

Summary
Despite the extreme diversity of T‐cell repertoires, many identical T‐cell receptor (TCR) sequences are found in a large number of individual mice and humans. These widely shared sequences, often referred to as “public,” have been suggested to be over‐represented due to their potential immune functionality or their ease of generation by V(D)J recombination. Here, we show that even for large cohorts, the observed degree of sharing of TCR sequences between individuals is well predicted by a model accounting for the known quantitative statistical biases in the generation process, together with a simple model of thymic selection. Whether a sequence is shared by many individuals is predicted to depend on the number of queried individuals and the sampling depth, as well as on the sequence itself, in agreement with the data. We introduce the degree of publicness conditional on the queried cohort size and the size of the sampled repertoires. Based on these observations, we propose a public/private sequence classifier, “PUBLIC” (Public Universal Binary Likelihood Inference Classifier), based on the generation probability, which performs very well even for small cohort sizes.
Keywords: inference, probability of generation, public sequences, TCR repertoires, TCR sharing
1. INTRODUCTION
The adaptive immune system relies on a diverse set of T‐cell receptors (TCR) to recognize pathogen‐derived peptides presented by the major histocompatibility complex. Each T cell expresses a distinct TCR that is created stochastically by V(D)J recombination. This process is very diverse, with the potential to generate up to 1061 different sequences in humans.1 The resulting “repertoire” of distinct TCRs expressed in an individual defines a unique footprint of immune protection. Despite this diversity, a significant overlap in the TCR response of different individuals to a variety of antigens and infections has been observed in humans,2, 3, 4 mice,5, 6, 7 and macaques8 (reviewed in 9, 10). This observation led to the notion of a “public” response shared by all, and a complementary “private” response specific to each individual.5 Since antigen‐specific TCRs have a restricted set of sequences,11, 12 and since there is no identified analog for T cells of B‐cell affinity maturation, a public response can only arise if the specific responding T cells are independently generated in each individual's T‐cell repertoire. It was proposed7, 8, 9 that these shared sequences can be explained by the biases inherent in the V(D)J recombination process, together with “convergent recombination,” the possibility to generate the same TCR sequence (especially the same CDR3 amino acid sequence) in independent recombination events. In this hypothesis, shared TCRs are simply those that have a higher‐than‐average generation probability and are thus more abundant in the unselected repertoire.13 The advent of high‐throughput sequencing of TCR repertoires14, 15, 16, 17 has largely confirmed this view through the analysis of shared TCR sequences between unrelated humans,18, 19, 20 monozygous human twins,21, 22 and mice.23 However, despite recent efforts to characterize the landscape of public TCRs,24 the contributions of V(D)J generation biases and convergent recombination relative to convergent selection remain to be elucidated and quantified. Selection effects include thymic selection25 by which receptors that bind too strongly or too weakly to self peptides are eliminated in the thymus, peripheral tolerance by clonal deletion or conversion, and clonal selection by which receptors proliferate upon recognizing specific antigens in the periphery.
In this review, we address the sharing phenomenon using quantitative models of the stochastic V(D)J recombination process that have been inferred from repertoire data.26, 27, 28, 29 These generative models, augmented by a simple one‐parameter model of selection, can be used to predict the number of sequences that will be shared between any number of individuals, each sampled to any sequencing depth. We make these predictions on the basis of stochastic simulations, but we also derive general mathematical formulas that allow us to calculate sharing from any recombination model. We show that these predictions are in excellent quantitative agreement with data from two recent T‐cell repertoire studies in humans30 and mice.23 The predictive power of our model points to convergent recombination as the leading factor for TCR publicness over sequence‐specific selection effects. Our results are consistent with arguments9, 31 that the dichotomy between public and private is misleading. Instead, we find a wide range of possible degrees of sharing, depending on sequencing depth of the individual repertoires, the number of individuals in the study, and the number of individuals between whom the sequence is shared. We propose “PUBLIC” (Public Universal Binary Likelihood Inference Classifier), a “publicness score” defined as the recombination probability predicted by our model. This score predicts the sharing status of any TCR with very high accuracy, irrespective of the definition for being public vs private.
2. PREDICTING SHARING BETWEEN REPERTOIRES
2.1. Spectrum of sharing numbers
We start with an operational definition of sharing in repertoire data obtained by high‐throughput sequencing from several individuals (one sample per individual), which closely follows that of Ref. 23. For each individual, we compile a list of unique TCR sequences (Figure 1A). Since the functional character of a T cell is thought to be largely determined by the amino acid sequence of the highly variable complementarity determining region 3, or CDR3 (to be more precisely defined later) of the beta chain protein, we record in our list just the unique CDR3 beta chain amino acid sequences found in a given biological sample of T cells. For each TCR amino acid sequence, we define the “sharing number” as the number of different samples in which that sequence was found (Figure 1B). The sharing number depends both on the number of samples and on the number of unique sequences in each sample. We note that more restricted definitions of sharing, based for example on the full nucleotide sequence, are possible, but the correspondingly reduced statistics make it harder to draw sharp conclusions. Counting the number of TCRs with each sharing number (Figure 1C), we obtain a distribution of sharing, from purely private sequences (sharing number 1) to fully public sequences (sharing number equal to the number of individuals), and everything in between. We will compare the distribution of sharing numbers obtained from the data sequences with predictions of our models.
Figure 1.
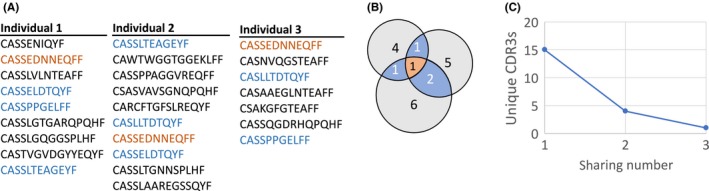
Cartoon representation of the pipeline for computing the distribution of shared sequences between samples. (A) Sharing between samples is analyzed by marking repeated CDR3s between K samples. (B) The overlapping sequences are counted and binned, and the number of CDR3s that were shared m times is computed. (C) Distribution of the number of sequences that are shared m times between the sample of K individuals
Early estimates of sharing of human TCRs7 showed that assuming a uniform distribution of TCR generation underestimates observed sharing by several orders of magnitude.18 Thus, having an accurate model for the non‐uniform distribution of TCR generation probabilities is crucial for making quantitative predictions of the sharing distribution. A simple non‐homogeneous model that assigns lower probability to TCR sequences with more non‐templated nucleotide insertions in the V(D)J recombination process is able to predict sharing between pairs of individuals within the correct order of magnitude.18 However, this estimate ignores the detailed structure of biases inherent to the recombination process and results in strong biases in the distribution of TCR sequences that, as we will show, influence the sharing spectrum.
2.2. TCR generation bias
T‐cell receptors are composed of an α and a β chain encoded by separate genes stochastically generated by the V(D)J recombination process.32 Each chain is assembled from the combinatorial concatenation of two or three segments (V as Variable, D as Diversity, and J as Joining for the β chain, and V and J for the α chain) picked at random from a list of germline template genes. Further diversity comes from random non‐templated nucleotide insertions between, together with random deletions from the ends of, the joined segments. The α chain is less diverse than the β chain and sharing analyses have mostly focussed on the latter. The germline gene usages are highly non‐uniform,14, 15, 33 due to differences in gene copy numbers34 as well as the conformation35 and processive excision dynamics36 of DNA during recombination. In addition, the distributions of the number of deleted and inserted base pairs, as well as the composition of N nucleotides, are also biased.37 Taken together, the biases imply that some recombination events are more likely than others. In addition, distinct recombination events can lead to the same nucleotide sequence, and many nucleotide sequences can lead to the same amino acid sequence. This convergent recombination further skews the distribution of TCRs, as some sequences can be produced in more ways than others.7, 9
The effects of recombination biases and convergent recombination can be captured by stochastic models of recombination. Given the probability distributions for the choice of gene segments, deletion profiles and insertion patterns, one can generate in silico TCR repertoire samples that mimic the statistics of real repertoires, and allow us to predict sharing statistics and the effects of convergent recombination.11, 20, 22, 23, 26, 38 To obtain accurate predictions, the distributions of recombination events used in the model must closely match repertoire data. This task is made difficult by the fact that, as a consequence of convergent recombination, the specific recombination event behind an observed sequence is not directly accessible. However, methods of statistical inference can be used to overcome this problem and learn accurate models of V(D)J recombination,26, 27, 29, 39 models which can in turn be used to predict sharing properties of sampled repertoires or of individual TCR sequences. These models have been shown to vary little between individuals, with small differences only in the germline gene usage and remarkable reproducibility in the insertion and deletion profiles.26 In our analysis we will assume a universal model, independent of the individual.
