocytes in an NF‐κB‐dependent manner. Transcriptionally Sost expression was also reduced to pre‐injury levels in injured joints treated with NF‐κB inhibitor and the TNFα antibody (Supplemental Fig. S5).
Overexpression of SOST reduced osteophyte formation in PTOA
Because Sost modulates bone formation,14, 16 we next examined osteoarthritic remodeling by quantifying the gain in osteophyte volume and the loss in subchondral trabecular bone at 6, 12, and 16 weeks after injury by μCT (Fig. 4). Consistent with the established catabolic role of Sost in bone, Sost‐/‐ injured joints proceeded to synthesize ∼50% and ∼28% more ectopic bone than WT by 12 and 16 weeks after injury, respectively (Fig. 4 A, B). Although no significant differences were observed between SOSTTG and WT joints at both 6 and 12 weeks post‐injury, ∼50% and ∼65% less osteophyte volume was measured in SOSTTG injured joints at 16 weeks post‐injury than in WT or Sost‐/‐, respectively (Fig. 4 B). Between 12 and 16 weeks post‐injury, both WT and Sost‐/‐ joints built significant amount of osteophytes, whereas SOSTTG injured joints acquired an insignificant amount of new osteophyte volume. Whereas both WT (27.2 ± 3%) and SOSTTG (21.5 ± 10%) injured joints lost significant subchondral bone volume in the femoral epiphysis relative to the uninjured contralateral joints, Sost‐/‐ injured joints were protected from bone loss (Fig. 4 C). These findings suggest that SOST overexpression protects the injured joint from excessive osteophyte formation, whereas lack of Sost protects the femur from bone loss resulting from disuse or injury mediated by elevated catabolic activity in the subchondral bone.
SOST inhibits the activity of cartilage matrix remodeling enzymes in injured joints
To determine whether transgenic SOST affects the activity of matrix metalloproteinases (MMPs), catabolic activity known to degrade articular cartilage, we visualized and quantified MMP activity using a fluorescent substrate of MMPs (MMPSense750) in vivo, 3 days post‐injury (Fig. 5 A). Although both WT (Fig. 5 D) and Sost ‐/‐ (Fig. 5 E) injured joints displayed similar levels of MMP activity, ∼2.65‐fold activation above the endogenous levels of the uninjured joint, activation in the SOSTTG injured joint was significantly lower (2‐fold less) (Fig. 5 B, F), suggesting that SOST inhibits the activation of proteolytic enzymes known to degrade the articular cartilage matrix. Similarly, when WT injured joints were dosed IA with recombinant mouse Sost protein (rmSost) immediately after injury (Fig. 5 A), a significant decrease in the levels of activated MMPs (35.8 ± 17%) was observed compared with PBS controls (Fig. 5 B, G, H). Because MMPSense is a broad‐range substrate for a wide range of MMPs, we assessed both mRNA mmp gene activation in response to injury by RNAseq (Supplemental Fig. S5S) and evaluated protein expression at 1 day after injury in injured and uninjured contralateral joints of WT, Sost‐/‐, and SOSTTG mice for MMPs 2, 3, 7, 9, 12, 13, 14, and furin. The expression levels of activated MMP2 and MMP3 were similar in injured and uninjured SOSTTG joints compared with WT and Sost‐/‐ (Supplemental Fig. S6), which had high levels in the injured joints, suggesting that elevated levels of SOST significantly reduced MMP activity and cartilage degradation after ACL tear. No other MMPs examined had a change either in mRNA expression or protein (Supplemental Figs. S6 and S7). Furthermore, MMP reduction was independent of β‐catenin because activated β‐catenin levels were the same in both WT and SOSTTG injured joints (Supplemental Fig. S8).
Figure 5.
