

Abstract
Aberrant activation of cancer‐derived mutants of the epidermal growth factor receptor (EGFR) is closely associated with cancer pathogenesis and is thought to be mediated through multiple tyrosine phosphorylations within the C‐terminal domain. Here, we examined the consequences of the loss of these C‐terminal phosphorylation sites on cellular transformation in the context of lung‐cancer‐derived L858R, exon 19 deletion and exon 20 insertion mutant EGFR. Oncogenic EGFR mutants with substitution of the 10 potential C‐terminal tyrosine autophosphorylation sites for phenylalanine (CYF10) were still able to promote anchorage‐independent growth in soft agar at levels comparable to the parental L858R or exon19 deletion or exon 20 insertion mutants with intact autophosphorylation sites. Furthermore, these CYF10 mutants retained the ability to transform Ba/F3 cells in the absence of IL‐3. Bead‐based phosphorylation and immunoprecipitation analyses demonstrated that key EGFR‐associated proteins—including Grb2 and PLC‐γ—are neither phosphorylated nor bound to CYF10 mutants in transformed cells. Taken together, we conclude that tyrosine phosphorylation is not required for oncogenic activity of lung‐cancer‐derived mutant EGFR, suggesting these mutants can lead to cellular transformation by an alternative mechanism independent of EGFR phosphorylation.
Keywords: epidermal growth factor receptor, lung cancer, autophosphorylation, cellular transformation, Grb2, Bcar1, Shc, Gab
Short abstract
What's new?
Mutations in the epidermal growth factor receptor (EGFR) are common cancer‐driving events that result in ligand‐independent receptor activation and autophosphorylation. Whether autophosphorylation of mutant EGFR is required for transformation, however, is unclear. In this study, lung‐cancer‐derived oncogenic mutant EGFRs lacking 10 potential C‐terminal tyrosine autophosphorylation sites were found to retain the ability to transform cells. Transformation occurred in the absence of IL‐3 ligand. The findings indicate that oncogenic EGFR mutants can be activated independently of autophosphorylation, making their mechanisms of activation distinct from wild‐type EGFR. The results provide mechanistic insight for the understanding of mutant EGFR‐mediated cellular transformation.
Epidermal growth factor receptor (EGFR) is a member of the ErbB family consisting of closely related receptor tyrosine kinases including ErbB2, ErbB3 and ErbB4.1 Aberrant activation of EGFR is among the most common oncogenic driving events in human cancer, which is mediated largely through different classes of genomic alterations within the EGFR gene.2 These genomic events include somatic EGFR mutations within regions of either the extracellular domain, in glioblastoma, or the kinase domain in lung adenocarcinoma,3, 4 as well as through gene amplification as observed in many other types of solid tumors.5, 6 In addition, several intragenic deletions within either the extracellular or C‐terminal domain of EGFR have also been reported to be oncogenic in a subset of glioblastoma and lung adenocarcinoma.7, 8, 9
The activation of EGFR is believed to be induced through sequential receptor dimerization occurring at two distinct sites of homo‐ or heterodimerization with other ErbB family members upon ligand stimulation; one between the extracellular domains of EGFR and one between the intracellular domains of the receptor.10, 11 Ligand‐induced dimerization of the receptor's extracellular domain leads to approximation of the intracellular domains, followed by asymmetric dimerization of the two kinase domains, where the C‐lobe of one kinase domain (the “activator” monomer) binds to the N‐lobe of the second kinase domain (the “receiver” monomer) and allosterically activates it.11 Following these receptor dimerization events, EGFR becomes fully activated and undergoes phosphorylation at multiple tyrosine residues within its C‐terminal tail. Signaling molecules containing Src homology 2 (SH2) domains and phosphotyrosine binding (PTB) domains are then recruited to specific phosphotyrosines of the receptor, subsequently priming the induction of downstream signaling cascades.12 As a consequence of the biochemical events described above, wild‐type EGFR has been observed to require C‐terminal phosphorylation to enable anchorage‐independent growth.13
Structural and biochemical studies have shown that oncogenic EGFR mutants, unlike wild‐type EGFR, can undergo constitutive receptor dimerization and consequent autophosphorylaton of the receptor in the absence of EGF or alternatively though activating mutations within the EGFR kinase domain that induce an active conformation not dependent on ligand‐induced asymmetric dimerization.14, 15, 16 Furthermore, accumulating data suggest that the downstream signaling pathways activated by oncogenic mutant EGFR differ from those activated by ligand stimulated wild‐type EGFR.17, 18, 19 Notably, C‐terminal deletion EGFR mutants, lacking some or all of the autophosphorylation sites that have been identified in GBM and lung adenocarcinoma, are able to induce cellular transformation.8, 9, 20 These observations have raised the question whether autophosphorylation of oncogenic mutant EGFR, which is a consequence of constitutive receptor dimerization, is required for oncogenic activation and induction of cellular transformation by cancer‐derived EGFR mutants.