2.3. Using TCR recombination models to predict sharing
We used the above‐described models of recombination to predict the distribution of sharing among cohorts of humans and mice. Specifically, we re‐analyzed published TCR β‐chain nucleotide sequences of 14 Black‐6 mice23 and 658 human donors30 (Section 7). Individual samples comprised 20 000‐50 000 unique sequences for mice, and up to 400 000 for humans. Sequences were translated into amino acid sequences, and trimmed to keep only the CDR3 loop, defined as the sequence between the last cysteine in the V gene and the first phenylalanine in the J gene.40 The sharing number of each observed CDR3 amino acid sequence, and the sharing number distribution, were then computed from the data. We chose to focus on the CDR3 amino acid sequences to get higher sharing numbers than would have been obtained for untrimmed nucleotide sequences, limiting the effects of sequencing errors and allowing for a better comparison to the model.
To obtain model predictions for humans, we used a previously described model for TCRβ sequence generation inferred by the software package IGoR29 from repertoire data of a single individual.30 IGoR infers the probability distribution of V(D)J recombination events from sequence data (see details in Section 7.1). The mouse model was inferred using IGoR from the repertoire data of one of the 14 mice.23 In both cases, the model is learned from unproductive rearrangements (ie, with a frameshift in the CDR3) since those sequences give us access to the raw result of recombination, without subsequent effects of selection.26 These unproductive sequences are only used to infer a generative model and are not used in the sharing analysis. A productive (in frame) sequence that is generated in a V(D)J recombination event will not necessarily survive thymic selection to become a functional T cell in the periphery. To model this effect, we assume that there is a probability q, independent of the actual sequence but dependent on the species under study, that any given generated sequence will survive thymic selection.41 Model sharing predictions are then obtained in two ways: (i) by simulating sequences and selecting them at random with probability q to generate samples of the same size as in the data (an important point about simulation is that, once a particular CDR3 amino acid sequence has been chosen to not pass thymic selection, any future recurrence of that sequence in the simulation is also discarded); (ii) by deriving analytical mathematical expressions for the expected value (Section 7). These predictions can then be directly compared to data.
2.4. Model predicts many degrees of publicness in the data
The comparison between data, model simulations, and mathematical predictions shows excellent agreement in mice (Figure 2A) and humans (Figure 2B). The predictions depend on the only free parameter of the model, the selection factor q. This parameter was not set simply by fitting the sharing curves to the data. Instead, it was obtained independently as a proportionality factor required to explain the number of observed unique amino acid CDR3 sequences given the number of unique nucleotide sequences (insets of Figure 2A and B). This convergent recombination curve depends on q in a predictable way (see Section 7 for mathematical expressions), making it possible to fit q to the data (insets of Figure 2A and B). This method yielded selection factors of q = 0.16 ± 0.03 for mice, and q = 0.037 ± 0.002 for humans, surprisingly close to the estimate of 3% for the fraction of human TCR that pass thymic selection.42 Comparison of the prediction with and without selection in mice (red and green lines and points in Figure 2A) shows that adding selection greatly improves the agreement, despite a slight overestimation of high sharing numbers. Adding selection yielded a similar improvement in the human sharing prediction of Figure 2B (the model prediction with no selection is not shown for figure clarity).
Figure 2.
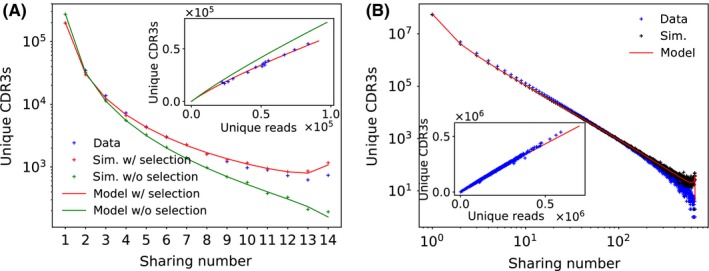
Distribution of sharing numbers. (A) Distribution of the number of sequences that are shared between m individuals (m = sharing number) for 14 mice. Data points (blue crosses) are compared to analytical model predictions (see Section 7.3.1) with selection (red curves) and without selection (green curve), and with simulations (see Section 7.2) based on the generation model with selection (red crosses) and without selection (green crosses). While the model without selection underestimates sharing, the prediction is improved by adding selection. The model predictions derived from analytical calculations and stochastic simulations agree well. The selection factor q, defined as the probability of a CDR3 to pass thymic selection, is inferred by least‐square regression from the relation between the number of unique CDR3 amino acid sequences with the number of unique nucleotide sequence reads (inset, see Section 7). (B) Distribution of sharing numbers in a cohort of 658 humans. The model prediction with selection (simulation: black crosses, analytics: red line) agrees well with the data (blue crosses). The selection factor is obtained as for mice (inset)
Humans have a much more diverse repertoire than mice,28 which should result in lower numbers of shared amino acid TCR sequences for equal sample and cohort sizes. However, the much larger cohort size in the human dataset allows us to illustrate a very wide range of sharing behaviors. In particular, we find a long‐tailed power‐law distribution in the distribution of sharing numbers (Figure 2B), a feature that is reproduced by the model. A very small fraction of sequences is shared between all individuals in the 658 donor cohort, while a large (>90%) fraction of TCRs is found in just one sample. This diversity of behaviors reflects the diversity of generation probabilities implied by the strong biases in the VDJ recombination process that are correctly captured by our model.
3. FROM SAMPLES TO FULL REPERTOIRES
3.1. Sampling depth affects sharing
While the sharing potential of a sequence depends just on its generation and selection probabilities, it is important to realize that actual sharing numbers will depend on the size of the cohort under study and the sampling depth of each individual T‐cell repertoire. To illustrate this effect, we downsampled both the cohort size and the number of sequences in the human dataset, and recalculated sharing. Figure 3A compares the distribution of sharing numbers in the original dataset, with the same distribution obtained from samples where a random half of the unique sequences was removed. The number of TCRs with each sharing number drops with downsampling, and this drop is more marked for high sharing numbers, as evidenced by the fraction of CDR3s with each sharing number (see inset of Figure 3A). In short, the more TCRs are captured in the repertoire samples, and the more likely sequences are to be shared. This effect is reproduced in detail by the model calculations. This result generalizes previous observations that the number of shared TCRs between a pair of individuals should scale approximately with the product of the numbers of unique TCRs in each sample20, 21, 26, 43 to arbitrary sharing numbers.
Figure 3.
