

Abstract
Scope
GTPs (green tea polyphenols) exert anti‐CRC (colorectal cancer) activity. The intestinal microbiota and intestinal colonization by bacteria of oral origin has been implicated in colorectal carcinogenesis. GT modulates the composition of mouse gut microbiota harmonious with anticancer activity. Therefore, the effect of green tea liquid (GTL) consumption on the gut and oral microbiome is investigated in healthy volunteers (n = 12).
Methods and results
16S sequencing and phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt) analysis of both fecal and saliva samples (collected before intervention, after 2 weeks of GTL (400 mL per day) and after a washout period of one week) in healthy volunteers show changes in microbial diversity and core microbiota and difference in clear classification (partial least squares‐discriminant analysis [PLS‐DA]). An irreversible, increased FIR:BAC (Firmicutes to Bacteroidetes ratio), elevated SCFA producing genera, and reduction of bacterial LPS synthesis in feces are discovered in response to GTL. GTL alters the salivary microbiota and reduces the functional pathways abundance relevance to carcinogenesis. Similar bacterial networks in fecal and salivary microbiota datasets comprising putative oral bacteria are found and GTL reduces the fecal levels of Fusobacterium. Interestingly, both Lachnospiraceae and B/E (Bifidobacterium to Enterobacteriacea ratio—markers of colonization resistance [CR]) are negatively associated with the presence of oral‐like bacterial networks in the feces.
Conclusion
These results suggest that GTL consumption causes both oral and gut microbiome alterations.
Keywords: colorectal cancer, Firmicutes, Bacteroidetes, green tea, gut microbiome, oral microbiome, short‐chain fatty acids
1. Introduction
Green tea (GT), which represents 20% of world consumption, is second only to water in terms of worldwide popularity. GT is characterized by the presence of large amounts of polyphenols, also known as catechins, such as epigallocatechin‐3‐gallate, epigallocatechin, epicatechin gallate, and epicatechin.1 Several health benefits have been claimed for GT extracts (GTE) and GT polyphenols (GTPs), including prevention of cardiovascular disease,2 metabolic syndrome (MS),3 and colorectal cancer (CRC),4 some of which are supported by evidence from randomized trials.5, 6 At the same time, conflicting results have been reported from epidemiological studies regarding the pathogenic effects of GT.7
The mechanism(s) underlying the CRC chemo preventative activity of GT is unclear. It has been shown in animal models that the intestinal microbiota may play a role in colorectal carcinogenesis based on the association of CRC with a specific intestinal microbiome profile, or so‐called dysbiosis, characterized by reduced FIR:BAC (Firmicutes to Bacteroidetes ratio),8, 9 depletion of SCFA‐producing members of Lachinospiracea and Ruminococacea such as Eubacterium and Roseburia,10, 11 as well as presence of putative pathobionts of oral origin such as Fusobacterium.12 One possibility is that modulation of the gut and oral microbiota may contribute to the cancer‐preventative properties of green tea liquid (GTL).
Research from animal models suggest that GTP intake is associated with differences in intestinal microbiota13, 14 and intestinal formation of SCFA.15 There have only been two clinical trial reports of the effect of GT capsules on targeted human intestinal microbiota.14, 16 Therefore, a plausible hypothesis is that GTL intake alters the human intestinal and oral microbiome relevant to intestinal dysbiosis associated with colorectal carcinogenesis.
To date, however, there are no animal and/or clinical trial data regarding the effect of GTL on oral microbiota. Moreover, there are no data regarding simultaneous analysis of changes in the gut and oral microbiome associated with GTL, the association between GTL‐altered oral and fecal microbiome changes, and high‐throughput 16S sequencing analysis of human microbiota changes induced by GTL. To address the above hypothesis, we studied the effect of GTL on the intestinal and oral microbiome of healthy volunteers aged between 27 and 50 years (a population relevant to CRC screening and chemoprevention), with an integrated “washout” period, with which to determine reversibility.
2. Experimental Section
2.1. Subjects, Study Protocol and Design, and GTL Preparation and Tea Polyphenols Evaluation
The subjects were all volunteers who were recruited by distributing leaflets at the Fourth Military Medical University in Xi'an, China, during the period from September to October, 2016. Subjects were healthy, normal weight (BMI 18–24 kg m2), or overweight/obese (BMI >24 kg m2) and aged between 27 and 46 years (Table S1 and Figure S1, Supporting Information). This study protocol was approved by the Ethical Committee of the First Affiliated Hospital of Fourth Military Medical University. Please refer to the supporting text in the Supporting Information for further details.
2.2. Fecal and Saliva Samples Collection
Baseline (BL), GTL intervention (GT), and washout (WO) period samples (both feces and saliva) were collected 3 days (day 4) before starting GTL intervention, after completing 2 weeks (day 21) of GTL consumption (400 mL per day) and after a washout period of 1 week (day 28), respectively (Figure S2, Supporting Information ). Desirable green tea intake is up to 1200 mL per day).7 Subjects in a study consumed 1000 mL per day and they found GT effects on Bifidobacterium species although GT was given only 10 days.14 Nevertheless, we decided to use moderate amount of GTL (400 mL per day) for 2 weeks to avoid adverse effects of GTP. For further details, please refer to the Supporting Information for online publication (Text S1C). Fecal samples (≈5 g) were collected in preweighed plastic stool collection tubes kept on ice for analyses of the gut microbiota. Subjects were advised to collect only one sample in a day although they were asked to record all the bowel movements they had for that day.
For the saliva collection, unstimulated saliva samples (≈10 mL) were collected between 7 a.m. and 8 a.m. with previously established protocols.17 Subjects were asked to refrain from eating, drinking, and smoking for at least 1 h before sample collection. To avoid the impact of oral hygiene habits on the oral microbiome, volunteers were not allowed to use toothpaste during the night before and during the day of the saliva collection. Subjects were instructed to rinse the mouth thoroughly with normal saline prior to the collection. Naturally outflowed saliva was allowed to accumulate in the floor of the mouth and the subject spat it out into the preweighed test tube every 60 s for a period of 10 min. Both fecal and saliva samples were collected by subjects at home, stored at −20 °C for a brief period of time and then delivered to the laboratory within 24 h. Both fecal and saliva samples were then immediately processed for genomic DNA (gDNA) extraction.
2.3. DNA Extraction
Bacterial gDNA was extracted from fecal samples using TIANamp DNA Stool Kit (TIANGEN, Beijing, China) according to the manufacturer's instructions (Text S2, Supporting Information).
2.4. 16S rRNA Library Preparation and DNA Sequencing
The V4, V5 regions of the bacteria 16S ribosomal RNA gene were amplified by PCR (95 °C for 2 min, followed by 25 cycles at 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s and a final extension at 72 °C for 5 min) using primers 515F 5′‐barcode‐TGCCAGCMGCCGCGG)‐3′ and 907R 5′‐CCGTCAATTCMTTTRAGTTT‐3′, where barcode is an eight‐base sequence unique to each sample. PCR reactions were performed in triplicate 20 μL mixture containing 4 μL of 5 × FastPfu buffer, 2 μL of 2.5 mm dNTPs, 0.8 μL of each primer (5 μm), 0.4 μL of FastPfu polymerase, and 10 ng of template DNA. Amplicons were extracted from 2% agarose gels and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) according to the manufacturer's instructions and quantified using QuantiFluor ‐ST (Promega, USA). Purified PCR products were quantified by Qubit 3.0 (Life Invitrogen, USA) and every 24 amplicons whose barcodes were different were mixed equally. The pooled DNA product was used to construct Illumina Pair‐End library following Illumina's genomic DNA library preparation procedure. Then the amplicon library was paired‐end sequenced (2 × 250) on an Illumina Hiseq platform (Beijing Capitalbio Technology Co., Ltd) according to the standard protocols. Raw fastq files were demultiplexed, quality‐filtered using QIIME (version 1.17) with the following criteria: (a) The 250 bp reads were truncated at any site receiving an average quality score <20 over a 10 bp sliding window, discarding the truncated reads that were shorter than 50 bp. (b) Exact barcode matching, two nucleotide mismatch in primer matching, reads containing ambiguous characters were removed. (c) Only sequences that overlap longer than 10 bp were assembled according to their overlap sequence. Reads which could not be assembled were discarded. Operational taxonomic units (OTUs) were clustered with 97% similarity cutoff using UPARSE (version 7.1 http://drive5.com/uparse/) and chimeric sequences were identified and removed using UCHIME. The phylogenetic affiliation of each 16S rRNA gene sequence was analyzed by RDP Classifier (http://rdp.cme.msu.edu/) against the Silva (SSU123)16S rRNA database using confidence threshold of 70%.18
Generation of α and β diversities and analysis and visualization of partial least squares‐discriminant analysis (PLS‐DA), principal coordinate analysis (PCoA), and principal component analysis (PCA) plots were performed using PAST19 and XLSTAT software. The α‐diversity of each group was calculated based on the annotated data using the diversity‐indices of the PAST version 2.17 software packages. Based on a non‐parametric two‐sample t‐test using the default number of Monte Carlo permutations (999), a comparative analysis of the group‐specific α‐diversity indices was performed at all the taxonomic levels. The relationship between the microbiota of the BL, GT, and WO time points was explored using a PLS‐DA method.20 PLS‐DA is a multivariate method used to eliminate the genera related to differences between groups. PLS‐DA model enhances predictive ability and simplifies interpretation. Following construction of the PLS‐DA model, the variable importance in projection (VIP) score of each taxon was calculated to select candidate taxa that reflected the difference between three time points. A VIP score higher than one is commonly utilized in multivariate analysis as the criterion for an important variable for driving the observed group separation. The similarity percentage analysis (SIMPER) was used to identify the specific genera with the greatest contribution to the differences observed between the groups identified.19 Ordinations are the dimensional‐reduction techniques which are commonly used to visualize complex relationships between communities between groups (β‐diversity). Dimensional reduction of the Bray–Curtis distance between microbiome samples using PCoA ordination method (PAST software) was done and significant differences among groups at all the taxonomic levels were tested with permutational multivariate analysis of variance (PERMANOVA), a multivariate non‐parametric one‐way ANOVA, which utilizes the sample‐to‐sample Bray–Curtis distance matrix directly. Differential abundance (non‐parametric ANOVA with Benjamini‐Hochberg false discovery rate [FDR] correction for multiple comparisons; p < 0.05) of taxon were identified using XLSTAT (Addinsoft, USA) software program, those with p < 0.05 were grouped, their relative abundance were shown by heat‐map with hierarchical clustering (HCN) analysis and their contribution to groups (between and within groups) were analyzed using PCA (variance‐covariance type) ordination method.21 To compare the RA of taxa between any two time points, a non‐parametric paired t‐test was used. Linear discriminant analysis effect size (LEfSe) is a biomarker discovery and explanation tool for high‐dimensional data.22 It couples statistical significance with biological consistency and effect size estimation. Microbiota‐based biomarker discoveries were done with LEfSe using online galaxy server (https://huttenhower.sph.harvard.edu/galaxy/) and the LDA scores derived from LEfSe analysis were used to show the relationship between taxon using a cladogram (circular hierarchical tree) of significantly increased or decreased bacterial taxa in the microbiota between groups. The relative abundance (RA) of each biomarker taxon across all samples was shown with straight and dotted lines that plot the means and medians, respectively, in each subgroup. Levels of the cladogram represent, from the inner to outer rings, phylum, class, order, family, and genus. Color codes indicate the groups, and letters indicate the taxa that contribute to the uniqueness of the corresponding groups at an LDA >2.0. Hierarchical Ward‐linkage clustering based on the Pearson correlation coefficients of the RA of genus level OTUs in fecal microbiota of 12 healthy subjects were performed using data from all 3 time points. Then, oral biofilm co‐abundance groups (CAGs) and oral pathogen CAGs were defined on the basis of recently published literature.12
2.5. Functional Capacity of Microbiota
The functional capacity of the microbiota present in each sample was inferred using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt), which reconstructs the functional composition of a microbial community sample using 16S rRNA phylogeny and a database of annotated reference genomes.23 For each functional pathway from the Kyoto Encyclopedia of Genes and Genomes (KEGG) that was putatively identified, comparisons were made between the time points using LEfSe, which identifies features that are statistically differentially abundant among biological classes (in this case time points) and then performs comparative tests between pairs of biological classes to identify where these features are significantly enriched or diminished. PLS‐DA plots were also generated to check whether the samples from three different time points are clustered according to predicted gene enrichments for microbial functions.