To elucidate these questions, isogenic cell lines were engineered to express tyrosine phosphorylation‐impaired mutant forms of lung cancer‐derived L858R, Ex19Del or Ex20Ins EGFR mutants. These double mutants were then used to assess the functional necessity for autophosphorylation to enable cellular transformation by mutant EGFR. Interestingly, we found that tyrosine phosphorylation of the carboxyl‐terminus is not required for the transforming activity of EGFR bearing activating mutations of the kinase domain. This result suggests that mutant EGFR may be able to transmit oncogenic downstream signals through an alternative mechanism independent of EGFR phosphorylation.
Materials and Methods
Expression constructs
Wild‐type EGFR, L747_E749del/A750P (Ex19Del), D770_N771insNPG (Ex20Ins) and L858R mutant EGFR expression vectors were prepared as previously described.15 For generating all mutant constructs described in this study, QuikChange site‐directed mutagenesis (Agilent Technology) was used with either wild‐type EGFR or the above mutant EGFR in pBabe‐puro as a template.
Cell culture and generation of cell lines by retroviral transduction
NIH‐3T3 cells were cultured according to the standard protocols provided by the American Type Culture Collection (ATCC). All NIH‐3T3 cell lines stably expressing EGFR mutants were generated by retroviral infections and pooled as described previously.15 Cultures were serum‐starved for 18 hr prior to EGF stimulation and harvesting. Epidermal growth factor (EGF, Invitrogen) stimulations were performed at 25 ng/ml for 5 min unless noted in the text. Ba/F3 cells were maintained in the presence of IL3.
Immunoblotting, immunoprecipitation and antibodies (Abs)
Cells were lysed in RIPA buffer (50 mM Tris‐HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% NP‐40, 0.5% mM sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitors (Roche) and phosphatase inhibitors (Calbiochem) prior to use according to the manufacturer's protocols. For immunoblotting, 50–100 µg of protein were separated by 8–10% SDS‐PAGE, transferred and probed with antibodies. For immunoprecipitation, 200–500 µg of protein lysates were incubated with protein A along with EGFR or Bcar1 antibodies for 3 hr. Anti‐EGFR antibody was purchased from Bethyl Laboratory (Montgomery, TX). Antibodies against p‐EGFR (Y1092, Y1110, Y1172, Y1197), p‐ErbB2 (Y1121/1222), ErbB2, p‐MET (Y1234/1235), MET, p‐Src (Y416), p‐Gab1 (Y307, Y627), Gab1, p‐Gab2 (S159, Y452), p‐Bcar1 (Y249), p‐Shc1 (Y317) and STAT3 were purchased from Cell Signaling Technology. Anti‐phospho‐tyrosine (4G10), Shc1 and Gab2 antibodies were from Millipore. Anti‐Grb2, anti‐PLCγ1, anti‐Bcar1 and antitubulin antibodies were from Santa Cruz Biotechnology.
Anchorage‐independent and cell proliferation growth assays
Soft agar assays were carried out in triplicate as previously described.15, 16 Photographed images of soft agar colonies taken after 2–3 weeks were quantified using Image J software (NIH). The data were normalized to the numbers of colonies formed by control cells (see figure legends). Each assay was repeated for at least two times with comparable results.