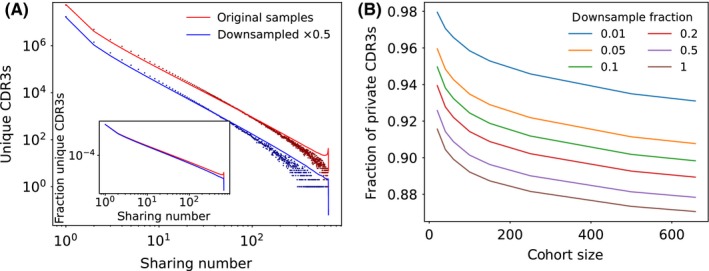
The sharing number depends on the sampling depth and cohort size. Downsampling the number of sequences in all individuals affects sharing, and decreases the observed probability to be public. (A) The number of sequences for each sharing number decreases as the repertoires of all individual are downsampled by a factor 0.5 (blue points) compared to the original sample (red points), as predicted by the model (red and blue lines). The normalized distribution of sharing numbers (inset) shows that downsampling affects larger sharing numbers more. (B) Model prediction of the fraction of sequences that are entirely private (ie, appearing in just one individual), as a function of the downsampling fraction and cohort size. Larger samples and cohorts result in fewer private sequences
To demonstrate the effects of varying cohort and sample size more clearly, we plot in Figure 3B the complementary quantity—the fraction of CDR3s which are purely “private” that is present in only one repertoire. This fraction decreases for large cohorts and large sample sizes. We note that cohort size and sample depth vary greatly from study to study; the data analyzed in this review go from a small cohort of mice (14 repertoires with a few tens of thousands TCRs each) to a very large cohort of humans (658 donors with 200,000 TCRs each). The strong dependence of the notion of privateness upon the parameters of the study cautions us against interpreting sharing numbers and public or private status of individual sequences too literally, and further emphasizes that publicness is not a binary but rather a continuous measure.
3.2. Cumulative diversity and extrapolation to full repertoires
As Figure 3B shows, most (more than 90%) amino acid TCRs are found in only one repertoire. This means that, when pooling repertoires, each newly added repertoire will contribute a brand new set of TCRs to the pool. To explore this idea, we define the “cumulative repertoire” obtained by pooling together the sampled repertoires of several individuals, and count the number of unique TCRβ amino acid sequences in it. This cumulative diversity grows almost linearly with the number of pooled samples (Figure 4A), both in the data and according to the model (see Section 7 for calculation of the model prediction). The ratio of unique to total sequences starts at 1 for small numbers of pooled individuals, and decreases to around 0.9 for high numbers of pooled individuals, consistent with the fraction of private sequences. It is interesting to ask whether this trend would continue for larger populations all the way up to the entire world population. Although we cannot answer this question directly by experiments, we can use the model to make predictions. Generating in silico repertoires for billions of individuals are of course impractical, but we can use mathematical expressions (Section 7) to calculate the expected diversity. Figure 4B shows the theoretical cumulative diversity as a function of the number of individuals for up to 1012 individuals. Even with numbers of individuals largely exceeding the number of humans having ever lived (1011), we are very far from saturating the space of observed TCRs.
Figure 4.
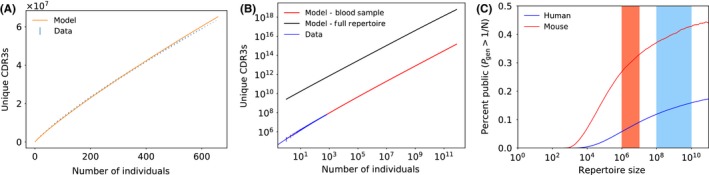
(A) Number of unique CDR3 amino acid sequences in the pooled repertoire of n individuals, as a function of n. This number does not depend strongly on the order in which individuals are added to the group (black error bars, obtained by measuring variations across 30 random orderings). The theoretical prediction (red line, see Section 7.3.4) agrees very well with the data. The model prediction was obtained using the mean sample size of the pooled repertoire across 30 random orderings. Each new individual adds ∼200 000 new CDR3 sequences. (B) Theoretical extrapolation to very large cohorts (red line). This model prediction is based on an average sample size. The same prediction can be done for the full repertoires contained in the human body (with 1011 unique recombination events), which yields much larger numbers of unique CDR3s (black line). (C) Model prediction for the fraction of sequences in each individual that are truly “public,” ie, have a generation probability larger than 1/N, where N is the number of unique TCRs in each individual (repertoire size). The red and blue stripes mark the possible range of repertoire sizes in mice and humans, according to current knowledge
The previous estimates rely on partial repertoires comprising a few hundred thousand unique TCRs obtained from small blood samples. However, the human body hosts 5 × 1011 T cells,44 and while the T‐cell population has a clonal structure, recent estimates of the number of clones, and thus of independent TCR recombination events, ranges from 108 (from indirect sampling using potentially inaccurate statistical estimators45), to 1010 (based on theoretical arguments46). The theoretical cumulative diversity based on that latter estimate of 1010 (Figure 4B, black curve) still shows no sign of saturation. These results are a consequence of the enormous potential diversity of VDJ recombination, and indicate that the diversity of TCRβ is not exhausted even by the pooled repertoire of the entire world population.
Extrapolating these considerations to the full TCR repertoire of an individual allows us to estimate the fraction of truly “public” TCRs, defined as the sequences that are present in almost all individuals. If we define a public TCR sequence as one that has a generation probability larger than 1/N, where N is the number of T‐cell clones in the body, then 1−e −1=63% of all individuals would be expected to have that sequence in their repertoire. With this definition, we can predict the percentage of public sequences as a function of repertoire size—ie, the number of T‐cell clones (Figure 4C). Interestingly, this fraction ranges from 10% to 20% for both humans and mice depending on estimates of the number of clones, despite their widely different TCRβ diversities and repertoire sizes. It is interesting to note that the lower diversity of the TCRβ repertoire in mice as compared to humans is matched in a proportional way to the ratio of the TCR repertoire sizes in the two species.
4. PREDICTING PUBLICNESS
4.1. Sharing and TCR generation probability
As we have seen, the sequence generation model correctly predicts the amount of sharing across individuals, as well as the fraction of public sequences. Underlying this prediction method is the idea that the likelihood that a given sequence will be shared is largely determined by the probability of generation of the sequence. Early versions of this argument9, 47 noted that sequences with a high number of N insertions have lower generation probability (because of the diversity of possible insertions, each reducing the generation probability by a factor ≈1/4), predicting that shared sequences would have fewer insertions than average. We have used recombination models inferred from data to refine this argument by accounting quantitatively for the effects of the generation biases and convergent recombination on the probability of generation of particular TCR sequences. As a further test of the underlying ideas, we compute the generation probability of TCR sequences and ask how this quantity correlates with the sharing numbers.
To calculate the generation probability of TCRs, one needs to sum the occurrence probabilities of all the possible recombination events leading to a given nucleotide sequence26, 29 and, since we choose to follow CDR3 amino acid sequences, sum the probabilities of all nucleotide sequences leading to the amino acid sequence of interest. This is a computationally hard task that can be rendered tractable using a dynamic programming approach (see Section 7). We find that the distribution of generation probabilities of all TCRβ CDR3 amino acid sequences (Figure 5, blue curves) is extremely broad, spanning many orders of magnitude. This observation is consistent with similar analyses at the level of nucleotide sequences in non‐productive26 and productive20 human TCRβ, in the α and β chains of monozygous twins,22 and mice.28 If we plot instead the generative probability distribution of sequences that are shared among two or more individuals in our dataset, we find that the distribution narrows and shifts toward higher generation probabilities20, 22, 26 as expected. This effect is displayed in more detail in a plot of the generative probability distribution for sequences in our dataset with different sharing numbers (Figure 5). On the same figure, we plot the predictions of the recombination model, following the same protocol used for predicting sharing numbers (see Section 7). There is a systematic shift between the predictions of the recombination model and the distribution of the data itself, for all sharing levels. This difference is due to the fact that the recombination model was inferred from non‐productive sequences, and does not account for selection effects. The data sequences, however, have passed thymic and possibly other kinds of peripheral selection, affecting their statistics. The sequence‐dependent nature of this effect was characterized and quantified in a previous work,20 with the general finding that selection favors sequences with high generation probability. This is qualitatively consistent with the positive sign of the shift (solid lines vs dotted lines) we see in Figure 5. Our sharing calculations ignore any possible sequence dependence of selection, and instead selects TCRs at random (with probability q), regardless of their sequence identity. The model prediction could in principle be improved by adding sequence‐dependent selection factors to match the distributions as was done previously.20 However, unlike the recombination model, such factors are expected to be specific to each individual, owing to their unique HLA type which is involved in thymic selection.