2.6. Statistical Analysis
Data are expressed as means ± standard errors of the means (SEM). To compare samples collected from any two time points, a non‐parametric paired Student's t‐test (Wilcoxon matched‐pairs signed rank test) was performed. PLS‐Canonical Correspondence Analysis (CCA), non‐parametric Mood test, multivariate analysis of variance (MANOVA), and generation of 3D views using plot scores were performed using XLSTAT (version 2017) and PAST software tools. All the univariate statistical analyses, including linear regression analysis, were performed using either with SPSS software (version 23.0) or GraphPad Prism (version 7.04) for windows. All statistical tests were two‐sided and differences were considered statistical significant if p < 0.05.
3. Results
3.1. GTL Consumption Alters the Gut Microbiome
To assess the impact of GTL consumption on gut microbiome (composition, abundance and function), we performed high‐throughput sequencing of the 16S rRNA gene V4‐V5 regions of the fecal samples. A total of 2 316 907 reads were generated from the V4‐V5 hypervariable region of 16S rRNA gene using 36 fecal samples. After strict quality and size filtering, a total of 6293 OTUs were detected across all samples. To reveal the most important microbial communities affected by GTL, we performed several high‐throughput multivariate analyses (Text S3a, Supporting Information). A multivariate statistical analysis approach is a powerful tool for integration and interpretation of such datasets toward sub‐phenotypes.24 First, SIMPER identified top 23 taxa, which were defined as those with >1% contribution to samples. Among them, the differences between the bacterial communities of samples are largely driven by the dominance of the taxa belonging to the phylum Bacteroidetes (family_Prevotellaceae; genus_Prevotella 9) with an average abundance of 16.2% in the BL samples, 7.65% in the GT samples and 10.5% in the WO sample (Figure 1A). Second, the RAs of core microbiome (CM),25 which were defined as those taxa with relative abundance >1.0% and shared taxa among three time points, are expressed in the form of heat map with HCN, a clustering technique for graphically summarizing the inter group relationships in the form of a dendrogram. In our study, 12 taxa belonging to phylum Firmicutes and Bacteroidetes were identified as establishing the CM although 16S sequencing found 13 phyla in total. HCN clearly separated the CM affected by GTL as a single cluster from the CM of BL and WO periods, which form two clusters within a clade (Figure 1B). Third, based on measurement of the RA of 559 microbial taxa in feces, a PLS‐DA (cross validation ANOVA with p‐value <0.05) (Text S3b, Supporting Information) was built to depict fecal microbial signature associated with BL, GT intervention and WO periods (Figure 1B) as described by Riba et al.20 A 3D PLS DA score plots revealed a clear discrimination of microbial profile for GT compared to BL. Moreover, WO samples were separated from the BL and this may indicate that GT‐induced global shift in microbial taxa might be persistent even after stopping the GTL. Associated VIP scores (Figure 1C) allowed to rank key microbial phylotypes (the top 25 taxa with high VIP scores has been shown) based on their importance in discrimination between three time points. Notably, SCFA‐producing Ruminococcaceae 10, 11 and anti‐inflammatory Bifidobacterium 26 deserved the highest VIP score (2.5 and 2.1, respectively) with PLS‐DA. Fourth, LEfSe analysis was performed to determine high‐dimensional biomarker bacterial taxa that significantly differed between BL, GT, and WO samples22 (Figure 1E). Then, a cladogram (Figure 1F) was generated from LEfSe analysis to show the relationship between biomarker taxa. This approach revealed a SCFA‐producing genus Dorea 10, 11 as one of the biomarkers bacteria, which is significantly higher with GTL consumption. Fifth, the differential abundance analysis (DAA)27revealed 7 taxa (FDR corrected p‐value <0.05) belong to phylum Firmicutes and Actinobacteria (Table S4, Supporting Information). In summary, these comprehensive multivariate analyses helped us to determine the overall microbiota changes (PLS‐DA and HCN) and taxa that largely (SIMPER), commonly (CM), and significantly (PLS‐DA and DAA) contributed to all three time points. In addition, this approach led us to further focus on those taxa which were revealed with multivariate analysis.
Figure 1.
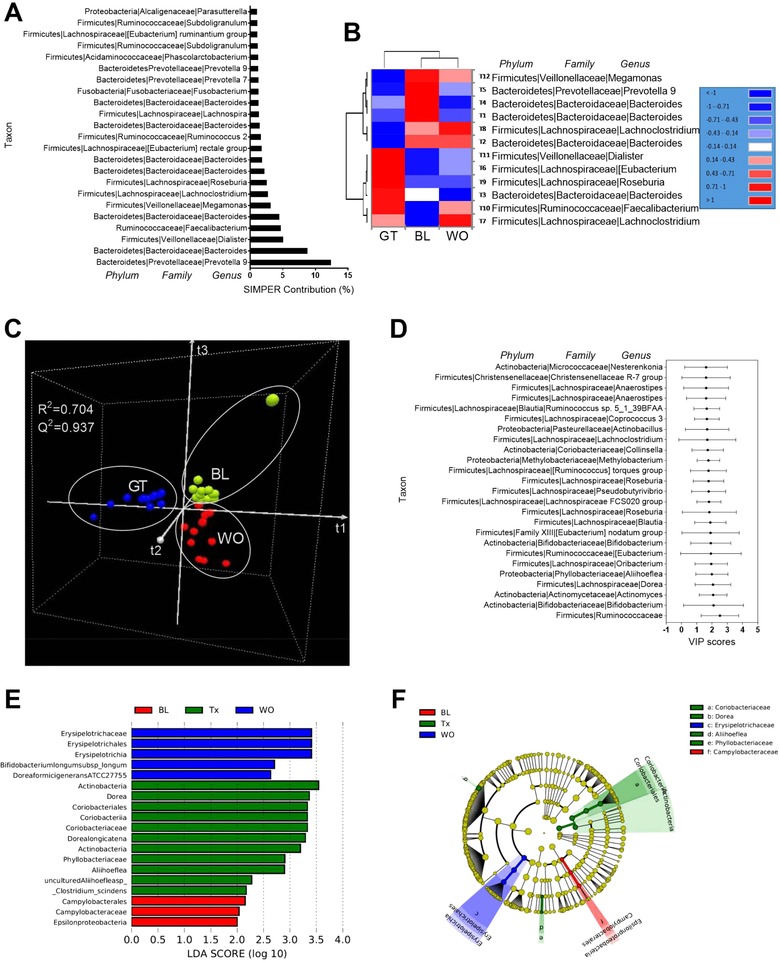
Green tea liquid consumption alters the overall gut microbiota composition. A) Similarity percentage (SIMPER) analysis using Bray–Curtis dissimilarity index showing the top 23 taxa with the greatest contribution (>1%) to the differences observed between 3 time points. B) Hierarchical clustering (HCN) with a heat map shows the relative abundances of core microbiota (relative abundance >1.0% and shared taxa among three time points). C) 3D view of score plots showing the results of supervised partial least squares‐discriminant analysis (PLS‐DA) with model fitness parameters of R 2(cum) = 0.937 and Q 2(cum) = 0.704, respectively. D) The top 25 taxa with variable importance in projection (VIP) scores (between 1.6 and 2.5) possibly responsible for discrimination of the GTL from BL and WO samples. The scores are given with upper bound (95%) and lower bound (95%). E) Linear discriminant analysis (LDA) scores (log 10) derived from LEfSe analysis, showing the biomarker taxa (LDA score of >2 and a significance of p <0.05 determined by the Wilcoxon signed‐rank test) for BL and Tx (GT) and WO. F) Cladogram generated from LEfSe analysis showing the relationship between taxon (the levels represent, from the inner to outer rings, phylum, class, order, family, and genus). Taxa are shown as phylum, family, and genus in (A), (B), and (D).