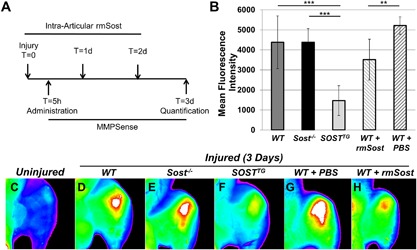
SOSTTG and WT injured joints treated with recombinant Sost (rmSost) protein have reduced levels of activated MMPs post‐injury. MMPSense was administered IV 5 hours post‐injury; rmSost was delivered IA post‐injury, and mean fluorescence intensity was measured 3 days post‐injury as depicted in A. SOSTTG‐ and rmSost‐treated injured joints displayed significantly less fluorescence than Sost‐/‐ or WT control joints (B). Representative ex vivo images of WT uninjured (C) and injured WT (D), Sost‐/‐ (E), and SOSTTG (F). Injured joints show reduced fluorescence in SOSTTG (F). Similarly, rmSost‐treated injured joints (H) show less fluorescence than PBS controls (G). **p < 0.01, ***p < 0.001.
Discussion
Significant evidence exists that implicates Wnt signaling to have opposing effects on bone and cartilage; hence, modulation of Wnt signaling in the musculoskeletal system can contribute to both osteoporotic (OP) and osteoarthritic (OA) outcomes.30 However, the multitude of Wnt signaling participants, including ligands, receptors, co‐receptors and inhibitors, has painted a picture of a pleiotropic Wnt signaling with many possibly redundant roles, hindering us from clearly delineating the role of specific Wnt molecules in the development of degenerative disorders like OA and OP. Since the discovery by Chan and colleagues17 that Sost is upregulated in focal areas of damaged cartilage in a sheep and mouse model of OA, conflicting reports by Roudier and colleagues28 have emerged about the impact loss of Sost has on the development of OA. Here, we argue that although Sost loss of function may slightly increase the severity of PTOA in described mouse models,6, 28 gain of function or ectopic administration of Sost has a significant beneficial effect on the progression and outcomes of PTOA.
In particular, elevated levels of SOST/Sost in the joint (either in transgenic mice or through intra‐articular administration) significantly reduce the expression and hinder the activity of catabolic enzymes (MMPs 2 and 3) known to degrade the cartilage extracellular matrix. High levels of Sost in the joint therefore help the articular cartilage maintain its integrity subsequent to trauma by opposing the normal upregulation of cartilage metabolic enzymes activated by inflammation. Because Wnt ligands have been shown to increase the expression of MMPs in the human synovium as well as to stimulate the chondrocyte metabolic action in rabbit models of OA,31 we propose a mechanism by which joint injury triggers the secretion of IL1, IL‐6, and TNFα by immune cells; these cytokines in turn activate the NF‐κB pathway to mediate the classic inflammatory response in many cell types,32 while simultaneously activating Sost expression in articular chondrocytes (Fig. 6). NF‐κB‐dependent activation of Sost has been previously demonstrated, where constitutively active IKK2 (IKK2ca) expression led to increased expression of Sost in osteoblasts and chondrocytes.33 Furthermore, consistent with the findings that IL1α stimulates Sost expression,17 we have identified several NF‐κB putative enhancers upstream of Sost that may be responsible for the transcriptional activation of Sost in cartilage (Table 1) and that injury‐mediated activation is NF‐κB dependent. High levels of Sost inhibit MMPs by an unknown mechanism; however, this inhibition may be upstream or independent of β‐catenin activation (Supplemental Fig. S8), consistent with Bouaziz and colleagues6 findings. Activated MMPs 2/3 protein levels were reduced in SOSTTG‐injured joints compared with WT and Sost‐/‐‐injured joints (Supplemental Fig. S6), suggesting that elevated levels of SOST in SOSTTG joints inhibit cartilage degradation after injury, through the selective inhibition of MMPs 2 and 3 expression (Fig. 5). A parallel mechanism of MMP activation, by inflammatory triggers, may also be repressed by a Sost feedback regulatory loop.34 Wehmeyer and colleagues recently proposed that sclerostin blocks TNFα signaling in fibroblast‐like synoviocytes;34 therefore, Sost may contribute to a feedback‐inhibitory loop in the joint. Consistent with their findings, we identified elevated expression levels of Relb and Fos, two transcription factors activated by TNFα signaling, in Sost‐/‐ joints compared with WT controls (Supplemental Fig. S9). Furthermore, inflammatory suppression of Wnt signaling suggests other antagonists may play a similar regulatory role in OA pathogenesis, including Dickkopf (DKK)‐1, secreted frizzled‐related protein I (sFRP‐1), WNT inhibitory factor 1 (WIF1), and Notum, which remains to be explored.