Figure 5.
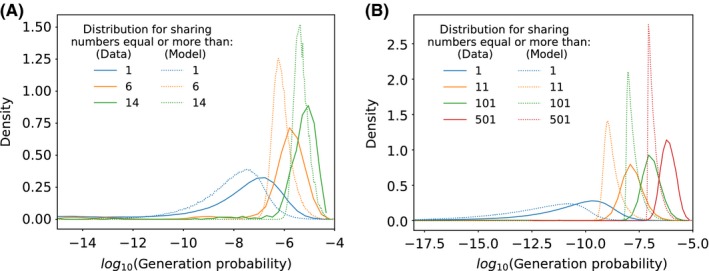
Distributions of the logarithm of the generation probability for different minimal sharing numbers, for (A) mice and (B) humans. For larger sharing numbers, the distribution shifts toward higher probabilities and becomes narrower. This shift enables the characterization of the sharing number, or the degree of publicness, using the generation probability. The model captures the right trend of the sharing numbers, despite predicting much narrower distributions
4.2. PUBLIC: Classifier of public vs private TCRs based on generation probability
The distributions of generation probabilities for the different sharing numbers suggest that the generation probability is a good proxy for the property of being public, regardless of the exact definition of publicness. We built a classifier called PUBLIC (Public Universal Binary Likelihood Inference Classifier), which is entirely based on the probability of generation computed as explained above (detailed in Section 7) for each amino acid sequence (Figure 6A). Before discussing the performance of this classifier, it is important to note that it is based on a model of recombination trained in a completely unsupervised way, ie, without using any information about the public status of the sequences. In fact, this training can be done with IGoR 29 from the repertoire of a single individual, without including any sharing information. Unlike previous approaches,23 we do not fit additional model features based on the catalog of sequences with their public or private status.
Figure 6.
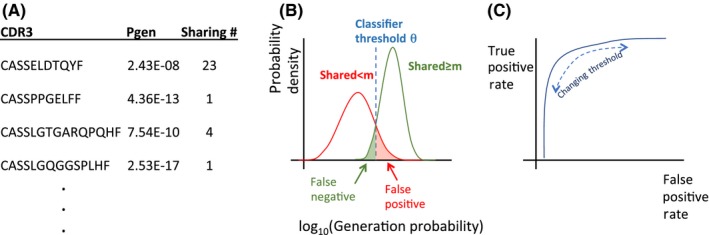
Cartoon representation of the pipeline for the PUBLIC classifier. (A) To each CDR3 sequence in the dataset we associate its generation probability (p gen), which PUBLIC uses to predict the empirical sharing number. (B) The p gen distributions of shared sequences depend on the sharing number m. We pick a classifier threshold value of P gen, θ, that separates public from private sequences for this sharing number value of m. The areas of the histograms that fall on the wrong side of the threshold are defined as the false positive and false negative rates. (C) For a given choice of the minimal sharing number m, we plot the true and false positive rates as a function of the classifier threshold θ to obtain a receiver operating characteristic
We arbitrarily define as “public” the TCRs that are found in at least m repertoire samples among a total pool of n individuals. The PUBLIC classifier calls a given TCR “public” if its generation probability is larger than a threshold θ, calling it “private” otherwise. Intuitively, the threshold should be set to separate reliably the peaks in the probability density function of Figure 5 corresponding to different sharing numbers, as schematized in Figure 6B. The general performance of the PUBLIC classifier can be estimated by plotting the receiver operating characteristic (ROC) curve, which represents the rate of false positives vs that of true positives as θ is varied (Figure 6C).
We plot ROC curves for a few different choices of m (the minimal number of individuals with the TCR in their sampled repertoire for the sequence to be called public) for mice (Figure 7A) and humans (Figure 7B). The classification accuracy improves as publicness is defined to be more restrictive (larger m), although it performs well even for small m. For mice, the dataset we used had few individuals, making the operational definition of publicness less reliable. However, for humans, we find highly public TCRs are predicted almost perfectly by PUBLIC, despite the larger diversity of human TCRs. This suggests that the lesser performance of PUBLIC for mice may be attributed to the small size of the cohort, rather than to limitations of the classifier itself.
Figure 7.
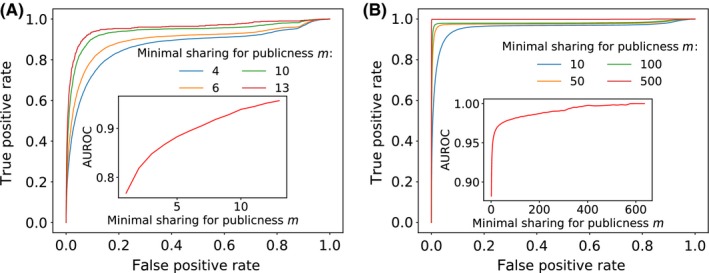
Performance of the PUBLIC classifier. Receiver operating characteristic (ROC) curves for (A) mice and (B) humans for different minimal sharing numbers m. Inset: the area under the ROC curve (AUROC) describes the probability of classifying a given sequence as public or private. Higher AUROC values correspond to a better a classifier. The AUROC score increases with the minimal sharing number m (inset), meaning that a more restrictive definition of publicness gives better classifiers
The performance of PUBLIC can be reduced to a single number by calculating the area under the ROC curve (AUROC). The AUROC corresponds to the probability that the classifier ranks a randomly chosen public sequence higher than a randomly chosen private one. The closer the AUROC score is to 1, the better the classifier. As was clear from the ROC curves themselves, the AUC improves as the degree of publicness is higher (insets of Figure 7A and B). As the minimal sharing number m increases, the classifying task becomes easier and the prediction better. In fact, having the minimal sharing number m close to the cohort size n will in general make publicness rarer, and the public sequences more extreme in their generation probabilities.
5. PUBLIC SPECIFIC RESPONSE
Sharing properties are interesting in their own right, but they also provide a basal expectation for the prevalence of certain TCRs. Using the sharing prediction, one can identify TCRs that are more shared in specific populations or subsets than expected according to the recombination model. When counting sharing in a population of individuals affected by a common condition, this “over‐sharing” can be indicative of a specific T‐cell response to the antigens associated with the condition. Such sharing of specific TCRs is expected from the relatively low diversity of antigen‐specific sequences revealed by in vitro multimer‐staining experiments.11, 12 A very similar idea has been exploited by several groups to identify TCRs specific to the Cytomegalovirus,30 Type‐1 diabetes,48, 49 arthritis50 and other immune diseases.51 In these studies, there is no theoretical expectation from the recombination model. Rather, the basal expectation for TCR sharing is given by a negative‐control cohort. However, this control can be efficiently replaced by the recombination model presented here, as was demonstrated before.41 In this analysis, specific TCRs emerge as outliers that are shared much more frequently than predicted by the model.