It has been shown that CRC patients showed changes in the bacterial diversity measures.9, 28 We next performed α and β diversity analysis on whole microbiota 16S profiling and at lower taxonomic levels as described by Collado et al.29 GTL consumption is associated with significantly elevated levels of all of these α‐diversity measures (Text S3c, Supporting information) only at the taxonomic level of genus and species (Figure 2A,B and Figure S2A–C, Supporting Information). Rarefaction curves show that a plateau of species richness was achieved in approximately 50 000 reads per sample (Figure S2D, Supporting Information). β‐diversity (between‐subject diversity), using PCoA with Bray–Curtis dissimilarity index (BCD) revealed a significant irreversible impact of GTL on overall and also between three time points only at the taxonomic level of phylum (Figure 2C), class and order (Figure S2E,F, Supporting Information). We next performed DAA and PCA at lower taxonomic levels to find out those bacterial groups significantly affected by GTL. Overall, 13 bacterial phyla were detected, with most OTUs affiliated with Bacteroidetes (54.6%), Firmicutes (39.4%), Proteobacteria (4.2%), Fusobacteria (1.4%), Cyanobacteria (0.18%), and Actinobacteria (0.1%) (Figure S2G, Supporting Information). GTL significantly increased Firmicutes and Actinobacteria, reduced Bacteroidetes and increased the FIR:BAC (Figure 2D–H). Decreased FIR and increased BAC have been shown in the CRC patients.10, 27, 30, 31 Most importantly, GTL‐induced phylum level changes were irreversible (Figure 2C–H). For each bacterial group in the PCA biplot diagram (Figure 2D), the length of the line is directly proportional to contribution and the direction of the line indicates the time point that bacterial group contributed to. At the level of family, GTL significantly increased the SCFA‐producing Lachinospiraceae and Ruminococcaceae,10, 11, 30, 32 elevated Erysipelotrichaceae and Bifidobacteriaceae and Coriobacteriaceae and decreased Prevotellaceae (Figure 2I–O).30 Notably, the GTL effect on Ruminococcaceae and Bifidobacteriaceae were irreversible (Figure 2K,M). At the taxonomic level of genus, increased levels of SCFA‐producing Roseburia, Feacalibacterium, Eubacterium, Blautia, Coprococcus, and Dorea (Figure 3A,B)10, 11, 30, 32 were found with GTL. Also, elevated levels of Bifidobacterium and B/E (Bifidobacterium to Enterobacteriacea ratio), which is a well‐established marker of colonization resistance (CR) against pathogenic bacteria,33 and decreased proinflammatory Fusobacterium genus (Figure 3A,B)3, 32, 34 were found with GTL. At the level of species, significantly elevated levels of uncultured (UCO) species from Roseburia, Faecalibacterium, Eubacterium rectale group, Blautia, Coprococcus, Dorea formicigenerans, Dorea longicatena, Clostridium scindens, and Bifidobacterium longum (Figure 3C,D) and lower levels of uncultured Prevotella 9 were associated with GTL (Figure 3C,D).
Figure 2.
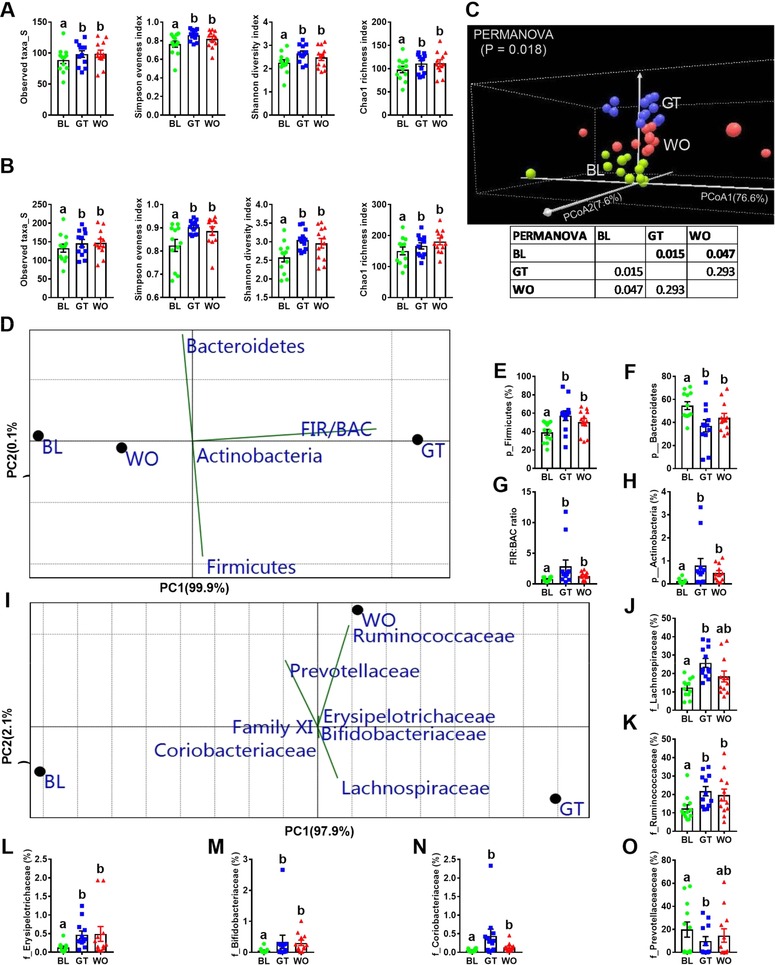
Green tea liquid (GTL) consumption is associated with altered gut microbiota diversity measurements. (A,B) α‐diversity (within‐subject diversity) measurements (observed taxa, Simpson Evenness Index, Shannon Diversity Index and Chao1 Richness Index) at the taxonomic level of genus and species for baseline (BL), GTL intervention (GT) and washout (WO) fecal samples. C) Principal‐coordinate analysis (PCoA) and PERMANOVA significance test (overall and pair wise comparisons) with Bray–Curtis dissimilarity index based on the relative abundance (RA) of phylum level OTUs identified in the feces at BL, GT, and WO samples. D) Differential abundance analysis (DAA) (non‐parametric one‐way ANOVA with FDR correction (p < 0.05) for multiple testing) followed by principal‐component analysis (PCA) (variance‐covariance type) showing the top three OTUs at phylum (p) level and Firmicutes (FIR) to Bacteroidetes (BAC) ratio included as vectors. The magnitude and direction correspond to the weights. (E–H) RA of FIR and BAC and FIR/BAC ratio. I) DAA with PCA analysis showing the top 7 OTUs at family level. J–O) RA of those families with FDR corrected p‐value <0.05. Data on the scatter plots with bar are expressed as means ± standard errors of the means (SEM). Data with different superscript letters are significantly different (p < 0.05) by Wilcoxon matched‐pairs signed rank test. n = 12. PERMANOVA, permutational multivariate analysis of variance; FDR, false discovery rate; OTUs, operational taxonomic units.
Figure 3.