Figure 6.
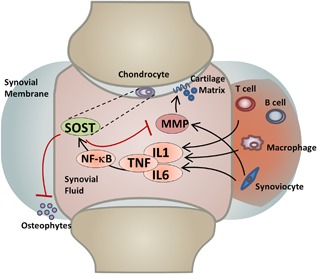
Model of Sost action in injured joints.
Although prior work had primarily referred to Sost as an exclusively osteocyte‐derived protein, our work further builds upon the findings that Sost has inducible activation in chondrocytes after injury. Here we show that Sost does not activate exclusively in damaged focal areas17 of articular cartilage, but rather turns on in the deep zone immediately after injury (Fig. 2). This discrepancy may be dependent on the injury model used. Although the tibial compression OA injury is noninvasive, this injury method is more severe than the surgical destabilization of medial meniscus (DMM) but slower in developing OA. This difference may account for the difference in Sost expression in chondrocytes. Moreover, our data suggest a uniform upregulation of Sost in injured cartilage (Fig. 2) with no obvious fibrillation or clefting of the cartilage. A limitation of the study is the limited number of time points examined (1 day, 6 weeks, 12 weeks, 16 weeks), intermediate time points (1∼4 weeks) post‐ACL injury may have consistent Sost expression and may reveal other aspects of Sost‐MMP relationship.
The opposing effects of Sost on bone and cartilage are further supported by clinical data that correlate high levels of plasma sclerostin with increased fracture risk35 and low levels of Sost in both OA patient‐derived plasma and synovial fluid.36 The mechanism by which sclerostin levels are reduced in circulation and in the synovial fluid of OA patients is unknown; nevertheless, our intra‐articular administration of Sost significantly reduced MMP activity shortly post‐injury, suggesting that Sost may represent a biomarker of OP and OA but may also have a therapeutic benefit in injured joints. The mechanism of Sost upregulation after injury may have two potential beneficial outcomes. First, NF‐κB upregulates Sost, and Sost in turn reduces MMP activity. The reduction in MMP activity may be direct or indirect, and one potential route is via a negative feedback loop where Sost tempers the immune responses. Second, Sost inhibits osteophyte formation most likely by inhibiting Wnt signaling in calcifying cartilage (Fig. 6). Inflammatory triggers have been previously demonstrated to upregulate MMPs,37, 38 suggesting that a negative feedback loop is the most likely mechanism of action. Overall, our observation of MMP reduction post Sost‐intra‐articular administrations suggests that Sost may greatly contribute to PTOA outcome, if administered to the synovium immediately after injury and after surgical stabilization of the injured joint. Furthermore, Sost administration to the injured joint may also prevent osteophyte chondrocyte formation and the accumulation of ectopic bone that may reduce mobility and increase pain in the joint, thus a balance between the anabolic role of Sost in cartilage and the catabolic role of Sost in bone may be beneficially manipulated to promote favorable outcomes in PTOA.
Disclosures
SH and ANE are employees of Regeneron Pharmaceuticals. All other authors state that they have no conflicts of interest.
Supporting information
Supporting Figures S1.
Acknowledgments
We thank the National Institutes of Health (NIH) Knock‐Out Mouse Program (KOMP) and Regeneron for providing the Sost knockout mice. JCC, CDB, BAC, NMC, and GGL were supported in part by DOD grant OR130220; DKM and GGL were supported in part by DK075730. GGL and NRH were also supported in part by LLNL LDRD ER (16‐ERD‐007). This work was performed under the auspices of the US Department of Energy by Lawrence Livermore National Laboratory under Contract DE‐AC52‐07NA27344.