We wondered whether such an approach could be useful for identifying tumor‐specific TCRs as sharing outliers among cancer patients. The T‐cell repertoire of tumor‐infiltrating cells has been studied to look for signatures of immunogenicity,52, 53, 54 and the overlap between the tumor and blood repertoires was shown to predict survival in glioblastoma patients.55 In addition, the tumor‐specific TCRs have been reported to be shared in the tumor‐infiltrating and blood T‐cell repertoires of breast cancer.56
We thus asked whether the blood repertoires of patients with bladder cancer contained TCRs with more sharing than would be predicted by our recombination model. We performed the sharing analysis on 30 patients with bladder cancer, on TCR repertoires sequenced from blood samples.54 We compared it with 30 healthy individuals, chosen at random among the individuals studied in 30 to have similar sample sizes. We then downsampled the reference repertoires of the healthy individuals to have the exact same sample sizes as the cancer patients to guarantee a fair comparison. We found that the numbers of shared sequences in the blood of bladder cancer patients are almost identical to those found in the healthy samples, and thus also in agreement with the recombination model (Figure 8). This is consistent with previous reports that did not find any signatures of TCR repertoire anomalies in the blood of bladder cancer patients, although some small differences could be seen in the tumors. There are many possible explanations for this observation: the tumor‐specific response may be statistically negligible amid the large number of other cells; or the response may not have propagated to the blood; or different patients generate responses against different neoantigens; or they generate very different responses against the same neoantigen; or the tumor does not generate any response at all. Tumor samples from larger cohorts would be needed to distinguish between these different hypotheses. Additionally, this result is only true for bladder cancer. Different tumor types that have a higher rate of infiltration to the blood may be more likely to result in detectable signatures in the blood.
Figure 8.
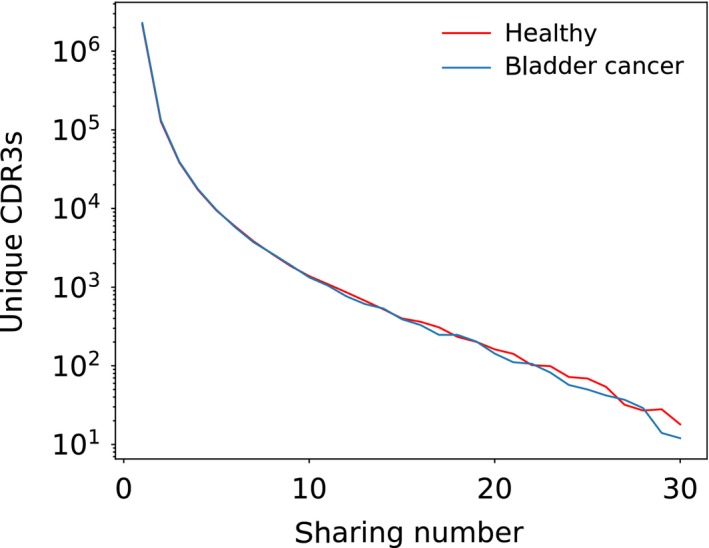
Distribution of sharing numbers in a cohort of 30 bladder cancer patients. The distribution is compared to a sub‐cohort of 30 healthy individuals downsampled to have the same sample sizes as the cancer samples. The distribution are the same in healthy and bladder cancer patients, indicating that there are no common significantly over‐represented TCRs in the blood repertoire of cancer patients
6. DISCUSSION
In this paper, we extensively tested and quantified the previously proposed hypothesis9, 31 that public TCRs owe their status to the ease of generating them through V(D)J recombination. Predicting and characterizing TCR sharing and publicness is important to identify universal features of the immune response across individuals. This knowledge can be useful when designing vaccines that have a high probability of eliciting an immune response, or for identifying candidate T‐cell clones in immunotherapeutic strategies.57
Our predictions, and their agreement with the data, support the notion that “publicness,” as it is usually defined, is context‐dependent.9 The public status of a TCR depends not only on its (intrinsic) generation probability, but also on (extrinsic) parameters including the number of individuals sampled, the sequencing depth of the samples, and the definition of publicness—the minimal number of individuals that need to share a TCR to call that TCR public. Instead, we have showed that we can define the potential for publicness, largely determined by the generation probability of the sequence, and use it to predict actual sharing numbers for any set of repertoire samples. At the same time, we proposed that an absolute notion of publicness can be defined based on the full repertoire of individuals. According to this definition, a TCR is public if its probability of occurrence is larger than the inverse of the number of unique TCRs hosted in the entire repertoire. While this definition is impossible to explore directly in humans, for whom only repertoire samples can be obtained, our data‐driven recombination model can make predictions about the public status of particular sequences, and the fraction of the repertoire that is public, using this specific definition (Figure 4).
We report a wide spectrum of publicness, which we show arises from the very wide distribution of TCR generation probabilities. The high‐end of the distribution holds sequences that will be included in any healthy repertoire, just by virtue of their high generation probability. Due to their publicness, it had been conjectured that some of these common TCRs might have a close to innate function.31 In this context, it should be noted that young, prebirth repertoires are known to be much less diverse both in humans22 and mice,28 due the late appearance of TdT, the enzyme responsible for insertions in the recombination process. Consequently, the prebirth repertoire is expected to be much more public that the adult one, and could be enriched in innate‐like TCRs. However, since no conclusive evidence has been provided about the functional role of these high probability sequences, we cannot rule out the possibility that they are just there by chance, without a specific function. The other end of the TCR distribution—the long tail of low generation probabilities—contributes to the private part of the repertoire, which makes up the majority of the repertoire according to our estimates. It would be interesting to explore whether these sequences have a functional role or are just by‐products of the recombination process.
High‐throughput TCR repertoire datasets contain abundance levels (number of reads) for each TCR. TCR abundances have be attributed to convergent recombination, implying a correlation between high abundance and publicness.9 However, this connection may be confounded by other processes affecting the abundance levels reported by high‐throughput sequencing. A big source of diversity in TCRs abundances is the peripheral proliferation of some TCRs, regardless of their generation probability. In addition, experimental or phenotypic noise, including PCR amplification noise58 and expression variability (for cDNA sequencing) also affect reported abundances. These various effects are expected to dilute the correlation between abundance and publicness. Note that our statistical models are constructed based only on unique sequences, circumventing clonal expansion dynamics, and ignoring abundance levels altogether.
The sharing analysis naturally leads to defining the PUBLIC score, which we show predicts sharing properties with high accuracy. The PUBLIC score is learned in an unsupervised manner, using a statistical model trained with no information about the sharing status of TCRs. Thus, sharing can be very well predicted with neither abundance nor sharing information. This success suggests that being public is a very basic property of the recombination process itself, and also provides a strong validation of the recombination model. It would be interesting to explore how using TCR sharing status and abundance levels in a supervised manner that refines the classifier could lead to better predictions.
Our prediction for sharing is mainly based on the generation model,29 which is sequence specific, attributing each sequence its own probability of generation. We have found that an overall selection factor is needed to predict sharing numbers correctly, but this simple and effective model is sequence independent. Previous work20 inferred a sequence‐specific selection process by comparing generation model results to observed sequences. In principle, such a model could be combined within our framework to yield refined sharing predictions. While the parameters of the generation process are largely invariant across individuals,29 selection is expected to be individual‐dependent and heritable due to the diversity of HLA types in the population.59 The large variability in the V and J genes selection pressures inferred previously20 is consistent with this notion, but in the same work some amino acid features of selection were found to be universal. Quantifying these universal features and including them in the model could both improve the predictions for the sharing numbers, and enable a better assessment of the potential publicness of specific sequences through an improved classifier.
The discussion in this work was focused on TCRβ chains, but in general can be applied to any recombined chain, including α, γ and δ TCR chains, as well as B‐cell receptor (BCR) light and heavy chains, or to paired chain combinations. The α chain, as part of the αβ receptor, contributes to antigen recognition. It is less diverse than the β chain, implying higher sharing numbers.22 Paired αβ data is becoming available as paired sequencing technologies improve,60, 61 but the resulting repertoires are currently too small or not yet available for analysis. As more paired sequencing data becomes available, it will be interesting to study the sharing properties of the αβ repertoire using recombination models for pairs.