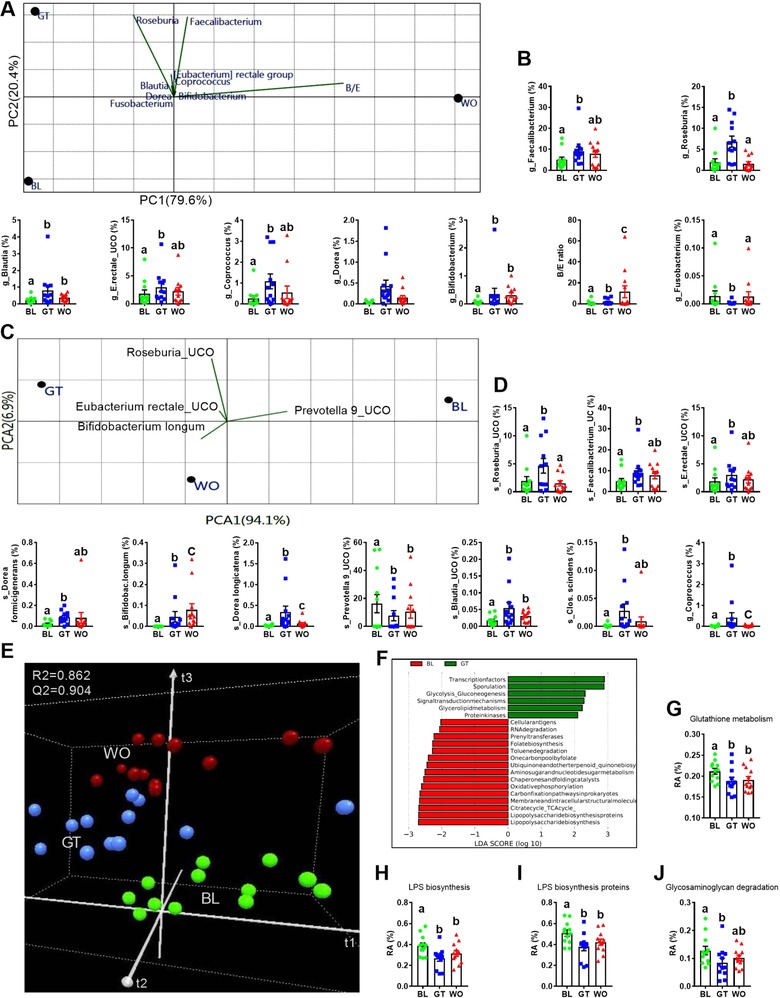
Green tea liquid consumption is associated with altered genus and species level microbiota composition and functions. A,B) Differential abundance analysis (DAA) followed by PCA (variance‐covariance type) showing score plots (A) and RA (B) of top eight OTUs at genus (g) level and Bifidobacterium (B) to Enterobacteriacea (E) ratio included as vectors for BL, GT and WO samples. The magnitude and direction correspond to the weights. C,D) DAA with PCA showing score plots (C) and RA (D) of top ten OTUs at species level for BL, GT, and WO samples. Species with invisible vectors were not mentioned in the PCA biplot. E,F) Functional prediction of microbial genes associated with BL, GT, and WO samples using PICRUSt followed by 3D projection of score plots (E) showing the results of PLS‐DA with model fitness parameters and LDA scores (F) of discriminating functional pathways between BL and GT derived from LEfSe analysis. G) Boxplots of RA of markers of microbial functional pathways relevance to inflammation. Data on the scatter plots with bar are expressed as means ± standard errors of the means (SEM). Data with different superscript letters are significantly different (p < 0.05) by Wilcoxon matched‐pairs signed rank test. n = 12. ANOVA, analysis of variance; RA, relative abundance; FDR, false discovery rate; OTUs, operational taxonomic units; LDA, linear discriminant analysis; LEfSe, LDA effect size.
Next, to determine whether the taxonomic differences between the groups' microbiota corresponded to functional changes, we performed a predictive functional analysis of the 16S rRNA sequences present. Using this validated pipeline—PICRUSt—we constructed a virtual metagenome for each of the samples'microbiomes.23, 35, 36 Analysis of functional pathways (as defined by KEGG, level 3) with PLS‐DA clearly classified all three time points (Figure 3E). Both PLS‐DA and LEfSe showed that BL microbiota is associated with increased LPS biosynthesis (VIP = 1.428), LPS biosynthesis proteins (VIP = 1.425) and glutathione metabolism (VIP = 1.426), which have been proved as markers of inflammation and inflammation‐induced carcinogenesis (Figure 3F–I).37, 38 Also, glycosaminoglycan degradation (VIP = 1.124), which may be associated with etiology of CRC,38, 39 was significantly lower in the GT compared to BL samples (Figure 3J). Analysis of microbiome data of healthy subjects with obesity (BMI > 24; n = 5) showed a clear classification between BL and GT samples with PLS‐DA, elevated levels of f_Ruminococcaceae, g_Faecalibacterium, and g_Roseburia, and a clear trend of either increase or decrease in other microbial parameters (major bacterial groups, predicted bacterial functions, α‐diversity measures and FIR/BAC ratio) altered with GT consumption (Figure S2H–L, Supporting Information). Combined, these results report the effect of GTL on the gut microbiota harmonious with anticancer activity.
3.2. GTL Consumption Alters the Salivary Microbiome
A total of 2 316 299 reads were produced for 36 saliva samples. After quality filtering, a total of 8399 operational taxonomic units (OTUs) were detected across all samples. SIMPER showed that Streptococcus, within the Firmicutes, is the most abundant genera across all samples (Figure 4A). Thirteen taxa belong to Firmicutes, Bacteroidetes, Actinobacteria, Proteobacteria, and Fusobacteria were established as core microbiome (Figure 4B) and heat map with HCN (Figure 4B) separated the core microbiome of GT from BL and WO samples that formed two individual clusters within a single clade. Next, PLS‐DA clearly classified all three time points (cross validation ANOVA with p‐value <0.05) (Figure 4C,D). Also, the LEfSe analysis (Figure 4E) and cladogram visualization (Figure 4F) revealed the high‐dimensional biomarkers genera associated with GTL (e.g., genus Alloprevotella) and all three time points were associated with different types of biomarker bacteria. Finally, DAA revealed 17 taxa for BL, GT, and WO samples (Table S4, Supporting Information).
Figure 4.
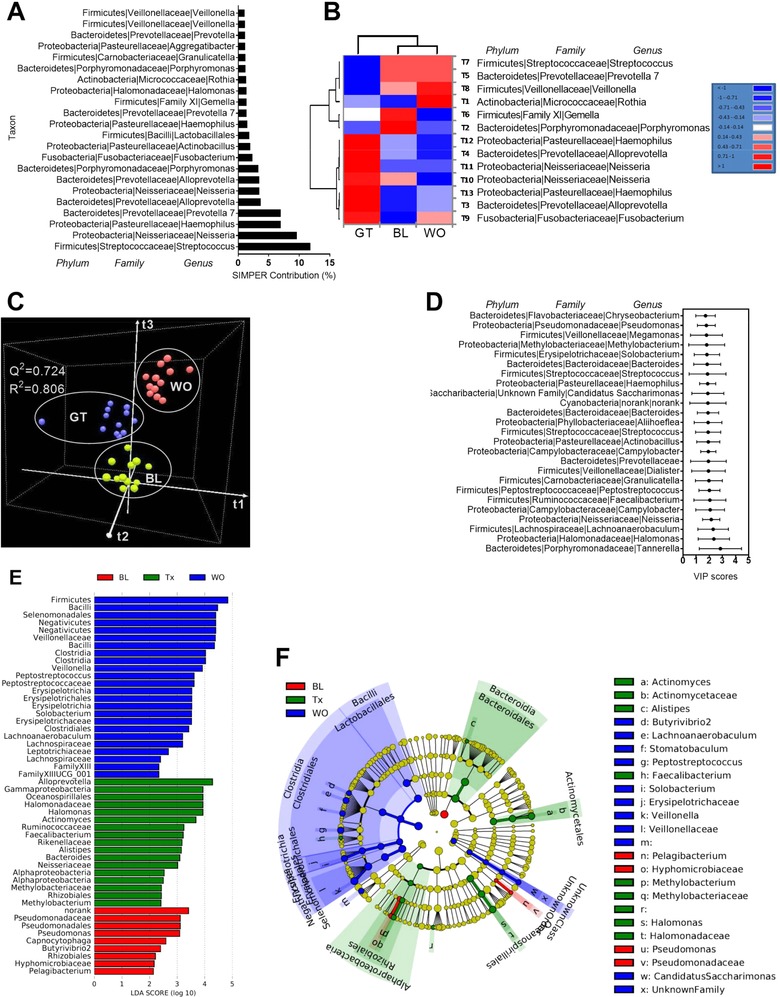
Green tea liquid consumption alters the overall salivary microbiota composition. A) Similarity percentage (SIMPER) analysis. B) Hierarchical clustering (HCN) with a heat map shows the relative abundances of core microbiota. C) 3D view of score plots showing the results of supervised PLS‐DA with model fitness parameters. D) The top 25 taxa with VIP scores (between 1.7 and 2.8) possibly responsible for discrimination of the GT from BL and WO samples. The scores are given with upper bound (95%) and lower bound (95%). E) LDA scores (log 10) derived from LEfSe analysis, showing the biomarker taxa for BL, Tx (GT) and WO. F) Cladogram generated from LEfSe analysis showing the relationship between taxon. LEfSe, LDA effect size.
GTL had no effect on α‐diversity measures derived either from whole 16S profile or at lower taxonomic levels (Figure S3A–F, Supporting Information). On the other hand, β‐diversity analysis distinctly clustered the GT samples at overall microbial community structure and also at the taxonomic level of class (Figure 5A,B). Interestingly, GTL effects on salivary microbiota looked reversible (Figure 5A,B). The ten most abundant bacterial genera across all saliva samples were Streptococcus (16.2% of all assigned reads), Neisseria (14.6%), Haemophilus (10.4%), Prevotella 7 (8.8%), Alloprevotella (8.03%), Porphyromonas (5.8%), Prevotella (4.2%), Fusobacterium (4.1%), Veillonella (3.8%), and Rothia (2.4%) (Figure S3H, Supporting Information). These genera covered almost 80% of the total RA and the proportional abundance values are similar to those reported in previous studies of the microbiota associated with the oral cavity.12, 40
Figure 5.
