

Abstract
Cardiovascular diseases are a leading cause of morbidity and mortality in most developed countries of the world. Pharmaceuticals, illicit drugs, and toxins can significantly contribute to the overall cardiovascular burden and thus deserve attention. The present article is a systematic overview of drugs that may induce distinct cardiovascular toxicity. The compounds are classified into agents that have significant effects on the heart, blood vessels, or both. The mechanism(s) of toxic action are discussed and treatment modalities are briefly mentioned in relevant cases. Due to the large number of clinically relevant compounds discussed, this article could be of interest to a broad audience including pharmacologists and toxicologists, pharmacists, physicians, and medicinal chemists. Particular emphasis is given to clinically relevant topics including the cardiovascular toxicity of illicit sympathomimetic drugs (e.g., cocaine, amphetamines, cathinones), drugs that prolong the QT interval, antidysrhythmic drugs, digoxin and other cardioactive steroids, beta‐blockers, calcium channel blockers, female hormones, nonsteroidal anti‐inflammatory, and anticancer compounds encompassing anthracyclines and novel targeted therapy interfering with the HER2 or the vascular endothelial growth factor pathway.
Keywords: dysrhythmia, heart failure, hypertension, myocardial infarction, stroke
ABBREVIATIONS
- ACEi
angiotensin‐converting enzyme inhibitor
- ADHD
attention‐deficit hyperactivity disorder
- ANT
anthracyclines
- AV
atrioventricular
- COMT
‐ catechol‐O‐methyltransferase
- EMA
The European Medicines Agency
- eNOS
endothelial isoform of NO‐synthase
- FDA
The Food and Drug Administration
- hERG
human ether‐a‐go‐go‐related gene—commonly used term for the channel responsible for I Kr current
- HER2
human epidermal growth factor receptor 2
- HRT
hormonal replacement therapy
- IKr
rapid component of delayed rectifier current
- LV
left ventricular
- MAO
monoamine oxidase
- MARTA
multiacting receptor targeted antipsychotics
- MDMA
3,4‐methylendioxymetamphetamine
- NSAIDs
nonsteroidal anti‐inflammatory drugs
- ROS
reactive oxygen species
- SSRi
selective serotonin reuptake inhibitors
- TCA
tricyclic antidepressant drug
- VEGF
vascular endothelial growth factor
1. INTRODUCTION
The cardiovascular toxicity of drugs and related agents attracts considerable attention from basic scientists to clinicians. Cardiovascular diseases are among the most significant determinants of morbidity and mortality in the developed countries; therefore, it is important that any negative impact of drugs or toxins on this system is not understated. Although there are many high‐quality pharmacology and toxicology review articles and textbooks,1, 2, 3, 4, 5, 6, 7 a comprehensive description of the cardiovascular toxicities induced by drugs and toxins is lacking. Naturally, the topic is broad and complex, as it integrates both clinical and experimental pharmacology and toxicology with projection to other clinical disciplines like cardiology. This monumental task is also complicated by several factors including the fact that many drugs have multiple and sometimes divergent effects on the cardiovascular system (e.g., cocaine, digoxin, tricyclic antidepressant drugs/TCAs, tobacco smoking). Similarly, the vascular system is functionally linked to the heart, thus its functions are interconnected (e.g., endothelial dysfunction and subsequent hypertension could result in a damage to the heart, and vice versa). Drugs primarily causing heart rhythm disturbances can ultimately result in impaired hemodynamic function of the heart, and so forth.
The aim of this review is to summarize the current knowledge regarding xenobiotic agents and toxins that cause cardiovascular toxicity. The authors were obliged to accept some compromises to achieve their purposes. In particular, for drugs with numerous cardiovascular actions, the classification was based on the assumed principal mechanism of cardiovascular toxicity, the mechanism commonly associated with severe cardiovascular effects, or the best‐documented (toxic) mechanism. Additional, therapeutic or toxic mechanisms of action are also discussed or mentioned in the corresponding section. The A‐type adverse effects, which may result in toxicity due to severe augmentation of the drug's therapeutic effects, are mentioned only when it is clinically relevant. Otherwise, they are considered predictable and beyond the scope of this review. Some toxicity is clinically important only in the patients with a specific disease, or as a result of interaction with other drug(s), or specific foods. These details are discussed only in typical and relevant cases. A similar rationale advocates for the inclusion of treatments and preventions of cardiovascular toxicity, especially when specific antidotes or protectants are available. This text focuses on the cardiovascular toxicity of drugs and related agents during the postnatal period and thus teratogenic effects involving deviations in the intrauterine development of cardiovascular structure and function are not discussed. Metabolic effects, such as lipid and glucose disorders, or toxicity that targets the cardiovascular system indirectly (e.g., kidney damage) exceed the scope of this review and are only briefly mentioned in cases where it is considered appropriate.
Indeed, even the definition of toxic effect may be a matter of debate and depending on the point of view, it may be either broad or strict. The broad definition refers to all harmful effects to living systems and it can be applied to all compounds, for example, pharmaceuticals, illegal and/or abused drugs, chemicals, toxins, and environmental pollutants. The strict definition of a toxic effect is related to an overdose, that is, the administration of doses/environmental exposures higher than recommended or allowed. This definition best fits pharmaceuticals used in clinical medicine. Even here, the difference between therapeutic doses and doses evoking deleterious effects might not be clear and the interindividual susceptibility is high for certain drugs.7, 8, 9 Thus, the clear‐cut difference between adverse effects observed at recommended doses and toxicity associated with overdose may be difficult to define. Therefore, in this review, toxic effects are understood and discussed in the broader meaning. There is additional reason for this approach. For instance, even small increases in blood pressure and/or heart rate caused by a drug in clinical studies should raise caution related to possible more severe consequences and, indeed, they are associated with higher cardiovascular mortality.10, 11 In general, the borderline zones between normal and toxic doses for some drugs affecting the cardiovascular system are often quite obscure. Nonetheless, the main emphasis is given to potentially severe effects on the cardiovascular system.
For systematic purposes, many classification systems have been adopted as the structure of this review. Some of them are quite practical and clinically oriented. These are usually based on main clinical presentations and its outcomes (Table 1). However, in a comprehensive review article such classification may be complicated by the fact that many drugs can evoke different effects at many different levels. For example, indirect sympathomimetics can produce tachycardia, hypertension, stroke, and also acute myocardial infarction, depending on the dose and other factors (Table 2). Furthermore, some effects are secondary rather than primary, for example, dysrhythmias can impair hemodynamic function and hypo/hypertension can result in acute myocardial infarction and/or heart failure.
Table 1.
Clinical classification of cardiotoxicity including known mechanisms
| 1. Toxicity to the heart | |
| 1.1 Rhythm disturbances | Mechanisms and drugs involved |
| 1.1.1 Bradydysrhythmias | Ca2+ channel blockade, Na+ channel blockade, blockade of If current, antagonism at β1‐receptors, agonism at M‐receptors, and increased vagal tone (e.g., digoxin), agonism at α2‐receptors, agonism at I1‐receptors, agonism at sphingosine‐1‐phosphate receptor, several anticancer drugs (e.g., thalidomide or paclitaxel) |
| 1.1.2 Tachydysrhythmias | Blockade of hERG channels function or intracellular trafficking, agonism at β1‐receptors (direct effects or indirect effect via endogenous catecholamines), antagonism at M‐receptors, inhibition of Na+/K+ pump, Na+ channel blockade, secondary effect (stimulation of sympathetic system, e.g., after hypotension) |
| 1.2 Myocardial ischemia | Agonism at β1‐receptors (direct effects or indirect effect via endogenous catecholamines), rebound phenomenon (nitrates, β‐blockers), and other secondary effects (arterial thromboembolism, hyperstimulation of the heart including tachycardia, pronounced hypotension), some anticancer drugs—complex mechanisms (5‐fluorouracil, capecitabine, or bevacizumab) |
| 1.3 Left ventricular (LV) dysfunction/heart failure | |
| 1.3.1 Mechanisms interfering with normal physiology of LV function | Ca2+ channel blockade, Na+ channel blockade, antagonism at β1‐receptors |
| 1.3.2 Anticancer drug‐induced LV dysfunction |
|
| 1.3.3 Myocarditis | Autoimmune reactions (e.g., clozapine), monoclonal antibodies targeting PD‐1 |
| 1.4 Impairment of cardiac valves | Agonism at 5‐HT2B receptors |
| 1.5 Induction of pericardial disease | Immune reaction (e.g., drugs inducing lupus erythematosus), hemorrhage |
| 2. Toxicity to the vascular system | |
| 2.1 Effect on arterial blood pressure | |
| 2.1.1 Systemic hypertension | Agonism at α1‐receptors, antagonism at α2‐receptors, rebound phenomenon (α2‐agonists and beta‐blockers), agonism at glucocorticoid receptors, inhibition of VEGF pathway, inhibition of monoamine oxidases (MAO), 11β‐hydroxysteroid dehydrogenase type 2 inhibition |
| 2.1.2 Systemic hypotension (first‐dose hypotension, orthostatic or symptomatic) | Antagonism at α1‐receptors, Ca2+ channel blockade, opening of K+ channels, inhibition or renin–angiotensin–aldosterone axis, agonism at β2‐receptors, agonism at α2‐receptors, agonism at I1‐receptors, stimulation of cGMP synthesis, inhibition of phosphodiesterase 5 |
| 2.1.3 Pulmonary hypertension | An effect on serotonergic system, for example, (dex)fenfluramine, benfluorex, other mechanism are largely unknown |
| 2.2. Thromboembolic complications | |
| 2.2.1 Arterial | Inhibition of cyclooxygenase 2, VEGF targeting, agonism at erythropoietin receptors |
| 2.2.2 Venous | Agonism at estrogenic receptors, VEGF targeting, agonism at erythropoietin receptors, unknown (thalidomide) |
Table 2.
Major groups of compounds with cardiovascular toxic effects
| Cardiac toxic effect | Toxic effect on vascular system | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Group of drugs or mechanism | Representatives | Tachydysrhythmias/tachycardia | Bradydysrrhythmias/bradycardia | Ischemiaa | Cardiomyopathy | Direct toxic effect on cardiomyocytes | Reversible effect on contractility | Hypotension | Hypertension | Venous thromboembolism | Strokes |
| Indirect sympatholytics | Reserpine | X | X | ||||||||
| Indirect sympathomimetics | Cocaine, amphetamines | X | X | X | X | X | |||||
| Tobacco smoking | X | X | X (acute) | X | |||||||
| Endogenous catecholaminesb | Adrenaline, noradrenaline | X | X | X | X | ||||||
| Nonselective beta agonists | Isoprenaline | X | AV blockc | X | Xc | Xc | X | ||||
| α1‐Mimetics | Phenylephrine | X (reflex) | X | X | |||||||
| α1‐Lytics | Doxazosin | X (reflex) | X | ||||||||
| α2‐Mimetics | Methyldopa, clonidine | X | X | ||||||||
| α2‐Lytics | Yohimbine | X | X | ||||||||
| β1‐Mimetics | Dobutamine | X | X ? | ||||||||
| β2‐Mimetics | Salbutamol, clenbuterol | X | X | Xc | Higher doses | ||||||
| β‐Blockers | Metoprolol | X (RP) | X | X(RP) | X | X | X (RP) | ||||
| PDE3 i | Milrinone | X | X | ||||||||
| PDE5 i | Sildenafil | X | |||||||||
| I kr inhibition | Ibutilide, arsenic trioxide | X | |||||||||
| Na+ channel activation | Aconitine | X | X | X | |||||||
| Neuronal Na+ channel activation | Grayanotoxins | X | X | ||||||||
| Na+ channel blockade | Local anesthetics | X | X | X | |||||||
| Ca2+ channel blockade | Verapamil | X | X | X | |||||||
| Dihydropyridines | X (reflex) | Possibly | Possibly | X | |||||||
| Calcium sensitizers | Levosimendan | X | X | ||||||||
| Blockade of Na+/K+ ATPase | Digoxin | X | X | ||||||||
| Cholinomimetics | Organophosphates | X | Lower dose | X | |||||||
| Parasympatholytics | Atropine | X | |||||||||
| Tricyclic antidepressant drugs (TCA) | Nortryptiline | X | X | X | X | ||||||
| Adenosine antagonists | Theophylline | X | X | ||||||||
| Anthracyclines (Type I cardiotoxicity) | Doxorubicin | Xd | X | X | |||||||
| HER2i and others (Type II cardiotoxicity) | Trastuzumab, lapatinib | X | X | ||||||||
| antiVEGF | Bevacizumab, sorafenib | X | ? | X | X | ? | X | ||||
| Androgens | Anabolic steroids | Not specified | X | mild | ? | ||||||
| Erythropoietin | X | X | X | ||||||||
| NSAID | Rofecoxib, ibuprofen | X | X | X | |||||||
| Estrogen + progestin | Combined oral contraception, HRTe | X | X | X | X | ||||||
| Glucocorticoids | Prednisone | X | |||||||||
| Ethanol | X | X | Acute | X | |||||||
RP, rebound phenomenon; reflex, secondary (reflex) autonomic reaction.
aSpasm of coronary arteries, increased platelet aggregation, or other reasons.
bGiven in the treatment in high doses.
cIn high, experimental doses.
dAcute toxicity unrelated to Type I cardiotoxicity.
eRisks for HRT are dependent on more factors—see main body of the article.
Due to the above‐mentioned facts and also because of the overwhelming number of articles (a search for the keyword “cardiotoxicity” in PubMed retrieves thousands of articles), it was not realistic to perform a systematic review of the literature. Furthermore, we have adopted a broader meaning of the term cardiovascular toxicity, which makes the situation is even more complex. Therefore, we have identified major types of toxic cardiovascular insults (disturbances in cardiac rhythm, functional and structural heart impairment, arterial and venous thromboembolism, effects on blood pressure) and the associated mechanisms/molecular targets. We then performed targeted searches for drugs, pharmaceuticals, illicit drugs, and toxins associated with the particular cardiotoxic event and/or mechanisms involved. For purposes of this review, the following classification of cardiotoxicity was used (Table 3).
Table 3.
Classification of cardiovascular toxicity of diverse agents used in this review
| 2. Drugs with possible toxic effects on both cardiomyocytes and the vascular system | 3 |
| 2.1 Indirect sympathomimetics | 3 |
| 2.1.1 Indirect sympathomimetics blocking noradrenaline‐reuptake mechanism | 8 |
| 2.1.2 Indirect sympathomimetics releasing monoamines and/or with mixed mechanisms | 11 |
| 2.2 Direct acting sympathomimetics | 14 |
| 2.2.1 Endogenous catecholamines | 14 |
| 2.2.2 Nonselective β‐agonists and β2‐agonists | 14 |
| 2.3 Nicotine and smoking | 15 |
| 2.4 Drugs affecting the adrenergic system via their action in the CNS | 17 |
| 2.5 Drugs influencing intracellular signalling downstream of adrenergic receptors with effects on both cardiomyocytes and the vascular system | 17 |
| 2.6 Other drugs causing sympathetic hyperactivity | 18 |
| 2.7 Ca2+ channel blockers and maitotoxin | 18 |
| 2.8 Tricyclic antidepressants (TCA) | 19 |
| 2.9 Ethanol | 19 |
| 2.10 Androgenic anabolic steroids | 20 |
| 3. Drugs with the major effects on the heart | 20 |
| 3.1 Drugs affecting the function of both the conduction system and the working myocardium | 20 |
| 3.1.1 β‐blockers | 21 |
| 3.2 Drugs with main toxic effects on cardiac electrophysiology—drugs causing dysrhythmias | 21 |
| 3.2.1 Sodium channels | 21 |
| 3.2.2 Potassium channels | 26 |
| 3.2.3 Sodium–potassium pump | 28 |
| 3.2.4 Acetylcholine receptors | 32 |
| 3.2.5 Adenosine receptors | 33 |
| 3.2.6 Other drugs causing rhythm disturbances | 33 |
| 3.3 Drugs with main toxic effects on the working myocardium | 33 |
| 3.3.1 Anticancer drug‐induced cardiac dysfunction and heart failure | 34 |
| 3.3.2 Others | 36 |
| 3.4 Drugs directly affecting cardiac valves | 37 |
| 3.5 Drugs causing pericarditis | 37 |
| 4. Drug affecting primarily the vascular system | 38 |
| 4.1 Drugs causing hypertension and arterial thrombosis | 38 |
| 4.1.1 NSAIDs | 38 |
| 4.1.2 Inhibitors of the VEGF pathway | 39 |
| 4.1.3 Erythropoietin and its analogues | 40 |
| 4.2 Drugs causing systemic arterial hypertension | 41 |
| 4.2.1 α1‐Adrenergic receptors agonists | 41 |
| 4.2.2 Glucocorticoids | 41 |
| 4.2.3 Licorice | 42 |
| 4.2.4 Calcineurin inhibitors | 42 |
| 4.2.5 Leflunomide and teriflunomide | 43 |
| 4.2.6 Others | 44 |
| 4.3 Drugs causing pulmonary hypertension | 44 |
| 4.4 Drugs causing hypotension due to direct action on the vascular system | 44 |
| 4.4.1 α1‐Adrenergic receptor antagonism | 44 |
| 4.4.2 Dihydropyridine calcium channel blockers | 45 |
| 4.4.3 Other antihypertensives | 45 |
| 4.4.4 Other drugs | 45 |
| 4.5 Drugs causing angioedema | 45 |
| 4.6 Drugs causing venous thromboembolism | 46 |
| 4.6.1 Female hormones and drugs acting at this level | 46 |
| 4.6.2 Thalidomide and lenalinomide | 49 |
| 4.6.3 Strontium ranelate | 49 |
2. DRUGS WITH POSSIBLE TOXIC EFFECTS ON BOTH CARDIOMYOCYTES AND THE VASCULAR SYSTEM
This category includes drugs with significant direct effects on both the myocardium/cardiomyocytes and the vascular system. The major representatives are indirect sympathomimetics and nonselective sympathomimetics. Other agents with miscellaneous mechanisms are also discussed.
2.1. Indirect sympathomimetics
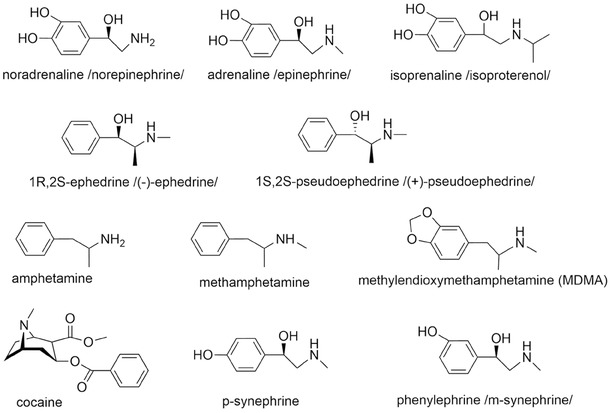
Indirect sympathomimetics (see Fig. 1) are very important from a toxicological point of view. Many illicit drugs, which are commonly abused, belong to in this category.12 This is particularly true for cocaine and amphetamines, which after cannabis, are the most commonly abused substances in Europe.13 Although the effects of all indirect sympathomimetics are similar, they are not entirely the same because there are marked differences in the major mechanisms of action and resulting effects on neurotransmitters (mainly with respect to dopamine and serotonin). Indirect sympathomimetic effects with clinical consequences are based on: (a) blockade of noradrenaline plasmalemmal synaptic transporters (see chapter 2.1.1), and (b) the release of catecholamines from the synaptic vesicles (see chapter 2.1.2, Fig. 2A–C). The latter effects are rather complex and usually involve inhibition of reuptake, as well as inhibition of neuronal monoamine oxidase‐A (MAO‐A). Indirect sympathomimetics affect both the vascular system via noradrenaline actions on α1‐adrenergic receptors and the heart due to noradrenaline actions on β1‐adrenoreceptors. The former manifests as an elevation in the blood pressure, while the latter is associated with increased contractility, heart rate, conduction velocity, and cardiac excitability. Such effects markedly increase myocardial oxygen consumption. Increased platelet aggregation due to sympathomimetics can be important as well.14
Figure 1.
Endogenous catecholamines (noradrenaline and adrenaline) and clinically + toxicologically important sympathomimetics.
Figure 2.
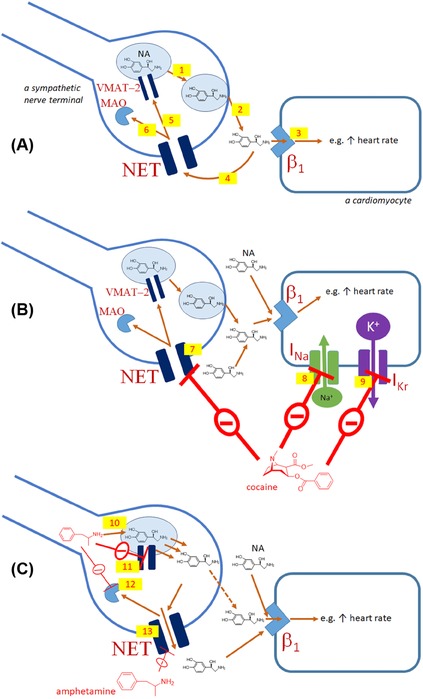
Noradrenaline release from sympathetic fibers at the synaptic cleft in the heart and the influence of indirect sympathomimetics. Physiological situation (A): upon stimulation of the sympathetic system, vesicles containing noradrenaline fuse with the cytoplasmic membrane of the synaptic cleft (1), noradrenaline (NA) is released (2), and stimulates β1‐adrenergic receptors (3). The effect of noradrenaline is, however, rapidly terminated (4) by the uptake (reuptake) mechanism via noradrenaline transporter (NET) and, thus, noradrenaline can be transported back to the vesicles (5) by vesicular monoamine transporter‐2 (VMAT‐2) or metabolized by MAO‐A (6, MAO). Effect of cocaine (B): cocaine blocks NET (7) and hence the half‐life and concentration of noradrenaline in the synaptic cleft is prolonged and increased, respectively. It also reduces the Na+ current (8, INa) and the rapid component of delayed rectifier current (9, I Kr). Effect of amphetamine (C): amphetamine replaces noradrenaline in vesicles (10) and blocks the uptake by VMAT‐2 (11) and metabolism by MAO‐A (12). This results in noradrenaline release into the cytosol. NET seems to work in the opposite direction (13) when the cytosolic concentration of noradrenaline is increased. On the other hand, NET is also partly blocked by amphetamine.
2.1.1. Indirect sympathomimetics blocking noradrenaline‐reuptake mechanism
There are many drugs that are able to block the noradrenaline‐reuptake transporter including cocaine, some antidepressants, sibutramine, and sympathomimetics/central nervous system (CNS) stimulants used for narcolepsy and attention‐deficit hyperactivity disorder (ADHD) treatment like methylphenidate and atomoxetine.15 In general, there are marked differences between these drugs regarding their cardiovascular effects and toxicity. One of the major reasons is the presence of additional mechanisms of action, which are very relevant, namely for cocaine and TCA. Therefore, these drugs need to be discussed separately.
Cocaine is not used clinically in developed countries, however, it remains an important drug of abuse, particularly in large cities.12 The molecular mechanisms underlying the cardiovascular actions of cocaine are largely based on its indirect sympathomimetic activity, but Na+ channel blockade may also be involved as an additional independent mechanism (see Fig. 2B). Cocaine is also a weak inhibitor of MAO, and this can contribute to the sympathomimetic effect.16 The most common first sign of acute overdose is generally chest pain caused by an increase in oxygen demand, vasoconstriction of coronary arteries, and/or thrombosis due to platelet activation. There is a risk of acute myocardial infarction within the first hours after cocaine abuse that can often occur in young men and in the absence of significant atherosclerosis. The risk is, however, higher in patients with atherosclerosis and in cigarette smokers. Electrocardiogram (ECG) may not be sufficient to make a clear diagnosis, unlike cardiac troponins. Cocaine roughly doubles the risk of both ischemic and hemorrhagic strokes, and seems to be a common cause of aortic dissection. The risk of dysrhythmias is also increased. These may range from sinus tachycardia due to indirect sympathomimetic effects, up to ventricular tachycardia. The local anesthetic properties of cocaine based on its Na+ channel blockade can impair impulse conduction in the ventricles, providing a substrate for reentrant ventricular dysrhythmias and potentially ventricular fibrillation, it may also be co‐responsible for cardiac arrest due to asystole. Cocaine also blocks K+ channels possibly resulting in QT interval prolongation. Since it is often difficult to identify a definitive cause of death at autopsy in cases of cocaine abuse, cocaine‐induced dysrhythmias might be the major cause of death. Furthermore, long‐term consequences of chronic cocaine abuse include cardiomyopathy and increased risk of heart failure. The best treatment to reduce the risks is obviously abstinence from cocaine use, which can also markedly improve the prognosis of cocaine‐induced heart failure. In cases of acute myocardial infarction, the standard treatment is somewhat different than that used for the general population and should start with benzodiazepines. Nitrates or phentolamine (an α1/2‐adrenergic receptor blocker) could be also useful. On the other hand, β‐blockers should be avoided, at least in the very acute phase, because β‐blockade will potentially leave α1‐receptors unopposed resulting in more severe coronary spasm, or arterial blood pressure increase. Data obtained in animal studies and after human administration confirm the risk posed by β‐blockers. Concerning dysrhythmias, hypertonic sodium bicarbonate serves as the first‐line treatment, while the use of lidocaine is controversial.17, 18, 19, 20, 21
As mentioned, there are other drugs that block the noradrenaline reuptake mechanism. This mechanism is common for many antidepressant drugs, namely TCA, serotonin–noradrenaline reuptake inhibitors (SNRI, e.g., venlafaxine), noradrenaline–dopamine reuptake inhibitors (NDRI, e.g., bupropion, Fig. 3), and relatively selective noradrenaline‐reuptake inhibitors (reboxetine). Regarding TCA, the potential cardiovascular toxic effects are well described, but the underlying mechanisms are complex and probably only weakly related to noradrenaline reuptake. In overdose, for example, due to suicide attempts, these drugs can induce life‐threatening dysrhythmias. These drugs cause hypotension, rather than hypertension, due to their α1‐adrenergic receptor blocking activity. They also possess significant antimuscarinic activity and their effects will be more thoroughly discussed in chapter 2.8. SNRI have a significant risk of blood pressure elevation10 and can increase heart rate.22 These effects are likely mediated by the inhibition of noradrenaline reuptake. Interestingly, bupropion does not seem to pose a risk of cardiovascular toxicity, but increases blood pressure without markedly effecting heart rate.22, 23, 24, 25 Reboxetine increases heart rate.22
Figure 3.
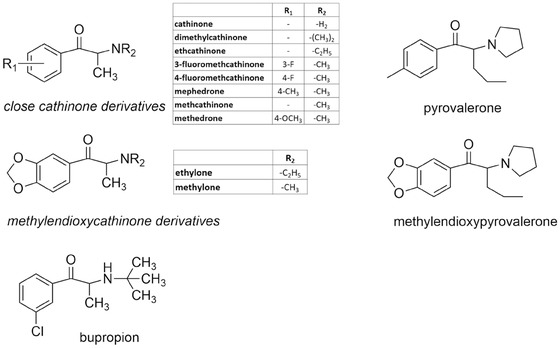
Chemical structure of cathinone derivatives “bath salts” and bupropion.