A similar analysis could be performed on BCRs. The problem is further complicated by somatic hypermutations, which add further diversity and are expected to reduce sharing as well as the ability to predict it. However, the role of the generation probability, for which the models have been trained,29, 39, 62 for sharing and publicness has not been explored. Machine‐learning approaches to predict publicness of BCR63 could be combined with estimates of the probability of generation and hypermutations profile29, 64, 65 to provide accurate predictions for the public status of BCRs. Such an analysis applied to the result of affinity maturation in different individuals infected with the same pathogens66 could be used to assess the impact of convergent recombination in the public response and better understand the evolution of specific antibodies, and guide vaccination strategies to facilitate the emergence of broadly neutralizing antibodies.67
7. METHODS
7.1. The probability of generating a TCR sequence
To evaluate TCR generation probabilities, we first constructed a probabilistic generation model of the recombination process.26 Such a model is parametrized by probabilities for each choice of V, D, and J gene, for each deletion length of the different genes, and for each insertion pattern of random nucleotide between the genes. Then the probability of a recombination scenario is a product of these probabilities:
| (1) |
The probability of a TCR sequence, whether it is a nucleotide of amino acid sequence, for the full sequence or just the CDR3, is obtained by summing the above probability over all the possible scenarios leading to the sequence of interest.
The different factors for gene choice, P(V,D,J), deletion patterns, P(delV|V), P(delD|D), and P(delJ|J), and insertion length and content, P(insVD) and P(insDJ), define the generation model. All these factors can be inferred efficiently using the IGoR software29 from non‐functional recombinations, which produce out‐of‐frame or stop codon containing sequences. Model training is done by finding model parameters that maximize the likelihood of the data, equal to the product of generation probabilities of the observed TCRs in the dataset. Here, we used IGoR to infer a generation model from the non‐functional reads in the datasets from which the productive reads used for the sharing analysis came, for human,30 and mice.23
To calculate the generation probabilities of CDR3 amino acid sequences, we used an efficient algorithm that avoids brute‐force summation of all possible scenarios using dynamic programming.
7.2. Evaluating the number of shared sequences using simulations
Once inferred, a generative model can be used to generate random in silico samples of any size. Recombination scenarios are generated using Monte Carlo sampling by drawing events such as gene choices, deletions, and insertions according to the model parameters. Each recombination scenario constructs a nucleotide sequence which is filtered for productivity (in‐frame, no stop codons or pseudogenes, and the conserved residues C and F are present). A productive nucleotide sequence is then trimmed to the CDR3β region and translated into an amino acid sequence. To model thymic selection only a random fraction q of the productive CDR3β sequences are considered. This is implemented using a hash function, keeping only sequences whose normalized hash values are less than q. This negative selection process is a random function of the sequence, which is consistent between any simulated individual sample, so that a given CDR3β will either pass or fail selection in all individuals. A simulated sample can thus be generated to match the cohort size and sequencing depth of the real data, and then analyzed with the same pipelines.
7.3. Analytical calculation of the number of shared sequences
7.3.1. Predicting sharing numbers from the distribution of generation probabilities
Given a collection of CDR3β sequences s ∈ S, a model that assigns probabilities p(s) for each sequence, and N independent sequences drawn from the model, the expected number of observed unique sequences M 0 is:
| (2) |
where we have made the Poisson approximation for small p(s). If there are n individuals, with sample sizes {N i}, then the expected number of sequences which will be found in exactly m individuals (sharing number m) is:
| (3) |
where J m is the collection of all possible combinations of m individuals. This can be computed more efficiently by use of the generating function G(x,{N i}):
| (4) |
where the M ms are the coefficients of the polynomial G(x,{N i}), and can be calculated just by expanding the polynomial in x and summing over s.
7.3.2. Density of states approximation
While the above equations are exact, summing over each individual sequence is intractable given the huge number of sequences. Instead, an integral approximation based on the “density of mstates” is used. Let us call E(s) = − ln p(s) the Shannon surprise of generating sequence s at random, also formally equivalent to an energy in physics according to the Boltzmann's law. The density of states, g(E)dE, counts the number of sequences between E and E+dE. Summation of an arbitrary function Φ(p(s)) = Φ(E(s)) over the states (sequences) in S can then be turned into an integral:
| (5) |
A numerical estimation of g(E) is required to compute this integral. Estimating g(E) is done by drawing large Monte Carlo samples of sequences (107 for humans and 106 for mice) from the generative model and calculating the generation probabilities of each sequence. Values of E(s)=− ln p(s) can then be histogrammed into bins of size dE and the resulting distribution normalized to integrate to 1. This yields a probability density, P(E) (shown Figure 5), which can be used to compute the density of states:
| (6) |
Equations 2 and 4 can now be rewritten in terms of integrals:
| (7) |
and
| (8) |
7.3.3. Sharing modified by selection
While the above analysis is general, it depends on the state or sequence space S (the collection of productive CDR3βs that pass selection) and on a model that assigns probabilities to each sequence. The preferred model to use will be the probability of generating a sequence (p gen(s)); however, this model is defined and normalized over a state space of all possible recombination events, many of which lead to non‐functional or negatively selected sequences. As a result, the model p(s) that will be used needs to be renormalized to reflect the reduced sequence space of productive, selected sequences. This introduces two factors. First factor, f, is the fraction of sequences which are functional (in‐frame, no stop codons or pseudogenes, conserved residues are present), and can be computed directly from the generative model (f = 0.236 for humans and f = 0.260 for mice). The second factor, q, is the fraction of productive sequences which pass selection and must be inferred (see below). These two factors provide the definition for the model that is used in the analysis:
| (9) |
The effect of renormalizing p gen(s) to p(s) on the density of states is that the energies are shifted by a constant ln f+ ln q and the density itself is everywhere reduced by a factor of f×q:
| (10) |
where g gen(E) is the density of states computed from p gen(s) and g(E) is derived from p(s).
7.3.4. Inferring the selection factor q
The selection factor q is inferred by running a least‐squares regression on the model predictions for the M 0(N) curve (Eq. (7)). This curve relates the number M 0 of unique amino acid CDR3 sequences observed to the number N of productive, selected recombinations generated. To determine the M 0(N) curve from the data, the number of productive selected recombinations must be determined for each sample. Fortunately, due to the limited sequencing depth, the number of unique productive nucleotide reads in each individual sample is very close to the actual number of selected recombinations. In practice, N was taken to be the number of unique nucleotide sequences of each repertoire, summed over a subset of the individuals, and M 0 was the number of unique amino acid sequences resulting from the translation of the aggregated repertoire of the same subset of individuals. The curve was obtained by adding more and more random individuals to the subset, and averaged over 100 realizations of that random addition process (Figure 4A). A least‐square regression of Equation (7) with Equation (10) to that empirical curve yielded a value for q of approximately q = 0.037 ± 0.002 for humans and q = 0.16 ± 0.03 for mice. Sequencing errors could produce spurious sequences which would be counted as distinct unique sequences, potentially biasing the estimation of q. In the datasets used the error rate was low enough to ignore this effect.
7.3.5. Analytic computation of public fraction of a repertoire
In Figure 4C, a sequence s in a repertoire of size N is defined as public if p(s)≥1/N. The fraction of the repertoire comprised of these sequences is computed by evaluating:
| (11) |
where the term in parenthesis corresponds to the probability that a given sequence with probability e −E is found in a repertoire of size N.
7.4. Sequence data
Mouse data was obtained from Friedman's lab, and published before in.23 Out of the 28 mice in that study, we analyzed 14 that had similar reads, with length of 52 bp. The sample sizes are summarized in Table 1.
Table 1.