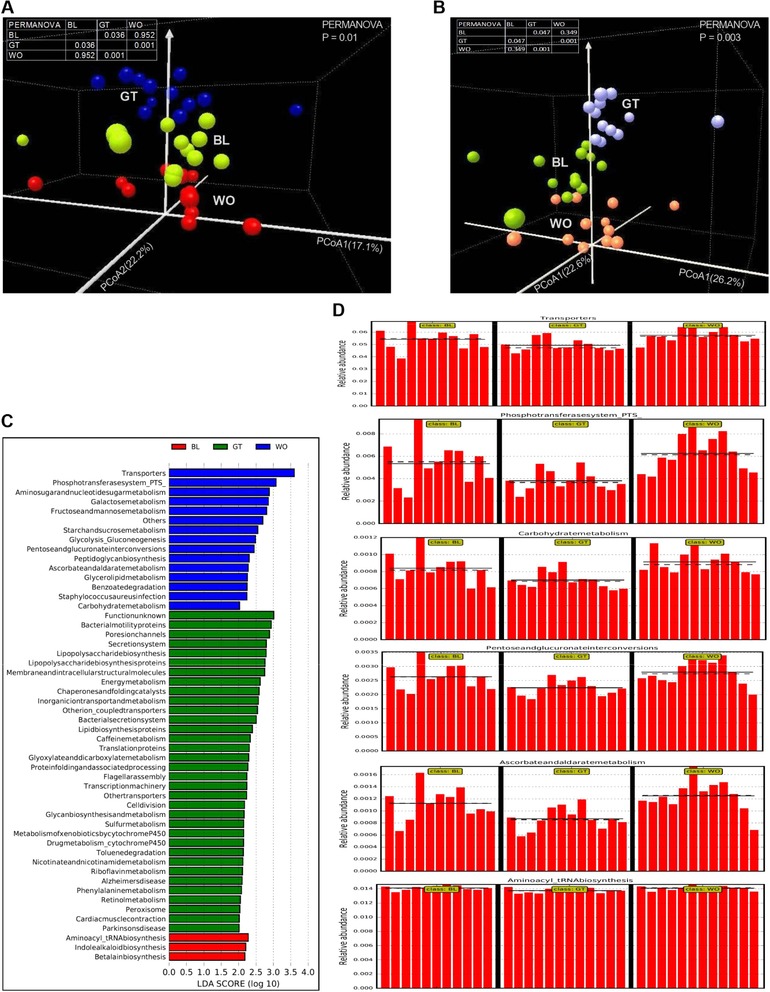
GTL consumption is associated with altered β‐diversity and functions of salivary microbiota. (A,B) 3D view of score plots showing the results of PCOA and PERMANOVA significance test (overall as well as pair wise comparisons) based on the relative abundance (RA) of whole microbiota (A) and class level (B) OTUs identified in the feces at BL, GT and WO periods. C) Functional prediction of microbial genes associated with BL, GT and WO samples using PICRUSt followed by LEfSe analysis showing LDA scores of discriminating functional pathways between 3 time points. D) RA of biomarker microbial functional pathways relevance to environmental information processing and carcinogenesis was shown in a panel where the straight and dotted lines plots means and medians of the RA, respectively, in each subgroup. n = 12. RA, relative abundance; FDR, false discovery rate.
Next, the PICRUSt analysis revealed a significantly decreased abundance of pathways relevance to environmental information processing41 and oral carcinogenesis42 with GTL consumption (Figure 5C,D). In comparison, GT samples showed increased levels of pathways (Figure S4A–D, Supporting Information), which were shown as salivary functional biomarkers in healthy control subjects compared to patients with oral cancer.42 Overall, these results reveal the impact of GTL consumption on the salivary microbiome.
Oral bacterial networks are detected in the feces of healthy subjects. Recently, oral bacterial networks were detected in colonic mucosa and are enriched in CRC.12 These are two co‐abundance groups (CAGs) comprised of oral pathogen CAG12 and oral biofilm CAG. Members of oral pathogen CAG are linked with late colonization of oral biofilms (e.g., Parvimonas, Solobacterium, Dialister, Gemella, Peptostreptococcus, and Fusobacterium). Members of oral biofilm CAG12 are dominant bacteria in early dental biofilm formation and associated with relatively healthy tooth pockets, including Granulicatella, Actinomyces, Haemophilus, Rothia, Streptococcus, and Veilonella. We followed the same method (hierarchical Ward‐linkage clustering based on the Pearson correlation coefficients of the RA of members of CAG) as Flemer B12 to identify and cluster the members of CAG in the feces. Interestingly, we found similar clusters of oral pathogen CAG and oral biofilm CAG in the fecal samples collected at three different time points (Figure 6A). However, a comparative analysis between saliva (Figure 6B) and feces (Figure 6B) showed a relatively lower presence these two CAGs in the feces our subjects except for the genera Dialister and Fusobacterium. Notably, the RA of Fusobacterium, which has extensively been studied in CRC,3, 22 was significantly lower after GTL consumption compared to BL (Figure 6B) although no changes were found with Fusobacterium in the saliva. Also, most of the members of these two CAGs (e.g., Streptococcus and Peptostreptococcus) were significantly lower in the saliva of GTL compared to baseline (Figure 6B). Interestingly, we found a negative correlation between oral pathogen CAG and Lachnospiraceae CAG in the feces (p < 0.001) (Figure 6C) of our subjects after combining all the samples regardless of time points. Moreover, we discovered a negative correlation between B/E ratio and oral pathogen CAG in the feces of same subjects (p < 0.001) (Figure 6D). Next, comparison between fecal and salivary microbiota profile showed a significantly higher α‐diversity measures in the saliva than feces (Figure 7A and Figure S4G, Supporting Information). Likewise, PCOA with PERMANOVA significance test performed at the genus level distinctly clustered the fecal and saliva samples (Figure 7B). Analysis of microbiome data of healthy subjects with obesity (BMI > 24; n = 5) showed a clear classification between BL and GT samples with PLS‐DA, decreased levels of specific CAG genera (Streptococcus, Dialister, Fusobacterium, Gemella, and Solobacterium), increased levels of predicted bacterial functions (e.g., peroxisome and inorganic ion transporters and metabolism) and unaltered α‐diversity measures with GT consumption (Figure S3H‐J, Supporting Information). Together, these results indicate the beneficial influence of GTL on the oral‐like bacterial networks detected in the feces of healthy subjects.
Figure 6.
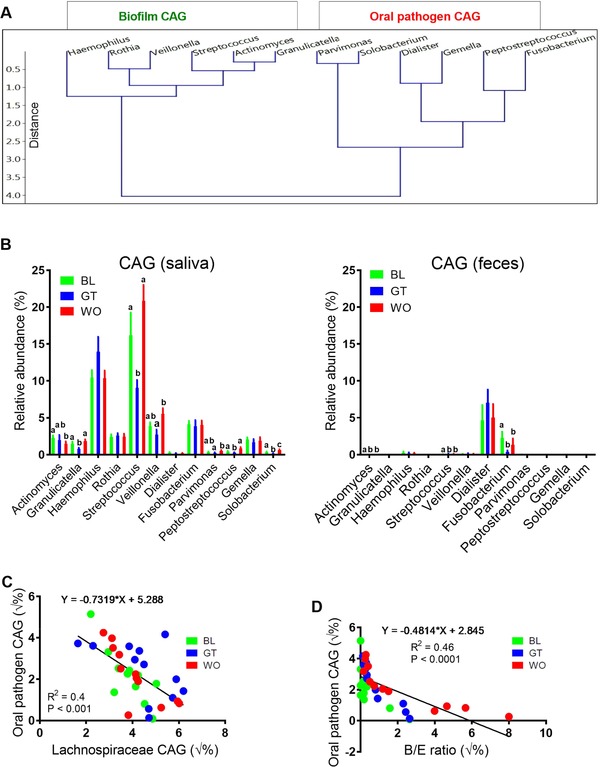
Higher fecal Lachnospiraceae and B/E ratio were negatively associated with colonization of gut with oral‐like bacterial networks. A) Hierarchical Ward‐linkage clustering based on the Pearson correlation coefficients of the RA of genus level OTUs in fecal microbiota of 12 healthy subjects. Data from all three time points were combined for this analysis. Oral biofilm co‐abundance groups (CAGs) and oral pathogen CAGs were defined on the basis of a recently published literature (please refer main text). B) Results showing comparative analysis performed between members of salivary biofilm and pathogen CAGs and oral‐like bacterial genera (members of both CAGs) present in feces using RA of genus level OTUs. C,D) Linear regression analysis showing an association between oral pathogen CAG and Lachnospiraceae CAG and between oral pathogen CAG and B/E ratio using the sqrt transformed RA of members these two CAGs identified in the whole fecal microbiota data profile. Data are expressed as means ± standard errors of the means (SEM). Data with different superscript letters are significantly different (p < 0.05) by Wilcoxon matched‐pairs signed rank test. n = 12. RA, relative abundance; OTUs, operational taxonomic units; CAGs, co‐abundance groups; B/E Bifidobacterium to Enterobacteriacea ratio; √%, square root transformed percentage values. p < 0.05 considered significant.
Figure 7.