Authors’ roles: GGL, JCC, and CDB designed research. JCC, NMC, CDB, BAC, DKM, and NRH performed research. JCC, AS, and GGL analyzed data. SH and ANE generated RNA‐seq data and contributed to analysis. JCC, AS, and GGL wrote the paper.
References
- 1. Christiansen BA, Anderson MJ, Lee CA, Williams JC, Yik JH, Haudenschild DR. Musculoskeletal changes following non‐invasive knee injury using a novel mouse model of post‐traumatic osteoarthritis. Osteoarthritis Cartilage. 2012; 20(7):773–82. [DOI] [PubMed] [Google Scholar]
- 2. Dejour H, Walch G, Deschamps G, Chambat P. Arthrosis of the knee in chronic anterior laxity. Orthop Traumatol Surg Res. 2014; 100(1):49–58. [DOI] [PubMed] [Google Scholar]
- 3. Loeser RF. Molecular mechanisms of cartilage destruction: mechanics, inflammatory mediators, and aging collide. Arthritis Rheum. 2006; 54(5):1357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lohmander LS, Englund PM, Dahl LL, Roos EM. The long‐term consequence of anterior cruciate ligament and meniscus injuries: osteoarthritis. Am J Sports Med. 2007; 35(10):1756–69. [DOI] [PubMed] [Google Scholar]
- 5. Ratzlaff CR, Liang MH. New developments in osteoarthritis. Prevention of injury‐related knee osteoarthritis: opportunities for the primary and secondary prevention of knee osteoarthritis. Arthritis Res Ther. 2010; 12(4):215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bouaziz W, Funck‐Brentano T, Lin H, et al. Loss of sclerostin promotes osteoarthritis in mice via beta‐catenin‐dependent and −independent Wnt pathways. Arthritis Res Ther. 2015; 17:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhu M, Tang D, Wu Q, et al. Activation of beta‐catenin signaling in articular chondrocytes leads to osteoarthritis‐like phenotype in adult beta‐catenin conditional activation mice. J Bone Miner Res. 2009; 24(1):12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hwang SG, Yu SS, Ryu JH, et al. Regulation of beta‐catenin signaling and maintenance of chondrocyte differentiation by ubiquitin‐independent proteasomal degradation of alpha‐catenin. J Biol Chem. 2005; 280(13):12758–65. [DOI] [PubMed] [Google Scholar]
- 9. Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013; 19(2):179–92. [DOI] [PubMed] [Google Scholar]
- 10. van Dinther M, Zhang J, Weidauer SE, et al. Anti‐Sclerostin antibody inhibits internalization of Sclerostin and Sclerostin‐mediated antagonism of Wnt/LRP6 signaling. PLoS One. 2013; 8(4): e22295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li X, Zhang Y, Kang H, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005; 280(20):19883–7. [DOI] [PubMed] [Google Scholar]
- 12. Balemans W, Patel N, Ebeling M, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002; 39(2):91–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Itin PH, Keseru B, Hauser V. Syndactyly/brachyphalangy and nail dysplasias as marker lesions for sclerosteosis. Dermatology. 2001; 202(3):259–60. [DOI] [PubMed] [Google Scholar]
- 14. Li X, Ominsky MS, Niu QT, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008; 23(6):860–9. [DOI] [PubMed] [Google Scholar]
- 15. Collette NM, Genetos DC, Economides AN, et al. Targeted deletion of Sost distal enhancer increases bone formation and bone mass. Proc Natl Acad Sci U S A. 2012; 109(35):14092–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Loots GG, Kneissel M, Keller H, et al. Genomic deletion of a long‐range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. 2005; 15(7):928–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chan BY, Fuller ES, Russell AK, et al. Increased chondrocyte sclerostin may protect against cartilage degradation in osteoarthritis. Osteoarthritis Cartilage. 2011; 19(7):874–85. [DOI] [PubMed] [Google Scholar]
- 18. Karlsson C, Dehne T, Lindahl A, et al. Genome‐wide expression profiling reveals new candidate genes associated with osteoarthritis. Osteoarthritis Cartilage. 2010; 18(4):581–92. [DOI] [PubMed] [Google Scholar]