Confocal immunofluorescent microscopy
Immunofluorescence analysis was conducted as previously described.16 In brief, cells were seeded on a 0.16‐mm‐thick 12 mm cover slip (Fisher Scientific) in 24 well plates. After serum starvation overnight, cells were fixed with paraformaldehyde (4% wt/vol) and permeabilized with Triton X‐100 (0.1% vol/vol in PBS). The fixed and/or permeabilized cells were incubated with anti‐EGFR‐FITC conjugated antibodies (Abcam) at 1:500 dilution for 1 hr, washed 2 times with PBG and incubated with anti‐DAPI antibodies (MP biomedicals, Solon, OH) for 10 min. Confocal immunofluorescence images were collected with 405 nm and 488 nm lasers (Andor Technology, South Windsor, CT) mounted on a Nikon Ti‐E inverted microscope (Nikon Instruments, Melville, NY). Images were acquired using a 100× Plan Apo objective lens with an Andor iXon 897 EMCCD camera (Andor Technology, South Windsor, CT). Acquisition parameters, shutters, filter positions and focus were controlled by Andor iQ software (Andor Technology, South Windsor, CT).
Luminex assay
Hundred microliters of each bead‐type of Luminex xMAP microspheres (Luminex Corporation, Austin, TX) were coupled separately to antibodies (EGFR, Grb2 and PLCγ) using the manufacturer's recommended procedure. Assays were performed as previously described.21 In brief, the data were acquired with a Luminex FlexMAP 3 D instrument (Luminex Corporation) according to the manufacturer's instructions. The background readings for each capture antibody were obtained using microspheres incubated with 1x cell lysis buffer (Cell Signaling Technology). Values were considered positive if they were threefold over the background. The data were normalized against unstimulated EGFR and graphed as a fold increase in relative phosphorylation. Data shown represent the average of three independent experiments.
Results
C‐terminal tyrosine phosphorylation is dispensable for mutant EGFR‐mediated cellular transformation
To investigate the role of tyrosine autophosphorylation on transformation by mutant EGFR, we established NIH‐3T3 derived cell lines expressing EGFR mutants where all 10 C‐terminal tyrosine residues were mutated to phenylalanine (CYF10) in L858R, Ex19Del and Ex20Ins mutant backgrounds as well as wild‐type EGFR (Fig. 1 a). The cancer‐derived EGFR mutants were previously shown to undergo high levels of constitutive dimerization and autophosphorylation in the C‐terminal domain.15, 16 Immunoblotting with the general antiphosphotyrosine antibody 4G10, as well as four anti‐phospho‐EGFR specific antibodies, revealed that EGF‐induced tyrosine phosphorylation on wild‐type EGFR was completely abolished in the CYF10 EGFR mutant (Fig. 1 b, lanes 2 and 4). Similarly, no detectable constitutive EGFR tyrosine phosphorylation of L858R/CYF10, Ex19Del/CYF10 or Ex20Ins/CYF10 EGFR mutants was found (Fig. 1 b, lanes 6, 8 and 10), confirming that all C‐terminal phosphorylation sites were mutated and that CYF10 EGFR mutants lack autophosphorylation. Furthermore, the absence of tyrosine phosphorylation was not required for activation of downstream oncogenic pathways including STAT3, and Src unlike what was observed in the WT/CYF10 EGFR mutant (Fig. 1 b, lanes 4, 6, 8 and 10).
Figure 1.
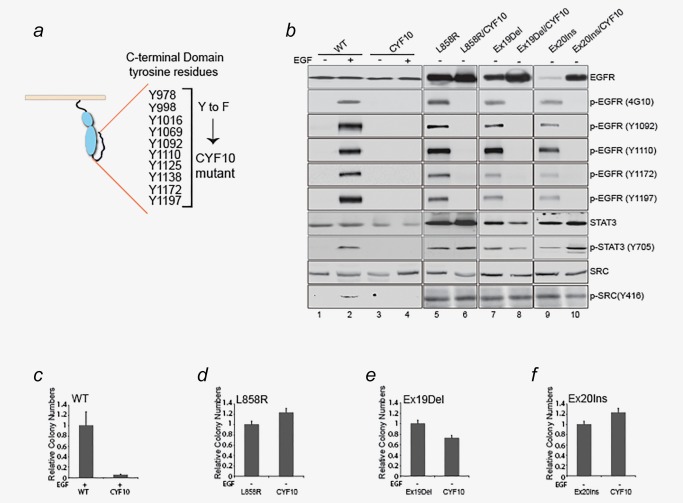
Lung adenocarcinoma‐driven EGFR mutants are oncogenic in the absence of tyrosine phosphorylation. (a) Schematic diagram of C‐terminal tyrosine residues and CYF10 mutants. (b) Tyrosine phosphorylation was not detectable in the CYF10 mutants. Whole‐cell lysates from NIH‐3T3 cells stably expressing wild‐type EGFR, L858R, Ex19Del or Ex20Ins mutants with or without the CYF10 mutation were subjected to immunoblotting with antibodies against phospho‐specific EGFR, phospho‐tyrosine (4G10), EGFR, p‐STAT3, STAT3, p‐Src and Src. (c–f) Transforming ability of mutant EGFR was not affected by abrogation of C‐terminal phosphorylation. Anchorage‐independent growth was assayed in the NIH‐3T3 cells used for immunoblotting in panel (b). The bar graph is depicted as relative number of colonies normalized to cell lines expressing the parental EGFR mutants (n = 3, mean + SD).