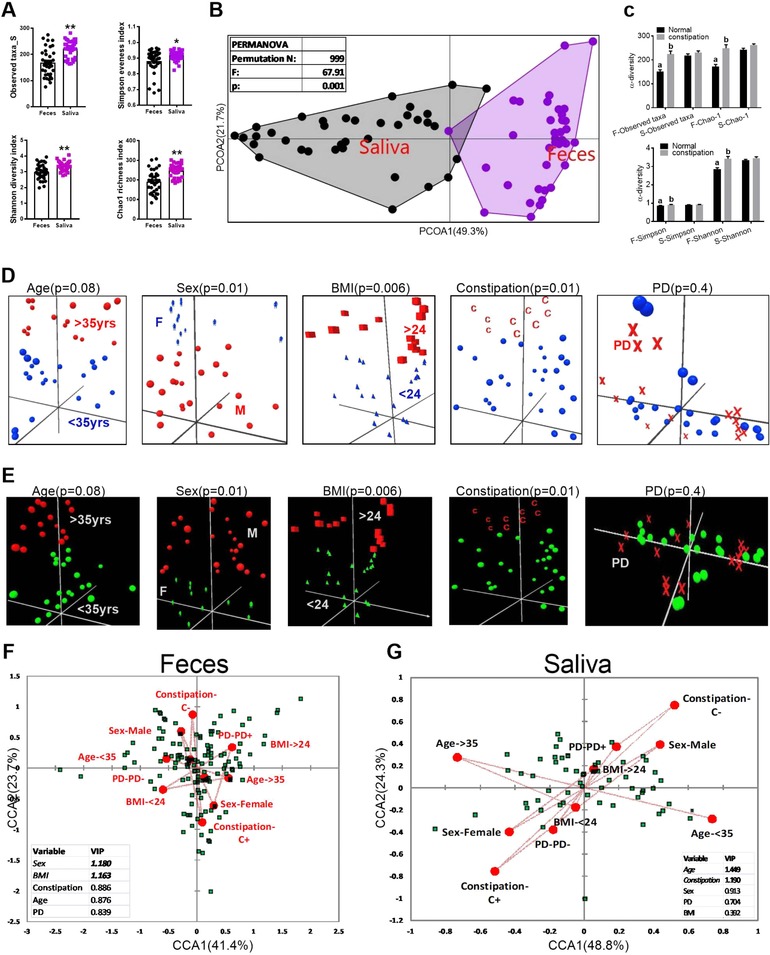
α and β diversity differences between fecal and salivary microbiota. A) Differences in the α‐diversity measurements of fecal and salivary microbiota. B) Genera shared between feces and saliva were identified and then PCOA and PERMANOVA significance test were performed based on the RA of OTUs identified at genus level. C) The effects of status of bowel movements (normal vs constipation) on the α‐diversity measurements of fecal and salivary samples. D,E) The influence of age, sex, BMI, bowel movements, periodontal disease (PD) status on the overall composition of fecal (D), and salivary microbiota (E). F,G) Results of PLS‐CCA showing the correlation between environmental variables and RA (>1%) of OTUs identified at genus level in fecal and salivary 16S profile. Variable biplot dotted lines indicate direction of environmental gradient. Inserts in the corresponding figures show the VIP scores derived from PLS‐CCA. Overall p < 0.0001 (1000 permutations) for both feces and saliva. Variables with VIP score close to 1 or >1 exert significant effects on the overall composition of microbiota.
3.3. Influence of Demographic Characteristics on the Overall Composition of Microbiota
There was no effect of age, sex, BMI, bowel movements, and periodontal disease (PD) status on fecal and salivary baseline microbiome profile or treatment effect of GTL intervention (Table S5, Supporting Information). On the other hand, analysis performed after combining all the samples showed elevated α‐diversity measures only in the feces of subjects with constipation compared to subjects with normal bowel movements (Figure 7C), which is consistent with a recent report,43 although age, sex, BMI, and PD status did not have any effect on the same measures both in the feces and saliva (Figure S5H‐O, Supporting Information). Both PCoA plots and PLS‐CAA showed that the overall compositions of the gut microbiota (all OTUs with >1%) were significantly shifted only by sex, BMI, and the status of bowel movements both in the feces (Figure 7D,F) and saliva (Figure 7E,G). Together, our results are consistent with recently published findings regarding the influence of age, sex, and BMI on gut microbiota.44, 45
4. Discussion
The purpose of this study was to explore the effect of GTL consumption on the fecal and oral microbiome and test our hypothesis that GTL alters the oral and gut microbiome harmonious with anti‐cancer activity and commonly studied in the context of intestinal dysbiosis associated with colorectal carcinogenesis. This is the first study to investigate whether GTL alters the gut and oral microbiome in humans using high‐throughput 16S rRNA gene sequencing, PICRUSt, and modern multivariate data analyzing software programs. We present for the first time that the combined analysis of gut and oral microbiota of healthy subjects with no metabolic alterations using samples collected before and after GTL consumption and after a WO period of 1 week. Indeed, only 2 weeks of GTL drinking, while keeping the energy and nutrients consumption unchanged between subjects and between three time points, caused changes in fecal and salivary microbiota. Considering the worldwide popularity about GTL consumption,1 the novel findings in this study are 1) GTL consumption induced irreversible shift of overall fecal microbiota structure; 2) GTL consumption was associated with irreversible elevation of α‐diversity measures at the taxonomic levels of genus and species in feces; 3) GTL induced irreversible elevation of Firmicutes, reduction of Bacteroidetes, and elevation of FIR/BAC; 4) GTL induced the growth of SCFA producing members of family Lachnospiraceae, Ruminococcaceae, and Bifidobacteriacea; 5) GTL induced a global functional shift related to microbiota metabolism. Notably, GTL significantly reduced the markers of inflammation and bacterial translocation37; 6) GTL consumption altered the composition (overall and at class level) and functions (e.g., pathways relevance to oral carcinogenesis) of salivary microbiota; 7) We detected similar bacterial networks in fecal and oral microbiota datasets comprising putative oral biofilm forming and pathogenic bacteria in these healthy subjects and GTL drinking was associated with reduction of most of these bacteria in the saliva; 8) GTL lowered the abundance of oral‐like genus fusobacterium in the feces; 9) Most importantly, GTL elevated the abundance of Lachnospiraceae and B/E ratio in the feces and both high abundance Lachnospiraceae and elevated B/E ratio were negatively associated with the presence of oral‐like bacterial networks in the feces.
The GTL‐induced elevated levels of SCFA‐producing so‐called “beneficial” bacterial genera such as Faecalibacterium, Blautia, Bifidobacterium, Roseburia, Eubacterium, and Coprococcus and reduction of functional markers of inflammation (e.g., LPS biosynthesis) may be relevant to one of the mechanisms underlying the CRC chemo preventative activity of GTL. Studies of CRC patients have reported elevated intestinal inflammation due to the reduction of SCFA‐producing bacteria46, 47 and increase of LPS‐producing bacteria10, 27 compared with healthy controls. An increase in SCFA‐producing and LPS‐suppressing bacteria such as Bifidobacterium, leading to increased mucosal SCFA exposure and decreased mucosal LPS‐induced intestinal inflammation, has been suggested to reduce mucosal inflammatory tone.48, 49 Therefore, our findings are harmonious with a hypothesis that GTL drinking is associated with gut microbiota changes driving increased luminal SCFA exposure and reduced intestinal inflammation. Interestingly, our findings are supported by both animal studies15, 50 and a recent in vitro study showing proliferation of certain SCFA‐producing genera with GTP and an elevated level of SCFA produced in cultures with tea polyphenols.16 Whether the changes in intestinal microbiota that we observed with GTL intervention increase in luminal SCFA levels is a question that will require a metabolomic approach.
We propose another mechanism by which the GTL drinking could maintain the normal gut homeostasis may be conferring CR to opportunistic pathogens growth due to intestinal inflammation and CRC‐associated oral taxa. Most recently, a causal role of oral bacteria ectopically colonizing the intestine has been shown, so it is important identifying members of the normal gut microbiota that can provide CR against orally derived bacteria that could induce chronic inflammation in the gut.51 Our hypothesis regarding the beneficial effect of GTL on CR is supported by GTL‐induced elevation of B/E ratio, a well‐established marker of colonization resistance against pathogenic bacteria33 and the abundance of Lachnospiraceae, which has recently been shown that it might confers CR to oral‐like bacterial network in the gut of both healthy and CRC patients.12 Interestingly, in agreement with Flemer et al.,12 we detected a negative association between the abundance of fecal Lachnospiraceae and oral pathogen present in the feces of healthy subjects in this study. Similarly and for the first time, we show a negative correlation between B/E ratio and oral pathogen CAG in healthy subjects. Moreover, we found similar bacterial taxa found in the oral cavity colonize in the colon of healthy subjects and form bacterial coabundance networks although the RA of these oral‐like bacterial taxa were found lower in the feces of our subjects. Among them, the genus Fusobacterium, a proinflammatory bacterium causatively linked with CRC,52 was relatively higher in the BL samples and GTL drinking was able to reduce the abundance of Fusobacterium in the feces. Similar to our results, the Fusobacterium genus has been detected in the feces of healthy controls12, 53, 54 although it was shown relatively higher in the CRC patients.
Decreased FIR/BAC has been observed in the gut of inflammatory bowel disease (IBD), one of the major causes of CRC, and CRC patients.8, 9 Based on the observed findings in the healthy subjects, we propose that GTL consumption may prevent the reduction of FIR/BAC If CRC patients consume GTL. A significant reduction of Bacteroidetes after GTL drinking in this study is supported by a most recent in vitro report that showed an inhibited Bacteroides‐Prevotella growth with GTP.16 Our results and hypothesis highlight the need for further studies with large number of CRC subjects to investigate the GTL effects on microbiota. On the other hand, the microbiome modifying effects of GTL should be investigated in large number of subjects with obesity and/or MS to confirm the GTL effects on FIR/BAC because an elevated FIR/BAC ratio has been found with obesity and associated MS.55
The strengths of this study include the utilization of high‐throughput 16S sequencing and PICRUSt‐based predicted functional analysis that allowed to examine both composition and functions of microbiota of fecal and saliva samples collected at the same time at three different time points, application of different types of multivariate analyses that allowed to find the most important taxa affected by GTL, bacterial diversity analysis performed at all the taxonomic levels, examination of the association between oral‐like bacteria and gut bacteria, and the effect of various demographic of the study on fecal and oral microbiota. It has been shown that differences in the diet were associated with inter‐individual and intra‐individual variations in microbiome composition between individuals. To minimize the effect of dietary and life style differences on the microbiome,56, 57 our study subjects, who had relatively similar lifestyle, were enrolled from a single university and instructed to consume the same diet during the 4 consecutive days before their visit on day 21 and 28 for samples collection according to the recorded days before the BL measurement. It is conceivable that GTL intervention in this study overcame the influences of diet and other factors influencing the microbiota based on the results that HCN separated the CM25 affected by GTL from baseline and washout periods. Methodological weaknesses were a small number of subjects and the lack of colorectal tissue to study the effect of GTL on the mucosa‐associated microbiome. The mucosa‐associated microbiome in intestinal tissue differs from the lumen,9 and these microbes also potentially play important roles. The results of this study seem to warrant further investigation using colorectal tissue to confirm the GT‐induced alterations in Firmicutes and Bacteroidetes. Also, the predictive functions should be examined with care as we are not sure whether the bacteria identified by 16S sequencing were alive and active. As a result, the future studies with GTL intervention will require a metatranscriptomic approach.