- 19. Collette NM, Genetos DC, Economides AN, et al. Targeted deletion of Sost distal enhancer increases bone formation and bone mass. Proc Natl Acad Sci U S A. 2012; 109(35):14092–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chang JC, Sebastian A, Murugesh DK, et al. Global molecular changes in a tibial compression induced ACL rupture model of post‐traumatic osteoarthritis. J Orthop Res. 2017; 35(3):474–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Glasson SS, Chambers MG, Van Den Berg WB, Little CB. The OARSI histopathology initiative—recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage. 2010; 18 Suppl 3:S17–23. [DOI] [PubMed] [Google Scholar]
- 22. Lockwood KA, Chu BT, Anderson MJ, Haudenschild DR, Christiansen BA. Comparison of loading rate‐dependent injury modes in a murine model of post‐traumatic osteoarthritis. J Orthop Res. 2014; 32(1):79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA‐S eq. Bioinformatics. 2009; 25(9):1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nat Protoc. 2012; 7(3):562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010; 28(5):511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods. 2001; 25(4):402–8. [DOI] [PubMed] [Google Scholar]
- 27. Ovcharenko I, Nobrega MA, Loots GG, Stubbs L. ECR Browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res. 2004; 32(Web Server issue):W280–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Roudier M, Li X, Niu QT, et al. Sclerostin is expressed in articular cartilage but loss or inhibition does not affect cartilage remodeling during aging or following mechanical injury. Arthritis Rheum. 2013; 65(3):721–31. [DOI] [PubMed] [Google Scholar]
- 29. Baek K, Hwang HR, Park HJ, et al. TNF‐alpha upregulates sclerostin expression in obese mice fed a high‐fat diet. J Cell Physiol. 2014; 229(5):640–50. [DOI] [PubMed] [Google Scholar]
- 30. Velasco J, Zarrabeitia MT, Prieto JR, et al. Wnt pathway genes in osteoporosis and osteoarthritis: differential expression and genetic association study. Osteoporos Int. 2010; 21(1):109–18. [DOI] [PubMed] [Google Scholar]
- 31. Kapoor M, Martel‐Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 2011; 7(1):33–42. [DOI] [PubMed] [Google Scholar]
- 32. Rigoglou S, Papavassiliou AG. The NF‐kappaB signalling pathway in osteoarthritis. Int J Biochem Cell Biol. 2013; 45(11):2580–4. [DOI] [PubMed] [Google Scholar]
- 33. Swarnkar G, Zhang K, Mbalaviele G, Long F, Abu‐Amer Y. Constitutive activation of IKK2/NF‐kappaB impairs osteogenesis and skeletal development. PLoS One. 2014; 9(3):e91421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wehmeyer C, Frank S, Beckmann D, et al. Sclerostin inhibition promotes TNF‐dependent inflammatory joint destruction. Sci Transl Med. 2016; 8(330): 330ra35. [DOI] [PubMed] [Google Scholar]
- 35. Reppe S, Noer A, Grimholt RM, et al. Methylation of bone SOST, its mRNA, and serum sclerostin levels correlate strongly with fracture risk in postmenopausal women. J Bone Miner Res. 2015; 30(2):249–56. [DOI] [PubMed] [Google Scholar]
- 36. Mabey T, Honsawek S, Tanavalee A, et al. Plasma and synovial fluid sclerostin are inversely associated with radiographic severity of knee osteoarthritis. Clin Biochem. 2014; 47(7‐8): 547–51. [DOI] [PubMed] [Google Scholar]
- 37. Goldring MB, Otero M, Plumb DA, et al. Roles of inflammatory and anabolic cytokines in cartilage metabolism: signals and multiple effectors converge upon MMP‐13 regulation in osteoarthritis. Eur Cell Mater. 2011; 21:202–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sokolove J, Lepus CM. Role of inflammation in the pathogenesis of osteoarthritis: latest findings and interpretations. Ther Adv Musculoskelet Dis. 2013; 5(2):77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Figures S1.