Pharmacological treatment of ADHD usually relies on drugs (stimulants) that interact directly with catecholaminergic systems, more specifically with dopamine or noradrenaline pathways. Stimulants are first‐line agents for ADHD and include methylphenidate and amphetamine salts, or the prodrug lisdexamfetamine. These drugs are considered equally effective. In general, they act on dopamine transporters (DAT) and noradrenaline transporters (NETs) given the drugs’ structural similarities to dopamine and noradrenaline. The first nonstimulant drug approved for ADHD treatment was atomoxetine. Atomoxetine is a selective NET inhibitor that increases dopamine and noradrenaline levels in prefrontal cortex, since the NET transporter clears both noradrenaline and dopamine in that brain region. Furthermore, α2‐agonists are also a therapeutic alternative for the treatment of ADHD symptoms. Clonidine and guanfacine mimic noradrenaline actions on the postsynaptic receptors (see chapter 2.5). In nonresponsive ADHD patients, a third‐line group of medication can be used, namely bupropion and TCA (see chapter 2.8).15, 26
Several studies have shown that drugs used to treat ADHD have a negative impact on the cardiovascular system. Stimulant medications for ADHD, including methylphenidate and amphetamine derivatives, are generally safe and well tolerated; however, small but statistically significant increases in blood pressure and heart rate are among the reported adverse events regardless of age. Methylphenidate usually increases systolic blood pressure and the increase in heart rate may be transient. Furthermore, children and adolescents treated with atomoxetine usually increase heart rate or blood pressure. Although uncommon, dysrhythmias and/or sudden death are also documented after ADHD medication.27, 28, 29, 30 In contrast, small decreases in mean systolic blood pressure, diastolic blood pressure, and heart rate have been observed with guanfacine‐extended release or clonidine‐extended release preparations, administered alone or in combination with psychostimulants to children and adolescents with ADHD.31
Sibutramine is effective in reducing body weight 32 and has been used for many years to treat obesity; however, it was withdrawn in 2010 by both the The European Medicines Agency (EMA) and The Food and Drug Administration (FDA).33 The drug is a monoamine uptake inhibitor with higher selectivity for noradrenaline and dopamine over serotonin. In line with other herein mentioned substances, the cardiovascular risk is clearly present. The drug increases arterial blood pressure and heart rate and there are higher risks of stroke and nonfatal myocardial infarction. The risk is greatest in patients with cardiovascular disease or cardiovascular risk factors.34, 35, 36
2.1.2. Indirect sympathomimetics releasing monoamines and/or with mixed mechanisms
This category includes primarily substances isolated from ephedra, their synthetic congeners (amphetamines, Fig. 1), and cathinone derivatives (Fig. 3). The mechanisms of action of ephedra alkaloids and amphetamines are complex and are based on several factors including lipophilicity, basicity, inhibition of monoamine transporters, and even a reduction in monoamine metabolism by inhibition of MAO (the simplified mechanism is depicted in Fig. 2C).37 Amphetamines are psychoactive substances derived from the core structure of β‐phenylethylamine. They possess a methyl group on the α‐carbon that increases their ability to cross‐membranes and that also largely contributes to their psychoactive proprieties. Their oxidative deamination by MAO is generally blocked by this substitution of the α‐carbon (amphetamines can suffer oxidative deamination, but the reaction is cytochrome P450 mediated).38 Moreover, biogenic catecholamines are usually metabolized by MAO or catechol‐O‐methyltransferase (COMT), while amphetamines are recognized for their MAO inhibitory properties. Therefore, amphetamines also increase the cytosolic content of monoamines in part through inhibition of their metabolism. Ephedrine is also a weak inhibitor of MAO, and that property may contribute to its sympathomimetic effects as well.16 There are some differences among these compounds, in particular regarding their affinities for monoamine transporters.39, 40 They increase the release of noradrenaline, dopamine, and serotonin in the synaptic cleft; however, their activity differs among compounds. In general, their effect on serotonin is lower with the exception of 3,4‐methylendioxymetamphetamine (MDMA),40 which will be discussed separately. Concerning their adrenergic activity, most of them are considered to be pure indirect sympathomimetics, but natural ephedrine (1R,2S‐ephedrine) also has significant direct agonistic activity on β1‐ and β2‐adrenergic receptors. Its isomer 2S,2R‐ephedrine is apparently free of direct β‐agonistic activity. There are data suggesting that alkaloids from ephedra are week antagonists at α2‐adrenergic receptors, which could contribute to their sympathomimetic activity.41, 42, 43 m‐Synephrine (Fig. 1), a compound structurally related to ephedra alkaloids, is mainly an α1‐adrenergic agonist and thus will be discussed in Section 4.2.1.
Ephedra and its natural alkaloids have been used as nasal decongestants, bronchodilators, CNS‐stimulants, antiobesitics, and for the improvement of athletic performance. The use of ephedra products, including synthetic mixtures of norephedrine and norpseudoephedrine, known as phenylpropanolamine, was banned in the United States in 2004 due to the high incidence of serious adverse reactions. Ephedrine is the dominant alkaloid (40–90%), isolated from different shrubs of the genus Ephedra grown in mild and subtropic areas. The term ephedra is used for a mixture of alkaloids isolated from dry branches of the plant. In addition to ephedrine, it also contains pseudoephedrine, norephedrine, norpseudoephedrine, and methylpseudoephedrine. It has also been sold under the Chinese name “ma huang,” which means “yellow adstringent” due to its color and sharp taste. The reported adverse reactions principally involve the cardiovascular system and are, in general, similar to other sympathomimetics. The most common side effect is hypertension with a risk of hemorrhagic stroke.44, 45, 46, 47 Also ischemic stroke due to vasoconstriction and likely platelet aggregation can occur after its consumption.46, 48 In the case of phenylpropanolamine, a higher incidence of hemorrhagic stroke in women is well documented and data show that increased risk also exists in men.47, 49, 50 Although the risk of hemorrhagic stroke with pseudoephedrine seems to be lower, it can occur and might result in death.47, 51 The adverse reactions after ephedra administration can more easily occur when it is used in combination with caffeine.52 This combination increases the effect of sympathomimetics, and the mechanisms will be discussed later.
Amphetamines are synthetic compounds, with important representatives being amphetamine, its dextrotatory form dexamphetamine, methamphetamine (also known as Pervitin), and MDMA (ecstasy). Only dexamphetamine and its prodrug lisdexamphetamine are used clinically in some countries for the treatment of narcolepsy and ADHD.15
The differences among amphetamines can be caused by different physicochemical properties and consequently related to the amount of drug transport into the cells. Lipophilicity, pKa, and protein binding determine the amount of drug present in each body compartment and its pharmacological/toxicological proprieties overall. Lipophilicity and polarity have been long studied regarding catecholamine‐related compounds. There are also some differences between endogenous catecholamines, but in comparison with amphetamines, the presence of hydroxyl groups in the catecholic ring decreases lipophilicity, while β‐hydroxylation contributes less to this property. The most important factor is alkylation, although the effect of that change on the physicochemical properties is dependent on the position and on the size of the alkyl group.53 In fact, until this day, these data strongly impact on the synthesis of new psychoactive illicit drugs. For instances, amphetamine and methamphetamine, which only differ in a methyl group, shows that the latter drug is considered more potent when used in comparable doses.54 They both share the basic nitrogen moiety and are weak bases with low plasma protein binding (usually under 20%); however, methamphetamine's relatively high lipophilicity (partition coefficient heptane/water of 5.14) may contribute to its fast entrance into brain (and cells in general), when compared to amphetamine, with a partition coefficient heptane/water of 1.88.38, 54, 55
Cardiovascular events ensuing from misuse or abuse of amphetamines include chest pain, tachycardia, dyspnoea, hypertension, dysrhythmias, acute myocardial infarction, aortic dissection, and sudden cardiac death. Data from emergency departments shows that tachycardia is a dominant feature upon methamphetamine admission. In about ⅓–½ of cases, hypertension was also observed and ¼ of patients had an acute coronary syndrome. Like cocaine, acute myocardial infarction can occur in patients with normal angiographic findings. The main culprit seems to be coronary artery vasospasm and enhanced oxygen demand, but atherosclerotic plaque rupture and/or enhanced platelet aggregation can also contribute. After essential hypertension, methamphetamine is the second most common cause of death from acute aortic dissection, probably because of the acute worsening of the hypertension. In chronic amphetamine abusers, coronary artery disease and/or dilated cardiomyopathy are relatively common.38, 56 Also amphetamine use in ADHD, at therapeutic doses, does not seem to be absolutely free of cardiovascular toxicity (see also above) and cases of sudden death mainly in children likely due to cardiac toxicity are probable.15, 57 Lisdexamphetamine seems to have a lower risk of adverse cardiac reaction due to the slow release of dexamphetamine.58
Inclusion of a methylenedioxy group to the aromatic ring of amphetamine and methamphetamine gives rise to hallucinogenic proprieties and both MDMA and MDA [(±)‐3,4‐methylenedioxyamphetamine] are also serotonin 2A‐receptor agonists.59, 60 Regarding pharmacokinetics, MDMA, like other amphetamines, has high oral bioavailability, high volume of distribution, and low plasma protein binding. It is a weakly basic drug with low molecular weight that easily enters cells. MDMA undergoes extensive metabolism and some of its metabolites cause cardiotoxic effects.38, 61, 62 MDMA abuse has a lower cardiovascular risk since it has less impact on noradrenaline release and a markedly higher increase in serotonin release compared to other sympathomimetics.40 However, studies in rats and in humans found prominent increases in heart rate and blood pressure, which were similar to other amphetamines.63, 64 Fatal outcomes based on cardiovascular and cerebrovascular complications, or due to accidents, can occur.65, 66 The identification of MDMA as the ultimate cause of death is commonly complicated by the presence of other drugs that are concomitantly taken (opioids, alcohol, cocaine, cannabis).66 Caffeine is commonly present in tablets of “ecstasy” and it is well documented that caffeine has pronounced effects on the acute toxicity of “ecstasy” in rats. In addition, lower concentrations of caffeine are needed to promote MDMA toxicity in contrast to amphetamine.67 However, MDMA can cause lethal cardiotoxicity in the absence of other substances.65, 68 The direct toxicity of the biogenic amines it releases, and the cardiotoxic actions of its metabolites (especially quinones and glutathione adducts) must be considered as important culprits of MDMA‐induced cardiotoxicity.62, 69 MDMA can cause serotonin syndrome with hyperthermia, but this is very rarely associated with death.66 Cardiomegaly or myocardial hypertrophy is common in chronic abusers of MDMA.56
Similar to cocaine, benzodiazepines are commonly the drugs of first choice in the management of acute amphetamine‐induced cardiovascular toxicity. They can sufficiently decrease sympathomimetic hyperactivity and thus, both tachycardia and hypertension. They can also alleviate hyperthermia. Alternatively, dexmedetomidine, an α2‐adrenoceptor agonist, can be useful. In resistant cases, a direct vasodilator such as nitroprusside should be administered. β‐blockers are not recommended for the same reason as in cocaine toxicity. However, anticipated positive effects were seen after administration of the mixed α‐ and β‐blockers, labetalol, and carvedilol. In the case of ventricular dysrhythmias, lidocaine is recommended.21, 70, 71, 72
The khat plant (Catha edulis) has been chewed for centuries because of its amphetamine‐like effects. The main active component of khat is cathinone. It is present in the leaves of khat along with its plant metabolite cathine, which is synonymous with 1S,2S‐norpseudoephedrine. Many synthetic congeners of cathinone were synthetized almost a century ago, but their large scale abuse began in approximately 2007. Synthetic cathinone derivatives (see Fig. 3 for chemical structure) are sold under a camouflage product classification like “bath salts” in America and “plant food” in Europe in order to bypass regulatory laws. Synthetic cathinones possess significant abuse risk with possible cardiovascular consequences. They are structurally similar to amphetamines. Their mechanism of action is complex and varies according to the derivative. Mostly, they possess amphetamine‐like monoamine vesicular releasing properties and they block monoamine synaptic reuptake. Interestingly, bupropion, the only cathinone compound used clinically, is selective for the noradrenaline/dopamine reuptake mechanism (see above). Synthetic cathinones are generally considered to be less pharmacologically active compared to amphetamines due to their higher hydrophilicity given the presence of the keto group. The cardiovascular effects of synthetic cathinones are similar to those of amphetamine/cocaine and include tachycardia, hypertension with a subsequent risk of myocardial infarction, stroke, and upon prolonged abuse, dilated cardiomyopathy. The effect on serotonin neurotransmission can also be substantial and hyperthermia and dehydration are observed. Fatal outcomes have been described, although the cause of death cannot be easily assessed because cathinone derivatives are often abused with other psychoactive components. The treatment of cathinone cardiotoxicity is similar to that of amphetamines or cocaine overdose. In cases of hyperthermia, extensive cooling is needed.73, 74, 75, 76, 77, 78 Similar to synthetic cathinones, some piperazines possess significant, complex sympathomimetic activity and other effects on monoamine receptors, in particular those associated with serotonin. For this reason, they are labeled as “legal ecstasy” and sometimes sold instead of “ecstasy” (MDMA). Although they have about 10% of amphetamine's potency, they may cause significant adverse cardiovascular effects. Fatalities are not common, they may appear in cases of attempted suicide.73, 79
The antiobesity drug phentermine shares its mechanism of action with amphetamines, but is selective for noradrenaline release.40 Interestingly, it does not increase blood pressure, but heart rate can rise. Its combination with topiramate, approved in 2012, showed similar outcomes. Moreover, likely due to body weight reduction, arterial blood pressure can even significantly decrease. Although heart rate can increase, no relevant cardiovascular risk has been noted to date.80
Tyramine is also an indirect sympathomimetic and its mechanism of action is analogous to that of amphetamines (e.g., it releases noradrenaline from the synaptic cleft). Physiologically, tyramine is ingested in the diet (e.g., aged cheese, soy sauce, red wine, and other fermented products) and is oxidatively deaminated in the small intestine by MAO A and B and thus it has no clinical effect. However, when MAO A and B are blocked by inhibitors, tyramine's oral bioavailability dramatically increases, leading to marked elevations in blood pressure (hypertensive crisis), tachydysrhythmias, and in rare cases, acute myocardial infarction.81, 82, 83 Currently, nonselective, irreversible MAO A/B blockers (phenelzine and tranylcypromine) are very rarely used in clinical psychiatry. However, some clinically used drugs (e.g., the antibiotic linezolid) are mild nonselective MAO blockers and thus they can also evoke this reaction, known as the “cheese effect.” This risk is nonetheless low.84, 85
2.2. Direct acting sympathomimetics
2.2.1. Endogenous catecholamines
The endogenous catecholamines, noradrenaline, adrenaline, and dopamine are commonly used in intensive care units to treat shock conditions associated with acute cardiovascular disorders. However, there are important differences between them in terms of their selectivity for adrenergic receptors. In general, adrenaline stimulates both α‐ and β‐adrenergic receptors, while noradrenaline has effects on α‐ and β1‐adrenergic receptors, but has little affinity for β2‐adrenergic receptors. Dopamin by stimulation of dopaminergic receptors on blood vessels produces vasodilation in physiological doses but it is also a weak agonist at adrenergic receptors. In principle, catecholamines may evoke different cardiotoxic reactions including dysrhythmias, cardiac ischemia, and hypertension. Clinically, the acute hemodynamic benefits on the heart outweigh the risks when they are used in appropriate indications, but careful monitoring of cardiovascular function should be performed. In comparison to other synthetic inotropes and vasopressors, these biogenic amines have short half‐lives, which may be an important advantage in terms of their potential to produce cardiotoxicity. Nonetheless, prolonged infusions of high doses of adrenaline or noradrenaline are not recommended, since it can cause direct cardiotoxic effects resulting in apoptosis/necrosis of cardiomyocytes.7, 86, 87 Cardiotoxicity may also be induced by rapid intravenous (i.v.) bolus administration of insufficiently diluted adrenaline solutions. This explains why the intramuscular (i.m.) route of administration, which is inherently safer for the heart, is often preferred in clinical practice (e.g., in anaphylactic reactions). When adrenaline is administered i.v. in clinical practice, it is often advised to use a “fractionated” administration approach, or to use slow injection, to avoid cardiotoxicity and acute hemodynamic complications. Also, the production of reactive oxygen species (ROS), either via adrenoceptors or via autoxidation cannot be overlooked in the cardiotoxicity inflicted by catecholamines, namely adrenaline.88, 89 Similar findings were also observed after noradrenaline administration, where mixed α‐and β‐blockade did not fully counteract oxidative stress.90
2.2.2. Nonselective β‐agonists and β2‐agonists
The nonselective β‐adrenoceptor agonist isoprenaline (also known as isoproterenol) has a large potential to induce myocardial damage. For this reason, it has been commonly used in experimental settings for induction of a pathological state similar to the acute myocardial infarction.91, 92, 93, 94 Its mechanisms of cardiotoxicity are complex and involve overstimulation of β‐adrenoceptors and ROS production. Excessive stimulation of β‐adrenoceptors leads to exaggerated myocardial energy demands. Additionally, due to its potent β2‐adrenoceptor agonistic effect, isoprenaline causes massive vasodilation in the periphery, significantly lowering diastolic blood pressure and hence reducing myocardial perfusion.95, 96, 97 Calcium overload is also a common finding due to stimulation of cardiac β1‐adrenoceptors.98, 99, 100 Enhanced platelet aggregation also likely contributes to toxicity.101, 102, 103 ROS can be formed directly by spontaneous or metal‐catalyzed oxidation of high levels of isoprenaline, or due to ischemia.104, 105, 106, 107, 108 Because of the complex pathophysiology, it is not surprising that no single agent can fully prevent/revert the injury caused by isoprenaline, or only works in low doses of isoprenaline.103, 109, 110, 111, 112, 113, 114 There are many reports showing that various natural compounds fully protect against isoprenaline cardiac injury in animal models,115, 116 but it is not clear whether these studies have been performed according to the appropriate experimental standards.117, 118
β2‐agonists, when inhaled at therapeutic doses to treat bronchoconstriction, are considered to be relatively safe from the cardiovascular point of view; however, their selectivity is not absolute and in higher doses they also bind to β1‐receptors. As a consequence, vasodilation due to β2‐receptor stimulation can be accompanied tachycardia due to either reflex sympathetic activation or nonselectivity. Indeed, palpitations and sinus tachycardia are relatively common.119, 120 Rarely, dysrhythmias including atrial fibrillation have been documented and in exceptional cases, acute myocardial infarction was observed.121, 122, 123, 124, 125 The latter is more relevant in the case of clenbuterol, a drug used to enhance performance in sports. The underlying mechanism can also involve spasm of coronary arteries and/or temporary thrombosis.125 Naturally, patients with angina pectoris, or with heart failure (where β2‐receptors may become more important due to the downregulation of β1‐receptors), are particularly sensitive to the cardiac effects of β2‐agonists. Transient hypokalemia may also precipitate the development of dysrhythmias.126, 127, 128 Pronounced hypotension with myocardial impairment can be achieved in very high doses under experimental conditions.95, 128, 129, 130 Similarly, elevations in biomarkers of cardiac injury have been observed in humans, but these seem to be transient.131, 132 Besides their use as bronchodilators, β2‐agonists are also used systemically in obstetrics for the management of premature labor. The systemic administration β2‐agonist in pregnancy is not without maternal and/or fetal side effects. Serious (albeit rare) maternal side effects have been reported. Terbutaline, which is often used in these settings, also binds to β1‐receptors at multiple sites, and may lead to maternal tachycardia and hypotension, leading to complaints of palpitations and symptomatic dysrhythmias. Rarely, pulmonary edema, myocardial infarction, and death have been reported.133 Interestingly, cardiac troponin levels (biomarkers of cardiac damage) increase during standard tocolysis in both maternal and neonatal cord blood.134, 135 Thus, terbutaline is contraindicated in women with heart disease, significant tachycardia, hemorrhage, or hypovolemia.133 The cardiovascular toxicity of β2‐agonists is one of the reasons why other tocolytic approaches (e.g., using nifedipine) are now gaining wider acceptance in the clinical community.
Mirabegron is a novel drug used for overactive bladder. Pharmacologically, it is a β3‐agonist that can dose dependently produce tachycardia. Studies have documented hypertension associated with its use; however, causality is lacking since hypertension was observed with a similar frequency in placebo and/or comparator arms of the studies.136, 137
2.3. Nicotine and smoking
In the 21st century, tobacco smoking represents a major worldwide health hazard. About 1 billion persons are estimated to smoke daily and millions of smokers die annually.138, 139 Current trends toward electronic cigarette use decrease somewhat the health risk; however, more data are required before definitive conclusions can be drawn.140, 141 Cigarette smoke contains both nicotine and a number of other chemicals formed by tobacco combustion, many of which are carcinogens and have the ability to produce ROS. Electronic cigarettes also contain nicotine. While the number of potentially toxic chemicals is diminished due to the lower temperature of thermal degradation, the compounds formed are also potential hazards to humans.140, 141 From the cardiovascular point of view, both nicotine and ROS explain the negative effects of smoking (Fig. 4). Also low exposure to cigarette smoke, in particular in nonsmokers (second hand smoke), represents an important cardiovascular risk. Smoking bans for public places in many countries have markedly decreased smoking‐related cardiovascular events and mortality.141, 142, 143, 144
Figure 4.
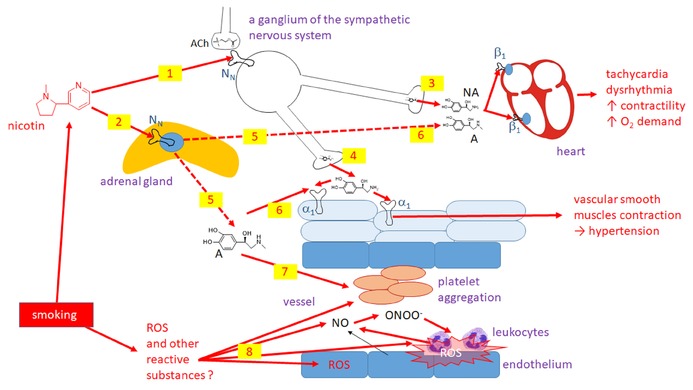
Simplified overview of the cardiovascular effect of cigarette smoke with separation of the effect of nicotine and other compounds generated by smoking. Nicotine stimulates nicotine receptors (NN) both in the sympathetic nervous system ganglia (1) and in the adrenal medulla (2). The former leads to release of noradrenaline (NA) from the sympathetic nerve terminals both in the heart (3) and vascular beds (4). The effect in the adrenal medulla leads mainly to the release of adrenaline (A) into the systemic blood circulation (5). The effect on the heart and majority of vessels is similar (6). In addition, adrenaline stimulates platelet aggregation (7). ROS (8) are formed and react with NO leading to the production of peroxynitrite (ONOO−), a highly toxic reactive species. Stimulation of platelet aggregation and activation of the immune system with subsequent inflammatory reaction is a consequence. ACh: acetylcholine.
Nicotine (a pyridine alkaloid of tobacco, Nicotiana tabacum) is an agonist at nicotinic acetylcholine receptors (N or nACh receptors) and these receptors are indirectly implicated in changes in cardiovascular function. The cardiovascular system is mainly influenced by the nicotine‐mediated release of adrenaline from the adrenal medulla and by stimulation of ganglionic N‐receptors in the sympathetic nervous system, leading to the amplification of sympathetic tone. The immediate consequences are well known (e.g., elevated blood pressure and tachycardia).140, 145 The incidence of ventricular and atrial dysrhythmias and the risk of sudden death is also increased in smokers. In particular, smoking doubles the incidence of atrial fibrillation.141 A very important effect of smoking is platelet aggregation.146, 147, 148 Indeed, there is a direct correlation between the urinary excretion of thromboxane A2 (an important stimulator of platelet aggregation) and the number of cigarettes smoked daily.149 The enhanced platelet aggregation is caused by both increased circulating catecholamines and other compounds from the cigarette smoke, since tobacco snuff does not produces the same biochemical effects, notwithstanding exposure to nicotine.14, 141, 149 Nonetheless, the effect of nicotine on platelet aggregation must be substantial, since smokeless nicotine (e.g., used as snuff) represents an elevated risk for fatal stroke. Interestingly in one study, the risk of fatal ischemic stroke was significant, while that of hemorrhagic stroke was not.150, 151, 152 Besides chronic abuse, nicotine may also be a source of acute intoxication, often due to the accidental oral intake by children—either as tobacco from cigarettes or liquid for e‐cigarettes. Besides profound vomiting and irritability, marked tachycardia and hypertension are most often observed.153
In addition to nicotine, other compounds from tobacco cigarettes are implicated in increased cardiovascular risk, mostly because they are ROS. ROS interact in a complex manner with the vascular system. The protective role of NO on endothelial function is quickly overcome when peroxynitrite (ONOO−) is formed, resulting in enhanced platelet activity (mentioned above), activation of leucocytes, and subsequent inflammation. Coagulation factors are consistently higher in smokers than in nonsmokers. The resulting endothelial dysfunction facilitates atherogenesis and thrombus formation and also complicates long‐term treatment of myocardial infarction. The electronic cigarettes do not seem to cause vascular dysfunction, at least on a short‐term basis, but long‐term studies are needed to assess the risk of chronic exposure.141
Current pharmacological alternatives to support smoking cessation involve nicotine replacement therapy, a partial agonist at one subtype of N‐receptors (varenicline) and an antidepressant (bupropion), which suppresses craving and ameliorates some withdrawal symptoms, even in nondepressed patients. Nicotine replacement therapy seems to represent a lower benefit, since smokeless nicotine is known to have mild, but significant risk of fatal myocardial infarction and fatal stroke.150 There were initial claims of varenicline's association with cardiovascular events, but recent network meta‐analysis of 18 randomized clinical trials, failed to find negative cardiovascular effects of varenicline.154 Similarly, the FDA Mini‐Sentinel Program showed no elevation in cardiovascular risk of varenicline versus bupropion.155 Interestingly, in the same network meta‐analysis, bupropion had a significant protective effect on major adverse cardiovascular effects in smoking cessation patients.154 Also other drugs can stimulate N‐receptors—see Section 5.2.1.
2.4. Drugs affecting the adrenergic system via their action in the CNS
Adrenergic α2‐receptors are localized mostly in the CNS and in the pelvic area. Stimulation of central α2‐adrenergic receptors is associated with inhibition of the central sympathetic tone. This has a clinical impact and the α2‐adrenoceptors agonists, clonidine and methyldopa, are sometimes used in the treatment of hypertension. Clonidine is also used in children for ADHD. In addition, some centrally acting α2‐agonists are used in vertebrogenic‐algic syndromes due to their significant skeletal muscle myorelaxant effects (e.g., tizanidine), or in veterinary anesthesia due to their sedative and myorelaxant effects (e.g., xylazine and medetomidine). α2‐Adrenoceptor agonist intoxication can cause hypotension and bradycardia, which are therapeutically manageable (i.v. fluid supplementation, atropine or catecholamines if needed, or administration of the α2‐antagonist, atipamezole).156, 157, 158, 159 The prognosis is generally good, even after high doses. However, combined use with other CNS depressant, including xylazine use as an adulterant (e.g., with heroin) can be fatal.160 At high doses, transient mild hypertension occurs due to stimulation of peripheral α2‐adrenergic receptors, but this response generally does not require any intervention.70, 161, 162, 163
Antagonists of α2‐adrenergic receptors can cause the opposite effects—an increase in sympathetic tone. They also cause vasodilation of the pelvic area by antagonism of peripheral α2‐adrenergic receptors, which can have a beneficial effect in erectile dysfunction. Previously, this condition has been treated using the natural alkaloid yohimbine, which can be isolated from the bark of West African tree Pausinystalia yohimbe. Yohimbine has been largely replaced by modern approaches to treat erectile dysfunction; however, it is still used for purposes of bodybuilding, improvement of athletic performance, and body weight loss, although data confirming the effectiveness of these uses are missing or conflicting. The use of yohimbine‐containing products is not negligible and many cases of intoxication are reported each year. Its cardiovascular effects are related to the above‐mentioned mechanisms and thus administration of yohimbine results in dose‐dependent elevations in the blood pressure and heart rate. The effect on the blood pressure is more pronounced in hypertensive patients. Fortunately, fatal cases after yohimbine intoxication are extremely rare and due to its short half‐life, most intoxications usually self‐resolve.164, 165
On the contrary, reserpine, an alkaloid isolated from the roots of Rauwolfia serpentina, decreases sympathetic tone and causes hypotension and bradycardia by blocking uptake of monoamines into synaptic vesicles.57, 166
2.5. Drugs influencing intracellular signaling downstream of adrenergic receptors with effects on both cardiomyocytes and the vascular system
In addition to direct effects on adrenergic receptors, or indirect effects due to prolongation of the half‐lives of endogenous catecholamines in the synaptic cleft, several drugs can also affect intracellular signaling downstream of adrenergic receptors in the heart. Common examples are inhibitors of phosphodiesterase‐3 and calcium sensitizers. Drugs that inhibit phosphodiesterase‐3 (milrinone, amrinone) bypass the β‐adrenergic receptors by prolonging the half‐life of intracellular cAMP. They may have important cardiovascular toxicity when given chronically. Oral daily milrinone treatment did not improve survival in patients with chronic heart failure in the PROMISE trial, instead a marked worsening of cardiovascular and overall mortality was observed when compared with placebo. The mechanisms behind this toxic effect are likely complex, but malignant ventricular dysrhythmias were implicated. As a result, these drugs are now indicated only for short treatment of acute heart failure with low cardiac output and hypotension. Nevertheless, the drug may impose cardiac risk particularly in the ischemic myocardium. Also, hypotension can be observed in high doses and thus careful cardiovascular monitoring is necessary during treatment.86, 167 Levosimendan, a direct inotrope with calcium‐sensitizing properties by virtue of its binding to troponin C, is linked to tachycardia and hypotension.86 The latter effect is, however, caused by activation of KATP channels in the blood vessels.