Following the approach used by Collado,29 performing both α and β diversity analysis at different taxonomic levels captured α‐diversity differences at lower taxonomic levels (e.g., species) when we did not find α‐diversity differences at higher taxonomic levels (e.g., OTUs in general). This could be possible because several interventions caused alteration in the abundance of microbiota only at lower taxonomic levels.58 Although the precise mechanism has yet to be clarified for not founding α‐diversity differences with salivary microbiota, it might be achieved with higher dose and/or longer duration of GTL. Higher α‐diversity with saliva samples compared to fecal samples in our study might be due to the impact of previous day(s) food timing condition before sampling the saliva and fecal samples. The food timing influences the daily rhythm in α‐diversity salivary microbiota although no significant effects of food timing were observed in α‐diversity fecal microbiota.29
Our findings suggest the possibility that GTL‐induced gut microbiome changes might be persistent for a period of time as we observed irreversible fecal microbiome changes after one week WO period although microbiota recovery depends on the dose and duration of a particular intervention. An interesting question, which needs further research, would be whether irreversible changes might be due to the effects of GTP on the activity several endogenous colonic anti‐microbial peptides that maintain the gut microbiota homeostasis. On the other hand, the reversible salivary microbiome changes could be due to the lack of prolonged contact between GTP and microbes because of the constant swallowing of saliva and oral hygiene habits.59 Indeed, to answer whether this will be persistent for longtime, we need further research with longer duration of WO period. From our findings, we could recommend that one could stop drinking the GTL for a week without losing some of the microbiome‐related benefits if there is a concern regarding the adverse effects of GTL drinking.7
In summary, we preliminarily identified that GTL given for 2 weeks is associated with significant alterations in gut and oral microbiota composition and functions although verification of the current findings in large cohorts is required. The increase in SCFA producing genera and FIR/BAC, elevated levels of CR markers such as B/E ratio and Lachnospiracea and reduction of oral‐like Fusobacterium genus in the gut are compatible with the known anti‐inflammatory and antineoplastic properties of GTL consumption.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Text S1. Study design and GT liquid preparation and tea polyphenols evaluation.
Text S2. DNA extration.
Text S3. Statistical analysis.
Figure. S1. Schedule of study visits and participant flow through the study and study protocol and determination of total tea polyphenols concentrations.
Figure. S2. α and β diversity measures and phylum level analysis of fecal samples.
Figure. S3. α‐diversity measures and relative abundance of top ten genera of saliva samples.
Figure. S4. Biomarker functional pathways and α‐diversity measures of saliva samples.
Table S1. Demographic information.
Table S2. Nutrients and energy consumption details of study subjects.
Table S3. Statistical analysis of nutrients and energy consumptions of study subjects.
Table S4. Differential abundance analysis performed on global fecal and salivary microbiota profile
Table S5. The influence of demographics factors on global fecal and salivary microbiome profile
Acknowledgements
The authors’ responsibilities were as follows. X.Y., Y.L., and Z.J. contributed equally to this article. Z.S., and Y.L., developed the study concept and theory; X.Y., Z.J., Z.S., and Y.L., designed and carried out the trial; X.Y., Z.J., L.Z., H.S., W.Z., X.W., Z.P., H.C., Y.W., X.G., and B.Y., helped to recruit subjects, instructed the subjects to complete the questionnaire and diary, performed dietary assessments and helped to collect samples; X.Y., J.G., M.Y., and T.F., carried out bacterial DNA extraction and quantification from fecal and saliva samples; K.K. performed the predicted functional analysis using PICRUSt pipeline; X.Y. and Z.J. drafted the initial version of the manuscript; K.K. carried out the analyses and interpretation of all the data and prepared the manuscript; K.K., X.Y., Z.J., Y.L., and Z.S. had primary responsibility for final content of the manuscript. All authors read and approved the final manuscript. This research was supported by a grant (81373058 and 81370082) from National Natural Science Foundation of China and a grant (2016SF‐001) from Shaanxi Provincial Natural Science Foundation of China to Z.S. as well as a grant (81773488) from National Natural Science Foundation of China to W.Z. This research was also supported by a grant from China Special Grant for the Prevention and Control of Infectious Disease (2017ZX10105011). The authors thank Yanan Wang at Capitalbio Technology Corporation and Yong Zhang at Genergybio Technology Corporation in Shanghai, China for commercial arrangement.
Yuan X., Long Y., Ji Z., Gao J., Fu T., Yan M., Zhang L., Su H., Zhang W., Wen X., Pu Z., Chen H., Wang Y., Gu X., Yan B., Kaliannan K., Shao Z., Mol. Nutr. Food Res. 2018, 62, 1800178 https://doi.org/10.1002/mnfr.201800178
Contributor Information
Kanakaraju Kaliannan, Email: kkaliannan@mgh.harvard.edu.
Zhongjun Shao, Email: 13759981783@163.com.
References
- 1. Stoner G. D., Mukhtar H., J. Cell Biochem. Suppl. 1995, 22, 169. [DOI] [PubMed] [Google Scholar]
- 2. Chen Z. M., Lin Z., J. Zhejiang. Univ. Sci. B 2015, 16, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tian C., Huang Q., Yang L., Legare S., Angileri F., Yang H., Li X., Min X., Zhang C., Xu C., Yuan J., Miao X., He M. A., Wu T., Zhang X., Sci. Rep. 2016, 6, 24353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chacko S. M., Thambi P. T., Kuttan R., Nishigaki I., Chin. Med. 2010, 5, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Toolsee N. A., Aruoma O. I., Gunness T. K., Kowlessur S., Dambala V., Murad F., Googoolye K., Daus D., Indelicato J., Rondeau P., Bourdon E., Bahorun T., Biomed. Res. Int. 2013, 2013, 412379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nogueira L. P., Nogueira Neto J. F., Klein M. R., Sanjuliani A. F., J. Am. Coll. Nutr. 2017, 36, 108. [DOI] [PubMed] [Google Scholar]
- 7. Boehm K., Borrelli F., Ernst E., Habacher G., Hung S. K., Milazzo S., Horneber M., Cochrane. Database Syst. Rev. 2009, https://doi.org/10.1002/14651858.CD005004.pub2, Cd005004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gao R., Kong C., Huang L., Li H., Qu X., Liu Z., Lan P., Wang J., Qin H., Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2073. [DOI] [PubMed] [Google Scholar]
- 9. Chen W., Liu F., Ling Z., Tong X., Xiang C., PLoS One 2012, 7, e39743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ahn J., Sinha R., Pei Z., Dominianni C., Wu J., Shi J., Goedert J. J., Hayes R. B., Yang L., J. Nat. Cancer Inst. 2013, 105, 1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peters B. A., Dominianni C., Shapiro J. A., Church T. R., Wu J., Miller G., Yuen E., Freiman H., Lustbader I., Salik J., Friedlander C., Hayes R. B., J. Ahn, Microbiome 2016, 4, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Flemer B., Warren R. D., Barrett M. P., Cisek K., Das A., Jeffery I. B., Hurley E., O'Riordain M., Shanahan F., O'Toole P. W., Gut 2017, https://doi.org/10.1136/gutjnl-2017-314814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jung E. S., Park H. M., Hyun S. M., Shon J. C., Singh D., Liu K. H., Whon T. W., Bae J. W., Hwang J. S., Lee C. H., PLoS One 2017, 12, e0187154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jin J. S., Touyama M., Hisada T., Benno Y., Microbiol. Immunol. 2012, 56, 729. [DOI] [PubMed] [Google Scholar]
- 15. Henning S. M., Yang J., Hsu M., Lee R. P., Grojean E. M., Ly A., Tseng C. H., Heber D., Li Z., Eur. J. Nutr. 2017, https://doi.org/10.1007/s00394-017-1542-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sun H., Chen Y., Cheng M., Zhang X., Zheng X., Zhang Z., J. Food Sci. Technol. 2018, 55, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Navazesh M., Ann. N. Y. Acad. Sci. 1993, 694, 72. [DOI] [PubMed] [Google Scholar]
- 18. Amato K. R., Yeoman C. J., Kent A., Righini N., Carbonero F., Estrada A., Gaskins H. R., Stumpf R. M., Yildirim S., Torralba M., Gillis M., Wilson B. A., Nelson K. E., White B. A., Leigh S. R., Isme. J. 2013, 7, 1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Motta V., Trevisi P., Bertolini F., Ribani A., Schiavo G., Fontanesi L., Bosi P., PLoS One 2017, 12, e0173029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Riba A., Olier M., Lacroix‐Lamande S., Lencina C., Bacquie V., Harkat C., Gillet M., Baron M., Sommer C., Mallet V., Salvador‐Cartier C., Laurent F., Theodorou V., Menard S., Gastroenterology 2017, 153, 1594. [DOI] [PubMed] [Google Scholar]