Next, we examined the oncogenic activity of various CYF10 EGFR mutants by assessing their ability to promote anchorage‐independent growth in soft agar, a hallmark of cellular transformation. When compared to wild‐type EGFR, the number of colonies formed by CYF10 EGFR mutant in the presence of EGF was significantly reduced, confirming the critical role of C‐terminal phosphorylation in the oncogenic activity of wild‐type EGFR (Fig. 1 c and Supporting Information, Fig. S1). In contrast, CYF10 double mutants with L858R, Ex19Del and Ex20Ins mutants were still able to form colonies in soft agar at levels comparable to either the L858R or the Ex19Del or the Ex20Ins mutants with intact autophosphorylation (Figs. 1 d–1 f and Supporting Information, Fig. S1), suggesting that C‐terminal phosphorylation of these cancer‐derived mutants is not required for their transforming capability.
To exclude the possibility that the phosphorylation‐independent transforming activity of mutant EGFR is limited to NIH‐3T3 cells, the oncogenic potential of the CYF10 EGFR mutants were tested in Ba/F3 cells. Consistent with previous reports,22 Ba/F3 cells expressing L858R, Ex19Del and Ex20Ins oncogenic EGFR mutants were able to grow in the absence of IL‐3. Ectopic expression of the double mutants in Ba/F3 cells also rendered these cells IL‐3 independent, and showed similar growth ability under the same conditions, thus confirming that the CYF10 EGFR mutants have the ability to induce cellular transformation (Fig. 2 a). As expected, we found that the phosphorylation of CYF10 EGFR mutants were not detectable, and the levels of all mutant EGFR in transformed Ba/F3 cells were equivalent to parental mutants with the exception of Ex20Ins/CYF10 EGFR mutant which is higher than Ex20Ins mutant (Fig. 2 b). Taken together, these results are consistent with reports showing that deletion of the entire EGFR C‐terminus does not disrupt the Ex19Del mutant EGFR activity.23, 24 Indeed, the current data extend these findings to indicate that even in the presence of an intact C‐terminus, tyrosine phosphorylation is still not necessary for the transforming activity of EGFR mutants.
Figure 2.
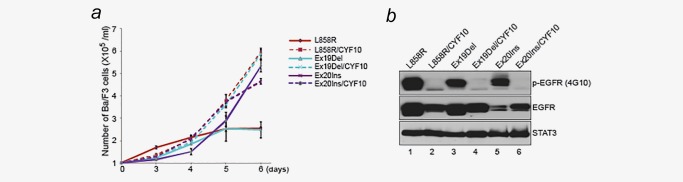
Ba/F3 cells stably expressing CYF10 mutants are able to grow in IL‐3 independent manner. (a) Cell growth transducing potential of mutant EGFR is not diminished by abrogation of C‐terminal phosphorylation. Cell proliferation ability of various transformed Ba/F3 cell lines used for panel (a) was assayed by counting cell numbers on 3, 4, 5 and 6 days later (0.2 × 106/ml each cell lines were seeded on day 0 after 2 weeks of IL‐3 withdrawal). The results are indicated as means ± SD of five cell counts. (b) Tyrosine phosphorylation was drastically reduced in CYF10 mutants. Whole‐cell lysates from Ba/F3 cells transformed with L858R, Ex19Del and Ex20Ins EGFR mutant with or without CYF10 mutation were subjected to immunoblotting with antibodies against phosphotyrosine (4G10), EGFR or STAT3. The level of STAT3 expression is shown as a loading control. Based on the molecular weight of EGFR, the lower bands detected by the antiphosphotyrosine antibody in the blot is not likely to be phospho‐EGFR, but an unknown phosphoprotein.