2.6. Other drugs causing sympathetic hyperactivity
The sympathetic system can be activated as a stress reaction after intoxication by different drugs, in spite of the fact that the drugs have little or no direct cardiovascular actions. An example is cannabinoids, which are generally considered quite safe from the cardiovascular point of view.168, 169, 170, 171 It should be mentioned that contaminants, including caffeine, can contribute for these cardiovascular hazards.73, 168
The serotonin syndrome, characterized by unusually high systemic levels of serotonin, can be produced by overdose, or due to pharmacokinetic interactions with drugs that increase serotonin levels (e.g., selective serotonin reuptake inhibitors and other antidepressants that increase levels of serotonin, several opioids of the tramadol type, the antibiotic linezolide, or MDMA). Autonomic hyperactivity associated with hypertension and tachycardia is commonly seen in moderate cases. Treatment involves benzodiazepines, which decrease adrenergic reactions and antagonists at 5‐HT2A receptors, like cyproheptadine.172, 173
2.7. Ca2+ channel blockers and maitotoxin
Pharmacologically, Ca2+ channel blockers are divided into nondihydropyridines, namely verapamil and diltiazem, and dihydropyridines. All of these drugs block vascular L‐type Ca2+ channels to different degrees, while the former group additionally blocks cardiac L‐type channels at therapeutic concentrations. However, during intoxication, this organ selectivity is largely lost; therefore these drugs will be discussed together. Ca2+ channel blockers are among the most common cause of intoxication and death by therapeutically used cardiovascular drugs. These on the other hand, they are considered quite safe. However when used in very small children, or when given in combination with similarly acting drugs (β‐blockers, digoxin, amiodarone), toxicity is more probable. During overdose, they can evoke bradydysrhythmias ranging from sinus bradycardia to complete atrioventricular (AV) block and from severe systemic hypotension to cardiovascular collapse.5, 174, 175, 176, 177, 178 Neonatal myocardium, at least in some species like rabbit, has fewer Ca2+ channels and this may be the reason for higher toxicity in very early phases of life.174, 179 In clinical pediatrics, verapamil is contraindicated in neonates and infants due to the high risk of severe bradycardia, hypotension, and cardiovascular collapse, although the justification in the latter pediatric population is a matter of debate.174 Nondihydropyridines, in particular verapamil, might also produce left ventricular heart failure, but this syndrome is usually mild and transient.5, 7 However, caution is necessary in patients with preexisting systolic dysfunction. Short‐acting dihydropyridines, like nifedipine, can produce reflex tachycardia as a result of their rapid and dominant peripheral vasodilator effect. Intoxication with nifedipine is commonly manifested as hypotension with tachycardia, which can convert to bradydysrhythmias due to a toxic direct effect on the heart.177 Therapy of severe Ca2+ blocker intoxication includes tools aimed to decrease drug absorption (e.g., activated charcoal and gastric lavage), particularly in the case of sustained‐release tablets. Intravenous calcium and/or high‐dose insulin‐glucose treatment are often the drugs of first choice in severe cases, while atropine may be used for treatment of bradydysrhythmias and i.v. fluid supplementation for hypotension, sympathomimetics in sustained bradydysrhythmias and/or hypotension, and glucagon for its positive chronotropic and inotropic activity (sometimes considered useful in verapamil overdose but convincing human data are missing). Pacing and lipid emulsion therapy might be required in life‐threatening cases if other means are ineffective.176, 177, 178, 180, 181, 182, 183
Maitotoxin is a large, very potent toxin found in the “red‐tide” dinoflagellate Gambierdiscus toxicus. It is one of the most potent ciguatera toxins known and is one cause of Ciguatera seafood poisoning.184 The underlying mechanism of maitotoxin toxicity is associated with several Ca2+‐dependent processes. Recent results suggest that it binds to the sarcolemmal Ca2+‐ATPase pump and converts it into a Ca2+‐permeable nonselective cation channel. This causes a massive increase in cytosolic free Ca2+ concentration with subsequent opening of large endogenous cytolytic pores.185, 186 Ciguatera poisoning can be caused not only by maitotoxin, but also by other ciguatera toxins. It is manifested as gastrointestinal, neurological, respiratory, and cardiovascular symptoms, including brady‐ and tachydyasrrhythmias and hypotension. Treatment of maitotoxin intoxication is the same as for ciguatera toxins in general and is symptomatic. In rare cases, when a patient presents early after ingestion, activated charcoal or gastric lavage may be of benefit. Atropine or i.v. fluid administration can be also useful. If hypotension persists, administration of dopamine or norepinephrine is employed.187, 188
2.8. Tricyclic antidepressants (TCAs)
TCAs are a specific class of drugs with multiple actions. Their antidepressant activity involves the inhibition of monoaminergic synaptic reuptake transporters. However, they also have significant activity as antagonist of α1‐, H1‐, H2‐, and M‐receptors and block Na+ and human ether‐a‐go‐go‐related gene (hERG) channels. Therefore, their cardiovascular effects are complex. The common symptom after TCA overdose is sinus tachycardia, but other atrial or ventricular dysrhythmias can be observed. They prolong the QT interval due to I Kr current blockade; however, owing to increases in heart rate, this rarely precipitates torsade de pointes (for more detail see chapter 3.2.2). Orthostatic hypotension is also a very common side effect with generally no tolerance. It is mainly caused by antagonism of α1‐adrenoceptors, but may also reflect decreased contractility/cardiac output due to cardiac Na+ channel inhibition. The latter is relevant only in overdose cases, since negative inotropic effects are not observed at therapeutic doses, even in patients with heart failure. At the beginning of treatment, hypertension due to indirect sympathomimetic activity can appear.23, 24, 25, 189, 190 Rare cases of myocardial infarction have also been described. After overdose, death can result from different etiologies, but cardiac dysrhythmias (QRS complex widening and ventricular arrhythmias) are usually the most important and prevalent and tend to occur within the first 24 hrs. TCAs have been commonly used for suicides, while other antidepressants are only rarely associated with death.25, 190, 191 Early administration of activated charcoal within 1 hr after TCA ingestion has a reasonable clinical effect. In cases of ventricular dysrhythmias, sodium bicarbonate or lidocaine can be useful. Sodium bicarbonate may be preferred since it also alleviates the acidosis produced by TCA intoxication. Administration of class Ia antiarrhythmic agents is contraindicated because they can potentiate Na+ channel blockade and cause heart block. Treatment of hypotension is based on fluid administration and vasoconstrictors counteract the α1‐adrenergic receptor blockade.182, 191, 192
2.9. Ethanol
Alcohol (ethanol) consumption in mild to moderate doses is generally considered to be cardioprotective, while in high doses, the opposite is true.7, 193, 194, 195, 196, 197 This is quite alarming since the current 1 year prevalence of alcohol use disorder in the US population over 18 years is 10% in women and 18% in men. Of these about one‐half are moderate or heavy drinkers.198 Acute alcohol overdose can be associated with hypotension and even a risk of cardiovascular collapse. Intravenous fluid administration is generally the first therapeutic measure.5 Even moderate chronic alcohol consumption can be associated with mild increases in arterial blood pressure. The effect seems to be dose dependent and about 5–10% of all hypertensive patients might result from alcohol consumption, while the prevalence of hypertension in younger persons with alcohol use disorder is approaching one‐half.7, 199, 200 The mechanisms of the blood pressure responses are not clear; however, at least in the early stages of abuse, adrenergic hyperactivity plays an active role.196 Chronic exposure to high doses of alcohol results in dysrhythmias and alcoholic cardiomyopathy. The first clinical sign in many cases is an isolated episode of atrial fibrillation. Atrial fibrillation is relatively common in alcohol consumers and might be responsible for about 15–40% of all idiopathic atrial fibrillation.6, 7 According to a large study, a daily alcohol intake of more than 60 g is associated with an increased risk of atrial fibrillation.201 There can be also other types of supraventricular or ventricular dysrhythmias mainly due to reentry mechanism. Ventricular tachydysrhythmias can result in death and, indeed, the risk of sudden cardiac death is higher after recent alcohol intake in chronic alcoholics. In general, chronic alcohol‐induced dysrhythmias are commonly resistant to pharmacotherapy or cardioversion.6, 7, 202 Alcoholic cardiomyopathy, which can account for up to about one‐half of idiopathic cardiomyopathies, shares some clinical signs with wet beriberi.6, 7, 203 It appears that at least 5 years of heavy drinking are needed. Alcoholic cardiomyopathy is a type of acquired dilated cardiomyopathy. Like other types of dilated cardiomyopathy, alcoholic cardiomyopathy is characterized by a dilated left ventricle, increased left ventricle mass, and left ventricular systolic dysfunction. Depending on the degree of alcohol consumption, it can be asymptomatic, or manifested as overt heart failure. The pathophysiology is complicated and not fully understood. It involves accumulation of lipids in the myocardium, increased levels of angiotensin II, mitochondrial dysfunction, and oxidative stress. Ethyl esters of fatty acids appear to contribute to cardiomyopathy development.204 Prognosis is poor if the patient continues to drink heavily. Treatment is thus based on cessation or reduction of alcohol consumption. There are no treatment guidelines. Common methods of heart failure treatment with emphasis on angiotensin‐converting enzyme inhibitor (ACEi) administration are efficient in alcoholic cardiomyopathy with reduced ejection fraction. Thiamine, the deficiency of which is the cause of beriberi, is also commonly administered, but its benefit has yet to be confirmed.6, 203, 204 Moderate and high alcohol consumption, in contrast to protective low consumption, is also associated with a higher incidence of both ischemic and hemorrhagic strokes and acute myocardial infarction.7, 196, 197
2.10. Androgenic anabolic steroids
The male hormone, testosterone is only sparingly used in therapy, but its close derivatives, the androgenic anabolic steroids, are misused at high doses to enhance physical performance. Data on their cardiovascular toxicities have not been systematically explored since their use is illegal in professional sport. In addition, androgenic anabolic steroids are commonly administered with other drugs such as β2‐mimetics, illicit sympathomimetics, diuretics, and/or psychoactive drugs.171, 205 Data on testosterone's effects on the cardiovascular system are mixed and they differ from those elicited by supraphysiologic doses of synthetic androgens.206, 207 Thus, it is not easy to decipher the cardiovascular impact of anabolic androgenic steroid abuse. The basis of their abuse is to increase lean striated muscle mass. However, similar effects are also observed in the heart with the most common cardiac finding of long‐term anabolic abuse being cardiac hypertrophy,171, 208 which is greater than that of exercise‐induced hypertrophy in professional athletes who do not abuse anabolic drugs.209 Maladaptive cardiac hypertrophy caused by these drugs is initially mostly concentric, but can be eccentric and can proceed to fibrosis and overt heart failure. Diastolic dysfunction is positively correlated to the dose and duration of anabolic steroid use. In addition, altered cardiac structure can predispose to dysrhythmia development that can result in sudden cardiac death.171, 210 Indeed, cardiac pathology is commonly observed in young people who died while abusing androgens.205, 211 Other effects of anabolic androgenic steroid abuse include a small increase in arterial blood pressure, which is expected due to the known physiological difference between men and women.207, 209 The cause can be increased sodium and water retention, or effects on red blood cell proliferation through increased expression of erythropoietin. Data also suggest facilitated vasoconstriction due to endogenous mediators. There are also reports of thrombotic events, possibly associated with increased erythropoietin mass, abnormalities in platelet aggregation, and atherosclerosis, which can be facilitated by the influence of androgens on lipid spectra.170, 171, 206, 207, 212, 213, 214 The cardiovascular effects of androgens are reversible and cessation is thus the most effective therapy, but it is not clear whether restoration of full cardiac function and structure can be achieved by the cessation of drug taking.208, 209, 215
3. DRUGS WITH THE MAJOR EFFECTS ON THE HEART
This category encompasses drugs with direct effects mainly targeted on the cardiomyocytes. These drugs selectively affect either the conduction system, the working myocardium, or both systems. Other cells in addition to the cardiomyocytes may also be involved in cardiotoxic responses and these will be briefly discussed below.
3.1. Drugs affecting the function of both the conduction system and the working myocardium
Drugs affecting β1‐receptors are the most common examples.
3.1.1. β‐Blockers
Drugs classified as β‐adrenoceptor blockers (antagonists) are considered to be quite safe in recommended doses mainly because of their large therapeutic indices. One of their indications is chronic heart failure with reduced ejection fraction. Although they are one of the cornerstones of current management of this disease, their introduction may cause transient worsening of heart failure symptoms (e.g., in too fast up‐titration) due to their negative inotropic action; therefore, the initiation of treatment is recommended after stabilization of heart failure symptoms.216 An increased risk of toxicity can be also the result of interactions with other drugs (Table 4). In general, the manifestations of β‐blockers overdose include bradycardia, atrioventricular (AV) blockade, hypotension, left ventricular failure, and cardiogenic shock. If death occurs, this is usually caused by asystole.5, 217 Besides the expected excessive blockade of adrenergic signaling, some β‐blockers also possess membrane stabilizing effects and intoxication with them is associated with more pronounced cardiovascular toxicity (e.g., propranolol, labetalol, acebutolol).218 In general, β‐blocker overdose is treated similarly to overdose with Ca2+ channel blockers. Bradydysrhythmias are treated with atropine (and adrenaline), sometimes pacing is needed. Glucagon is often very useful and recommended as a first‐line treatment for its positive chronotropic and inotropic effects with i.v. calcium salts, high‐dose insulin‐glucose therapy, and phosphodiesterase 3 inhibitors as potential alternatives. Lipid emulsion therapy is discussed in severe cases poorly responding to other treatment. In contrast to Ca2+ channel blocker overdose, hemodialysis or hemoperfusion can be effective for some hydrophilic β‐blockers. In addition to the adverse and toxic effects described above, rebound phenomenon, which stems from rapid discontinuation of their chronic use, should be noted and can be associated with worsening of coronary artery disease together with a risk of tachyarrhythmia, acute myocardial infarction, sudden cardiac death, or hypertensive crisis.7, 57, 176, 217
Table 4.
Selected clinically relevant interactions leading to possibly severe cardiovascular toxicity
| Drug 1 | Drug 2 | Type | Relevant cardiovascular risk | Mechanism |
|---|---|---|---|---|
| ACEi | Antagonists at AT1 receptors for angiotensin II (sartans) | PD | Hypotension and hyperkalemia | Activity at consequent steps of renin–angiotensin–aldosterone cascade |
| ACEi | Neprylisin inhibitors (sacubitril) | PD | Angioedema | Bradykinin accumulation |
| Disulfiram | Alcohol | PK | Hypotension, tachycardia | Disulfiram inhibits alcohol metabolism |
| Amphetamines | Cocaine | PD | All risks described in relevant sections of this manuscript | Synergistic effects on adrenergic system |
| Amiodarone | Nondihydropyridine Ca2+ channel blockers | PD (+? PK) | Sinus arrest | Potentiation of negative chronotropic and inotropic effect |
| Amiodarone | Some quinolones | PD | Torsade de pointes | Additive effect on QT interval ? |
| Drugs prolonging QT | Drugs lowering plasma potassium concentration (amphotericin B, β2‐agonists, corticosteroids, loop and thiazide diuretics, theophylline, misuse or overuse of laxatives) | PD (in some cases also + PK) | Torsade de pointes | Synergistic effect |
| Amiodarone | Some? β‐blockers (metoprolol, carvedilol) | PD + PK | Hypotension, bradycardia, asystole, possibly ventricular fibrillation | Additive effect on the heart and inhibition of CYP2D6 by amiodarone |
| Amiodarone | Sotalol | PD | Torsade de pointes, hypotension | Similar effect on the heart, excessive bradycardia can facilitate torsade de pointes |
| β‐Blockers | Cholinomimetics | PD | Bradycardia, AV blocks, and hypotension | Synergistic negative chronotropic effect |
| β‐Blockers | Nondihydropyridine Ca2+ channel blockers | PD | Bradycardia, asystole, sinus arrest | Additive effect on the heart |
| β‐Blockers | Digoxin | PD | Bradycardia, AV block | Additive effect |
| β‐Blockers | Dronedarone | PD + PK | Bradycardia | Both drugs slow heart rate and dronedarone can inhibit CYP2D6 altering metabolism of some β‐blockers |
| β‐Blockers | Antipsychotics‐phenothiazines | PD | Hypotension | Additive effect |
| β‐Blockers | Propafenone | PD + PK | Profound hypotension and cardiac arrest | Similar effect on the heart, propafenone can inhibit metabolism of some β‐blockers through inhibition of CYP2D6 |
| Some β‐blockers | Some SSRi | PK | Bradycardia, AV blocks, hypotension | Fluoxetine and paroxetine are inhibitors of CYP2D6 and thus slow metabolism of some β‐blockers |
| Calcium channel blockers | Azoles, clarithromycin, some HIV‐protease inhibitors | PK | Hypotension and/or bradycardia | Mentioned drugs inhibit metabolism of Ca2+ channel blockers |
| Digoxin | Amiodarone | PK + ? PD | Dysrhythmias, also torsade de pointes | Amiodarone blocks P‐glycoprotein, torsade de pointes might by facilitated by bradycardia caused by digoxin |
| Digoxin | Azoles, clarithromycin, some HIV‐protease inhibitors | PK | Dysrhythmias | Inhibition of P‐glycoprotein |
| Digoxin | Nondihydropyridine Ca2+ channel blockers | PK + PD | Bradycardia, asystole, sinus arrest | Inhibition of P‐glycoprotein, synergistic effect on the heart |
| Digoxin | Loop or thiazide diuretics, amphotericin B, corticosteroids | PD | Dysrhythmias | Hypokalemia potentiates digoxin toxicity |
| Digoxin | i.v. calcium | PD | Dysrhythmias | Hypercalcemia increases effect of cardiac glycosides |
| Digoxin | Propafenone | PK ? | Dysrhythmias | Probably inhibition of P‐glycoprotein by propafenone |
PD, pharmacodynamic; PK, pharmacokinetic.
Note that while these drug combinations have the potential to produce cardiovascular toxicity, the contraindications are not absolute (e.g., ACE inhibitors with antagonists of angiotensin II receptors or β‐blockers with Ca‐channel inhibitors can be given for certain indications).
3.2. Drugs with main toxic effects on cardiac electrophysiology—Drugs causing dysrhythmias
Besides effects on adrenergic receptors and Ca2+ channels, the major drug targets associated with cardiac dysrhythmias are Na+ and K+ channels, the Na+‐K+ pump (Na+‐K+ adenosine triphosphatase), acetylcholine, and adenosine receptors. Since many channels or receptors, can under different conditions, cause both tachy‐ and bradydysrhythmias, the classification will be based on the mechanism/target of cardiotoxicity.
3.2.1. Sodium channels
There are drugs and natural toxins that inhibit Na+ channel function or promote their activation (i.e., Na+ channel openers)—see Figure 5. Both mechanisms can contribute to dysrhythmogenicity. Prevention of Na+ channel closing prolongs their open state and this is clinically relevant for accidental or intentional aconitine poisoning. Inhibition of Na+ channel function, by local anesthetics, is a possible complication of therapy resulting from inappropriate administration of local anesthetic into the systemic circulation. Furthermore, Na+ channel blockade is a mechanism of action of many antiepileptics and class I antidysrhythmics, known for their prodysrhythmic effects due to the narrow therapeutic window between antidysrhythmic and prodysrhytmic doses.
Figure 5.
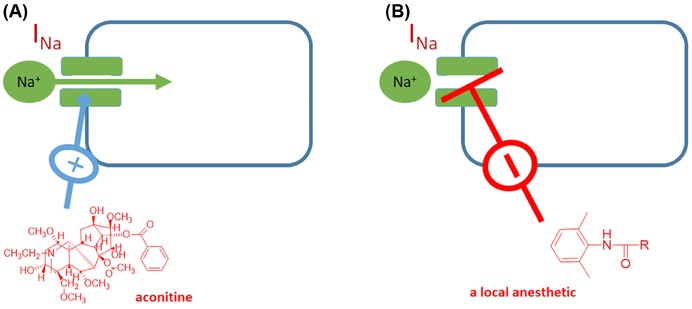
Drugs acting on Na+ channels. (A) Aconitine binds to the open state of the channel and blocks channel closing, therefore Na+ can cross the plasmatic membrane continuously, (B) an amide local anesthetic blocks channel opening and thus Na+ influx necessary for action potential generation and conduction.
In Europe and North America, aconitine intoxication most often results from the accidental ingestion of the Aconitum plant (mainly Aconitum napellus, monkshood or devil´s helmet). However, in Asia, aconitine root, after processing, which reduces about 90% of the alkaloid content, is still used in traditional Chinese medicine as an analgesic, anti‐inflammatory, or cardiotonic agent. Importantly, faulty preparation is a common reason for intoxication.219, 220 The last reported incidence of intoxication in Hong Kong is 0.28 cases per 100,000 population, with the great majority of these cases resulting from the faulty preparation of decoction of Aconitum roots.220 Aconitine is a highly toxic diterpenoid alkaloid that has long been associated with severe cardiovascular toxicities including tachyarrhythmia and hypotension. The main causes of fatal outcomes are refractory ventricular dysrhythmias and asystole. Aconitine binds to the alpha‐subunit of the Na+ channel, which remains as a consequence in the open form, resulting in delayed repolarization or early after depolarization. By this mechanism, ectopic beats, ventricular tachycardia, torsade de pointes, or even ventricular fibrillation may develop. Aconitine also stimulates the vagus nerve, potentially producing bradycardia. The intoxication is generally rapid and cardiovascular symptoms are accompanied by neurological and gastrointestinal manifestations. A specific treatment of aconitine poisoning is not available. It would seem logical to administer class I antidysrhythmic agents to block Na+ channels; however, this treatment has a low success rate, similarly electrocardioversion is generally unsuccessful. The most, but not always, successful approach seems to be the administration of amiodarone.219, 221 There are also other natural toxins with similar mechanism of action, in particular batrachotoxin, which is considered to be a full activator of Na+ channels, in contrast to the partial activators, aconitine, or veratridine.222, 223, 224 Veratridine and other derivatives of this class of steroid alkaloids are present in Veratrum album, known also as white hellebore or false helleborine. Cases of intoxication with this plant are very rare, but the plant can be easily confused with Gentiana lutea that is used as a digestive agent.225
Also diterpenic grayanotoxins prevent Na+ channel inactivation, but their effect seems to be limited to neuronal Na+ channels. Intoxication with these toxins is quite common in the Black Sea region of Turkey, where so called “mad honey” is sold in local markets for treatment of different illnesses (e.g., gastrointestinal /GIT/ disorders, hypertension or as a sexual stimulant). Mad honey is produced by honeybees from the nectar of Rhododendron genus flowers. Also direct consumption of the plant or the drinking of tea prepared from Rhododendron species (e.g., Labrador tea) can lead to intoxication, but such cases are rare. Symptoms of intoxication (also known as “mad honey disease”) include cardiovascular symptoms such as sinus bradycardia, different degrees of AV block, and hypotension. Fatalities are rare and patients are discharged from hospital usually within 1 or 2 days. Treatment involves mainly atropine and saline i.v.226, 227, 228
As mentioned, class I antidysrhythmic drugs block Na+‐channels. However, many of these drugs also possess additional activities producing a variety of cardiovascular actions/toxicities. For example, class Ia drugs also commonly block the I Kr‐current causing QT prolongation and torsades de pointes (e.g., quinidine). Quinidine was widely used for decades as a broad‐spectrum antidysrhythmic drug, but its toxicity, in particular its cardiovascular toxicities severely limited its clinical use. Beyond the significant risk of torsade de pointes (see chapter 3.2.2), it can cause or worsen other dysrhythmias and heart failure. Drugs from the class Ia antidysrhythmic drugs are rarely used today.7
Class Ic antidysrhythmic drugs also have a poor record of safety, which stems from their proarrhythmic potential taking place under certain circumstances. This is based mainly on the CAST I and CAST II trials, where flecainide, encainide, and moricizine were used to test the hypothesis that these agents could protect postmyocardial‐infarction patients from premature ventricular beats and hence from ventricular dysrhythmias and death. Unexpectedly, there were significant reductions in survival in those patients treated with the class Ic drugs.229, 230, 231 Propafenone, the most commonly used class Ic drug, also possess significant β‐blocking properties. Besides β‐adrenergic related side effects, it can as a Ia class antidysrhythmic also accelerate ventricular rate in patients with atrial flutter.7, 232, 233 From the clinical point of view, these agents should be avoided in patients with ischemic heart disease and/or significant structural heart disease that are considered to predispose patients to negative outcomes of the treatment.234
Some local anesthetics are also used as class 1b antidysrhythmic agents. Their toxicity can follow both type of indications. They are considered to be more selective for Na+ channels; however, at higher concentrations, they can also block other ion channels.235, 236 There are marked differences between local anesthetics in terms of toxicity. The racemic bupivacaine has the lowest therapeutic index, followed by ropivacaine and levobupivacaine. Lidocaine and mepivacaine are considered safer.235, 237 The toxicity of local anesthetics after systemic absorption most often begins with signs of CNS toxicity (e.g., sedation, lethargy, confusion) with cardiac and vascular system effects occurring at higher concentrations. The typical signs of cardiac toxicity are bradydysrhythmias with possible cardiac arrest and hypotension, but ventricular tachydysrhythmias can also be observed.238, 239 Hypotension results from both decreased cardiac contractility and direct peripheral vasodilator activity due to effects on smooth muscle Na+ channels. While the incidence of systemic local anesthetic‐induced cardiac toxicity is relatively low, and since there are no clinical trials, the treatment still represents an important challenge. Conventional methods of intoxication treatment have limited utility. Vasopressors like adrenaline are not always useful, in particular adrenaline in higher doses can promote tachydysrhythmias. Interestingly, lipid emulsions represent a relatively novel pharmacological first‐line treatment after standard resuscitation. There is still some controversy, for example, there is no definite clinical proof that they are the most effective treatment. Paradoxically, this treatment was first used to analyze the cardiotoxic mechanisms of local anesthetics under the assumption that it would worsen the toxicity; however, the emulsions had the opposite effect and reduced the toxicity. The mechanism behind this observation is not clear. The first theory of a “lipid sink,” postulated the redistribution of local anesthetic from tissues into the circulation by dissolving them into lipid micelles, with subsequent elimination in the liver. This mechanism, also referred to as pharmacokinetic effects, is questionable, since the concentration of local anesthetic in the plasma decreases after the lipid emulsion is administered. Therefore, other mechanism(s) is/are involved. The local anesthetics also impair cardiac energy metabolism at the level of mitochondria, which further worsens contractile function. Hence the lipid emulsions may act as a direct energy source and this pharmacodynamics effect probably participates in the alleviation of local anesthetic toxicity. Moreover, the lipid emulsion treatment is generally well tolerated.182, 236, 237, 239, 240, 241 Hypoxia, coronary ischemia and/or acidosis are known to amplify cardiotoxicity of local anesthetics. As these complications can be induced, or significantly worsened by the toxicity itself, it can result in a potentially fatal vicious cycle. Hence, treatment of systemic toxicity induced by local anesthetics should include appropriate airway maintenance, oxygenation, and/or ventilation.242
Lidocaine and class Ib antidysrhythmics were also reported to be effective in prophylaxis of serious ventricular dysrhythmias after acute myocardial infarction. However, a meta‐analysis of randomized, controlled trials showed that there were only insignificant trends toward reduced ventricular fibrillation, but significantly increased mortality.243 The increased mortality was attributed to asystole, resulting from the suppression of a ventricular escape rhythm that normally occurs if complete heart block develops. Due to the suggestion of possible harm and unsure benefit, the routine prophylactic administration of lidocaine postinfarction is not recommended.244
There are also natural toxins from the guanidinium class that are powerful blockers of Na+ channels. The most well‐known of these is tetrodotoxin, which exists in pufferfish and many other animals including crabs, frogs, and gastropods. It is a very toxic compound with assessed human lethal doses of 1–2 mg. However, relative to its cardiovascular toxicity, its risk is rather marginal, because tetrodotoxin has a very high affinity (nanomolar concentrations) for neural and skeletal muscle fibers (tetrodotoxin‐sensitive channels) and quite low affinity (micromoles) for cardiac channels (tetrodotoxin resistant). Cardiovascular symptoms might occur in very severe intoxications, but by far the most common cause of death is respiratory failure.245, 246 Other natural toxins from the same class with similar actions are saxitoxin and zetekitoxin AB.247
Aluminum (pesticide) and zinc phosphide (rodenticide) are common methods for suicide in India. They share the same mechanism of action: they release phosphine. The mechanism of toxicity is not known, but symptomatically it resembles Brugada syndrome, which is typically characterized by impaired function of Na+ channels in the heart. Thus, phosphine might have a negative impact on Na+ channel function. Currently a specific treatment has not been identified.248, 249, 250
Taxines—These alkaloids are found together with the more commonly known taxanes (e.g., paclitaxel, see Fig. 6) in the yew tree (Taxus baccata). Intoxication was shown to be the most common method of suicide attempt using plant materials in a recent study. Taxines blocks both Na+ and Ca2+ channels in cardiomyocytes. Thus, intoxication with yew produces brady‐ or tachycardia followed by ventricular tachydysrhythmias and ventricular fibrillation. Yew‐mediated cardiac dysrhythmias can be fatal, despite the best supportive treatment.251, 252, 253
Figure 6.
