

Abstract
Machines learn chemistry: An artificial intelligence algorithm has learned to predict the outcomes of C−N coupling reactions from a few thousand nanomole‐scale experiments. This Highlight discusses this work in the context of other state‐of‐the‐art approaches for predicting the yields of organic reactions and explains the significance of the results.
Keywords: Buchwald–Hartwig reaction, high-throughput synthesis robot, machine learning, nanomole-scale reactions
The ability to predict the outcome of complex chemical transformations has been a long‐standing challenge for chemists. The development of quantum‐chemical approaches has already opened some opportunities in this direction, and in many cases, the outcomes of experiments can be efficiently modeled in silico.1, 2, 3, 4, 5, 6 The advent of artificial intelligence (AI) algorithms to automatize, improve, and generalize predictions is gaining importance in this field, and several recent studies have been published in this area. For example, in 2016, Aspuru‐Guzik and co‐workers reported their attempt to apply neural networks to basic reactions of alkenes and alkyl halides, and they were able to identify the correct reaction type for the majority of a set of textbook problems.7 In 2017, Gambin and co‐workers tested AI algorithms to predict a large set (450 000 cases) of manifold organic reactions, emphasizing that it might be essential to identify new chemoinformatic descriptors for future developments.8 Among other important attempts to predict and optimize organic reactions on the basis of AI, recent studies by the group of Zare9 as well as Jensen, Green, and co‐workers are noteworthy examples.10 Although the predictions had some limitations, in general, the AI algorithms showed an encouragingly good performance even for sophisticated organic systems.
A recent study by the groups of Doyle and Dreher11 now demonstrates how the yields of a Buchwald–Hartwig coupling (Scheme 1) with a large set of different substrates can be accurately predicted with an AI algorithm, in this case a so‐called random forest. The particularity of the study is that the data from which the algorithm learns are generated experimentally with a nanomole‐scale high‐throughput robot. The AI predictions substantially outperformed many previous works.
Scheme 1.
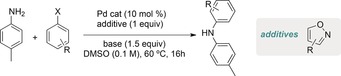
Buchwald–Hartwig coupling investigated in the study by the groups of Doyle and Dreher.11
The procedure is as follows: First, the random forest model is trained. Here, molecular properties of the reactants, for example, their vibrational frequencies or dipole moments, are calculated by quantum chemistry. These properties serve as “descriptors”, that is, as inputs for the random forest algorithm. The reaction yield with a given set of reactants is then determined experimentally with the high‐throughput robot, and is fed into the machine learning algorithm. The algorithm learns to generate these yields as outputs when provided with the corresponding inputs generated from quantum‐chemistry calculations. After this training step, the random forest algorithm is able to predict the reaction yield of other, previously untested reactant combinations, whereby the procedure could be summarized in an oversimplified manner as: “If the reactants feature these vibrational frequencies and these dipole moments, then the reaction yield will be that number.” In this regard, it is interesting to consider that machine learning algorithms (which have been employed for decades) think differently to an experimental organic chemist, who would probably not take properties such as the vibrational spectrum of a reactant or its dipole moment into detailed account to estimate whether a reaction involving that reactant shall result in a high or a low yield. The work of Doyle and Dreher is a very promising breakthrough as they managed to obtain an excellent prediction accuracy, and it opens a range of opportunities for both theoretical and experimental chemists. It holds promise to dramatically accelerate the reaction optimization process in modern organic synthesis.
A particularly interesting outcome of the study relates to the conspicuous problems encountered when palladium‐catalyzed Buchwald–Hartwig coupling procedures are applied to the preparation of complex drug‐like products, namely the strong limitations observed for substrates containing heteroatom–heteroatom bonds, such as isoxazoles (Scheme 1). The authors sought to probe this in their model by concurrently screening several structurally diverse oxazole additives by using the fragment additive approach proposed by Glorius and co‐workers.12 The results of the study and the predictive model that it afforded (whereby certain properties of the oxazole additives were found to strongly correlate with the yield of the Buchwald–Hartwig coupling) ultimately guided the mechanistic discovery that Pd0 competitively inserts into the N−O bond of isoxazoles, as demonstrated in a series of guided experiments. Two isoxazole fragments with dramatically different C3 NMR shifts (13C NMR shifts being one of the top 10 descriptors of the trained random forest model) were shown to behave rather differently when exposed to a prototypical Pd0 precatalyst (Figure 1). As the authors themselves point out, such a mechanistic assumption would certainly not have been unconceivable without the machine learning process, and it also hints at a more “human” intuitive dimension that must still accompany the development of such AI‐generated algorithms.