Cellular transformation by mutant EGFR does not involve Grb2 nor PLC‐γ activation
Next, we sought to examine which critical oncogenic signaling proteins downstream of EGFR were involved and potentially contributing to cellular transformation by CYF10 mutants in a phosphorylation‐independent manner. Among known signaling molecules, we focused on Grb2, which is a critical adaptor protein that is specifically recruited to phosphorylated EGFR to mediate numerous downstream signaling pathways including MAPK and PI3K/AKT pathways.25 Through immunoprecipitation with anti‐EGFR antibodies, we found that Grb2 is constitutively associated with all kinase‐domain mutants and, in an EGF‐dependent manner with wild‐type EGFR (Fig. 3 a, lanes 2, 5, 7 and 9). However, Grb2 failed to associate with any of the CYF10 mutants under the same conditions (Fig. 3 a, lanes 4, 6, 8 and 10), which is consistent with previous reports that phosphorylation (pY1092 and pY1110) on EGFR is required for binding of Grb2 to the receptor.26 Furthermore, we examined the phosphorylation status of EGFR, Grb2 and PLC‐γ using a Luminex assay (see details in Materials and Methods) and the phosphorylation of these proteins was abolished in the CYF10 double mutants irrespective of EGF stimulation (Fig. 3 b, p‐EGFR and Supporting Information, Fig. S2). These results confirm previous findings that the association of signaling proteins with EGFR is mediated by tyrosine phosphorylation within its C‐terminal domain and the binding may be required for phosphorylation of these proteins.26 Given that the phosphorylation‐impaired CYF10 EGFR mutants are able to transform the cells at comparable levels to phosphorylation‐competent EGFR mutants without binding to EGFR and/or activation of Grb2 and PLC‐γ, we concluded that these proteins are unlikely to be the essential signaling mediators for the oncogenic EGFR mutants‐mediated cellular transformation.
Figure 3.
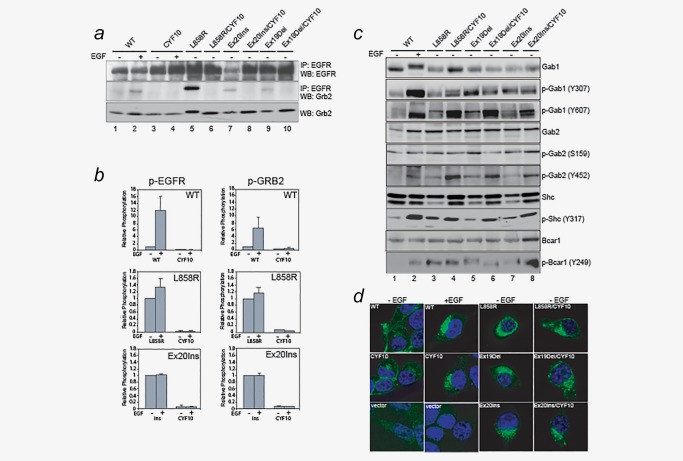
Shc1 and Bcar1, but not Grb2, may play a crucial role in induction of oncogenic signal activation by CYF10 mutants. (a) Grb2 adaptor proteins fail to associate with CYF10 mutants. Cell lysates prepared from NIH‐3T3 cells expressing L858R, Ex19Del, EX20Ins mutants or wild‐type EGFR with or without the CYF10 mutation were subjected to immunoprecipitation with anti‐EGFR antibody followed by immunoblotting for Grb2. The same blot was stripped and reprobed with anti‐EGFR antibody. The level of Grb2 expression was similar in all samples. (b) Phosphorylation of EGFR and Grb2 is abolished in CYF10 mutants. The levels of phospho‐EGFR and phospho‐Grb2 in CYF10 mutants were quantified by Luminex assays (see Experimental Procedures) in the presence or absence of EGF. The bar graph shows the relative levels of phosphorylation of EGFR and Grb2 in each CYF10 mutant normalized with to the respective EGF‐stimulated wild‐type EGFR, L858R or Ex20Ins mutants (n = 3, mean + SD). (c) Gab1/2, Shc1 and Bcar1 adaptor proteins were constitutively phosphorylated by mutant EGFR irrespective of C‐terminal phosphorylation. Cell lysates used in (a) were subjected to immunoblotting with antibodies against p‐Gab1/2, Gab1/2, p‐Shc1, Shc1 and p‐Bcar1. (d) The pattern of subcellular localization of kinase domain mutant EGFR was similar to that of EGF‐stimulated wild‐type EGFR, which is not affected by receptor C‐terminal phosphorylation. Confocal microscope images were acquired from the NIH‐3T3 stable cells described above after being fixed, permeablized and stained with FITC‐conjugated anti‐EGFR antibody.