Chemical differences between taxanes and taxines. (A) Taxan taxol and (B) taxine B. Blue shows the basic core for the most common taxines and taxanes, while red highlights differences between taxanes and taxines.
3.2.2. Potassium channels
There are several types of cardiac K+ channels in the heart that are responsible for different phases of the action potential. Three of them are involved in (late) repolarization. One of these channels, known as hERG (human ether‐a‐go‐go‐related gene,254 or Kv11.1 or KCNH2, the latter is the name of the gene) is extremely sensitive to inhibition by many compounds. It mediates the so‐called rapid component of the delayed rectifier current (I Kr current). If this current is suppressed, repolarization is slowed and QT interval prolongation is observed in the ECG (Fig. 7A).255, 256 Because the synthesis of the hERG channels is particularly complicated (Fig. 7B), the I Kr current can be suppressed not only by direct inhibition of the hERG channels, but also by any interference in their synthesis and/or intracellular trafficking.255 Most drugs that cause QT interval prolongation are direct inhibitors of the channel, but there are many compounds that block their synthesis/trafficking or interfere at both levels.257, 258, 259 Good examples are the anticancer drugs arsenic trioxide and geldanamycin and the antiprotozoal agent pentamidine. The first two drugs inhibit hERG trafficking at the level of heat shock proteins. These proteins are necessary to protect misfolding or degradation of the hERG polypeptide chain as it is processed in the endoplasmic reticulum. Pentamidine, on the other hand, blocks the transport of hERG protein from the endoplasmic reticulum.259
Figure 7.
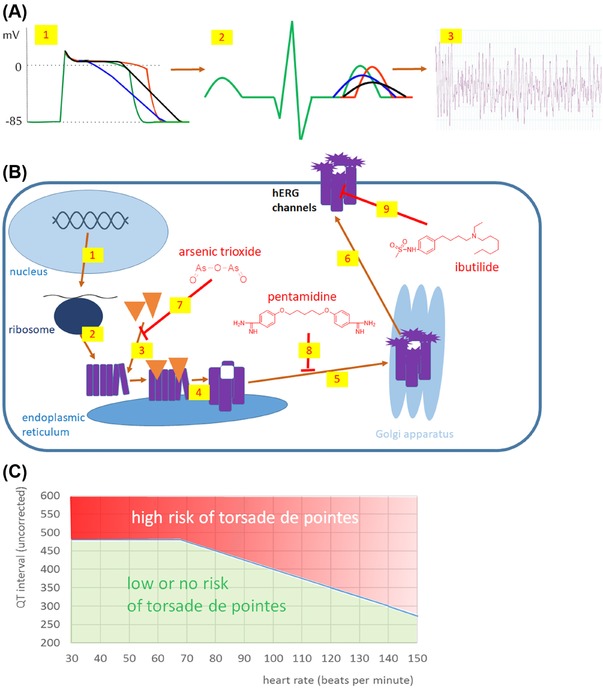
QT interval and torsade de pointes. (A) Different scenarios of QT prolongation. (1) Action potential. Normal action potential is shown in green, the prolongation of QT mainly due to plateau prolongation is shown in red, QT prolongations where rapid repolarization phase is also prolonged in black and blue (here the triangulation is apparent). (2) Corresponding QT prolongation on ECG. Marked QT prolongation, in particular with triangulation, might result in torsade de pointes (3). (B) Complicated synthesis and trafficking of hERG channels. After transcription to mRNA (1), primary the polypeptide is formed in ribosomes (2). In endoplasmic reticulum it is first associated with chaperons (3, heat shock proteins 70 and 90) and then four units are assembled into a tetramer forming the K+ channel (4). Thereafter, the immature channel is transported into the Golgi apparatus for final glycosylation (5). This mature protein is trafficked to the plasma membrane (6). Drugs can interfere with this process in different ways: arsenic trioxide hinders the formation of the chaperon–hERG polypeptide complex (7), pentamidine binds to the tetramer and blocks the transport from the endoplasmic reticulum (8), or ibutilide is a representative of many drugs that directly block the active channel (9). (C) QT interval nomogram according to Isbister270 and Isbister et al.286
Since direct inhibition of the hERG channel is the most common cause of drug‐induced QT prolongation, it is mandatory that the potential for hERG channel antagonism be investigated at the preclinical stage of drug development before clinical investigation. Interestingly, 75–86% of potential new drugs may carry some risk of interfering with hERG channels in early phases of preclinical development.260 Although this number is clearly overstated by false‐positive results, there is no question that a large number of clinically used drugs, at least in high concentrations, possess this property. Studies have shown that the intracellular cavity of the hERG channel contains a promiscuous binding site formed by polar and aromatic amino acids. Lipid solubility is an important factor and lipophilic drugs have a higher probability of interfering with hERG channels than hydrophilic drugs.255, 256, 261 There are also some natural toxins (some scorpion toxins, sea anemone toxin APETx1), which can extracellularly block this channel, but the inhibition is incomplete.255 Prolongation of the QT interval is an important risk factor for the polymorphic ventricular tachydysrhythmia called “torsade de pointes.” The word comes from French and it is translated as "twisting of the points,” because there is a gradual change in the amplitude and twisting of the QRS complexes around the isoelectric line (Fig. 7A). Torsade de pointes may result in ventricular fibrillation and sudden cardiac death. Several types of drugs have been withdrawn from the market due to their ability to induce torsade de pointes, including antihistamines (terfenadine, astemizole), prokinetic agents (cisapride), spasmolytics (terodiline), antipsychotics (thioridazine), and quinolone antibiotics (grepafloxacine).256, 262, 263, 264 In some cases, in particular the mentioned antihistamines, inhibition of their CYP3A4‐mediated metabolism by several drugs (e.g., macrolides like erythromycin, azole antimycotics such as itrakonazole and ketokonazole) increased the plasma concentration of them with subsequent blockade of the I Kr current and markedly increased risk of torsade de pointes. A similar situation is encountered in persons with reduced metabolic capacity due to liver diseases.265, 266, 267, 268
The relationship between QT interval prolongation and the incidence of torsade de pointes is not direct.261, 262, 269 Also, QT prolongation is not a uniform condition (see Fig. 7A). In fact, some drugs from the antidysrhythmic class III are used clinically for treatment of different tachydysrhythmias due to prolongation of the action potential/QT as this also prolongs the effective refractory period of cardiomyocytes. There are many factors influencing the risk of torsade de pointes development and many conditions, which can worsen or reduce it. Several factors can be crucial:
Torsade de pointes is a reentry dysrhythmia likely induced by an early afterdepolarization due to a Ca2+ current through persistently open L‐type Ca2+ channels, or possibly due to sodium inward current. This can be supported by several facts: (i) some Ca2+ channels blockers (e.g., verapamil) are potent direct inhibitors of hERG channels, but are not associated with torsade de pointes. 255, 261 (ii) Mentioned class III antidysrhythmic drugs slow down delayed repolarization and these drugs block hERG channels. Logically, all these drugs are prolonging the QT interval and thus the risk of torsade de pointes is expected and clinically well documented.263 However, amiodarone, the keystone of this category, only rarely causes torsade de pointes. This is explained by its ability to also block Ca2+ channels and possibly Na+ channels. 262, 270, 271 (iii) Intravenous magnesium sulfate is the best treatment modality to stop torsade de pointes. Its mechanism of action is explained by physiological antagonism between Mg2 and Ca2+ and hence probably associated with inhibition of Ca2+ channels. Moreover, magnesium sulfate stops dysrhythmias, but has no effect on QT interval duration.255, 271 Dispersion in action potential/QT interval duration among different ventricular cells might enable or facilitate the development of dysrhythmias.269, 272, 273
Plasma potassium levels are another crucial factor. Hypokalemia leads to downregulation of hERG channels and hence to QT interval prolongation. It should also be mentioned that some clinically used drugs induce hypokalemia, namely the majority of diuretics and the chronic abuse of contact laxatives.255, 259 Also hypomagnesaemia increases the risk of torsade de pointes.255, 262
Heart rate is a determinant of the cardiac cycle duration. When the heart rate is slow, the cardiac cycle and hence QT/action potential duration are logically longer.269, 270, 274 As mentioned in chapter 2.8, TCAs prolong the QT interval, but rarely cause torsade de pointes. This is explained by the fact that TCAs cause tachycardia.
Women have physiologically longer QT intervals than men and thus are at a higher risk of torsade de pointes. In the elderly, the QT interval is also prolonged.255, 261, 270, 275
Several cardiovascular diseases or their risk factors modify the cardiac conduction pathways. Pharmacovigilance data from Sweden showed that cardiac disease (e.g., heart failure, ischemic heart disease, cardiomyopathies, and diabetes mellitus) was the most common factor in reported cases of torsade de pointes.255, 259, 261, 262, 263, 275
Notwithstanding these variances, the QT interval remains the most commonly used predictor of torsade de pointes, although it is far from being optimal and torsade de pointes can even occur when the action potential is shortened.263, 269 As mentioned, the QT interval is dependent on heart rate. Thus, several mathematical formulas calculating corrected QT interval have been proposed, but none is ideal. Other possible assessment of the risk of torsade de pointes represents the so‐called QT nomogram—Fig. 7C.270, 274 Large animal experimentation confirmed that instability (beat to beat QT interval differences), triangulation (change in the shape of action potential to a more triangular pattern, see Fig. 7A), and reverse use dependence (with resulting excessive prolongation of action potential at a slow heart rate) are much better predictors of torsade de pointes than QT interval prolongation.269, 272 A list of drugs with increased risk of torsade de pointes is shown in Table 5 and a detailed list can be found online from the Arizona Center for Education and Research on Therapeutics (AZCERT) program.276 As mentioned above, the major treatment modality for torsade de pointes is i.v. magnesium sulfate.255, 273 Temporary rapid atrial or ventricular pacing, or administration of the β‐agonist isoprenaline, or electrical cardioversion can be used in resistant cases.266, 271, 273
Table 5.
List of selected drugs able to induce torsade de pointes through QT interval prolongation
| Class (compounds) | Comment |
|---|---|
| Antidysrhythmic drugs of class III (sotalol, ibutilide, dofetilide, almokalant, amiodarone, dronedarone) | Amiodarone and dronedarone have only a small risk even if they prolong QT. |
| Antidysrhythmic drugs of class I (quinidine, disopyramide, procainamide) | |
| Some antipsychotic drugs (amisulpiride,a droperidol, haloperidol, chlorpromazine, pimozide, quetiapine, sertindole, thioridazine,b ziprasidone) | This risk varies within this group. Clozapine is a potent hERG blocker, but likely due to interference with other ion channels, it has a very low risk of dysrhythmic activity. Aripiprazole, olanzapine, and zotepine are not associated with torsade de pointes. |
| Some opioids—methadone in higher doses and levacetylmethadol, buprenorfin | Morphine and codeine are considered to be very safe from this point of view. |
| Some antidepressant drugs (TCA, SSRi) | The risk of TCAs is referred to the main text. The risk with SSRi's is very low considering the frequency at which these drugs are used—citalopram has the highest risk in this group but in absolute numbers, the risk is very low. |
| Pentamidine, arsenic trioxide | |
| Antihistamines terfenadine and astemizole, prokinetic cisapride | The relative risk is low but the actual number of cases was considerable due to the widespread use of these drugs. For example, the risk of terfenadine was assessed to be 1 in 50,000 patients and 1 in 120,000 patients for cisapride. These data are likely underestimated, but still the risk is clearly far below 1%. Other antihistaminics with an exception of diphenhydramine do not possess the risk or the risk is very low. |
| Quinolones | The risk is a class effect but varies within the group. Grepafloxacin and sparfloxacin had higher risk, while ofloxacin, levofloxacin, ciprofloxacin seem to have much lower risk, moxifloxacin is in between. |
| Macrolides erythromycin and clarithromycin; antifungals ketoconazole and fluconazole, halofanthrine, artemether, chloroquine, bedaquiline, delamanid, bedaquiline | |
| 5‐HT3 antagonists (dolasetron and ondasetron) | Palonosetron, granisetron, and tropisetron are probably free of this risk. |
| Vandetanib, probucol, domperidone, inhibitors of phosphodiesterase 5 (sildenafil and vardenafil) |
In bold are drug that were withdrawn or restricted at least in some countries due to the risk of torsade de pointes.
aA high risk for amisulpride is documented in overdose.
bThioridazine is considered to possess a higher risk than haloperidol; torsade de pointes can develop with low doses, but in overdose torsade de pointes is infrequent.
Data from the following sources were used: 262–268,270,273‐275,277–290.
3.2.3. Sodium–potassium pump
Another drug target that can lead to dysrythmogenicity is the sodium–potassium adenosine triphosphatase (Na+/K+‐ATPase, also called the “sodium–potassium pump,” or “sodium pump”). A group of drugs named the cardioactive steroids bind to this Na+/K+‐ATPase. Cardioactive steroids are generally known as cardiac glycosides, but not all cardioactive steroids contain a sugar moiety. Such exceptions include compounds in toad venoms.291 Cardioactive steroids were long used as the first‐line treatment of chronic heart failure; however, their current use is limited due to the better safety and efficacy of other drugs, in particular ACE inhibitors/angiotensin receptor antagonists and beta‐blockers.292 Nevertheless, digoxin, the only clinically available representative of the class, is still used today in clinical practice in combination with other drugs to further improve symptoms and reduce hospitalization in congestive heart failure with reduced ejection fraction.216 In addition, it is used as a rate‐control agent to control the rapid ventricular response in supraventricular tachydysrhythmias (atrial fibrillations in particular).234 As such, digoxin is and has been an important source of intoxication.293 Digoxin toxicity may originate, or may be exacerbated by drug interactions 294, 295—see Table 4. For example, inhibitors of P‐glycoprotein such as verapamil, amiodarone, or macrolide antibiotics can enhance oral bioavailability of digoxin by decreasing its efflux from the enterocytes into the lumen of the intestine and decrease its active tubular secretion into the urine in the kidney. As a result, plasma concentrations of digoxin may significantly increase to toxic levels. Other very significant sources of cardiac glycoside poisoning occur after the ingestion of some plants, in particular yellow oleander, which is a relatively common tool for suicides in southern Asia, especially in Sri Lanka and India.296, 297, 298
A major drawback of the clinical use of cardioactive steroids is their low therapeutic index; that is to say, the therapeutic concentrations are very close to the toxic concentrations.299 Thus, a recommended window of therapeutic concentrations is quite narrow (0.8–2.0 ng/mL) and more recent recommendations suggest even lower and more narrow range (0.5–1.0 ng/mL).294 The pharmacological characteristics of cardioactive steroids are quite homogenous, but there are some differences regarding their pharmacodynamics and pharmacokinetic properties.300, 301 The knowledge of the latter can be advantageous in the treatment of poisoning, for example, the slow absorption of oleander glycosides allows higher efficacy of activated charcoal.296 Interestingly, although being rationally used since the 18th century, there is still discussion regarding the overall mechanism of action. The accepted theory is that cardiac glycosides block Na+/K+‐ATPase (Fig. 8). This leads to a higher intracellular Na+ concentration, which alters the activity of the Ca2+/Na+ exchanger, resulting in a lower efflux of Ca2+. Higher intracellular Ca2+ concentration results in improved myocardial contraction. It is thought that for therapeutic purposes, only a partial inhibition of ATPase has to be achieved, while some studies suggest that lower doses activate Na+/K+‐ATPase; however, this is still a topic of debate.300, 302 At high concentrations of these drugs, or under some circumstances the Ca2+/Na+ exchanger may work in reverse to actively increase the intracellular Ca2+ load. Signs of toxicity may occur even when appropriate doses of the digoxin are administered, due to a sudden reduction in elimination (e.g., kidney for digoxin) and/or hypokalemia.294 The latter is likely to occur in severe congestive heart failure requiring intensive diuretic treatment. The mechanisms whereby hypokalemia sensitizes the heart to digoxin cannot be explained solely by the prodysrhythmic effect of this electrolyte imbalance. Interestingly, digoxin reportedly binds to the extracellular domain of the Na+/K+‐ATPase, where K+ normally binds for its transport. Thus hypokalemia enhances the efficacy of the interaction of the drug with its molecular target.303 Notwithstanding, strong or complete blockade of the Na+/K+‐pump is considered to be the cause of toxicity, since this is associated with intracellular Na+ and Ca2+ overload. Marked accumulation of these ions determines the positive bathmotropic effects of higher doses. Furthermore, at toxic doses, this ion imbalance enhances the repolarization phase due to the faster K+ current that makes the action potential duration shorter with subsequent shortening of the effective refractory period. The toxicity is complicated by the fact that digoxin and likely all other cardiac glycosides, increase vagal tone, likely via central actions.
Figure 8.
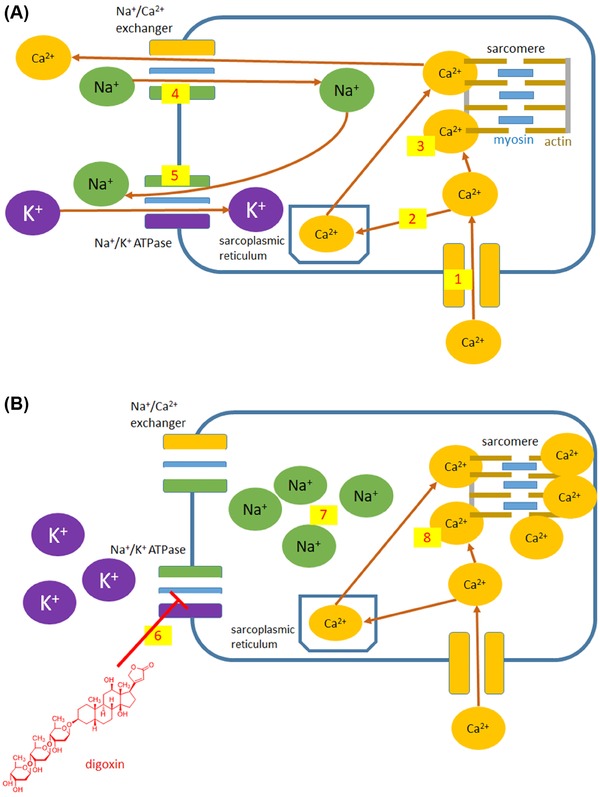
Calcium trafficking in cardiomyocytes under physiological conditions (A) and mechanism of action of digoxin (B). Ca2+ is transported into the cell mainly through voltage gated L‐type Ca2+ channels (1). An increase in intracellular Ca2+ levels triggers Ca2+ release from sarcoplasmic reticulum (2). Ca2+ is needed for interaction with actin‐myosin resulting in muscle contraction (3). Ca2+ is then returned back to sarcoplasmic reticulum (not shown) and to the extracellular space by the Na+/Ca2+ exchanger (4). By this transporter, Na+ is transported inside the cells and its level on both sides must be recovered by activity of sodium–potassium adenosine triphosphatase (Na+/K+‐ATPase, 5). Digoxin and other cardioactive steroids (B) block Na+/K+‐ATPase (6) and thus increase the intracellular Na+ levels (7). Higher intracellular Na+ levels block the passive and thus concentration‐dependent exchange of Ca2+ and Na+ through the Ca2+‐Na+ exchanger. This results in higher intracellular level of Ca2+ (8) available for myocardial contraction.
In summary, cardiac glycosides might cause very different types of dysrhythmias. In “milder” intoxication, bradycardia and AV blocks of different degrees are predominant, due to the excessive stimulation of vagal tone. In more severe intoxications, atrial and also ventricular tachydysrhythmias could appear, likely due to the delayed afterdepolarization leading to increased automaticity and/or ectopic activity. The treatment of these dysrhythmias is not simple and mortality is still a serious issue. There are several treatment approaches that depend on the symptoms.4, 304 Activated charcoal is used to avoid further absorption of the cardiac glycoside and interrupt the enterohepatic circulation. Atropine is preferred for bradycardia or AV blocks and temporal pacing can also be used.305 The digoxin‐specific antibody fragments are very important in the treatment of moderate or severe intoxication. As a result of their use, the mortality from digoxin intoxication dropped from 20–30% to 5–8%.306 In addition, these antibodies have lower, but still significant affinity in vitro to other structurally similar cardiac glycosides (see Fig. 9 for chemical differences) 307, 308, 309, 310, 311 and were successfully employed for the treatment of oleander, Indian hemp, and toad venom intoxications.296, 312, 313, 314, 315 In addition, a placebo‐controlled clinical study confirmed their effectiveness against yellow oleander intoxication.316 However, the digoxin‐specific antibody fragments do not seem to be useful for the intoxication with glycosides from the lily‐of‐the‐valley (Convallaria majalis).317 Limited shelf‐life and their higher cost make these antidotes available only at special cardiology, or toxicology centers. Correction of possible hypokalemia should be performed as soon as possible and ventricular dysrhythmias may require additional symptomatic treatment.
Figure 9.
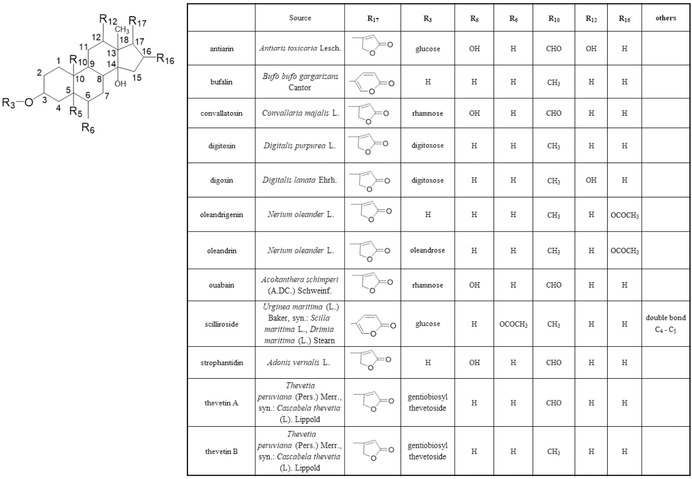
Chemical structures and sources of the major natural inhibitors of Na+/K+‐ATPase.
Na+/K+‐ATPase is also the target of palytoxin, which represents one of the most toxic nonpeptidic substances, with an oral acute reference dose suggested to be 64 μg for a person with an average weight of 60 kg.318, 319 It is produced by cnidarian Palythoa toxica (seaweed like coral, Limu‐make‐o‐Hana = Seaweed of Death from Hana) in the tropical areas of the Pacific Ocean.320, 321 The majority of palytoxin poisonings in humans are a result of seafood consumption. Cases of death and near‐death illness resulting from palytoxin ingestion have been reported after consumption of contaminated crabs in the Philippines, sea urchins in Brazil, and fish in Japan, Madagascar, and the United States.322, 323, 324, 325 Structurally, it is a polyalcohol consisting of a very long, partially unsaturated aliphatic chain, interspersed with five sugar moieties.326, 327 It binds selectively and with high affinity to the Na+/K+‐pump and transforms the pump into a channel permeable to monovalent cations. As a result, different gastrointestinal, neurological, and cardiovascular effects including bradycardia, hypertension, and cardiac dysfunction have been described.328 Death occurs due to myocardial injury.329 Treatment is symptomatic and supportive.330
3.2.4. Acetylcholine receptors
There are two types of acetylcholine receptors, the nicotinic (N) receptors and muscarinic (M1–M5) types. Stimulation of N‐receptors was partially described in the section concerning nicotine (see chapter 2.3). In the heart, muscarinic acetylcholine receptors are limited mainly to atria and AV conduction pathways. Thus, in contrast to adrenergic receptors, the effects of muscarinic receptors are limited to sinus or atrial dysrhythmias. On the other hand, stimulation of N‐receptors leads also to the activation of the sympathetic nervous system through both vegetative ganglia and the adrenal medulla. Thus, the effect of parasympathomimetics/lytics (drugs acting only on M‐receptors) and cholinomimetics (drugs affecting both M‐ and N‐receptors) are different.