- 21. Needell J. C., Dinarello C. A., Ir D., Robertson C. E., Ryan S. M., Kroehl M. E., Frank D. N., Zipris D., PLoS One 2017, 12, e0173968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ren Z., Jiang J., Xie H., Li A., Lu H., Xu S., Zhou L., Zhang H., Cui G., Chen X., Liu Y., Wu L., Qin N., Sun R., Wang W., Li L., Wang W., Zheng S., Oncotarget 2017, 8, 95176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Langille M. G., Zaneveld J., Caporaso J. G., McDonald D., Knights D., Reyes J. A., Clemente J. C., Burkepile D. E., Vega Thurber R. L., Knight R., Beiko R. G., Huttenhower C., Nat. Biotechnol. 2013, 31, 814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wheelock A. M., Wheelock C. E., Mol. Biosyst. 2013, 9, 2589. [DOI] [PubMed] [Google Scholar]
- 25. Ogawa T., Hirose Y., Honda‐Ogawa M., Sugimoto M., Sasaki S., Kibi M., Kawabata S., Ikebe K., Maeda Y., Sci. Rep. 2018, 8, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tirandaz H., Mohammadi E., Med. Hypotheses. 2013, 80, 675. [DOI] [PubMed] [Google Scholar]
- 27. Hale V. L., Chen J., Johnson S., Harrington S. C., Yab T. C., Smyrk T. C., Nelson H., Boardman L. A., Druliner B. R., Levin T. R., Rex D. K., Ahnen D. J., Lance P., Ahlquist D. A., Chia N., Cancer Epidemiol. Biomarkers Prev. 2017, 26, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shazali N., Foo H. L., Loh T. C., Choe D. W., Abdul Rahim R., Gut. Pathog. 2014, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Collado M. C., Engen P. A., Bandin C., Cabrera‐Rubio R., Voigt R. M., Green S. J., Naqib A., Keshavarzian A., Scheer Fajl, Garaulet M., Faseb. J. 2017, https://doi.org/10.1096/fj.201700697RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Borges‐Canha M., Portela‐Cidade J. P., Dinis‐Ribeiro M., Leite‐Moreira A. F., Pimentel‐Nunes P., Rev. Esp. Enferm. Dig. 2015, 107, 659. [DOI] [PubMed] [Google Scholar]
- 31. Russo E., Bacci G., Chiellini C., Fagorzi C., Niccolai E., Taddei A., Ricci F., Ringressi M. N., Borrelli R., Melli F., Miloeva M., Bechi P., Mengoni A., Fani R., Amedei A., Front Microbiol. 2017, 8, 2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun J., Kato I., Genes Dis. 2016, 3, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Si J. M., Yu Y. C., Fan Y. J., Chen S. J., World J. Gastroenterol. 2004, 10, 1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zackular J. P., Rogers M. A., th Ruffin M. T., Schloss P. D., Cancer Prev. Res. (Phila) 2014, 7, 1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kanehisa M., Goto S., Nucleic Acids Res. 2000, 28, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Carroll I. M., Chang Y. H., Park J., Sartor R. B., Ringel Y., Gut. Pathog. 2010, 2, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vazquez‐Castellanos J. F., Serrano‐Villar S., Latorre A., Artacho A., Ferrus M. L., Madrid N., Vallejo A., Sainz T., Martinez‐Botas J., Ferrando‐Martinez S., Vera M., Dronda F., Leal M., Del Romero J., Moreno S., Estrada V., Gosalbes M. J., Moya A., Mucosal. Immunol. 2015, 8, 760. [DOI] [PubMed] [Google Scholar]
- 38. Zeller G., Tap J., Voigt A. Y., Sunagawa S., Kultima J. R., Costea P. I., Amiot A., Bohm J., Brunetti F., Habermann N., Hercog R., Koch M., Luciani A., Mende D. R., Schneider M. A., Schrotz‐King P., Tournigand C., Tran Van Nhieu J., Yamada T., Zimmermann J., Benes V., Kloor M., Ulrich C. M., von Knebel Doeberitz M., Sobhani I., Bork P., Mol. Syst. Biol. 2014, 10, 766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Feng Q., Liang S., Jia H., Nat. Commun. 2015, 6, 6528. [DOI] [PubMed] [Google Scholar]
- 40. Bassis C. M., Erb‐Downward J. R., Dickson R. P., Freeman C. M., Schmidt T. M., Young V. B., Beck J. M., Curtis J. L., Huffnagle G. B., MBio. 2015, 6, e00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang S. F., Huang H. D., Fan W. L., Jong Y. J., Chen M. K., Huang C. N., Chuang C. Y., Kuo Y. L., Chung W. H., Su S. C., Oral. Oncol. 2018, 77, 1. [DOI] [PubMed] [Google Scholar]
- 42. Wolf A., Moissl‐Eichinger C., Perras A., Koskinen K., Tomazic P. V., Thurnher D., Sci. Rep. 2017, 7, 5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Parthasarathy G., Chen J., Chia N., O'Connor H. M., Gaskins H. R., Bharucha A. E., Neurogastroenterol. Motil. 2017, 29, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Haro C., Rangel‐Zuniga O. A., Alcala‐Diaz J. F., Gomez‐Delgado F., Perez‐Martinez P., Delgado‐Lista J., Quintana‐Navarro G. M., Landa B. B., Navas‐Cortes J. A., Tena‐Sempere M., Clemente J. C., Lopez‐Miranda J., Perez‐Jimenez F., Camargo A., PLoS One 2016, 11, e0154090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yacoub R., Nugent M., Cai W., Nadkarni G. N., Chaves L. D., Abyad S., Honan A. M., Thomas S. A., Zheng W., Valiyaparambil S. A., Bryniarski M. A., Sun Y., Buck M., Genco R. J., Quigg R. J., He J. C., Uribarri J., PLoS One 2017, 12, e0184789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Miao H., Wu N., Luan C., Yang X., Zhang R., Lv N., Zhu B., Wei Sheng Wu Xue Bao 2014, 54, 1228. [PubMed] [Google Scholar]
- 47. Chen H. M., Yu Y. N., Wang J. L., Lin Y. W., Kong X., Yang C. Q., Yang L., Liu Z. J., Yuan Y. Z., Liu F., Wu J. X., Zhong L., Fang D. C., Zou W., Fang J. Y., Am. J. Clin. Nutr. 2013, 97, 1044. [DOI] [PubMed] [Google Scholar]
- 48. Singh N., Gurav A., Sivaprakasam S., Brady E., Padia R., Shi H., Thangaraju M., Prasad P. D., Manicassamy S., Munn D. H., Lee J. R., Offermanns S., Ganapathy V., Immunity 2014, 40, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. van der Beek C. M., Dejong C. H. C., Troost F. J., Masclee A. A. M., Lenaerts K., Nutr. Rev. 2017, 75, 286. [DOI] [PubMed] [Google Scholar]
- 50. Yuan Z. H., Wang J. P., Zhang K. Y., Ding X. M., Bai S. P., Zeng Q. F., Xuan Y., Su Z. W., Biol. Trace. Elem. Res. 2016, 174, 419. [DOI] [PubMed] [Google Scholar]
- 51. Atarashi K., Suda W., Science 2017, 358, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bullman S., Pedamallu C. S., Sicinska E., Clancy T. E., Zhang X., Cai D., Neuberg D., Science 2017, 358, 1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Eklof V., Lofgren‐Burstrom A., Zingmark C., Edin S., Larsson P., Karling P., Alexeyev O., Rutegard J., Wikberg M. L., Palmqvist R., Int. J. Cancer 2017, 141, 2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gao R., Zhu C., Li H., Yin M., Pan C., Huang L., Kong C., Wang X., Zhang Y., Qu S., Qin H., Obesity (Silver Spring) 2017, 26, 351. [DOI] [PubMed] [Google Scholar]
- 55. Koliada A., Syzenko G., Moseiko V., Budovska L., Puchkov K., Perederiy V., Gavalko Y., Dorofeyev A., Romanenko M., Tkach S., Sineok L., Lushchak O., Vaiserman A., BMC Microbiol. 2017, 17, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tyakht A. V., Kostryukova E. S., Popenko A. S., Belenikin M. S., Pavlenko A. V., Larin A. K., Karpova I. Y., Selezneva O. V., Semashko T. A., Ospanova E. A., Babenko V. V., Maev I. V., Cheremushkin S. V., Kucheryavyy Y. A., Shcherbakov P. L., Grinevich V. B., Efimov O. I., Sas E. I., Abdulkhakov R. A., Abdulkhakov S. R., Lyalyukova E. A., Livzan M. A., Vlassov V. V., Sagdeev R. Z., Tsukanov V. V., Osipenko M. F., Kozlova I. V., Tkachev A. V., Sergienko V. I., Alexeev D. G., Govorun V. M., Nat. Commun. 2013, 4, 2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. David L. A., Maurice C. F., Carmody R. N., Gootenberg D. B., Button J. E., Wolfe B. E., Ling A. V., Devlin A. S., Varma Y., Fischbach M. A., Biddinger S. B., Dutton R. J., Turnbaugh P. J., Nature 2014, 505, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Watson H., Mitra S., Croden F. C., Taylor M., Gut 2017, pii: gutjnl‐2017‐314968. [Google Scholar]
- 59. Koopman J. E., Hoogenkamp M. A., Buijs M. J., Brandt B. W., Keijser B. J., Crielaard W., Ten Cate J. M., Zaura E., Arch. Oral. Biol. 2017, 73, 79. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Text S1. Study design and GT liquid preparation and tea polyphenols evaluation.
Text S2. DNA extration.
Text S3. Statistical analysis.
Figure. S1. Schedule of study visits and participant flow through the study and study protocol and determination of total tea polyphenols concentrations.
Figure. S2. α and β diversity measures and phylum level analysis of fecal samples.
Figure. S3. α‐diversity measures and relative abundance of top ten genera of saliva samples.
Figure. S4. Biomarker functional pathways and α‐diversity measures of saliva samples.
Table S1. Demographic information.
Table S2. Nutrients and energy consumption details of study subjects.
Table S3. Statistical analysis of nutrients and energy consumptions of study subjects.
Table S4. Differential abundance analysis performed on global fecal and salivary microbiota profile
Table S5. The influence of demographics factors on global fecal and salivary microbiome profile