Phosphorylation of major EGFR adaptor proteins are maintained during CYF10 mutant‐induced cellular transformation
To further investigate the potential key proteins involved in oncogenic signaling cascade, we examined the phosphorylation status of major EGFR adaptor proteins including Gab1/2, Shc1 and Bcar1, which were reported to be highly phosphorylated in cells that express mutant EGFR.18, 19, 27 We observed that the phosphorylation of these proteins in cells expressing wild‐type EGFR are induced by EGF stimulation (Fig. 3 c, lanes 1 and 2). However unlike Grb2, these adaptor proteins are constitutively phosphorylated at levels comparable or higher between cells expressing the CYF10 mutants as either the L858R or the Ex19Del or the Ex20Ins mutant EGFR (Fig. 3 c, lanes 3–8). These results indicate that these adaptor proteins are able to be activated in an EGFR phosphorylation‐independent manner.
Heterodimerization of CYF10 mutants with other endogenous ErbB family members and/or Met receptors could be a possible explanation for the constitutive phosphorylation of these adaptor proteins.23 To rule this out, phospho‐specific immunoblotting of ErbB2 and MET was performed; we did not observe detectable levels of total or phosphorylated forms of ErbB2, ErbB3, ErbB4 or Met in cells transformed by CYF10 mutants (Supporting Information, Figs. S3a and S3b and data not shown). Furthermore, differences in subcellular localization were also not associated with phosphorylation‐independent oncogenic activation of mutant EGFR as we did not observe any difference in receptor localization between kinase domain mutant EGFR and CYF10 mutants (Fig. 3 d).
Taken together, our results suggest that major EGFR adaptor proteins such as Gab1/2, Shc1 and Bcar1 proteins, but not Grb2, can be activated through an alternative mechanism independent of EGFR phosphorylation.
Discussion
Ligand‐induced tyrosine phosphorylation on specific residues within the C‐terminal domain of EGFR following asymmetric receptor dimerization of its kinase domain has been well characterized.28 Specifically, this posttranslational modification on EGFR is known to provide a docking site for various signaling proteins which subsequently induce the canonical downstream signaling cascade, leading to various biological effects such as proliferation, migration and cell survival.12 Consistent with previous studies,24 our functional analysis demonstrated that disruption of autophosphorylation on EGFR impaired the ability of EGF‐stimulated cellular transformation of wild‐type EGFR, demonstrating the essential requirement for this mechanism in the activation of downstream signaling pathways leading to cellular transformation. Intriguingly, unlike wild‐type EGFR, our data clearly demonstrate that tyrosine phosphorylation‐impaired CYF10 EGFR mutants are still able to transform NIH‐3T3 and Ba/F3 cells, suggesting that constitutive phosphorylation on mutant EGFR may be dispensable for their transforming potential. These paradoxical results are consistent with our reports that a subset of mutant EGFR is capable of cellular transformation irrespective of asymmetric dimerization16 and that various C‐terminal intragenic deletion mutants identified in glioblastomas and lung adenocarcinoma are oncogenic.29, 30 Furthermore, an Ex19Del mutant was shown to retain its oncogenic activity in the absence of the C‐terminal domain or autophosphorylation,23, 24 putatively through heterodimerization of phosphorylated ErbB3 with C‐terminal domain deleted Ex19Del. However, under our experimental conditions, we did not detect any phosphorylated ErbB family members including EGFR in the cell lysates prepared from transformed cells by CYF10 mutants, suggesting that the cellular transforming ability of these mutants may be mediated by the other mechanism.