Indirect cholinomimetics include carbamates and organophosphates. Historically, the consumption of Calabar beans (Physostigma venenosum) with substantial content of physostigmine and related carbamate alkaloids in trial by ordeals led to considerable mortality even in the 19th century.331 Currently, apart from wars and terrorist attacks, accidental intoxication with subsequent death is still possible (e.g., by agricultural insecticides).332, 333 At low doses, the muscarinic effects on the heart predominate and these include, bradycardia and other dysrhythmias. In higher doses, stimulation of N‐receptors occurs and this might result in paradoxical hypertension and tachycardia due to increased sympathetic tone. Death is usually associated with respiratory muscle paralysis, but dysrhythmias can also contribute.5
Administration of muscarine (a parasympathomimetic) produces muscarinic effects, but the intoxication is not very relevant since (1) this drug is not used as a therapeutic agent and (2) although discovered in the red fly agaric mushroom (Amanita muscaria), poisoning after its consumption, or of the similar Amanita species, is not associated with muscarinic effects due to very low content of muscarine. In fact, the intoxication by this mushroom is associated with antimuscarinic effects (atropine‐like)—see below.334, 335 Accidental intoxication with the parasympathomimetic pilocarpine, or even its local application can result in hypotension and bradycardia, or tachycardia (probably due to the stimulation of the sympathetic system).336, 337
Atropine is the most well‐known parasympatholytic drug. It is present naturally in a mixture with other parasympatholytics (e.g. hyoscyamine /its levorotatory isomer/, and scopolamine) in Atropa bella‐donna (also known as deadly nightshade) and Datura stramonium (known also as jimson weed, devil's snare, thornapple, and moon flower). Voluntary ingestion of these plants is relatively frequent both for their psychotomimetic effects and in suicide.251 Tachycardia is common, but may not be observed in all cases. The direct and indirect effects in skin capillaries resulting into vasodilation and a red appearance are among the typical clinical signs of overdose, but the mechanism(s) is(are) not fully understood. In general, while CNS derangement is dominant, deaths are rare.338, 339, 340, 341, 342
In addition to the clinically used parasympatholytics, many other drugs possess substantial anticholinergic (i.e., antimuscarinic) activity. This is particularly important for several antipsychotics and TCA. There are many antipsychotic drugs that differ in their mechanism of action, but many of them are able to block muscarinic receptors. Basal antipsychotic (low‐potency phenothiazines) and MARTA (multiacting receptor targeted antipsychotics) are commonly associated with an increase in heart rate due to their antimuscarinic activity, particularly in overdose.192
3.2.5. Adenosine receptors
Adenosine receptors respond to the endogenous agonist adenosine. This compound has a very short half‐life and is therefore sometimes used for pharmacological cardioversion of paroxysmal supraventricular tachycardia (AV nodal re‐entrant tachycardia in particular) to sinus rhythm with a very low risk of toxicity. Because its mechanisms of action are based on brief interference with AV node conduction, it is understandable that the drug is not suitable for patients with higher degree AV blocks. Other potential adverse effects in cardiovascular systems are typically short‐lived and involve vasodilation (including flushing), hypotension, and chest pain. Methylxanthines, like caffeine and theophylline, are more frequently used; the later as a bronchodilator in the treatment of chronic obstructive pulmonary disease/bronchial asthma and the former as the component of tea and coffee. Their mechanism of action is complex, but clearly involves antagonism at adenosine receptors. Theophylline and much less caffeine, induce catecholamine release by an unknown mechanism.4, 343 They also block phosphodiesterases, but only in high, clinically unachievable concentrations.344 In general, caffeine's toxicity occurs at higher concentrations when compared with theophylline, which is considered to be a drug with a very narrow therapeutic index. Theophylline overdose is potentially life threatening and affects mainly the gastrointestinal tract, the cardiovascular system, and the CNS. Sinus tachycardia can occur at therapeutic doses and various, potentially fatal dysrhythmias can appear after theophylline intoxication. The management of toxicity is mainly supportive and includes fluid supplementation, or sympathomimetics in case of hypotension and sometimes mild K+ supplementation. Dysrhythmias are treated according to their type, by adenosine, lidocaine, or β‐blockers. For caffeine intoxication, which is rare, the treatment is similar. Although patients with propensity to certain types of tachyarythmias (such as paroxysmal atrial fibrillation) have traditionally been advised to avoid caffeine containing beverages, recent data suggest that this restriction is not entirely justified.345 Long‐term high caffeine consumption is positively correlated with hypertension. However, mild to moderate intake of caffeine has been found to be free of any serious cardiovascular risk.4, 5
3.2.6. Other drugs causing rhythm disturbances
Fingolimod was registered as the first orally active agent to treat multiple sclerosis. It is a modulator of the sfingosine‐1‐phosphate receptors at which its initial agonistic activity leads to pronounced decreases in heart rate (about nine beats per min after 5 hrs) and can cause AV blockade. These first‐dose effects are infrequent, mostly asymptomatic and resolve spontaneously in the majority of cases. ECG monitoring during the first 6 hrs after drug administration is considered to be the standard.346, 347 Interestingly, fingolimod can acutely cause slight transient hypotension due to the activation of the eNOS pathway, but this is sometimes followed by a small increase in blood pressure (+3/1 mmHg of systolic/diastolic blood pressure).348
Other drugs, such as lithium and carbamazepine might also cause rhythm disturbances, but the underlying mechanisms are not known. Lithium reversibly targets the sinus node, while carbamazepine affects several parts of the action potential conducting system.349
3.3. Drugs with main toxic effects on the working myocardium
Direct toxic effects on the working (contractile) myocardium are a serious situation, which limit the clinical utility of those drugs. However, there are exceptions, such as the antineoplastic anthracyclines (ANT), where use of the drug is required for survival. Other examples of drugs that adversely affect the working myocardium are abused drugs (androgenic anabolic steroids) and/or compounds given in high does for a very long time (alcohol), or drugs whose risk of toxicity is very low (clozapine). The complex cardiovascular actions of ethanol and androgenic anabolic steroids were discussed in chapters 2.9 and 2.10, respectively.
3.3.1. Anticancer drug‐induced cardiac dysfunction and heart failure
Modern anticancer drugs have significantly contributed to the striking improvement in survival in many types of malignant neoplasm. Unfortunately, besides the typical adverse effects of anticancer drugs (e.g., nausea, vomiting, myelosuppression), several of these drugs may severely affect the heart with potentially life‐threatening consequences.350 Numerous detrimental effects to the heart may happen, including morphological alterations resulting in cardiomyopathy and/or changes in cardiac function (dysrhythmias or systolic/diastolic dysfunction). Clinically, heart failure/left ventricular dysfunction ranks among the most important cardiotoxic effects of some anticancer drugs.350, 351 Other cardiotoxic effects of anticancer drugs are discussed only briefly.
3.3.1.1. Drugs inducing heart failure/cardiac dysfunction
There are two distinct types of anticancer drug‐induced cardiac dysfunction—termed Type I and Type II (Table 6).352 Type I is induced by classic (old) anticancer chemotherapeutic agents, typically the ANT, and to a certain extent, high‐dose cyclophosphamide or taxanes. This type of toxicity depends on the cumulative dose of the drug administered and is exemplified histopathologically by loss of myofibrils and vacuolization of the cytoplasm of the cardiomyocytes and is deemed to be largely irreversible. In contrast, Type II is mostly associated with the new biologically targeted drugs—such as the monoclonal antibody trastuzumab, which targets the human epidermal growth factor receptor 2 (HER2)—and is likely largely reversible. Type II toxicity is also associated with small molecule tyrosine kinase inhibitors (like, e.g., lapatinib, sunitinib) and proteasome inhibitors (e.g., bortezomib). It is of particular note, that the risk of cardiotoxicity posed by all of these agents was not recognized during preclinical toxicology testing. Instead, it was found during advanced clinical evaluation, or even later after the approval for clinical use. This suggests that routine toxicological testing in experimental animals is currently not sufficient to predict clinical risks.
Table 6.
Anticancer drugs inducing heart failure/left ventricular cardiac dysfunction
| Heart failure/LV dysfunction induced by anticancer drugs | |
|---|---|
| Type I | Anthracyclines (doxorubicin, daunorubicin, epirubicin) |
| Alkylating agent (cyclophosphamide) | |
| Taxanes (docetaxel, paclitaxel) | |
| Type II | HER2 targeting antibodies (trastuzumab, pertuzumab, adotrastuzumab) |
| HER2 targeting tyrosine kinase inhibitors (lapatinib) | |
| (Tyrosine) kinase inhibitors (sunitinib, sorafenib, dobrafenib, ponatinib) | |
| Proteasome inhibitors (bortezomib, karfilzomib) | |
3.3.1.2. Type I cardiac dysfunction induced by anticancer drugs—Anthracycline cardiotoxicity
The first ANT, daunorubicin, was isolated from Streptomyces peucetius, the other clinically used members of this class (doxorubicin, epirubicin, or idarubicin) are semisynthetic derivatives. These agents are still heavily used in oncological practice to treat numerous types of hematological and solid tumors. While intercalation into DNA strands has long been believed to be the main mechanism of their anticancer effect, it is now generally accepted that ANT inhibit the enzyme topoisomerase IIα (Top2α) in cancer cells, as so‐called Top2α‐poisons, to induce double‐strand breaks in DNA, resulting in and programmed cell death.353
3.3.1.2.1. Types of ANT cardiotoxicity
ANT elicits both acute and chronic cardiotoxicity, which differ in their onset, manifestation, and clinical importance.354 Acute forms are generally represented by subclinical changes in cardiovascular function soon after drug administration (this often concerns electrophysiological changes) and are only rarely a significant clinical issue. Subchronic cardiotoxicity associated with myocarditis–pericarditis syndrome has been seldom reported. In contrast, chronic forms, developing months or years after chemotherapy (early and delayed type), are particularly feared because they are associated with irreversible dilated cardiomyopathy and the resulting heart failure poorly responds to standard treatment.
3.3.1.2.2. Mechanisms of ANT cardiotoxicity
Despite many experimental and clinical studies performed throughout past 40 years, the molecular basis for ANT‐induced cardiotoxicity remains elusive and is still a matter of debate and controversy.350, 351, 352 The prevailing mechanistic explanation centers around ANT‐induced and iron‐catalyzed formation of ROS, resulting in direct oxidative damage to the myocardium.355 Other hypotheses point to ANT‐induced impairment of mitochondrial bioenergetics, damage to mitochondrial DNA with subsequent perturbations in the expression of mitochondrial DNA‐encoded proteins, disruption of mitochondrial and cellular Ca2+ homeostasis, and alterations in the expression and stability of cardiac myofilaments.350, 355 Recently, a new and particularly interesting concept of ANT cardiotoxicity emerged when Yeh et al. proposed that ANT binds to topoisomerase IIβ (Top2β), which is the predominant Top2 isoform in the heart, to induce cardiomyocyte DNA damage, apoptosis, and impairment of mitochondrial biogenesis with secondary formation of ROS and mitochondrial dysfunction.356
3.3.1.2.3. Prevention and management of anthracycline cardiotoxicity
Effective management of established ANT cardiotoxicity is difficult and relies largely on pharmacological intervention with ACE inhibitors and β‐adrenergic blockers to control heart failure symptoms.350, 351 Hence, there is a great emphasis to prevent the onset of cardiotoxicity. A widely employed approach is to limit the total cumulative dose of ANT (usually below 300–450 mg/m2 of doxorubicin).352 Another option is to employ drug targeting strategies (such as encapsulation of the drug into liposomes), which reduces distribution of ANT into the heart and thus reduces the risk of cardiotoxicity. A lot of research has been focused on pharmacological cardioprotection.355 Interestingly, while different natural and synthetic antioxidants have proven effective in providing protection in acute high‐dose experimental cardiotoxicity models (e.g., vitamin E, acetylcysteine, or flavonoids), the treatments have largely failed in clinically relevant chronic models and in the few clinical trials that have been attempted.355 The only drug with well‐documented efficacy in both experimental and clinical settings is dexrazoxane, a prodrug that is thought to be metabolized into an iron‐chelating active metabolite that then shields catalytically active iron, thus preventing ANT‐induced ROS formation in the heart.355 However, stronger and more selective iron chelators were unable to provide comparable protection in the same models. Recent data suggest that dexrazoxane inhibits and depletes Top2β in the heart, thereby preventing the onset of ANT‐induced cardiotoxicity.350, 355, 357 Current clinical use of dexrazoxane is unfortunately limited mainly because of concerns about its adverse effects. It was suggested that dexrazoxane might have some impact on anticancer effects and increase the risk of secondary malignancies. However, neither of these claims has been supported by strong clinical evidence and most of available data argues against them.358, 359, 360
While both ACE inhibitors/angiotensin receptor antagonists and β‐blockers are indicated in the treatment of systolic dysfunction induced by ANT, it has been proposed that both agents could be useful prophylactically in cardioprotective settings. Although there is a significant amount of experimental data on the possible cardioprotective effects of ACE inhibitors/angiotensin receptor antagonists, it is not clear whether these agents provide true long‐lasting protection against chronic ANT cardiotoxicity similarly as dexrazoxane. The β‐blocker, carvedilol showed significant cardioprotection against chronic ANT cardiotoxicity, while atenolol did not.361, 362 Clinical trials with these drugs were performed quite recently and/or are currently on the way. Several findings suggested cardioprotection in ACE inhibitors/angiotensin receptor antagonists and β‐blockers, but strong conclusion cannot be made because several of these studies suffered from significant limitations (e.g., borderline significance of the findings, small declines in systolic function in the ANT‐only group, and examinations performed under effect of drugs with significant direct hemodynamic activity). Indeed, some trials yielded mixed or negative outcomes with these agents.361, 363
3.3.1.3. Type II cardiac dysfunction induced by anticancer drugs
3.3.1.3.1. HER2 targeted therapeutics—trastuzumab, ado‐trastuzumab emtansine, pertuzumab, and lapatinib
Overexpression of HER2 is a significant negative prognostic marker, particularly in breast cancer. HER2 targeting with the monoclonal antibody trastuzumab was the first clinically successful use of biologically targeted anticancer therapy for solid tumors. Using specifically targeted antibodies focused on tumor cells, these agents should generally be less toxic to other cells in the body. Interestingly, despite these expectations, cardiotoxicity was soon observed with this drug in clinical settings.352 However, the type of cardiotoxicity observed was different from that produced by ANT in terms of the lack of clear dose dependency, absence of histopathological hallmarks, and at least partial reversibility. Thus, it has been proposed that this form of cardiotoxicity be distinguished as a Type II cardiotoxicity.352 It is of note, that the risk of heart failure is particularly important and irreversible in patients receiving both trastuzumab and ANT. Concomitant treatment with both drugs has very high risk (up to 27%) of cardiotoxicity. Later it was discovered that the risk of toxicity could be markedly reduced if the drugs were given separately with a significant gap between them.352 The mechanisms of trastuzumab cardiotoxicity are still under investigation, but it has been proposed that HER2 mediates important prosurvival paracrine stimulation of cardiomyocytes via neuregulin‐1 and this may be perturbed by trastuzumab.350, 352 In this regard, trastuzumab may inhibit the recovery of cardiomyocytes stressed by ANT treatment. Interestingly, lower cardiotoxicity has been associated with other selective HER2 targeting drugs like the complex antibody ado‐trastuzumab emtansine and the small drug—tyrosine kinase inhibitor lapatinib, but further studies are needed to clarify this point.
3.3.1.3.2. Other tyrosine kinase inhibitors and other agents
Tyrosine kinase inhibitors are modern small‐molecule anticancer agents that rank among the fastest growing drug classes. Several drugs, like imatinib that inhibits bcr‐abl kinase, revolutionized the field of cancer treatment by offering a strikingly improved prognosis. However, multiple agents of this class induce Type II cardiac dysfunction and symptomatic congestive heart failure.350, 351, 352 The first drug associated with the risk of cardiac dysfunction was imatinib, but later analysis showed that the incidence of its toxicity is very rare (<1%) and was largely reversible. A higher risk of cardiac dysfunction has been attributed to multikinase inhibitors, particularly those affecting among others, the vascular endothelial growth factor (VEGF)/platelet‐derived growth factor receptors (PDGFR) pathway, like sunitinib and sorafenib.350, 351 While earlier studies implicated direct damage to cardiomyocytes via impaired mitochondrial function and/or AMPK inhibition, new data suggest that cardiomyocyte function can be affected indirectly through damage to pericytes–endothelial interactions, with subsequent induction of chronic microvascular dysfunction.350, 364 Further research is needed to shed light on the mechanisms of the cardiotoxicity and thus aid the development of safer drugs and strategies.
Another class of biologically targeted anticancer drugs are the proteasome inhibitors (e.g., reversible bortezomib or irreversible carfilzomib), which have markedly improved the prognosis during treatment of refractory multiple myeloma and some other types of cancer. Indeed, the proteasome system is now considered essential for the maintenance of cellular protein homeostasis, this is particularly true for cardiomyocytes that have minimal regenerative potential and face constant intensive mechanical, metabolic, and oxidative burden. A risk of cardiac dysfunction was found during clinical evaluation and reported in multiple case reports.351, 365 The risk appears to be more significant for carfilzomib; however, the overall risk is not entirely clear due to the lack of appropriately designed clinical trials. Interestingly, marked synergy in anticancer effects have been noted in combination with ANTs in certain protocols, but the cardiovascular safety of this combination remains undetermined.
3.3.2. Others
Clozapine is able to induce myocardial damage in animals and myocarditis with an estimated frequency of 1:500 to 1:10,000 patients. This complication occurs mostly within the first 2 months of treatment and it can progress to dilated cardiomyopathy, with a fatality rate of at least 10%. The mechanism seems to be associated with type 1 IgE‐mediated hypersensitivity and there are some indications that other antipsychotic drugs also present some risk. In the management, discontinuation of clozapine is needed. Reinitiation of therapy will relapse the disorder in the majority of patients. The therapeutic utility of corticosteroids is ambiguous, while ACEi and diuretics are useful.366, 367
3.4. Drugs directly affecting cardiac valves
Drug‐induced valvulopathy, also known as valvular heart disease, is an uncommon, but serious reaction. Its diagnosis is complicated and causality is not easily established, since previous valvular damage can be present. Also in preclinical animal studies, analysis of the heart valves is not routinely performed. Likely because of this oversight, drug‐induced valvulopathy was first described in the 1960s. Currently, we know that the serotonin 5‐HT2B receptor is the major player in this pathology and explains why some drugs from very different pharmacological groups (ergot derivatives and some anorexigens or recreational drugs with similar effects) cause this adverse toxic reaction. Stimulation of 5‐HT2B receptors initiates a signaling cascade resulting in the upregulation of genes involved in the proliferation and regulation of valvular interstitial cells, with thickening of valvular surface and the subvalvular apparatus. This process is associated with increased valvular stiffness and reduced mobility of the valvular leaflets, leading to regurgitation. The effect is largely dependent on the cumulative dose of the drug. Little is known about the reversibility of the process, but it seems to be limited. In severe cases, the only treatment available is valve replacement surgery. Dexfenfluramine and pergolide were withdrawn from the market because of their high risk of valvulopathy. Indeed, the anorexigenic combination of fenfluramine and phentermine had an unacceptably high incidence of valvulopathy in patients (6 to 30%). Similarly, a high incidence was observed with pergolide and cabergolide administration. On the other hand, similar dopaminergic agonists, like lisuride, pramipexol, and ropinirol, apparently do not pose a risk. The data on bromocriptine are not conclusive, but some risk can be associated with its use, since it has partial agonist activity at 5‐HT2B receptor. The list of valvulopathic drugs is short and can be seen in Table 7. According to a recent analysis, other drugs, in particular guanfacine, might possess some risk, but clinical data are yet not available.368, 369, 370
Table 7.
Drugs that can cause valvulopathy
| Ergot alkaloids and their derivatives | Sympathomimetics/anorexigens |
|---|---|
| Ergotamine, methysergide, pergolide, cabergoline, ergonovine, methylergonovine | (Dex)fenfluramine,a benfluorex, MDMA, 3,4‐methoxyamphetamine |
aReports of increased incidence of valvular disease in patients taking fenfluramine and phentermine are largely attributed to the former, since phentermine alone has never been associated with valvulopathy.
3.5. Drugs causing pericarditis
Pericarditis is itself a minor cardiovascular problem, but it might lead to life‐threatening cardiac tamponade.371 It is very rarely elicited by drugs and causality is not easily established.372 The connection is usually based on case reports. Currently, it seems that hypersensitive reactions, in particular development of lupus erythematosus, and excessive anticoagulant treatment leading to intrapericardial bleeding are among the most common causes of drug‐developed pericarditis.373, 374, 375, 376, 377, 378, 379, 380, 381, 382, 383 Several anticancer drugs have a less clear etiology, but hypersensitivity can play a role.384, 385, 386 Similarly, pericardial effusion caused by specific bcr‐abl tyrosine kinase inhibitors can have an immune background.387 Moreover, cancer itself is a frequent cause of pericarditis 373. Also sympathomimetics are associated with pericarditis, but in this case, the injury also involves the myocardium and thus may be a consequence of the above‐reported sympathomimetic effects on the heart.388 Constrictive pericarditis can also develop due to fibrosis produced by dopaminergic agonists with an ergot scaffold.389, 390, 391, 392 A list of drugs potentially associated with pericarditis is seen in Table 8. Treatment of all types of pericarditis is based on drug discontinuation. Pericardial effusion is treated mainly with nonsteroidal anti‐inflammatory drugs (NSAIDs).393 Colchicine and glucocorticoids can be also employed. Cardiac tamponade is mostly treated with pericardiocentesis.371
Table 8.
Drugs that can cause pericarditis
| Mechanism or group | Drugs |
|---|---|
| Drug‐induced lupus erythematosus | Isoniazid, hydralazine, procainamide, sulfasalazine, carbamazepine |
| Other immune reactions | TNFα‐antibodies (infliximaba), natalizumab, 5‐aminosalicylic acid (mesalazine) and its prodrugsb (balsalazide), methotrexate, penicillins, cephalosporins, interferon α, minoxidil |
| Anticancer drugs (hypersensitivity cannot be fully excluded) | Cytostatics (cytarabine, treosulfan, cyclophosphamide), bcr‐abl tyrosine kinase inhibitors (dasatinib, imatinib, nilotinib), methyltransferase inhibitors (azacitidine, decitabine) |
| Ergot derivatives | Cabergoline, pergolide, bromocriptine, methysergide |
| Anticoagulants | Heparin, warfarin, rivaroxaban |
| Others | Sympathomimetics (methylphenidate), minoxidil, tramadol |
Data obtained from 374–392,394–404.
aInfliximab can induce pericarditis due to drug‐induced lupus erythematosus.
bSulfasalazine can induce pericarditis due to drug‐induced lupus erythematosus.
4. DRUG AFFECTING PRIMARILY THE VASCULAR SYSTEM
4.1. Drugs causing hypertension and arterial thrombosis
4.1.1. NSAIDs
NSAIDs are the most used treatments for pain relief. Their mechanism of action involves the blockade of cyclooxygenases 1 and 2 (COX‐1, COX‐2, respectively). The most widely reported problem of nonselective, sometimes designated as traditional, NSAIDs is the risk of peptic ulcers and gastrointestinal bleeding mainly due to blockade of COX‐1. Indeed, the discovery of selective COX‐2 inhibitors, called coxibs, reduced the risk of gastrointestinal side effects.405, 406 However, later it was found that coxibs might cause different kinds of cardiovascular side effects.407 Since that discovery, other NSAIDs have been analyzed and the mechanism of their cardiovascular complications revealed. There are apparently two mechanisms associated with these cardiovascular side effects: (A) an imbalance between the production of proaggregatory thromboxane A2 formed by platelets via COX‐1 and antiaggregatory active prostacyclin formed via endothelial cyclooxygenases with a decisive involvement of COX‐2,406, 408 (B) blockade of the synthesis and consequent lack of prostaglandin E2 effect in the kidney. It is suggested that prostaglandin E2 in the kidney is formed mainly by COX‐2, but both COX enzymes are constitutively expressed there and the data regarding which types of prostaglandin(s) are formed is not very clear.408, 409, 410 Nonetheless, the involvement of COX‐2 in the cardiovascular effects is unambiguous, since this pathway is potentially blocked by all known NSAIDs with an exception of low dose of acetylsalicylic acid (aspirin).411 Therefore, all NSAIDs are able to increase blood pressure and also to cause/worsen heart failure. 10, 405, 412, 413, 414 On the other hand, the imbalance between the thromboxane A2 and the prostacyclin system is influenced differently based on the degree of inhibition of COX 1/2, duration of action, and reversibility of inhibition. Regarding NSAIDs, only acetylsalicylic acid in low doses (and rarely indobufen) is used clinically for protection against atherothrombosis.415, 416, 417 This is because low‐dose acetylsalicylic acid irreversibly blocks platelet COX‐1 without having a significant effect on endothelial COX‐2.411 Conversely, coxibs (which are selective for the COX‐2) decrease prostacyclin production, without having an important effect on COX‐1‐mediated thromboxane A2 production (Fig. 10).
Figure 10.
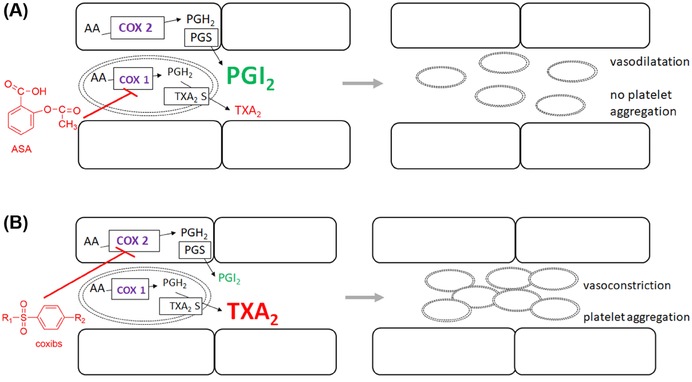
Different effects of low dose of acetylsalicylic acid and coxibs on the vascular system. (A) Acetylsalicylic acid (ASA) at low doses blocks mainly platelet cyclooxygenase 1, resulting in a relative excess of prostacyclin (PGI2) over thromboxane A2 (TxA2), vasodilation, and inhibition of platelet aggregation. (B) Coxibs are selective for cyclooxygenase 2. Therefore, their administration leads to excess of TxA2 with subsequent risk of vasoconstriction and platelet aggregation.
The unbalanced ratio of thromboxane A2 to prostacyclin is considered to be the major culprit for the higher risk of major vascular events, including the more prominent coronary events.405, 406 There is considerable discussion whether there are differences between coxibs. Currently, it appears that the differences are not very significant, although it is thought that low doses of celecoxib are likely to have a lower risk. Interestingly, the NSAIDs diclofenac and to some extent ibuprofen, have the same cardiovascular risk as coxibs (e.g., 418). The likely explanation is that thromboxane A2 production must be almost completely inhibited (around 95%) to block platelet aggregation 419, 420 and most nonselective NSAIDs are not able to achieve such a high degree of inhibition, or the inhibition is reversible and transient. Only high dose naproxen (2 × 500 mg daily) has a neutral effect on coronary events likely due its long half‐life and corresponding persistent inhibition of both COXs.405 It should not be forgotten that there is a possible important interaction between naproxen and low dose of aspirin and that naproxen, like all other NSAIDs, increases the risk of blood pressure elevation and hospitalization for heart failure, as mentioned above.421
4.1.2. Inhibitors of the VEGF pathway
VEGF plays an important role in angiogenesis, thus it is not surprising that drugs blocking this pathway (Fig. 11) might cause disturbances in the vascular system. Most available data refer to the oldest representative of this group, which is the monoclonal antibody against VEGF, bevacizumab. However, data regarding intracellular multikinase inhibitors are also increasing rapidly. These drugs block the tyrosine kinase activity of VEGF receptors and include the clinically used sunitinib, sorafenib, regorafenib, pazopanib, axitinib, cediranib, vandetanib, and lenvatinib. Tyrosine kinase blockers are generally nonselective, and this is valid as well for ponatinib, designated mainly as the inhibitor of bcr‐abl tyrosine kinase. The latter is also markedly active toward the VEGF pathway in contrast to other drugs from this group.422 Recently, the fusion protein aflibercept from the extracellular domains of human VEGF receptors and the Fc immunoglobulin portion was registered. All these drugs block the VEGF pathway, which can result in marked arterial blood pressure elevation, arterial thromboembolism, and can also lead to bleeding. As noted above, heart failure is observed with several of these drugs, but the mechanism(s) of its development may be independent of hypertension, although it can ultimately contribute to this process.423, 424, 425, 426 Data on venous thromboembolism are ambiguous, claiming increased risk, neutral effects, or a tendency to decrease.424, 427, 428 The major mechanism seems to be the blockade of vascular NO production through eNOS because, VEGF binding to the VEGFR‐2 receptor in vascular endothelium leads to eNOS upregulation. Also decreases in NO production in the kidney are documented and alterations in kidney function caused by inhibitors of the VEGF pathway can contribute to the pathophysiology. In general, since NO has antiaggregatory, vasodilator, and antimitotic properties, this explanation seems to be logical. Hence, blockade of NO production by these drugs might lead to vasoconstriction and endothelial dysfunction, with increased risk of arterial thrombus formation. A loss of capillaries (rarefaction) has also been implicated. In addition, some researchers documented decreases in prostacyclin production by the above‐mentioned drugs,423, 426 while others highlighted increased expression and secretion of endothelin‐1.429
Figure 11.
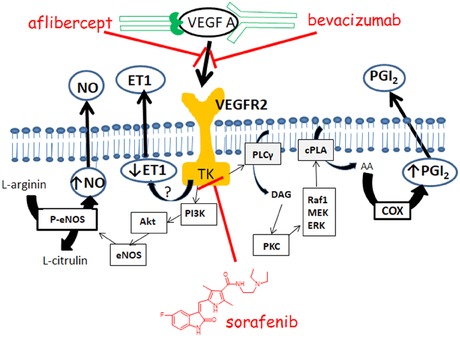
Examples of drugs acting on the VEGF pathway.
This figure shows drugs that can inhibit the VEGF pathway and hence decrease the production of NO and prostacyclin (PGI2). The role of endothelin 1 (ET‐1) is less known. Alflibercept and bevacizumab bind directly to vascular endothelial growth factor A (VEGF A), while small drugs such as sunitinib block the tyrosine kinase domain (TK) of the vascular endothelial growth factor receptor 2 (VEGFR2).