Mice dataset sample sizes
| Mouse id | Unique reads | Unique CDR3 amino acid |
|---|---|---|
| 1 | 22 118 | 18 257 |
| 2 | 46 116 | 32 469 |
| 3 | 55 124 | 38 669 |
| 4 | 74 026 | 49 165 |
| 5 | 53 019 | 37 419 |
| 6 | 23 676 | 17 032 |
| 7 | 50 672 | 33 347 |
| 8 | 52 607 | 34 629 |
| 9 | 51 377 | 35 803 |
| 10 | 83 641 | 54 711 |
| 11 | 29 764 | 21 557 |
| 12 | 66 632 | 44 269 |
| 13 | 40 428 | 27 658 |
| 14 | 26 350 | 18 704 |
Human dataset was used from the CMV study in.30 658 individual samples were used, with mean number of reads ≈200 000, and mean number of unique CDR3s ≈180 000.
Bladder cancer data came from a published immunotherapy study.54 We used the blood samples from the first time point of each patient in the study. Samples were generally smaller than those in the CMV study, with a mean number of unique CDR3s ≈100 000, and a smaller spread.
7.5. Code availability
Code for the PUBLIC classifier is available at github.com/yuvalel/PUBLIC.
ACKNOWLEDGEMENTS
The work of TM and AMW was supported in part by grant ERCCOG n. 724208. The work of ZS and CC was supported in part by NSF grant PHY‐1607612. The work of YE was supported by a fellowship from the V Foundation. The work of CC was performed in part at the Aspen Center for Physics, which is supported by National Science Foundation grant PHY‐1607611.
CONFLICTS OF INTEREST
The authors have no conflicts of interest.
Elhanati Y, Sethna Z, Callan CG Jr, Mora T, Walczak AM. Predicting the spectrum of TCR repertoire sharingwith a data‐driven model of recombination. Immunol Rev. 2018;284:167–179. https://doi.org/10.1111/imr.12665
Elhanati and Sethna contributed equally.
Mora and Walczak contributed equally.
This article is part of a series of reviews covering Characterization of the Immunologic Repertoire appearing in Volume 284 of Immunological Reviews.
Contributor Information
Thierry Mora, Email: tmora@lps.ens.fr.
Aleksandra M. Walczak, Email: awalczak@lpt.ens.fr
References
- 1. Mora T, Walczak A. Quantifying lymphocyte receptor diversity. In: Das J, Jayaprakash C, eds. Systems Immunology: An Introduction to Modeling Methods for Scientists. Boca Raton, FL: CRC Press, Taylor and Francis; 2018;1–10. [Google Scholar]
- 2. Moss PA, Moots RJ, Rosenberg WM, et al. Extensive conservation of alpha and beta chains of the human T‐cell antigen receptor recognizing HLA‐A2 and influenza A matrix peptide. Proc Natl Acad Sci USA. 1991;88:8987–8990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Casanova JL, Cerottini JC, Matthes M, et al. H‐2‐restricted cytolytic T lymphocytes specific for HLA display T cell receptors of limited diversity. J Exp Med. 1992;176:439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Argaet VP, Schmidt CW, Burrows SR, et al. Dominant selection of an invariant T cell antigen receptor in response to persistent infection by Epstein‐Barr virus. J Exp Med. 1994;180:2335–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cibotti R, Cabaniols JP, Pannetier C, et al. Public and private V beta T cell receptor repertoires against hen egg white lysozyme (HEL) in nontransgenic versus HEL transgenic mice. J Exp Med. 1994;180:861–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bousso P, Casrouge A, Altman JD, et al. Individual variations in the murine T cell response to a specific peptide reflect variability in naive repertoires. Immunity. 1998;9:169–178. [DOI] [PubMed] [Google Scholar]
- 7. Venturi V, Kedzierska K, Price DA, et al. Sharing of T cell receptors in antigen‐specific responses is driven by convergent recombination. Proc Natl Acad Sci USA. 2006;103:18691–18696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Venturi V, Chin HY, Price DA, Douek DC, Davenport MP. The role of production frequency in the sharing of simian immunodeficiency virus‐specific CD8+ TCRs between macaques. J Immunol. 2008;181:2597–2609. [DOI] [PubMed] [Google Scholar]
- 9. Venturi V, Price DA, Douek DC, Davenport MP. The molecular basis for public T‐cell responses? Nat Rev Immunol. 2008;8:231–238. [DOI] [PubMed] [Google Scholar]
- 10. Li H, Ye C, Ji G, Han J. Determinants of public T cell responses. Cell Res. 2012;22:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dash P, Fiore‐Gartland AJ, Hertz T, et al. Quantifiable predictive features define epitope‐specific T cell receptor repertoires. Nature. 2017;547:89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Glanville J, Huang H, Nau A, et al. Identifying specificity groups in the T cell receptor repertoire. Nature. 2017;547:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Quigley MF, Greenaway HY, Venturi V, et al. Convergent recombination shapes the clonotypic landscape of the naive T‐cell repertoire. Proc Natl Acad Sci USA. 2010;107:19414–19419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Robins HS, Campregher PV, Srivastava SK, et al. Comprehensive assessment of T‐cell receptor beta‐chain diversity in alphabeta T cells. Blood. 2009;114:4099–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Freeman JD, Warren RL, Webb JR, Nelson BH, Holt Ra. Profiling the T‐cell receptor beta‐chain repertoire by massively parallel sequencing. Genome Res. 2009;19:1817–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Benichou J, Ben‐Hamo R, Louzoun Y, Efroni S. Rep‐Seq: uncovering the immunological repertoire through next‐generation sequencing. Immunology. 2012;135:183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Six A, Mariotti‐Ferrandiz ME, Chaara W, et al. The past, present and future of immune repertoire biology ‐ the rise of next‐generation repertoire analysis. Front Immunol. 2013;4:413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Robins HS, Srivastava SK, Campregher PV, et al. Overlap and effective size of the human CD8+ T cell receptor repertoire. Sci. Transl Med. 2010;2:47ra64–47ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Venturi V, Quigley MF, Greenaway HY, et al. A mechanism for TCR sharing between T cell subsets and individuals revealed by pyrosequencing. J Immunol. 2011;186:4285–4294. [DOI] [PubMed] [Google Scholar]
- 20. Elhanati Y, Murugan A, Callan CG, Mora T, Walczak AM. Quantifying selection in immune receptor repertoires. Proc Natl Acad Sci USA. 2014;111:9875–9880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zvyagin IV, Pogorelyy MV, Ivanova ME, et al. Distinctive properties of identical twins’ TCR repertoires revealed by high‐throughput sequencing. Proc Natl Acad Sci USA. 2014;111:5980–5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pogorelyy MV, Elhanati Y, Marcou Q, et al. Persisting fetal clonotypes influence the structure and overlap of adult human T cell receptor repertoires. PLoS Comput Biol. 2017;13:e1005572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Madi A, Shifrut E, Reich‐Zeliger S, et al. T‐cell receptor repertoires share a restricted set of public and abundant CDR3 sequences that are associated with self‐related immunity. Genome Res 2014;24:1603–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Madi A, Poran A, Shifrut E, et al. T cell receptor repertoires of mice and humans are clustered in similarity networks around conserved public CDR3 sequences. eLife. 2017;6:e22057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Furmanski AL, Ferreira C, Bartok I, et al. Public T cell receptor‐chains are not advantaged during positive selection. J Immunol. 2008;180:1029–1039. [DOI] [PubMed] [Google Scholar]