We believe that these findings reveal several interesting and previously unknown mechanisms that contribute to mutant EGFR mediated cellular transformation. First, autophosphorylation of EGFR can have either a positive or a negative role in the regulation of EGFR activity depending upon the site and temporal–spatial balance. We hypothesized that absence of two major negative regulatory phosphorylation sites at Y1016 and Y1069, in the L858R/CYF10, Ex19Del/CYF10 or Ex20Ins/CYF10 EGFR mutants are sufficient to induce oncogenic signaling cascades through aberrant activation of the Ras‐ERK signaling and loss of EGFR ubiquitination and subsequent receptor degradation, respectively.31, 32 However, this is not the case in wild‐type EGFR/CYF10 mutants, suggesting that ligand induced multiple autophosphorylation is required for cellular transformation in wild‐type EGFR. Second, we found that the adaptor proteins Gab1/2, Shc1 and Bcar1 (p130 Cas), but not Grb2 and PLC‐γ, were still activated by L858R/CYF10, Ex19Del/CYF10 or Ex20Ins/CYF10 mutants. Given that Grb2 is well known to be a key player in initiation of signaling cascade by EGFR such as in the RAS/RAF/MEK/MAP kinase pathway, the finding that a lack of Grb2 association to the receptor does not affect oncogenic transformation by CYF10 mutants is somewhat intriguing. We hypothesize that unlike wild‐type EGFR, mutant EGFR may possess an ability to interact directly or indirectly with key signaling molecules and regulatory proteins responsible for induction and maintenance of oncogenic signaling networks in an autophosphorylation‐independent way. This may be caused by either unique structural configuration of mutant EGFR and/or increased enzymatic activity by structural changes of C‐terminal tail. A recent proteomic study has identified several proteins that are differentially phosphorylated between wild‐type and mutant EGFR.18 Thus, one possibility is that Gab1/2, Shc1 and Bcar1 adaptors which are found to be constitutively phosphorylated in CYF10 mutants expressing cells may associate with and are activated in a mutant‐EGFR‐specific manner to function as crucial players in mediating constitutive oncogenic activation of various signaling pathways independently of C‐terminal phosphorylation. The detailed mechanism and functional significance of activated adaptor proteins in CYF10 mutants need to be further explored in future studies.
In summary, our functional and biochemical data clearly demonstrate that C‐terminal phosphorylation is not required for oncogenic transformation by mutant EGFR. In contrast, a phosphorylation‐independent activation mechanism leading to cellular transformation appears to exist for kinase domain activating mutant EGFR found in lung cancer.
Supporting information
Supporting Information
Supporting Information
Competing Interests: Matthew Meyerson is an inventor on patent for EGFR mutation analysis in lung cancer, licensed to LabCorp. The other authors have no relevant competing interests.
Contributor Information
Jeonghee Cho, Email: jeonghee.cho@dankook.ac.kr.
Matthew Meyerson, Email: matthew_meyerson@dfci.harvard.edu.
References
- 1. Yarden Y, Pines G. The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer 2012;12:553–63. [DOI] [PubMed] [Google Scholar]
- 2. Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy‐number alteration across human cancers. Nature 2010;463:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee JC, Vivanco I, Beroukhim R, et al. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med 2006;3:e485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Castellanos E, Feld E, Horn L. Driven by mutations: the predictive value of mutation subtype in EGFR‐mutated non‐small cell lung cancer. J Thorac Oncol 2016. [DOI] [PubMed] [Google Scholar]
- 5. Hirono Y, Tsugawa K, Fushida S, et al. Amplification of epidermal growth factor receptor gene and its relationship to survival in human gastric cancer. Oncology 1995;52:182–8. [DOI] [PubMed] [Google Scholar]
- 6. Al‐Kasspooles M, Moore JH, Orringer MB, et al. Amplification and over‐expression of the EGFR and erbB‐2 genes in human esophageal adenocarcinomas. Int J Cancer 1993;54:213–9. [DOI] [PubMed] [Google Scholar]
- 7. Gan HK, Cvrljevic AN, Johns TG. The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. FEBS J 2013;280:5350–70. [DOI] [PubMed] [Google Scholar]