AA: arachidonic acid; Akt: protein kinase B (serine/threonine‐specific protein kinase); cPLA: cytosolic phospholipase A2; DAG: diacylglycerol; ERG: transcription factor; eNOS: endothelial NO‐synthase; MEK: mitogen‐activated protein kinase kinase; P‐eNOS: phosphorylated eNOS; PI3K: phosphoinositide 3‐kinase; PKC: protein kinase C; PLAγ: phospholipase γ1 (also known as phospholipase C); Raf: a serine/threonine‐specific protein kinase.
On one hand, the risk of hypertension is quite high, generally ranging from 20% to 40% or more depending on the drug and type of tumor. In many studies, elevations in blood pressure were found to be a good predictor of the success of the treatment and an even better indicator of prognosis.423, 425, 430 Cediranib seems to have an exceptional risk of hypertension (up to 80–87% of patients) and similarly, a very high risk is associated with the combination of bevacizumab with a tyrosine kinase inhibitor affecting the VEGF pathway.423, 426, 431 Treatment of the hypertension caused by these drugs with common antihypertensive drugs generally has a high success rate. In particular, an ACEi, due to its positive effects on the kidney, is a reasonable first‐line drug. However, there is still a certain population of patients with severe hypertension who do not respond sufficiently to antihypertensive treatment and changes in the type of anticancer drug administered, or their dosing may be necessary. Due to the underlying pathophysiology, donors of NO are currently being examined as therapeutic options. Prevention of arterial thromboembolism can be achieved by acetylsalicylic acid, but one must take into consideration the increased risk of bleeding.423, 425, 426 Other kinases inhibitors, such as MEK and BRAF inhibitors (trametinib and its combination with dabrafenib) could also cause hypertension.432 For many of the agents listed above, LV dysfunction has also been observed, with hypertension thought to contribute to the development of cardiotoxicity.
4.1.3. Erythropoietin and its analogues
Erythropoietin or its analogues are essential in the management of several types of anemia, but they have also been misused as doping agents in sport. The effect of erythropoietin is slow and dose dependent, leading to an increase of erythrocyte mass. Excessive treatment can, however, lead to increased blood viscosity, which can increase arterial blood pressure and thrombotic episodes including pulmonary embolism. Also a higher risk of arterial thrombosis has been documented. There is still some controversy regarding the underlying mechanisms, which can be complicated by concomitant patient disease (e.g., renal failure) and treatments (e.g., hemodialysis). Enhanced platelet aggregation, impaired endothelial function, and erythropoietin antibodies have been proposed as contributing mechanisms for its prothrombotic effects, but the results are contradictory. New onset hypertension or worsening of existing hypertension is relatively common and is associated with increased plasma volume.7, 170, 171, 433, 434, 435, 436, 437, 438
It should also be mentioned that female hormones (e.g., oral contraception, hormone replacement therapy) can increase blood pressure and cause arterial thrombosis, but because of their higher incidence of venous thrombotic events, they will be discussed in the chapter 3.6.1.
4.2. Drugs causing systemic arterial hypertension
4.2.1. α1‐Adrenergic receptors agonists
Agonists at α1‐adrenergic receptors are commonly used for the treatment of nasal congestion and in ophthalmology, due to their ability to cause vasoconstriction. While local vasoconstriction is the desired therapeutic goal, systemic vasoconstriction is a potentially serious side effect. These drugs are mostly applied locally in the form of nasal drops/sprays and eye drops; however, even such local administration is associated with possible short‐term elevations in the blood pressure and risk of ischemic stroke, even in young sensitive patients.47, 439 Oral administration is somewhat less frequent, but still widely used. As noted previously, the indirect sympathomimetic agent pseudoephedrine (see chapter 2.1.2) is often used in this setting,440, 441 but because of its frequent use for illegal synthesis of methamphetamine, there has been an increase in the use of the more selective α1‐adrenoceptor agonist, phenylephrine (also known as m‐synephrine, Fig. 1), in over‐the‐counter oral decongestant preparations. In a recent meta‐analysis, the administration of a 10 mg dose of phenylephrine did not significantly increase blood pressure, probably because of its low oral bioavailability.442 This is supported by available case reports on hemorrhagic strokes, which occur mostly after parenteral or local administration, and only one case after oral administration is documented.443 On the other hand, more pronounced effects of phenylephrine after higher oral doses are likely.442 Phenylephrine is commonly combined with paracetamol, which doubles the bioavailability of phenylephrine, and thus likely increase the hypertensive effect of phenylephrine, but more data are needed.444, 445
Ischemic stroke is uncommon, but can occur even in young people after year‐long misuse of nasally applied α1‐adrenoceptor agonists.47, 446, 447 Similarly, local application of these drugs in order to stop bleeding can provoke hypertension with reflex bradycardia and severe rebound hypotension.448 Bradycardia and mild hypotension were also observed after oral ingestion of a relatively large amount (0.1 g in a 5‐year old boy) of naphazoline.449
p‐Synephrine, an isomer of phenylephrine, was considered to be an alternative for ephedra products after their ban. It is a natural compound found in Citrus aurantium subsp. amara, commonly known as bitter orange, and was proposed for the management of obesity and improvement in sport performance. The alleged effectiveness of synephrine‐containing supplements is attributed to the thermogenic effects arising from its adrenergic stimulation, namely of β3‐receptors.450 p‐Synephrine is considered to be an α1‐adrenoreceptor agonist, but it has much lower affinity for α1‐adrenoreceptors than does phenylephrine. It can also have antagonistic effects at α2‐adrenergic receptors,42 although the clinical relevance of this effect is not clear. Despite controversies, the drug does not seem to have clinically relevant effects on the blood pressure, but adverse cardiac events have been reported after its consumption.450, 451, 452, 453 Moreover, the coadministration of synephrine and caffeine in dietary supplements has the same potential to cause adverse cardiac events (e.g., dysrhythmias, hypertension, heart attacks, and strokes) as does the similar combination with ephedra.454
4.2.2. Glucocorticoids
Glucocorticoids are well known to increase arterial blood pressure and in conjunction with abnormalities in glucose metabolism, can also cause cardiovascular complications.455 Interestingly, the incidence of hypertension in Cushing's syndrome ranges from 55% to 100% depending on the cause, but exogenous administration of glucocorticoids is associated with hypertension only in about 20% of cases.455, 456 The reason is not known, but possible differences in the mineralocorticoid effect between the synthetic glucocorticoids and endogenous cortisol may be responsible. Similarly, there is not clear agreement about the precise pathophysiology of glucocorticoid‐induced hypertension. The most common explanation is that the excess of glucocorticoids can also stimulate mineralocorticoid receptors and lead to increased plasma volume with increased systemic resistance as a consequence. This has been documented, but it does not explain the rapid induction of hypertension. Moreover, spironolactone, an antagonist at mineralocorticoid receptors, is not able to revert hypertension in these conditions,456, 457, 458 suggesting that stimulation of ubiquitous glucocorticoid receptors apparently plays an important role. Data showing involvement of 11β‐hydroxysteroid dehydrogenase type 1, which in contrast to the type 2 (see below in chapter 4.2.3), recovers active glucocorticoids, in hypertension supports this assumption.459 In glucocorticoid‐induced hypertension, derangement of vasodilator mediators (prostaglandin E2, prostacyclin, NO, and kinin‐kallikrein system) and variable responsiveness of the blood vessels to vasoconstrictors (catecholamines and angiotensin II) were observed. The effect of glucocorticoids on prostaglandin synthesis is understandable due to their ability to inhibit phospholipase A2. However, there is no definite link to other vascular abnormalities, some of which may be secondary to the induced hypertension (e.g., vascular remodeling and hypertrophy). There are ambiguous data on renin–angiotensin–aldosterone system activation. In general, an ACEi (captopril), or an older antagonist at angiotensin II receptors (saralasin) provided insufficient, or no protection. The best treatment of glucocorticoid hypertension is achieved by drug discontinuation or, at least, by dose reduction. From the pharmacological point of view, Ca2+ channel antagonists or other conventional vascular relaxants are indicated, possibly accompanied by diuretics.456, 457, 458
4.2.3. Licorice
Licorice—Glycyrrhiza glabra,460, 461 depending on the extraction process, can contain up to 20% glycyrrhizic acid (known also as glycyrrhizin). Glycyrrhizic acid is metabolized in vivo into glycyrrhetinic acid,462 which is a very high‐affinity inhibitor of 11β‐hydroxysteroid dehydrogenase type 2. This enzyme is crucial for a specific pattern of mineralocorticoid effects in humans. In tissues that are dependent on mineralocorticoid (aldosterone) activity, like the distal/collecting tubule cells in the kidney, this enzyme converts cortisol (hydrocortisone) into cortisone, which does not have affinity for the mineralocorticoid receptor (Fig. 12). When glycyrrhetinic acid blocks this enzyme, cortisol is not inactivated and binds to the mineralocorticoid receptor, producing the same effects normally mediated by aldosterone. Due to excessive stimulation of this pathway by cortisol, patients consuming large quantities of licorice will develop hypertension and hypokalemia. A similar condition called apparent mineralocorticoid excess is observed with nonfunctional mutations of the enzyme.463 The mineralocorticoid antagonist, spironolactone, is an effective treatment option, but the best treatment is the discontinuation of licorice intake, which restores physiological function of the system.464
Figure 12.
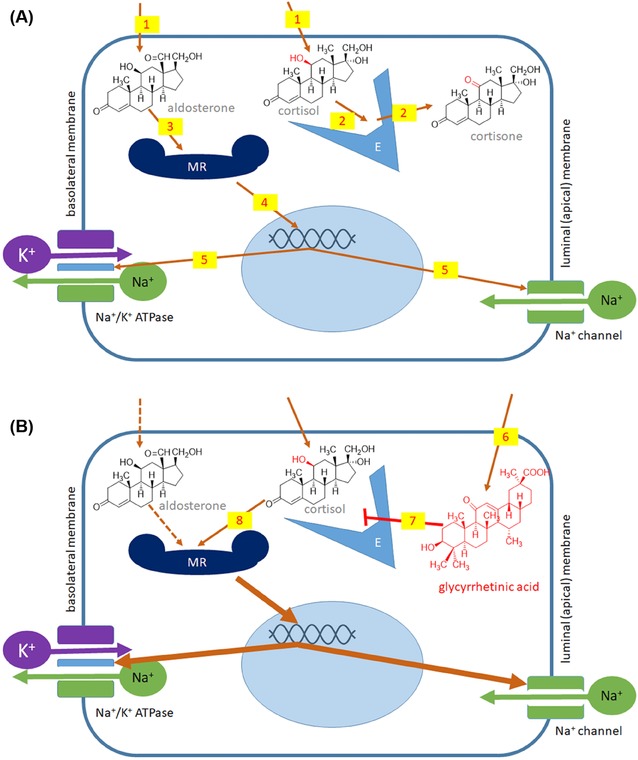
Aldosterone/cortisol activity in a mineralocorticoid sensitive kidney tubule cell under physiological conditions (A), or after inhibition by glycyrrhetinic acid (B). (1) Both aldosterone and cortisol are lipophilic enough to enter the cells, (2) cortisol is, however, immediately metabolized by the enzyme 11‐β‐hydroxysteroid dehydrogenase type 2 (E) into cortisone, which does not have affinity for the mineralocorticoid receptor (MR), (3) binding of aldosterone to MR begins the cascade. Through DNA binding (4), the expression of Na+ channels and Na+/K+‐ATPase (5) is upregulated. When glycyrrhetinic acid is present in plasma, it can penetrate into cells (6), where it has a high affinity for the enzyme (7). As a consequence of enzyme inhibition, cortisol is not inactivated and binds to MR (8) and the same cascade (part A, 4–5) occurs without the need for aldosterone.
4.2.4. Calcineurin inhibitors
The calcineurin inhibitors, cyclosporine and tacrolimus (FK506), are commonly used in organ transplant cases and less frequently in the treatment of autoimmune diseases. They can cause acute and chronic kidney damage. However, cyclosporine is frequently associated with hypertension, which ranges from 20% to 50% in autoimmune patients, to 100% in transplant patients. Kidney derangement can be the cause of the hypertension, but the pathophysiology is far more complex. In particular, at least acutely, hyperactivity of the adrenergic system plays an important role. Moreover, abnormalities in vessel reactiveness to vasoconstrictors and vasodilators, together with imbalances in their production (e.g., NO), and a blunted baroreceptor reflex have been described.465, 466, 467, 468 Cyclosporine and tacrolimus bind to different intracellular molecules before they block calcineurin.469 There are both experimental and clinical differences between cyclosporine and tacrolimus, since the latter has a lower incidence of hypertension, likely due to the lower systemic vasoconstriction caused by tacrolimus.465, 470, 471, 472 Ca2+ channel antagonists are considered to be the best treatment. When kidney damage is present, an ACEi has to be employed.465 Interestingly, in a recent study about one‐third of heart transplant patients, mostly treated with calcineurin inhibitors, did not achieve target blood pressure values after antihypertensive measures.473 Thus, the current management of hypertension seems suboptimal. Sirolimus, another immunosuppressant usually considered as an alternative to calcineurin inhibitors, also has the potential to increase blood pressure, but generally, it causes fewer cases of hypertension.466
4.2.5. Leflunomide and teriflunomide
When leflunomide is used for rheumatoid arthritis, hypertension (increases in systolic and diastolic blood pressure) can develop 2–4 weeks after initiating drug treatment with an overall incidence of 2–4.7%. Increases in heart rate can also occur.474, 475 Its active metabolite, teriflunomide, used to treat multiple sclerosis, increases the risk of hypertension as well.348, 476 The underlying mechanisms are not clear, but may involve dysregulation of the sympathetic nervous system.
4.2.6. Others
Female hormones can cause hypertension, but in this overview they were placed in the category of drugs causing venous thromboembolism as their major mechanism of cardiotoxicity.
4.3. Drugs causing pulmonary hypertension
Some drugs that act on the serotoninergic system can also cause hypertension, including pulmonary hypertension. Elevated levels of serotonin in patients with pulmonary arterial hypertension may cause vasoconstriction of pulmonary vessels and act as a growth factor for pulmonary artery smooth muscle cells.477 Aminorex (amphetamine‐like anorectic agent) administration increases 100‐fold the risk of pulmonary hypertension and its use in the 1960s led to an “epidemic” increase in pulmonary hypertension with fivefold increase in its incidence. Ten years after this epidemic, half of the patients had died, usually of right heart failure.477 A similarly acting anorectic (dex)fenfluramine also increased the risk of pulmonary hypertension; however the incidence of pulmonary hypertension produced was much less than in the case of aminorex.478, 479, 480 Overall, about 10% of the total number of cases of pulmonary arterial hypertension in the 2000s were due to anorectic agents, with the majority of cases associated with fenfluramine derivatives. Importantly, pulmonary hypertension can occur after even as little as 3 months of treatment.481 Also another drug, benfluorex, is metabolized into norfenfluramine (Fig. 13), which is thought to produce pulmonary hypertension and valvulopathy (see chapter 3.4). This drug was also withdrawn from the market due to its cardiotoxicity.482
Figure 13.
Norfenfluramine, the common metabolite of fenfluramine and benfluorex.
Pulmonary hypertension is also a documented adverse effect of dasatinib; however, pulmonary hypertension is not, or is rarely seen during treatment with other bcr/abl inhibitors, such as imatinib.422, 483 There is a hypothesis that dasatinib‐induced pulmonary hypertension may be due to the inhibition of tyrosine kinase Src, which plays a critical role in smooth muscle cell proliferation and vasoconstriction, thus altering the equilibrium of proliferation/antiproliferation between the endothelia and smooth muscle of pulmonary arteries.477
Also IFN‐α‐ and ‐β‐based treatments may be associated with an increased risk for the development of pulmonary hypertension. This is currently under study.477
4.4. Drugs causing hypotension due to direct action on the vascular system
4.4.1. α1‐Adrenergic receptor antagonism
α1‐Adrenoceptor blockers like doxazosin, used for the treatment of hypertension may induce hypotension following their initial dose. Subsequently, hypotension, in particular orthostatic hypotension may be observed in normotensive patients with benign prostatic hyperplasia. Considerably less orthostatic hypotension is associated with the use of alfuzosin and tamsulosin, which are preferred in the urology field, because they have much less affinity for α1B‐receptors located in the smooth muscle of vessels. However, there are many other drugs that cause hypotension due to antagonism of α1‐adrenergic receptors. These drugs include mainly psychiatric drugs used for the treatment of depression and psychosis, namely TCA and several antipsychotic drugs including some low‐potent antipsychotic drugs like chlorpromazine, and newer drugs from MARTA and dopamine/serotonin drugs. Hypotension, especially orthostatic hypotension, can have serious consequences, such as falls (producing fractures and lacerations), or cardiovascular effects including myocardial infarction and sudden death. TCAs are particularly potent in this sense.23, 190, 366
4.4.2. Dihydropyridine calcium channel blockers
Ca2+ channel blockers and their toxicity were discussed in chapter 2.7.
4.4.3. Other antihypertensives
Basically, all antihypertensive agents may produce hypotension as type A adverse effects and dose titration may be necessary as a preventive measure, but this is beyond the scope of the present review. Some frequently used drugs, such as ACEi and antagonist at type 1 angiotensin receptors, are known to produce hypotension after the first dose. This is particularly common in patients with hypovolemia due to diuretic treatment. In such cases, dose reduction of the diuretics or temporary withdrawal of treatment may be necessary. Serious systemic hypotension may also result from drug interactions. While, in most of cases, interactions between antihypertensive drugs are predictable and easily managed, there are some specific cases where it may cause serious consequences—for example, combination of phosphodiesterase 5 inhibitors, like sildenafil, with nitrates. This can evoke a large build‐up of cGMP (Cyclic guanosine monophosphate), marked vasodilation, and severe hypotension in some patients. Such combinations therefore represent absolute contraindications.7, 484
4.4.4. Other drugs
Parasympathomimetics, such as muscarine, can produce hypotension (see chapter 3.2.4).
4.5. Drugs causing angioedema
Angioedema (syn. angioneurotic edema) is characterized by excessive vasodilation and permeability of vessels in the deepest layers of the skin and in subcutaneous and submucosal tissue. Postcapillary venules are the primary target.485 Angioedema can arise from allergic reactions to different drugs (i.e., histamine‐mediated angioedema with urticaria), but this is beyond the scope of this review. More relevant cases are drug‐induced angioedema without urticaria, which is usually kinin‐mediated. It is typically associated with ACE inhibitor therapy, where it is attributed to excessive effects induced by bradykinin, which is physiologically degraded by the ACE enzyme.485, 486 Angioedema often affects soft tissue of the head such as lips, face, tongue, periorbital tissue and may cause obstruction of oropharyngeal airways that can be dangerous.487 Swelling is accompanied by characteristic erythema (without itching) and pain/discomfort. The symptoms typically last for 24–72 hr, followed by spontaneous remission. The incidence of ACEi‐induced angioedema is relatively low (approximately 0.2–0.7%) with higher chance in older patients and in females as compared to males.486, 487 Importantly, African Americans are four to five times more likely than Caucasians to experience angioedema. Genetic polymorphism in aminopeptidase P, which is a major degrading enzyme for bradykinin during ACE‐inhibition, seems to play a role.485, 488 Common treatments for histamine‐mediated angioedema (such as antihistamines and glucocorticoids) are not effective in this setting. A risk of angioedema has been also described for angiotensin receptor blockers, but seems to be significantly lower (0.1%) with a low incidence of cross‐reactivity (<10%).486 The mechanisms are thus much less clear. Of note, dual inhibitors of ACE and neutral endopeptidase—neprilysin, such as omapatrilate, have shown an unacceptably high incidence and severity of angioedema 489 and the development of such agents was stopped. The increased risk is attributable to the fact that both ACE and neprilysin are important for bradykinin degradation. In contrast, very promising are recent data on a combination of neprilysin inhibition with antagonists at angiotensin II receptors in the treatment of chronic heart failure with reduced ejection fraction. The first agent sacubitril–valsartan complex showed both remarkable efficacy and safety, with a relatively low risk of angioedema.490 However, sacubitril–valsartan cannot be combined with ACE inhibitors and 36 hrs washout period is necessary when switching patients from ACEi to this new treatment.216 Other drugs inducing nonhistamine angioedema are NSAIDs with an incidence 0.1–0.3% and the mechanisms seems to be based on COX inhibition.487
4.6. Drugs causing venous thromboembolism
4.6.1. Female hormones and drugs acting at this level
4.6.1.1. Hormonal contraception
Hormonal contraception is frequently employed. It is estimated that about 80% of women in some developed countries have used oral contraception, at least once, during their lives.491, 492 Besides reliably hindering conception, combined oral contraception has several other advantages with respect to women's health, including lower dysmenorrhoea, reduced acne, hirsutism, and reduced risk of endometrial and ovarian cancers.493 However, this therapy can also lead to serious side effects. Data are not always consistent, but currently, it is well documented that elevated blood pressure, increased risk of acute myocardial infarction, and serious thromboembolism with a risk of ischemic stroke highlight the cardiovascular risks associated with hormonal contraceptive use.494, 495, 496, 497, 498, 499 The risk is generally low, especially since hormonal contraception is used mainly by young, “healthy” women. However, in combination with other concomitant cardiovascular disease/risk factors, including elevated blood pressure, obesity, hyperlipidemia, thrombophilia, and smoking, it might produce serious consequences.500, 501, 502, 503 In particular, venous thromboembolism seems to be the most important relevant cardiovascular risk in this “healthy” group of women.7 Smoking was found in the majority of, but not in all studies, to be a crucial enhancer of negative cardiovascular outcomes in women taking oral contraceptives.495, 504 Moreover, although an increase in arterial blood pressure is generally low, it cannot be ignored since in a recent study contraceptives were associated with hypertension in 19% of women below the age of 40.505 In general, the cardiovascular risk is present only in current users, while past use confers a markedly diminished risk.494, 495, 498 It is not clear whether decreasing the dose of estrogen, mostly ethinyl estradiol, and the selection of a progestin, could reduce risk. Decreasing the dose of estrogen from 50 to 20 μg decreased the risk of myocardial infarction and tended to decrease the risk of thrombotic stroke (see Fig. 14). However, vaginal rings with 15 μg of ethinyl estradiol, or oral contraception with 20 μg can still have negative cardiovascular effects.495, 506 In addition, lowering the dose of estrogen does not seem to diminish the effect on blood pressure.498, 507 The selection of progestin remains controversial, since progestins can have different additional effects. For example, they can act as agonists or antagonists at androgen receptors, they can have estrogenic potential and the relatively new compound, drospirenone, has an antagonistic effect at mineralocorticoid receptors.508, 509 It has been suggested that the third‐generation progestins such as desogestrel and gestodene might have lower risk than the second‐generation drugs levonorgestrel and norgestimate, but so far results are mixed.504, 510 A few years ago, mentioned drospirenone, coined a fourth‐generation progestins, was considered to be the ideal choice, due to its neutral or reducing effect on body weight and blood pressure. However, recent studies showed that drospirenone has a higher risk of thromboembolism and acute myocardial infarction than does levonorgestrel. These data markedly changed prescription recommendations particularly in high‐risk populations.494, 511, 512
Figure 14.
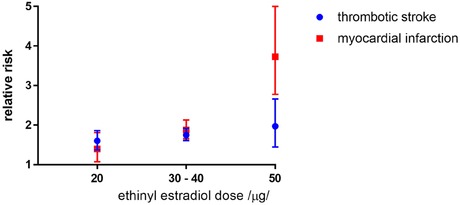
Relative risk of stroke and myocardial infarction in relation to the dose of estrogen (ethinyl estradiol). The term thrombotic stroke means cerebral infarction due to cerebral thrombosis or embolism. Figure shows the relative risk with 95% confidence intervals. The figure was prepared using data from the clinical study of Lidegaard et al.494
While it is believed that contraceptive preparations containing only progestins (pills, implants, intrauterine device) are devoid of clinically negative effects on the cardiovascular system,494, 497 very little is known about progestin's effects on the cardiovascular system. Progestins seem to oppose or modify some of the cardiovascular effects of estrogens in combined preparations.513 It is well known that estrogens, in physiological doses/concentrations, have positive effects on the vascular system likely due to activation of an endothelial isoform of NO synthase (eNOS), decreasing low density lipoprotein (LDL) cholesterol and increasing High density lipoprotein (HDL) cholesterol. On the other hand, they may increase triglyceride levels and modify the ratio between procoagulatory/anticoagulatory mediators in favor of the former.495, 509, 510, 514 Progestins do not appear to influence the coagulation cascade, but rather antagonize the positive lipid effect of estrogens. Notwithstanding, mixed results on lipid spectra were found in human studies.510, 515, 516 There must also be other positive effects of estrogens on the vascular system, because even though oral contraception had a very negative effect on HDL cholesterol in macaques, it did not worsen atherosclerosis. Moreover, pure estrogen reduced the extent of atherosclerosis.513, 517 In addition, estrogens increase the synthesis of angiotensinogen and subsequent activation of the renin–angiotensin–aldosterone system, which likely represents the basis of their blood pressure elevating properties.509, 518 However, pure estrogens also block renin activity, so pure estrogens can even decrease blood pressure, in contrast to combined contraceptive preparations.518, 519, 520
4.6.1.2. Hormonal replacement therapy (HRT)
Another clinical use of female hormones was HRT in postmenopausal women were the goal of treatment was to replace the missing estrogens with the aim of obtaining positive effects on the vascular system. In this setting, lower doses of estrogens (generally 2.5–5 μg of ethinylestradiol equivalents daily) were employed.495 HRT, likely due to the lower hormone doses, also demonstrated a positive effect on certain risk factors of coronary heart disease: decreases in heart rate, arterial blood pressure, LDL‐cholesterol, and increases in HDL‐cholesterol.519, 520, 521, 522, 523 Observational studies also suggested its positive effect and thus HRT was initially considered to be a very useful therapeutic tool for protecting against coronary artery disease, which seems to be reasonable due to the relationship between the onset of atherosclerosis and menopause.524 However, the large prospective Women´s Health Initiative study, focusing on older postmenopausal women (over age 60 years), was a huge disappointment for the field because it identified an increased risk of breast cancer with combined estrogen–progesterone HRT, and an increased risk of stroke and venous thromboembolism. Furthermore, protection against coronary heart disease was not observed in either the combined HRT, or the estrogen replacement group.525 There has been extensive research regarding the cardiovascular outcomes observed after HRT and it appears that the combination of conjugated equine estrogen, which is a mixture of estrogens from horse urine that also contains progestins, androgens, and other substances, with progestin medroxyprogesterone acetate, is not ideal.522, 524 Differences between estradiol and conjugated equine estrogen were found.521 Recent studies with estradiol used in HRT showed decreased mortality from coronary heart disease, or a decrease in combined cardiovascular endpoints.523, 526 Another important factor for cardiovascular toxicity is the time of HRT initiation, since a very late start, or treatment several years after menopause is associated with a worsening of coronary heart disease in contrast to treatment initiated in younger women.524, 525, 527 On the other hand, “rebound” risk of death from coronary heart disease is present within the first year after HRT is stopped.526 After 1 year, the increased risk disappears. Either way, it must be reiterated that HRT, like oral contraception, can increase the risk of venous thromboembolism, which can be fatal. It is not clear if the risk of thromboembolism can be modulated by selection of a specific progestin or estrogen.522, 527, 528
4.6.1.3. Selective estrogen receptor modulators (SERM)
SERM comprise a group of compounds that selectively stimulate estrogen receptors in one tissue and block them in another. These drugs have a combination of estrogen agonist, partial agonist, or antagonist activities depending on the tissue and they are not fully homogenous in terms of their activity within the group. Tamoxifen and raloxifen are the most well studied of these drugs. Both of them, like estrogen, unambiguously increase the risk of venous thromboembolism; however, data regarding the risk of stroke and acute myocardial infarction are divergent. There are data showing that tamoxifen can increase the risk of stroke, while raloxifen seems rather to decrease it.529, 530, 531, 532
4.6.1.4. Tibolone and other estrogenic medications
Another specific drug is tibolone, which is metabolized into three metabolites that act as agonists at estrogen, progestogen, and androgen receptors and also influence estrogen metabolism. Surprisingly, tibolone does not increase the risk of venous thromboembolism, but one clinical study was stopped prematurely due to a higher incidence of stroke.533
As expected, the cytostatic agent estramustin phosphate, which contains the estradiol moiety in its structure, possesses mild, but significant risk of deep venous thrombosis.534
4.6.2. Thalidomide and lenalinomide
Thalidomide and lenalinomide are well documented to increase the risk of venous thromboembolism. Interestingly, the risk is not elevated when they are used as monotherapy, but increases in combination with other cytostatics (in particular with doxorubicin) and corticoids. Moreover, the risk of venous thromboembolism is expressed only in patients with multiple myeloma. The risk of venous thromboembolism in combined regimens can reach 30% and there is apparently no difference between lenalinomide and thalidomide. The implicit mechanism is not yet clear, but elevated levels of Factor VIII and von Willebrand factor have been observed.535, 536, 537, 538
4.6.3. Strontium ranelate
The question of whether strontium confers a higher risk of venous thromboembolism is unresolved. In addition, the drug can potentially cause acute myocardial infarction; however, causality has not been clearly confirmed.539, 540
5. CONCLUSION
Cardiovascular toxicity represents an important risk associated with the administration of several classes of clinically and illegally used drugs and can present even after a single administration, or after an overdose. The most clinically important case of cardiovascular toxicities and their first line therapies are shown in Table 9. Acute poisoning is mostly overt, in contrast to the chronic negative cardiovascular effects of these agents. Chronic effects may not be apparent for a long time and early diagnosis of these toxicities is challenging. Our current knowledge of these cardiovascular toxic effects is mostly based on long‐term clinical trials, or clinical experience and hence largely unknown in the case of novel drugs. Since mild changes in arterial blood pressure and heart rate might be later associated with higher cardiovascular mortality, all drugs causing such cardiovascular effects must be recognized for their potential cardiac risk to the patient. This comprehensive review summarizes our current understanding of the cardiovascular toxicities of known substances and we suppose that such information can be helpful both for medical doctors, pharmacists, toxicologists, and pharmacologists who conduct research in this field.