- 26. Murugan A, Mora T, Walczak AM, Callan CG. Statistical inference of the generation probability of T‐cell receptors from sequence repertoires. Proc Natl Acad Sci USA. 2012;109:16161–16166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Elhanati Y, Marcou Q, Mora T, Walczak AM. RepgenHMM: a dynamic programming tool to infer the rules of immune receptor generation from sequence data. Bioinformatics. 2016;32:1943–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sethna Z, Elhanati Y, Dudgeon CS, et al. Insights into immune system development and function from mouse T‐cell repertoires. Proc Natl Acad Sci USA. 2017;114:2253–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marcou Q, Mora T, Walczak AM. High‐throughput immune repertoire analysis with IGoR. Nat Commun. 2018;9:561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Emerson RO, DeWitt WS, Vignali M, et al. Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA‐mediated effects on the T cell repertoire. Nat Genet. 2017;49:659–665. [DOI] [PubMed] [Google Scholar]
- 31. Venturi V, Rudd BD, Davenport MP. Specificity, promiscuity, and precursor frequency in immunoreceptors. Curr Opin Immunol. 2013;25:639–645. [DOI] [PubMed] [Google Scholar]
- 32. Hozumi N, Tonegawa S. Evidence for somatic rearrangement of immunoglobulin genes coding for variable and constant regions. Proc Natl Acad Sci USA. 1976;73:3628–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roldan EQ, Sottini A, Bettinardi A, Albertini A, Imberti L, Primi D. Different TCRBV genes generate biased patterns of V‐D‐J diversity in human T cells. Immunogenetics. 1995;41:91–100. [DOI] [PubMed] [Google Scholar]
- 34. Luo S, Yu JA, Song YS. Estimating copy number and allelic variation at the immunoglobulin heavy chain locus using short reads. PLoS Comput Biol. 2016;12:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ndifon W, Gal H, Shifrut E, et al. Chromatin conformation governs T‐cell receptor J gene segment usage. Proc Natl Acad Sci USA. 2012;109:15865–15870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Warmflash A, Dinner AR. A model for TCR gene segment use. J Immunol. 2006;177:3857–3864. [DOI] [PubMed] [Google Scholar]
- 37. Gauss GH, Lieber MR. Mechanistic constraints on diversity in human V(D)J recombination. Mol Cell Biol. 1996;16:258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Greenaway HY, Ng B, Price DA, Douek DC, Davenport MP, Venturi V. NKT and MAIT invariant TCR?? sequences can be produced efficiently by VJ gene recombination. Immunobiology. 2013;218:213–224. [DOI] [PubMed] [Google Scholar]
- 39. Ralph DK, Matsen FA. Consistency of VDJ rearrangement and substitution parameters enables accurate B cell receptor sequence annotation. PLoS Comput Biol. 2016;12:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lefranc MP, Giudicelli V, Ginestoux C, et al. IMGT®, the international ImMunoGeneTics information system®. Nucleic Acids Res. 2009;37:1006–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pogorelyy MV, Minervina AA, Chudakov DM, et al. Method for identification of condition‐associated public antigen receptor sequences. Elife. 2018;7:e33050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shortman K, Egerton M, Spangrude GJ, Scollay R. The generation and fate of thymocytes. SeminImmunol. 1990;2:3–12. [PubMed] [Google Scholar]
- 43. Pogorelyy MV, Minervina AA, Chudakov DM, et al. Method for identification of condition‐associated public antigen receptor sequences. eLife. 2018;7:e33050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jenkins MK, Chu HH, McLachlan JB, Moon JJ. On the composition of the preimmune repertoire of T cells specific for Peptide‐major histocompatibility complex ligands. Annu Rev Immunol. 2009;28:275–294. [DOI] [PubMed] [Google Scholar]
- 45. Qi Q, Liu Y, Cheng Y, et al. Diversity and clonal selection in the human T‐cell repertoire. Proc Natl Acad Sci USA. 2014;111:13139–13144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lythe G, Callard RE, Hoare R, Molina‐París C. How many TCR clonotypes does a body maintain? J Theor Biol. 2015;389:214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fazilleau N, Cabaniols JP, Lemaitre F, Motta I, Kourilsky P, Kanellopoulos JM. V and V public repertoires are highly conserved in terminal deoxynucleotidyl transferase‐deficient mice. J Immunol. 2004;174:345–355. [DOI] [PubMed] [Google Scholar]
- 48. Seay HR, Yusko E, Rothweiler SJ, et al. Tissue distribution and clonal diversity of the T and B cell repertoire in type 1 diabetes. JCI Insight. 2016;1:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fuchs YF, Eugster A, Dietz S, et al. CD8+ T cells specific for the islet autoantigen IGRP are restricted in their T cell receptor chain usage. Sci Rep. 2017;7:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Faham M, Carlton V, Moorhead M, et al. Discovery of T cell receptor β motifs specific to HLAB27 positive ankylosing spondylitis by deep repertoire sequence analysis. Arthritis Rheumatol. 2017;69:774–784. [DOI] [PubMed] [Google Scholar]
- 51. Zhao Y, Nguyen P, Ma J, et al. Preferential use of public TCR during autoimmune encephalomyelitis. J Immunol. 2016;196:4905–4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li B, Li T, Pignon JC, et al. Landscape of tumor‐infiltrating T cell repertoire of human cancers. Nat Genet. 2016;48:725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pasetto A, Gros A, Robbins PF, et al. Tumor‐ and neoantigen‐reactive T‐cell receptors can be identified based on their frequency in fresh tumor. Cancer Immunol Res. 2016;4:734–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Snyder A, Nathanson T, Funt SA, et al. Contribution of systemic and somatic factors to clinical response and resistance to PD‐L1 blockade in urothelial cancer: an exploratory multi‐omic analysis. PLoS Med. 2017;14:e1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hsu MS, Sedighim S, Wang T, et al. TCR sequencing can identify and track glioma‐infiltrating T cells after DC vaccination. Cancer Immunol Res. 2016; 4:412–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Munson DJ, Egelston CA, Chiotti KE, et al. Identification of shared TCR sequences from T cells in human breast cancer using emulsion RT‐PCR. Proc Natl Acad Sci USA. 2016;113:201606994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sadelain M, Rivière I, Riddell S. Therapeutic T cell engineering. Nature. 2017;545:423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Best K, Oakes T, Heather JM, Shawe‐Taylor J, Chain B. Computational analysis of stochastic heterogeneity in PCR amplification efficiency revealed by single molecule barcoding. Sci Rep. 2015;5:14629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rubelt F, Bolen CR, McGuire HM, et al. Individual heritable differences result in unique cell lymphocyte receptor repertoires of naïve and antigen‐experienced cells. Nat Commun. 2016;7:11112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Howie B, Sherwood AM, Berkebile AD, et al. High‐throughput pairing of T cell receptor a and b sequences. Sci Transl Med. 2015;7:301ra131. [DOI] [PubMed] [Google Scholar]
- 61. Grigaityte K, Carter JA, Goldfless SJ, et al. Single‐cell sequencing reveals αβ chain pairing shapes the T cell repertoire. bioRxiv. 2017:213462. [Google Scholar]
- 62. Elhanati Y, Sethna Z, Marcou Q, Callan CGJ, Mora T, Walczak AM. Inferring processes underlying B‐cell repertoire diversity. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Greiff V, Weber CR, Palme J, et al. Learning the high‐dimensional immunogenomic features that predict public and private antibody repertoires. bioRxiv. 2017. 127902. [DOI] [PubMed] [Google Scholar]
- 64. Yaari G, Kleinstein SH. Practical guidelines for B‐cell receptor repertoire sequencing analysis. Genome Med. 2015;7:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mccoy CO, Bedford T, Minin VN, Bradley P, Robins H, Iv FAM. Quantifying evolutionary constraints on B‐cell affinity maturation. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Georgiou G, Ippolito GC, Beausang J, Busse CE, Wardemann H, Quake SR. The promise and challenge of high‐throughput sequencing of the antibody repertoire. Nat Biotechnol. 2014;32:158–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Corti D, Lanzavecchia A. Broadly neutralizing antiviral antibodies. Annu Rev Immunol. 2013;31:705–742. [DOI] [PubMed] [Google Scholar]