Figure 1.
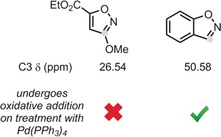
Simplified diagram depicting isoxazole additives to Buchwald–Hartwig coupling reactions, compared in terms of the C3 13C NMR chemical shift descriptor and the experimentally confirmed propensity to undergo N−O oxidative addition upon exposure to a Pd0 precatalyst.11
This milestone achievement immediately leads to several questions, such as: How generalizable is this approach, that is, is it possible to use the method for other classes of organic reactions? Can the predictions be made even more efficiently? And, for all organic chemists reading this article, how far ahead is the (dystopian?) scenario of machine‐learning algorithms combined with synthesis robots effectively replacing them?
One of the next likely steps is the improvement of the computational approach employed to obtain the descriptors. Indeed, other classes of organic reactions are likely to require the consideration of more structurally flexible and branched molecular systems. For these systems, it might not be enough to calculate only one conformational minimum. This is perhaps best illustrated with an example: Consider two structurally similar reactants, each with two possible stable conformations A and B. A single quantum‐chemical minimization of each reactant might find conformation A for the one reactant and conformation B for the other reactant. The two reactants might thus be recognized as being very different by the AI algorithm, resulting in different reaction yields being predicted although they may be similar in practice.
Furthermore, the actual quantum‐chemistry method employed (mostly B3LYP/6‐31G* in this case) can be discussed. Just hearing this acronym might trigger a flurry of suggestions for improvement from quantum chemists; nevertheless, one should bear in mind that the AI algorithm only needs to learn about the similarities of the reactants and their reactions (which can also be obtained from similarly wrong results for similar reactants). It is therefore imaginable that semi‐empirical methods might provide similar, satisfactory results at reduced computational cost.
The “age of automation”13 thus appears to hold the potential to advance organic synthesis in a revolutionary way. We can finally ask provocatively, as in the title of this manuscript: Are robots replacing chemists? Looking at the possible pitfalls of the methods discussed above, we believe that we are not there yet. Overall, the main problem remains a lack of generality. However, the rapid development of AI approaches in combination with modern organic and quantum chemistry might change this situation in the near future. Additionally, the “human intuition” factor alluded to previously should provide some comfort—at least until AI algorithms are capable of mechanistic inferences.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
We thank the University of Vienna for continued support of our research programs and the European Research Council (ERC CoG VINCAT 682002).
B. Maryasin, P. Marquetand, N. Maulide, Angew. Chem. Int. Ed. 2018, 57, 6978.
References
- 1. Hie L., Fine Nathel N. F., Shah T. K., Baker E. L., Hong X., Yang Y. F., Liu P., Houk K. N., Garg N. K., Nature 2015, 524, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bess E. N., Guptill D. M., Davies H. M. L., Sigman M. S., Chem. Sci. 2015, 6, 3057–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Margrey K. A., Mcmanus J. B., Bonazzi S., Zecri F., Nicewicz D. A., J. Am. Chem. Soc. 2017, 139, 11288–11299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kromann J. C., Jensen J. H., Kruszyk M., Jessing M., Jørgensen M., Chem. Sci. 2018, 9, 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peng Q., Duarte F., Paton R. S., Chem. Soc. Rev. 2016, 45, 6093–6107. [DOI] [PubMed] [Google Scholar]
- 6. Sperger T., Sanhueza I. A., Schoenebeck F., Acc. Chem. Res. 2016, 49, 1311–1319. [DOI] [PubMed] [Google Scholar]
- 7. Wei J. N., Duvenaud D., Aspuru-Guzik A., ACS Cent. Sci. 2016, 2, 725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Skoraczyński G., Dittwald P., Miasojedow B., Szymkuć S., Gajewska E. P., Grzybowski B. A., Gambin A., Sci. Rep. 2017, 7, 3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou Z., Li X., Zare R. N., ACS Cent. Sci. 2017, 3, 1337–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coley C. W., Barzilay R., Jaakkola T. S., Green W. H., Jensen K. F., ACS Cent. Sci. 2017, 3, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ahneman D. T., Estrada J. G., Lin S., Dreher S. D., Doyle A. G., Science 2018, 359, 1–9. [Google Scholar]
- 12. Collins K. D., Gensch T., Glorius F., Nat. Chem. 2014, 6, 859–871. [DOI] [PubMed] [Google Scholar]
- 13. Milo A., Isr. J. Chem. 2018, 58, 131–135. [Google Scholar]