Table 9.
Cardiovascular toxicity associated with acute intoxications—overview of selected clinically important cases and their treatment
| Class of drugs | Typical representatives | Clinical presentation | Treatment |
|---|---|---|---|
| CNS‐stimulants/indirect sympathomimetic agents | Cocaine, amphetamine, and related agents | Tachycardia, hypertension and hypertensive crises, acute coronary syndrome, aortic artery dissection, QT prolongation, and dysrhythmias including ventricular fibrillation | Benzodiazepines and targeted symptomatic treatment as necessary (e.g., nitrates in acute coronary syndrome), avoid β‐blockers, lidocaine administration is controversial in dysrhythmias |
| Calcium channel blockers | Nondihydropyridines (verapamil, diltiazem) and dihydropyridines (such as nifedipine or amlodipine) | Severe bradycardia, transient reflex tachycardia (with dihydropyridines), AV blocks, cardiac arrest, hypotension, left ventricular dysfunction | Prevention of GIT absorption—particularly in sustained release formulations (incl. whole bowel irrigation). i.v. calcium, high‐dose insulin‐glucose therapy, i.v. isotonic fluids, atropine, temporal pacing, glucagon (disputed), vasopressors (e.g., noradrenaline), lipid emulsions (in resistant cases) |
| β‐Blockers | Propranolol, metoprolol, carvedilol, and others | Bradycardia, cardiac arrest, LV dysfunction, hypotension | Atropine, glucagon, isotonic fluids, adrenaline, high‐dose insulin‐glucose therapy, i.v. calcium salts, lipid emulsion therapy |
| Cardioactive steroids (glycosides) | Digoxin, digitoxin, lanatosides, oubain, and herbs and animal toxins containing such agents (see Fig. 9) | Almost any dysrhythmia can occur. Often presented as sinus bradycardia, AV‐blocks, ventricular extrasystoles, ventricular tachycardia, or fibrillations | Atropine for bradycardia and temporal pacing may be necessary, correction of hypokalemia, lidocaine for ventricular tachydysrhythmias, digoxin‐specific antibody fragments for moderate to severe cases (effective against digoxin and some plant poisoning—see chapter 3.2.3) |
| Local anesthetics | Bupivacaine, mepivacaine, ropivacaine, lidocaine, etc. | Bradycardia, asystole or ventricular tachydysrhythmias, palpitations, hypotension | Oxygenation, lipid emulsion therapy for cardiac arrest adrenaline (but not vasopressin), amiodarone may be used in ventricular tachydysrhythmias |
| Drug‐induced long QT syndrome | Miscellaneous drugs—see Table 5 | QT prolongation, ventricular ectopic beats, torsade de pointes, ventricular fibrillation, sudden death | Correct hypokalemia, i.v. magnesium (first‐line approach bolus or infusion), temporary transvenous overdrive pacing if magnesium is ineffective (at rates ≈ 100), or isoprenaline |
| Tricyclic antidepressants | Amitriptyline, desimipramine, imipramine, nortriptyline, dosulepin, or doxepin | Life‐threatening dysrhythmias—typically wide QRS complex tachycardia, hypotension | Sodium bicarbonate (targeted arterial pH should be a in a range of 7.45–7.55), lipid emulsion therapy is being evaluated |
ACKNOWLEDGMENTS
This article was supported by TOX‐OER project No. 2015‐1‐ES01‐KA203‐015957 and Centre for the Study of Toxic and Protective Effects of Drugs on Cardiovascular System of Charles University (UNCE 204019/304019/2012). Authors have been supported by Research Projects Progres Q40 and Progres Q42. Authors would like to thank to Prof. Radomír Hrdina for his critical comments.
Biographies
Přemysl Mladěnka is Associate Professor, Head of the Group of Cardiovascular and Respiratory Pharmacology and Toxicology, Department of Pharmacology and Toxicology, Faculty of Pharmacy, Charles University, Czech Republic. He received MSc/PharmD in Pharmacy in 2003/2009 and PhD in Pharmacology and Toxicology in 2008. Since 2014 he has been an Associate Professor of Human and Veterinary Pharmacology — all at the Faculty of Pharmacy in Hradec Králové, Charles University. His primary research focus encompasses cardiovascular pharmacology and toxicology of both natural and synthetic drugs with main emphasis on their positive and negative effects on the heart, arterial blood pressure, and platelets. He received the prize of the Minister of the Czech Republic Education, Youth and Sports for remarkable results during doctoral study (2009) and The young scientist award of the Danone institution for the best work in the field of nutrition (2011). He is author of 39 peer‐reviewed publications mainly focused on the cardiovascular system and transition metals that have been cited more than 480 times.
Lenka Applová is making her PhD at the department of Pharmacology and Toxicology, at the Faculty of Pharmacy, Charles University, under the supervision of Assoc. Prof. Mladěnka. The topic of her dissertation work focuses on platelets. As part of her doctoral study, she has been teaching selected chapters from toxicology.
Jiri Patocka, Professor of toxicology, Institute of Radiology, Toxicology and Civil Protection, Faculty of Health and Social Studies, University of South Bohemia Ceske Budejovice and researcher in Biomedical Research Center, University Hospital, Hradec Kralove, Czech Republic. Graduated in chemistry and physics, PhD in biochemistry and DrSc in toxicology. He has published more than 400 scientific papers (with more than 3000 citations, H‐index 28) and presented more than 200 lectures and posters on scientific meetings. Author and co‐author of 20 books and/or textbooks mainly in the area of toxicology.
Vera Marisa Costa is a researcher in the Toxicology Group of the Applied Molecular Biosciences (UCIBIO, REQUIMTE) Research Unit and a lecturer at the Faculty of Pharmacy of University of Porto, Portugal. She graduated in Pharmaceutical Sciences from the latter institution in 2004 and she obtained her PhD in Toxicology in 2009. Her primary research focus is cardiovascular pharmacology and toxicology of catecholamines, anticancer drugs, and drugs of abuse. She belongs to the editorial Board of Cardiovascular Regenerative Medicine since 2015. She is author of 39 peer‐reviewed international publications mainly focused on cardiotoxicity that have been cited more than 600 times.
Fernando Remião is Associate Professor at the Faculty of Pharmacy of University of Porto, Portugal. He graduated in Pharmaceutical Sciences at the same institution in 1993 where he also obtained his PhD in Toxicology in 2002. He is also a Senior Researcher in the Toxicology Group of the Applied Molecular Biosciences (UCIBIO, REQUIMTE) Research Unit. He is the Associated Editor of the journal Toxicology and Applied Pharmacology and the author/co‐author of more than 100 papers published in international journals.
Jana Pourová a senior lecturer in Pharmacology at the Faculty of Pharmacy, Charles University. In 1995, she graduated from the same faculty, and in 1999, she received there her PhD in Pharmacology. She is author of 9 publications addressing the respiratory and cardiovascular pharmacology that have been cited more than 200 times.
Aleš Mladěnka, M.D., is the head of the Gynecologic inpatient department, Clinic of Gynecology and Obstetrics, University hospital Ostrava, Czech Republic. He takes care primary of oncogynecologic patients during clinical studies.
Jana Karlíčková is a senior lecturer, member of the Group of Cardiovascular and Respiratory Pharmacology and Toxicology, Department of Pharmaceutical Botany, Faculty of Pharmacy, Charles University, Czech Republic. She received MSc/Pharm.D. in Pharmacy in 1991/1996 and PhD in Toxicology of natural products in 2002. Her research focuses on biological activity of natural compounds.
Luděk Jahodář is Professor of pharmacognosy. He was the Dean of the Faculty of Pharmacy in Hradec Králové, Charles University, Czech Republic (1994‐1997). Educated at the Comenius University in Bratislava (1967/72 MrSc/RNDr), he has carried out postgraduate studies at the Faculty of Pharmacy, Charles University (1978‐1983, Ph.D.), he received grades of Associate Professor (1988) and Professor (1995) at the same University. He is author of 81 peer‐ reviewed publications on plant secondary metabolities and their biological activities (880 citations) and author/coauthor four monographs focused on plant toxins and human plant poisoning. Prof Jahodá? is a member of several professional and academic societies. Among his numerous awards are the Gold Medal of the Charles University in Prague; the Silver Medal of Comenius University in Bratislava, Slovak Republic; the Rudolf Skarnitzl Medal of Czech Pharmaceutical Society etc.
Marie Vopršalová is the senior lecturer in Toxicology at the department of Pharmacology and Toxicology, Faculty of Pharmacy, Charles University, Czech Republic. She received her MSc./PharmD in Pharmacy in 1982 and PhD in Pharmacology in 1992. Her research focus is on respiratory and cardiovascular pharmacology, both in vivo and ex‐vivo conditions. She has been teaching toxicology since 1993.
Kurt J. Varner is the Lederle Laboratories/David R. Bethune Professor and Head of the Department of Pharmacology and Experimental Therapeutics at the Louisiana State University Health Sciences Center (LSUHSC) in New Orleans, USA. He received his PhD in Pharmacology and Toxicology from Michigan State University in 1987. He was a postdoctoral fellow in the Department of Pharmacology and the Cardiovascular Center at the University of Iowa from 1988‐1991, before taking a position as an Assistant Professor in the Department of Pharmacology and Experimental Therapeutics at LSUHSC. His primary research focus centers on the cardiac and cardiovascular pharmacology and toxicology of pharmaceutical agents, drugs of abuse and environmental pollutants. In 2016, he was recognized as the Kenneth Moore, Distinguished Alumnus by the Department of Pharmacology and Toxicology at Michigan State University. Is an Associate Editor for Cardiovascular Toxicology and has over 60 peer reviewed publications.
Martin Štěrba is Associate Professor, Deputy Head of the Department, Group Leader of the Laboratory for in vivo study of the cardiovascular toxicity of anticancer drugs, Senior Investigator of the Research Center of Charles University (Cardiotox HK), Department of Pharmacology, Faculty of Medicine in Hradec Králové, Charles University, Czech Republic. He received his MSc/PharmD in Pharmacy in 2002/2007 from the Faculty of Pharmacy in Hradec Králové, Charles University and PhD in Medical Pharmacology in 2008 from the Faculty of Medicine in Hradec Králové, Charles University. He was appointed as an Associate Professor of Medical Pharmacology at Faculty of Medicine in Hradec Králové, Charles University, Czech Republic. His primary research focus is on the cardiovascular toxicity of anticancer drugs (since 2000)—molecular mechanisms, monitoring and biomarkers, risk factors, and pharmacological cardioprotection. Among others he has been a winner of the Prize for Pharmacy 2009 awarded by the French Embassy in Prague. He is a member of the editorial board of ESC Heart Failure (Wiley, since 2015, Associate Editor since 2017) and Cardio‐Oncology (BioMed Central, since 2015). He is the author of 44 publications mainly related to the cardiovascular toxicity of anticancer drugs that have been cited more than 1000 times.
Mladěnka P, Applová L, Patočka J, et al. Comprehensive review of cardiovascular toxicity of drugs and related agents. Med Res Rev. 2018;38:1332–1403. https://doi.org/10.1002/med.21476
Contributor Information
Přemysl Mladěnka, Email: mladenkap@faf.cuni.cz.
Martin Štěrba, Email: sterbam@lfhk.cuni.cz.
REFERENCES
- 1. Klaassen CD. Casarett and Doull's Toxicology: The Basic Science of Poisons. 7th ed New York: McGraw‐Hill; 2008. [Google Scholar]
- 2. Reichel F‐X, Ritter L. Illustrated Handbook of Toxicology. 4th ed Stuttgart: Thieme; 2011. [Google Scholar]
- 3. Olson KR, Anderson, IB , Benowitz NL, Poisoning & Drug Overdose. 5th ed New York: McGraw‐Hill; 2006. [Google Scholar]
- 4. Shannon MW, Borron SW, Burns M. Haddad and Winchester´s Clinical Management of Poisoning and Drug Overdose. 4th ed Philadelphia: Sunders/Elsevier; 2007. [Google Scholar]
- 5. Bryson PD. Comprehensive Review in Toxicology for Emergency Clinicians. 3rd ed Washington: Taylor & Francis; 1997. [Google Scholar]
- 6. Ellenhorn MJ, Schonwald S, Ordog G, Wasserberger J. Ellenhorn´s Medical Toxicology. 2nd ed Baltimore: Williams & Wilkins; 1997. [Google Scholar]
- 7. Brunton L, Chabner B, Knollman B. Goodman & Gilman's The Pharmacological Basis of Therapeutics. 12th ed New York: Mc Graw‐Hill; 2011. [Google Scholar]
- 8. Lewis RA. Lewis´ Dictionary of Toxicology. Boca Raton, FL: CRC Press LLC; 1998. [Google Scholar]
- 9. Loomis TA, Wallace HA. Loomis´s Essentials of Toxicology. 4th ed San Diego: Academic Press; 1996. [Google Scholar]
- 10. Blankfield RP, Iftikhar IH. Food and drug administration regulation of drugs that raise blood pressure. J Cardiovasc Pharmacol Ther. 2015;20:5–8. [DOI] [PubMed] [Google Scholar]
- 11. Borer JS. Heart rate: From risk marker to risk factor. Eur Heart J Suppl. 2008;10:F2–F6. [Google Scholar]
- 12. Been F, Lai FY, Kinyua J, Covaci A, van Nuijs AL. Profiles and changes in stimulant use in Belgium in the period of 2011–2015. Sci Total Environ. 2016;565:1011–1019. [DOI] [PubMed] [Google Scholar]
- 13. European Drug Report 2015: Trends and Developments. In: European Drug Report Lisbon: European Monitoring Centre for Drugs and Drug Addiction (EMCDDA) 2015.
- 14. Ahtee L, Michal F. Proceedings: Effects of sympathomimetic amines on rabbit platelet aggregation in vitro. Br J Pharmacol. 1972;44:363P–364P. [PMC free article] [PubMed] [Google Scholar]
- 15. Sharma A, Couture J. A review of the pathophysiology, etiology, and treatment of attention‐deficit hyperactivity disorder (ADHD). Ann Pharmacother. 2014;48:209–225. [DOI] [PubMed] [Google Scholar]
- 16. Sjoqvist F. Psychotropic drugs (2). Interaction between monoamine oxidase (MAO) inhibitors and other substances. Proc R Soc Med. 1965;58:967–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bauman JL, Grawe JJ, Winecoff AP, Hariman RJ. Cocaine‐related sudden cardiac death: A hypothesis correlating basic science and clinical observations. J Clin Pharmacol. 1994;34:902–911. [DOI] [PubMed] [Google Scholar]
- 18. O'Leary ME, Hancox JC. Role of voltage‐gated sodium, potassium and calcium channels in the development of cocaine‐associated cardiac arrhythmias. Br J Clin Pharmacol. 2010;69:427–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schwartz BG, Rezkalla S, Kloner RA. Cardiovascular effects of cocaine. Circulation. 2010;122:2558–2569. [DOI] [PubMed] [Google Scholar]
- 20. Stankowski RV, Kloner RA, Rezkalla SH. Cardiovascular consequences of cocaine use. Trends Cardiovasc Med. 2015;25:517–526. [DOI] [PubMed] [Google Scholar]
- 21. Amsterdam EA, Wenger NK, Brindis RG, et al. AHA/ACC guideline for the management of patients with non‐ST‐elevation acute coronary syndromes: Executive summary: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;130:2354–2394. [DOI] [PubMed] [Google Scholar]
- 22. Khawaja IS, Feinstein RE. Cardiovascular effects of selective serotonin reuptake inhibitors and other novel antidepressants. Heart Dis. 2003;5:153–160. [DOI] [PubMed] [Google Scholar]
- 23. Roose SP, Glassman AH, Giardina EGV, et al. Cardiovascular effects of imipramine and bupropion in depressed‐patients with congestive‐heart‐failure. J Clin Psychopharmacol. 1987;7:247–251. [PubMed] [Google Scholar]
- 24. Roose SP. Considerations for the use of antidepressants in patients with cardiovascular disease. Am Heart J. 2000;140:584–588. [DOI] [PubMed] [Google Scholar]
- 25. Glassman AH, Preud'homme XA. Review of the cardiovascular effects of heterocyclic antidepressants. J Clin Psychiatry. 1993;54(Suppl):16–22. [PubMed] [Google Scholar]
- 26. Arnsten AF, Pliszka SR. Catecholamine influences on prefrontal cortical function: Relevance to treatment of attention deficit/hyperactivity disorder and related disorders. Pharmacol Biochem Behav. 2011;99:211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arcieri R, Germinario EA, Bonati M, et al. Cardiovascular measures in children and adolescents with attention‐deficit/hyperactivity disorder who are new users of methylphenidate and atomoxetine. J Child Adolesc Psychopharmacol. 2012;22:423–431. [DOI] [PubMed] [Google Scholar]
- 28. Hennissen L, Bakker M, Banaschewski T, et al. Cardiovascular effects of stimulant and non‐stimulant medication for children and adolescents with ADHD: A systematic review and meta‐analysis of trials of methylphenidate, amphetamines and atomoxetine. CNS Drugs. 2017;31:199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Munk K, Gormsen L, Kim WY, Andersen NH. Cardiac arrest following a myocardial infarction in a child treated with methylphenidate. Case Rep Pediatr. 2015;2015:905097. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30. Shin JY, Roughead EE, Park BJ, Pratt NL. Cardiovascular safety of methylphenidate among children and young people with attention‐deficit/hyperactivity disorder (ADHD): Nationwide self controlled case series study. Brit Med J. 2016;353:i2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Martinez‐Raga J, Knecht C, Szerman N, Martinez MI. Risk of serious cardiovascular problems with medications for attention‐deficit hyperactivity disorder. CNS Drugs. 2013;27:15–30. [DOI] [PubMed] [Google Scholar]
- 32. Garcia Diaz E, Martin Folgueras T. Systematic review of the clinical efficacy of sibutramine and orlistat in weight loss, quality of life and its adverse effects in obese adolescents. Nutr Hosp. 2011;26:451–457. [DOI] [PubMed] [Google Scholar]
- 33. Krentz AJ, Fujioka K, Hompesch M. Evolution of pharmacological obesity treatments: Focus on adverse side‐effect profiles. Diabetes Obes Metab. 2016;18:558–570. [DOI] [PubMed] [Google Scholar]
- 34. James WP, Caterson ID, Coutinho W, et al. Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects. N Engl J Med. 2010;363:905–917. [DOI] [PubMed] [Google Scholar]
- 35. Hayes JF, Bhaskaran K, Batterham R, Smeeth L, Douglas I. The effect of sibutramine prescribing in routine clinical practice on cardiovascular outcomes: A cohort study in the United Kingdom. Int J Obes (Lond). 2015;39:1359–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou YH, Ma XQ, Wu C, et al. Effect of anti‐obesity drug on cardiovascular risk factors: A systematic review and meta‐analysis of randomized controlled trials. PLoS One. 2012;7:e39062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Panenka WJ, Procyshyn RM, Lecomte T, et al. Methamphetamine use: A comprehensive review of molecular, preclinical and clinical findings. Drug Alcohol Depend. 2013;129:167–179. [DOI] [PubMed] [Google Scholar]
- 38. Carvalho M, Carmo H, Costa VM, et al. Toxicity of amphetamines: An update. Arch Toxicol. 2012;86:1167–1231. [DOI] [PubMed] [Google Scholar]
- 39. Yamamoto BK, Moszczynska A, Gudelsky GA. Amphetamine toxicities: Classical and emerging mechanisms. Ann N Y Acad Sci. 2010;1187:101–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rothman RB, Baumann MH, Dersch CM, et al. Amphetamine‐type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse. 2001;39:32–41. [DOI] [PubMed] [Google Scholar]
- 41. Vansal SS, Feller DR. Direct effects of ephedrine isomers on human beta‐adrenergic receptor subtypes. Biochem Pharmacol. 1999;58:807–810. [DOI] [PubMed] [Google Scholar]
- 42. Ma G, Bavadekar SA, Schaneberg BT, Khan IA, Feller DR. Effects of synephrine and beta‐phenethylamine on human alpha‐adrenoceptor subtypes. Planta Med. 2010;76:981–986. [DOI] [PubMed] [Google Scholar]
- 43. Ma G, Bavadekar SA, Davis YM, et al. Pharmacological effects of ephedrine alkaloids on human alpha(1)‐ and alpha(2)‐adrenergic receptor subtypes. J Pharmacol Exp Ther. 2007;322:214–221. [DOI] [PubMed] [Google Scholar]
- 44. Andraws R, Chawla P, Brown DL. Cardiovascular effects of ephedra alkaloids: A comprehensive review. Prog Cardiovasc Dis. 2005;47:217–225. [DOI] [PubMed] [Google Scholar]
- 45. Morgenstern LB, Viscoli CM, Kernan WN, et al. Use of Ephedra‐containing products and risk for hemorrhagic stroke. Neurology. 2003;60:132–135. [DOI] [PubMed] [Google Scholar]
- 46. Haller CA, Benowitz NL. Adverse cardiovascular and central nervous system events associated with dietary supplements containing ephedra alkaloids. N Engl J Med. 2000;343:1833–1838. [DOI] [PubMed] [Google Scholar]
- 47. Cantu C, Arauz A, Murillo‐Bonilla LM, Lopez M, Barinagarrementeria F. Stroke associated with sympathomimetics contained in over‐the‐counter cough and cold drugs. Stroke. 2003;34:1667–1672. [DOI] [PubMed] [Google Scholar]
- 48. Brust JC. Editorial comment—Over‐the‐counter cold remedies and stroke. Stroke. 2003;34:1673. [DOI] [PubMed] [Google Scholar]
- 49. Kernan WN, Viscoli CM, Brass LM, et al. Phenylpropanolamine and the risk of hemorrhagic stroke. N Engl J Med. 2000;343:1826–1832. [DOI] [PubMed] [Google Scholar]
- 50. Fleming GA. The FDA, regulation, and the risk of stroke. N Engl J Med. 2000;343:1886–1887. [DOI] [PubMed] [Google Scholar]
- 51. Kernan WN, Viscoli CM, Brass LM, Horwitz RI. Phenylpropamolamine and hemorrhagic stroke—Author reply to comments of Ernst and Hartz. N Engl J Med. 2001;344:1095. [PubMed] [Google Scholar]
- 52. Haller CA, Jacob P, Benowitz NL. Short‐term metabolic and hemodynamic effects of ephedra and guarana combinations. Clin Pharmacol Ther. 2005;77:560–571. [DOI] [PubMed] [Google Scholar]
- 53. Mack F, Bonisch H. Dissociation constants and lipophilicity of catecholamines and related compounds. Naunyn Schmiedebergs Arch Pharmacol. 1979;310:1–9. [DOI] [PubMed] [Google Scholar]
- 54. National Institute on Drug Abuse . Methamphetamine abuse and addiction In: NIH Publication No. 06–4210. Rockville, MD: National Clearinghouse on Alcohol and Drug Information; 2006. (September, revised). [Google Scholar]
- 55. Vree TB, Muskens AT, van Rossum JM. Some physico‐chemical properties of amphetamine and related drugs. J Pharm Pharmacol. 1969;21:774–775. [DOI] [PubMed] [Google Scholar]
- 56. Kaye S, McKetin R. Cardiotoxicity associated with methamphetamine use and signs of cardiovascular pathology among methamphetamine users. In: Technical report No. 238. University of New South Wales, Sydney: National Drug and Alcohol Research Center (NDARS); 2005.