- 8. Cho J, Pastorino S, Zeng Q, et al. Glioblastoma‐derived epidermal growth factor receptor carboxyl‐terminal deletion mutants are transforming and are sensitive to EGFR‐directed therapies. Cancer Res 2011;71:7587–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Imielinski M, Berger AH, Hammerman PS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012;150:1107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dawson JP, Berger MB, Lin CC, et al. Epidermal growth factor receptor dimerization and activation require ligand‐induced conformational changes in the dimer interface. Mol Cell Biol 2005;25:7734–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang X, Gureasko J, Shen K, et al. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006;125:1137–49. [DOI] [PubMed] [Google Scholar]
- 12. Schlessinger J, Lemmon MA. SH2 and PTB domains in tyrosine kinase signaling. Sci STKE 2003;2003:re12. [DOI] [PubMed] [Google Scholar]
- 13. Decker SJ. Transmembrane signaling by epidermal growth factor receptors lacking autophosphorylation sites. J Biol Chem 1993;268:9176–9. [PubMed] [Google Scholar]
- 14. Yun CH, Boggon TJ, Li Y, et al. Structures of lung cancer‐derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007;11:217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Greulich H, Chen TH, Feng W, et al. Oncogenic transformation by inhibitor‐sensitive and ‐resistant EGFR mutants. PLoS Med 2005;2:e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cho J, Chen L, Sangji N, et al. Cetuximab response of lung cancer‐derived EGF receptor mutants is associated with asymmetric dimerization. Cancer Res 2013;73:6770–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen YR, Fu YN, Lin CH, et al. Distinctive activation patterns in constitutively active and gefitinib‐sensitive EGFR mutants. Oncogene 2006;25:1205–15. [DOI] [PubMed] [Google Scholar]
- 18. Guha U, Chaerkady R, Marimuthu A, et al. Comparisons of tyrosine phosphorylated proteins in cells expressing lung cancer‐specific alleles of EGFR and KRAS. Proc Natl Acad Sci USA 2008;105:14112–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Park E, Kim N, Ficarro SB, et al. Structure and mechanism of activity‐based inhibition of the EGF receptor by Mig6. Nat Struct Mol Biol 2015;22:703–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Park AK, Francis JM, Park WY, et al. Constitutive asymmetric dimerization drives oncogenic activation of epidermal growth factor receptor carboxyl‐terminal deletion mutants. Oncotarget 2015;6:8839–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Du J, Bernasconi P, Clauser KR, et al. Bead‐based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat Biotechnol 2009;27:77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiang J, Greulich H, Janne PA, et al. Epidermal growth factor‐independent transformation of Ba/F3 cells with cancer‐derived epidermal growth factor receptor mutants induces gefitinib‐sensitive cell cycle progression. Cancer Res 2005;65:8968–74. [DOI] [PubMed] [Google Scholar]
- 23. Rothenberg SM, Engelman JA, Le S, et al. Modeling oncogene addiction using RNA interference. Proc Natl Acad Sci USA 2008;105:12480–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maegawa M, Arao T, Yokote H, et al. Epidermal growth factor receptor lacking C‐terminal autophosphorylation sites retains signal transduction and high sensitivity to epidermal growth factor receptor tyrosine kinase inhibitor. Cancer Sci 2009;100:552–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lowenstein EJ, Daly RJ, Batzer AG, et al. The SH2 and SH3 domain‐containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 1992;70:431–42. [DOI] [PubMed] [Google Scholar]
- 26. Batzer AG, Rotin D, Urena JM, et al. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol Cell Biol 1994;14:5192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Song X, Fan PD, Bantikassegn A, et al. ERBB3‐independent activation of the PI3K pathway in EGFR‐mutant lung adenocarcinomas. Cancer Res 2015;75:1035–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kovacs E, Zorn JA, Huang Y, et al. A structural perspective on the regulation of the epidermal growth factor receptor. Annu Rev Biochem 2015;84:739–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ekstrand AJ, Sugawa N, James CD, et al. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N‐ and/or C‐terminal tails. Proc Natl Acad Sci USA 1992;89:4309–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. TCGA . Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008;455:1061–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Agazie YM, Hayman MJ. Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol Cell Biol 2003;23:7875–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Levkowitz G, Waterman H, Ettenberg SA, et al. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c‐Cbl/Sli‐1. Mol Cell 1999;4:1029–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information