- 57. Brayfield A. Martindale: The Complete Drug Reference. 38th ed London: Pharmaceutical Press; 2014. [Google Scholar]
- 58. Hutson PH, Pennick M, Secker R. Preclinical pharmacokinetics, pharmacology and toxicology of lisdexamfetamine: A novel d‐amphetamine pro‐drug. Neuropharmacology. 2014;87:41–50. [DOI] [PubMed] [Google Scholar]
- 59. Capela JP, Ruscher K, Lautenschlager M, et al. Ecstasy‐induced cell death in cortical neuronal cultures is serotonin 2A‐receptor‐dependent and potentiated under hyperthermia. Neuroscience. 2006;139:1069–1081. [DOI] [PubMed] [Google Scholar]
- 60. Rosecrans JA, Glennon RA. The effect of MDA and MDMA (“ecstasy”) isomers in combination with pirenpirone on operant responding in mice. Pharmacol Biochem Behav. 1987;28:39–42. [DOI] [PubMed] [Google Scholar]
- 61. Costa VM, Carvalho F, Duarte JA, Bastos Mde L, Remiao F. The heart as a target for xenobiotic toxicity: The cardiac susceptibility to oxidative stress. Chem Res Toxicol. 2013;26:1285–1311. [DOI] [PubMed] [Google Scholar]
- 62. Shenouda SK, Varner KJ, Carvalho F, Lucchesi PA. Metabolites of MDMA induce oxidative stress and contractile dysfunction in adult rat left ventricular myocytes. Cardiovasc Toxicol. 2009;9:30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Badon LA, Hicks A, Lord K, et al. Changes in cardiovascular responsiveness and cardiotoxicity elicited during binge administration of Ecstasy. J Pharmacol Exp Ther. 2002;302:898–907. [DOI] [PubMed] [Google Scholar]
- 64. Burgess C, O'Donohoe A, Gill M. Agony and ecstasy: A review of MDMA effects and toxicity. Eur Psychiatry. 2000;15:287–294. [DOI] [PubMed] [Google Scholar]
- 65. Sano R, Hasuike T, Nakano M, Kominato Y, Itoh H. A fatal case of myocardial damage due to misuse of the “designer drug” MDMA. Leg Med (Tokyo). 2009;11:294–297. [DOI] [PubMed] [Google Scholar]
- 66. Kaye S, Darke S, Duflou J. Methylenedioxymethamphetamine (MDMA)‐related fatalities in Australia: Demographics, circumstances, toxicology and major organ pathology. Drug Alcohol Depend. 2009;104:254–261. [DOI] [PubMed] [Google Scholar]
- 67. Vanattou‐Saifoudine N, McNamara R, Harkin A. Caffeine provokes adverse interactions with 3,4‐methylenedioxymethamphetamine (MDMA, ecstasy') and related psychostimulants: Mechanisms and mediators. Brit J Pharmacol. 2012;167:946–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hua YS, Liang R, Liang L, Huang GZ. Contraction band necrosis in two ecstasy abusers: A latent lethal lesion associated with ecstasy. Am J Forensic Med Pathol. 2009;30:295–297. [DOI] [PubMed] [Google Scholar]
- 69. Carvalho M, Remiao F, Milhazes N, et al. Metabolism is required for the expression of ecstasy‐induced cardiotoxicity in vitro. Chem Res Toxicol. 2004;17:623–632. [DOI] [PubMed] [Google Scholar]
- 70. Spiller HA, Hays HL, Aleguas A, Jr. Overdose of drugs for attention‐deficit hyperactivity disorder: Clinical presentation, mechanisms of toxicity, and management. CNS Drugs. 2013;27:531–543. [DOI] [PubMed] [Google Scholar]
- 71. Richards JR, Albertson TE, Derlet RW, et al. Treatment of toxicity from amphetamines, related derivatives, and analogues: A systematic clinical review. Drug Alcohol Depend. 2015;150:1–13. [DOI] [PubMed] [Google Scholar]
- 72. Fitzgerald KT, Bronstein AC. Adderall (R) (amphetamine‐dextroamphetamine) toxicity. Top Companion Anim Med. 2013;28:2–7. [DOI] [PubMed] [Google Scholar]
- 73. Kersten BP, McLaughlin ME. Toxicology and management of novel psychoactive drugs. J Pharm Pract. 2015;28:50–65. [DOI] [PubMed] [Google Scholar]
- 74. Ross EA, Watson M, Goldberger B. “Bath salts” intoxication. New Engl J Med. 2011;365:967–968. [DOI] [PubMed] [Google Scholar]
- 75. Hall C, Heyd C, Butler C, Yarema M. “Bath salts” intoxication: A new recreational drug that presents with a familiar toxidrome. Can J Emerg Med. 2014;16:171–176. [DOI] [PubMed] [Google Scholar]
- 76. Coppola M, Mondola R. Synthetic cathinones: Chemistry, pharmacology and toxicology of a new class of designer drugs of abuse marketed as “bath salts” or “plant food.” Toxicol Lett. 2012;211:144–149. [DOI] [PubMed] [Google Scholar]
- 77. Coppola M, Mondola R. 3,4‐Methylenedioxypyrovalerone (MDPV): Chemistry, pharmacology and toxicology of a new designer drug of abuse marketed online. Toxicol Lett. 2012;208:12–15. [DOI] [PubMed] [Google Scholar]
- 78. Prosser JM, Nelson LS. The toxicology of bath salts: A review of synthetic cathinones. J Med Toxicol. 2012;8:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wood DM, Button J, Lidder S, et al. Dissociative and sympathomimetic toxicity associated with recreational use of 1‐(3‐trifluoromethylphenyl) piperazine (TFMPP) and 1‐benzylpiperzine (BZP). J Med Toxicol. 2008;4:254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jordan J, Astrup A, Engeli S, et al. Cardiovascular effects of phentermine and topiramate: A new drug combination for the treatment of obesity. J Hypertens. 2014;32:1178–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Alkhouli M, Mathur M, Patil P. Revisiting the “cheese reaction”: More than just a hypertensive crisis? J Clin Psychopharmacol. 2014;34:665–667. [DOI] [PubMed] [Google Scholar]
- 82. Blackwell B, Mabbitt LA. Tyramine in cheese related to hypertensive crises after monoamine‐oxidase inhibition. Lancet. 1965;1:938–940. [DOI] [PubMed] [Google Scholar]
- 83. Sathyanarayana Rao TS, Yeragani VK. Hypertensive crisis and cheese. Indian J Psychiatry. 2009;51:65–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. French G. Safety and tolerability of linezolid. J Antimicrob Chemother. 2003;51(Suppl 2):ii45–53. [DOI] [PubMed] [Google Scholar]
- 85. Ramsey TD, Lau TT, Ensom MH. Serotonergic and adrenergic drug interactions associated with linezolid: A critical review and practical management approach. Ann Pharmacother. 2013;47:543–560. [DOI] [PubMed] [Google Scholar]
- 86. Overgaard CB, Dzavik V. Inotropes and vasopressors: Review of physiology and clinical use in cardiovascular disease. Circulation. 2008;118:1047–1056. [DOI] [PubMed] [Google Scholar]
- 87. Costa VM, Carvalho F, Bastos ML, et al. Contribution of catecholamine reactive intermediates and oxidative stress to the pathologic features of heart diseases. Curr Med Chem. 2011;18:2272–2314. [DOI] [PubMed] [Google Scholar]
- 88. Costa VM, Silva R, Ferreira LM, et al. Oxidation process of adrenaline in freshly isolated rat cardiomyocytes: Formation of adrenochrome, quinoproteins, and GSH adduct. Chem Res Toxicol. 2007;20:1183–1191. [DOI] [PubMed] [Google Scholar]
- 89. Costa VM, Silva R, Ferreira R, et al. Adrenaline in pro‐oxidant conditions elicits intracellular survival pathways in isolated rat cardiomyocytes. Toxicology. 2009;257:70–79. [DOI] [PubMed] [Google Scholar]
- 90. Neri M, Cerretani D, Fiaschi AI, et al. Correlation between cardiac oxidative stress and myocardial pathology due to acute and chronic norepinephrine administration in rats. J Cell Mol Med. 2007;11:156–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mladěnka P, Hrdina R, Bobrovová Z, et al. Cardiac biomarkers in a model of acute catecholamine cardiotoxicity. Hum Exp Toxicol. 2009;28:631–640. [DOI] [PubMed] [Google Scholar]
- 92. Rona G. Catecholamine cardiotoxicity. J Mol Cell Cardiol. 1985;17:291–306. [DOI] [PubMed] [Google Scholar]
- 93. Rona G, Chappel CI, Balazs T, Gaudry R. An infarct‐like myocardial lesion and other toxic manifestations produced by isoproterenol in the rat. AMA Arch Pathol. 1959;67:443–455. [PubMed] [Google Scholar]
- 94. Heather LC, Catchpole AF, Stuckey DJ, et al. Isoproterenol induces in vivo functional and metabolic abnormalities: Similar to those found in the infarcted rat heart. J Physiol Pharmacol. 2009;60:31–39. [PubMed] [Google Scholar]
- 95. Filipský T, Zatloukalová L, Mladěnka P, Hrdina R. Acute initial haemodynamic changes in a rat isoprenaline model of cardiotoxicity. Hum Exp Toxicol. 2012;31:830–843. [DOI] [PubMed] [Google Scholar]
- 96. Blasig IE, Zipper J, Muschick P, Modersohn D, Lowe H. Absolute and relative myocardial ischemia by isoproterenol overdosage. Biomed Biochim Acta. 1985;44:1641–1649. [PubMed] [Google Scholar]
- 97. Dhalla NS, Dent MR, Arneja AS. Pathogenesis of catecholamine‐induced cardiomyopathy In: Acosta D, Jr, ed. Cardiovascular Toxicology, Vol. 25 (Target Organ Toxicology Series, Hayes AW, Thomas JA, Gardner DE, eds). 4th ed New York: Informa Health Care; 2008:207–262. [Google Scholar]
- 98. Azuma J, Hamaguchi T, Ohta H, et al. Calcium overload‐induced myocardial damage caused by isoproterenol and by adriamycin: Possible role of taurine in its prevention. Adv Exp Med Biol. 1987;217:167–179. [DOI] [PubMed] [Google Scholar]
- 99. Tappia PS, Hata T, Hozaima L, et al. Role of oxidative stress in catecholamine‐induced changes in cardiac sarcolemmal Ca2+ transport. Arch Biochem Biophys. 2001;387:85–92. [DOI] [PubMed] [Google Scholar]
- 100. Nichols CB, Rossow CF, Navedo MF, et al. Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of L‐type calcium channels. Circ Res. 2010;107:747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Pinelli A, Trivulzio S, Tomasoni L, et al. Isoproterenol‐induced myocardial infarction in rabbits. Protection by propranolol or labetalol: A proposed non‐invasive procedure. Eur J Pharm Sci. 2004;23:277–285. [DOI] [PubMed] [Google Scholar]
- 102. von Kanel R, Mills PJ , Ziegler MG, Dimsdale JE. Effect of beta2‐adrenergic receptor functioning and increased norepinephrine on the hypercoagulable state with mental stress. Am Heart J. 2002;144:68–72. [DOI] [PubMed] [Google Scholar]
- 103. Pinelli A, Trivulzio S, Tomasoni L, et al. Myocardial infarction non‐invasively induced in rabbits by administering isoproterenol and vasopressin: Protective effects exerted by verapamil. Fundam Clin Pharmacol. 2004;18:657–667. [DOI] [PubMed] [Google Scholar]
- 104. Remiao F, Carmo H, Carvalho F, Bastos ML. The study of oxidative stress in freshly isolated Ca(2+)‐tolerant cardiomyocytes from the adult rat. Toxicol In Vitro. 2001;15:283–287. [DOI] [PubMed] [Google Scholar]
- 105. Remiao F, Carvalho M, Carmo H, Carvalho F, Bastos ML. Cu2+‐induced isoproterenol oxidation into isoprenochrome in adult rat calcium‐tolerant cardiomyocytes. Chem Res Toxicol. 2002;15:861–869. [DOI] [PubMed] [Google Scholar]
- 106. Hašková P, Koubková L, Vávrová A, et al. Comparison of various iron chelators used in clinical practice as protecting agents against catecholamine‐induced oxidative injury and cardiotoxicity. Toxicology. 2011;289:122–131. [DOI] [PubMed] [Google Scholar]
- 107. Hašková P, Kovaříková P, Koubková L, et al. Iron chelation with salicylaldehyde isonicotinoyl hydrazone protects against catecholamine autoxidation and cardiotoxicity. Free Radic Biol Med. 2011;50:537–549. [DOI] [PubMed] [Google Scholar]
- 108. Bindoli A, Rigobello MP, Deeble DJ. Biochemical and toxicological properties of the oxidation products of catecholamines. Free Radic Biol Med. 1992;13:391–405. [DOI] [PubMed] [Google Scholar]
- 109. Zatloukalová L, Filipský T, Mladěnka P, et al. Dexrazoxane provided moderate protection in a catecholamine model of severe cardiotoxicity. Can J Physiol Pharmacol. 2012;90:473–484. [DOI] [PubMed] [Google Scholar]
- 110. Mladěnka P, Kalinowski DS, Hašková P, et al. The novel iron chelator, 2‐pyridylcarboxaldehyde 2‐thiophenecarboxyl hydrazone, reduces catecholamine‐mediated myocardial toxicity. Chem Res Toxicol. 2009;22:208–217. [DOI] [PubMed] [Google Scholar]
- 111. Mladěnka P, Semecký V, Bobrovová Z, et al. The effects of lactoferrin in a rat model of catecholamine cardiotoxicity. Biometals. 2009;22:353–361. [DOI] [PubMed] [Google Scholar]
- 112. Mladěnka P, Zatloukalová L, Šimůnek T, et al. Direct administration of rutin does not protect against catecholamine cardiotoxicity. Toxicology. 2009;255:25–32. [DOI] [PubMed] [Google Scholar]
- 113. Říha M, Hašková P, Martin J, et al. Protective effects of D‐penicillamine on catecholamine‐induced myocardial injury. Oxid Med Cell Longev. 2016;2016:5213532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wexler BC, McMurtry JP. Allopurinol amelioration of the pathophysiology of acute myocardial infarction in rats. Atherosclerosis. 1981;39:71–87. [DOI] [PubMed] [Google Scholar]
- 115. Raja S, Ramya I, Ravindranadh K. A review on protective role of phytoconstituents against isoproterenol induced myocardial necrosis. Int J Pharmacogn Phytochem Res. 2016;8:848–864. [Google Scholar]
- 116. Upaganlawar A, Gandhi H, Balaraman R. Isoproterenol induced myocardial infarction: Protective role of natural products. J Pharmacol Toxicol 2011;6:1–17. [Google Scholar]
- 117. Říha M, Vopršalová M, Pilařova V, et al. Oral administration of quercetin is unable to protect against isoproterenol cardiotoxicity. Naunyn Schmiedebergs Arch Pharmacol. 2014;387:823–835. [DOI] [PubMed] [Google Scholar]
- 118. Mladěnka P, Hrdina R, Filipský T, Říha M, Palicka V. Is a highly linear relationship between the dose of quercetin and the pharmacological effect possible? A comment on Liu, et al. Evaluation of antioxidant and immunity activities of quercetin in isoproterenol‐treated rats, Molecules 2012:17:4281–4291. Molecules. 2014;19:9606–9609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Sarnaik SM, Saladino RA, Manole M, et al. Diastolic hypotension is an unrecognized risk factor for beta‐agonist‐associated myocardial injury in children with asthma. Pediatr Crit Care Med. 2013;14:e273–279. [DOI] [PubMed] [Google Scholar]
- 120. Wisecup S, Eades S, Hashmi SS, Samuels C, Mosquera RA. Diastolic hypotension in pediatric patients with asthma receiving continuous albuterol. J Asthma. 2015;52:693–698. [DOI] [PubMed] [Google Scholar]
- 121. Beach C, Marcuccio E, Beerman L, Arora G. Accelerated idioventricular rhythm in a child with status asthmaticus. Pediatrics. 2015;136:e527–529. [DOI] [PubMed] [Google Scholar]
- 122. Carson MP, Fisher AJ, Scorza WE. Atrial fibrillation in pregnancy associated with oral terbutaline. Obstet Gynecol. 2002;100:1096–1097. [DOI] [PubMed] [Google Scholar]
- 123. Fisher AA, Davis MW, McGill DA. Acute myocardial infarction associated with albuterol. Ann Pharmacother. 2004;38:2045–2049. [DOI] [PubMed] [Google Scholar]
- 124. Wills BK, Kwan C, Bailey M, Johnson L, Allan N. Recalcitrant supraventricular tachycardia: Occult albuterol toxicity due to a factitious disorder. J Emerg Med. 2015;49:436–438. [DOI] [PubMed] [Google Scholar]
- 125. Kierzkowska B, Stanczyk J, Kasprzak JD. Myocardial infarction in a 17‐year‐old body builder using clenbuterol. Circ J. 2005;69:1144–1146. [DOI] [PubMed] [Google Scholar]
- 126. Tucker C, Villanueva L. Acute hypokalemic periodic paralysis possibly precipitated by albuterol. Am J Health Syst Pharm. 2013;70:1588–1591. [DOI] [PubMed] [Google Scholar]
- 127. Hartman S, Merkus P, Maseland M, Roovers L, van Setten P. Hypokalaemia in children with asthma treated with nebulised salbutamol. Arch Dis Child. 2015;100:970–972. [DOI] [PubMed] [Google Scholar]
- 128. Petruska JM, Beattie JG, Stuart BO, et al. Cardiovascular effects after inhalation of large doses of albuterol dry powder in rats, monkeys, and dogs: A species comparison. Fundam Appl Toxicol. 1997;40:52–62. [DOI] [PubMed] [Google Scholar]
- 129. Burniston JG, Tan LB, Goldspink DF. Relative myotoxic and haemodynamic effects of the beta‐agonists fenoterol and clenbuterol measured in conscious unrestrained rats. Exp Physiol. 2006;91:1041–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Whitehurst VE, Joseph X, Alleva FR, et al. Enhancement of acute myocardial lesions by asthma drugs in rats. Toxicol Pathol. 1994;22:72–76. [DOI] [PubMed] [Google Scholar]
- 131. Carroll CL, Coro M, Cowl A, Sala KA, Schramm CM. Transient occult cardiotoxicity in children receiving continuous beta‐agonist therapy. World J Pediatr. 2014;10:324–329. [DOI] [PubMed] [Google Scholar]
- 132. Kalyanaraman M, Bhalala U, Leoncio M. Serial cardiac troponin concentrations as marker of cardiac toxicity in children with status asthmaticus treated with intravenous terbutaline. Pediatr Emerg Care. 2011;27:933–936. [DOI] [PubMed] [Google Scholar]
- 133. Haas DM, Benjamin T, Sawyer R, Quinney SK. Short‐term tocolytics for preterm delivery—Current perspectives. Int J Womens Health. 2014;6:343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Adamcova M, Kokstein Z, Palicka V, et al. Cardiac troponin T in pregnant women having intravenous tocolytic therapy. Arch Gynecol Obstet. 1999;262:121–126. [DOI] [PubMed] [Google Scholar]
- 135. Adamcova M, Kokstein Z, Palicka V, et al. Cardiac troponin T in neonates after acute and long‐term tocolysis. Biol Neonate. 2000;78:288–292. [DOI] [PubMed] [Google Scholar]
- 136. Rossanese M, Novara G, Challacombe B, et al. Critical analysis of phase II and III randomised control trials (RCTs) evaluating efficacy and tolerability of a beta(3)‐adrenoceptor agonist (Mirabegron) for overactive bladder (OAB). BJU Int. 2015;115:32–40. [DOI] [PubMed] [Google Scholar]
- 137. Vij M, Drake MJ. Clinical use of the beta3 adrenoceptor agonist mirabegron in patients with overactive bladder syndrome. Ther Adv Urol. 2015;7:241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Forouzanfar MH, Alexander L, Anderson HR, et al. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks in 188 countries, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;386:2287–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Ng M, Freeman MK, Fleming TD, et al. Smoking prevalence and cigarette consumption in 187 countries, 1980–2012. J Am Med Assoc. 2014;311:183–192. [DOI] [PubMed] [Google Scholar]
- 140. Nelluri BK, Murphy K, Mookadam F. Electronic cigarettes and cardiovascular risk: Hype or up in smoke? Future Cardiol. 2015;11:271–273. [DOI] [PubMed] [Google Scholar]
- 141. Morris PB, Ference BA, Jahangir E, et al. Cardiovascular effects of exposure to cigarette smoke and electronic cigarettes: Clinical perspectives from the prevention of cardiovascular disease section leadership council and early career councils of the American College of Cardiology. J Am Coll Cardiol. 2015;66:1378–1391. [DOI] [PubMed] [Google Scholar]
- 142. Law MR, Wald NJ. Environmental tobacco smoke and ischemic heart disease. Prog Cardiovasc Dis. 2003;46:31–38. [DOI] [PubMed] [Google Scholar]
- 143. Barone‐Adesi F, Vizzini L, Merletti F, Richiardi L. Short‐term effects of Italian smoking regulation on rates of hospital admission for acute myocardial infarction. Eur Heart J. 2006;27:2468–2472. [DOI] [PubMed] [Google Scholar]
- 144. Cesaroni G, Forastiere F, Agabiti N, et al. Effect of the Italian smoking ban on population rates of acute coronary events. Circulation. 2008;117:1183–1188. [DOI] [PubMed] [Google Scholar]
- 145. Watts DT. The effect of nicotine and smoking on the secretion of epinephrine. Ann N Y Acad Sci. 1960;90:74–80. [DOI] [PubMed] [Google Scholar]
- 146. Nowak J, Murray JJ, Oates JA, Fitzgerald GA. Biochemical‐evidence of a chronic abnormality in platelet and vascular function in healthy‐individuals who smoke cigarettes. Circulation. 1987;76:6–14. [DOI] [PubMed] [Google Scholar]
- 147. Levine PH. An acute effect of cigarette smoking on platelet function. A possible link between smoking and arterial thrombosis. Circulation. 1973;48:619–623. [DOI] [PubMed] [Google Scholar]
- 148. Fuster V, Chesebro JH, Frye RL, Elveback LR. Platelet survival and the development of coronary‐artery disease in the young‐adult—Effects of cigarette‐smoking, strong family history and medical therapy. Circulation. 1981;63:546–551. [DOI] [PubMed] [Google Scholar]
- 149. Wennmalm A, Benthin G, Granstrom EF, et al. Relation between tobacco use and urinary‐excretion of thromboxane‐a2 and prostacyclin metabolites in young men. Circulation. 1991;83:1698–1704. [DOI] [PubMed] [Google Scholar]
- 150. Boffetta P, Straif K. Use of smokeless tobacco and risk of myocardial infarction and stroke: Systematic review with meta‐analysis. Brit Med J 2009;339:b3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Hergens MP, Lambe M, Pershagen G, Terent A, Ye WM. Smokeless tobacco and the risk of stroke. Epidemiology. 2008;19:794–799. [DOI] [PubMed] [Google Scholar]
- 152. Hansson J, Galanti MR, Hergens MP, et al. Snus (Swedish smokeless tobacco) use and risk of stroke: Pooled analyses of incidence and survival. J Intern Med. 2014;276:87–95. [DOI] [PubMed] [Google Scholar]
- 153. Gill N, Sangha G, Poonai N, Lim R. E‐cigarette liquid nicotine ingestion in a child: Case report and discussion. CJEM. 2015;17:699–703. [DOI] [PubMed] [Google Scholar]
- 154. Mills EJ, Thorlund K, Eapen S, Wu P, Prochaska JJ. Cardiovascular events associated with smoking cessation pharmacotherapies: A network meta‐analysis. Circulation. 2014;129:28–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Toh S, Baker MA, Brown JS, et al. Rapid assessment of cardiovascular risk among users of smoking cessation drugs within the US Food and Drug Administration's Mini‐Sentinel Program. JAMA Intern Med. 2013;173:817–819. [DOI] [PubMed] [Google Scholar]
- 156. Karhuvaara S, Kallio A, Salonen M, Tuominen J, Scheinin M. Rapid reversal of alpha 2‐adrenoceptor agonist effects by atipamezole in human volunteers. Br J Clin Pharmacol. 1991;31:160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Langoi DL, Mwethera PG, Abelson KS, Farah IO, Carlsson HE. Reversal of ketamine/xylazine combination anesthesia by atipamezole in olive baboons (Papio anubis). J Med Primatol. 2009;38:404–410. [DOI] [PubMed] [Google Scholar]
- 158. Gallanosa AG, Spyker DA, Shipe JR, Morris DL. Human xylazine overdose: A comparative review with clonidine, phenothiazines, and tricyclic antidepressants. Clin Toxicol. 1981;18:663–678. [DOI] [PubMed] [Google Scholar]
- 159. Malanga G, Reiter RD, Garay E. Update on tizanidine for muscle spasticity and emerging indications. Expert Opin Pharmacother. 2008;9:2209–2215. [DOI] [PubMed] [Google Scholar]
- 160. Ruiz‐Colon K, Chavez‐Arias C, Diaz‐Alcala JE, Martinez MA. Xylazine intoxication in humans and its importance as an emerging adulterant in abused drugs: A comprehensive review of the literature. Forensic Sci Int 2014;240:1–8. [DOI] [PubMed] [Google Scholar]
- 161. Eddy O, Howell JM. Are one or two dangerous? Clonidine and topical imidazolines exposure in toddlers. J Emerg Med. 2003;25:297–302. [DOI] [PubMed] [Google Scholar]
- 162. Zarifis J, Lip GY, Ferner RE. Poisoning with anti‐hypertensive drugs: Methyldopa and clonidine. J Hum Hypertens. 1995;9:787–790. [PubMed] [Google Scholar]
- 163. Shnaps Y, Almog S, Halkin H, Tirosh M. Methyldopa poisoning. J Toxicol Clin Toxicol. 1982;19:501–503. [DOI] [PubMed] [Google Scholar]
- 164. Cimolai N, Cimolai T. Yohimbine use for physical enhancement and its potential toxicity. J Diet Suppl. 2011;8:346–354. [DOI] [PubMed] [Google Scholar]
- 165. Tam SW, Worcel M, Wyllie M. Yohimbine: A clinical review. Pharmacol Ther. 2001;91:215–243. [DOI] [PubMed] [Google Scholar]
- 166. Pfeifer HJ, Greenblatt DK, Koch‐Wester J. Clinical toxicity of reserpine in hospitalized patients: A report from the Boston Collaborative Drug Surveillance Program. Am J Med Sci. 1976;271:269–276. [DOI] [PubMed] [Google Scholar]
- 167. Packer M, Carver JR, Rodeheffer RJ, et al. Effect of oral milrinone on mortality in severe chronic heart failure. The PROMISE Study Research Group. N Engl J Med. 1991;325:1468–1475. [DOI] [PubMed] [Google Scholar]
- 168. Fitzgerald KT, Bronstein AC, Newquist KL. Marijuana poisoning. Top Companion Anim Med. 2013;28:8–12. [DOI] [PubMed] [Google Scholar]
- 169. Bachs L, Morland H. Acute cardiovascular fatalities following cannabis use. Forensic Sci Int. 2001;124:200–203. [DOI] [PubMed] [Google Scholar]
- 170. Deligiannis A, Bjornstad H, Carre F, et al. ESC study group of sports cardiology position paper on adverse cardiovascular effects of doping in athletes. Eur J Cardiovasc Prev Rehabil. 2006;13:687–694. [DOI] [PubMed] [Google Scholar]
- 171. Deligiannis AP, Kouidi EI. Cardiovascular adverse effects of doping in sports. Hell J Cardiol. 2012;53:447–457. [PubMed] [Google Scholar]
- 172. Boyer EW, Shannon M. The serotonin syndrome. New Engl J Med. 2005;352:1112–1120. [DOI] [PubMed] [Google Scholar]
- 173. Bonnet U. Moclobemide: Therapeutic use and clinical studies. CNS Drug Rev. 2003;9:97–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Lapage MJ, Bradley DJ, Dick M, 2nd . Verapamil in infants: An exaggerated fear? Pediatr Cardiol. 2013;34:1532–1534. [DOI] [PubMed] [Google Scholar]
- 175. Richards TR, Tobe SW. Combining other antihypertensive drugs with beta‐blockers in hypertension: A focus on safety and tolerability. Can J Cardiol. 2014;30:S42–46. [DOI] [PubMed] [Google Scholar]
- 176. Ojetti V, Migneco A, Bononi F, De Lorenzo A, Gentiloni Silveri N. Calcium channel blockers, beta‐blockers and digitalis poisoning: management in the emergency room. Eur Rev Med Pharmacol Sci. 2005;9:241–246. [PubMed] [Google Scholar]
- 177. Pearigen PD, Benowitz NL. Poisoning due to calcium‐antagonists—Experience with verapamil, diltiazem and nifedipine. Drug Saf. 1991;6:408–430. [DOI] [PubMed] [Google Scholar]
- 178. Arroyo AM, Kao LW. Calcium channel blocker toxicity. Pediatr Emerg Care. 2009;25:532–538; quiz 539–540. [DOI] [PubMed] [Google Scholar]
- 179. Wetzel GT, Chen F, Klitzner TS. L‐ and T‐type calcium channels in acutely isolated neonatal and adult cardiac myocytes. Pediatr Res. 1991;30:89–94. [DOI] [PubMed] [Google Scholar]
- 180. Costello M, Syring RS. Calcium channel blocker toxicity. J Vet Emerg Crit Care. 2008;18:54–60. [Google Scholar]
- 181. St‐Onge M, Dube PA, Gosselin S, et al. Treatment for calcium channel blocker poisoning: A systematic review. Clin Toxicol (Phila). 2014;52:926–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Truhlar A, Deakin CD, Soar J, et al. European Resuscitation Council Guidelines for Resuscitation 2015: Section 4. Cardiac arrest in special circumstances. Resuscitation. 2015;95:148–201. [DOI] [PubMed] [Google Scholar]
- 183. Rietjens SJ, de Lange DW, Donker DW, Meulenbelt J. Practical recommendations for calcium channel antagonist poisoning. Neth J Med. 2016;74:60–67. [PubMed] [Google Scholar]
- 184. Armstrong P, Murray P, Nesdale A, Peckler B. Ciguatera fish poisoning. N Z Med J. 2016;129:111–114. [PubMed] [Google Scholar]
- 185. Sinkins WG, Estacion M, Prasad V, et al. Maitotoxin converts the plasmalemmal Ca(2+) pump into a Ca(2+)‐permeable nonselective cation channel. Am J Physiol Cell Physiol. 2009;297:C1533–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Wisnoskey BJ, Estacion M, Schilling WP. Maitotoxin‐induced cell death cascade in bovine aortic endothelial cells: Divalent cation specificity and selectivity. Am J Physiol Cell Physiol. 2004;287:C345–356. [DOI] [PubMed] [Google Scholar]
- 187. Legrand AM, Galonnier M, Bagnis R. Studies on the mode of action of ciguateric toxins. Toxicon. 1982;20:311–315. [DOI] [PubMed] [Google Scholar]
- 188. Puente Puente S, Cabrera Majada A, Lago Nunez M, Azuara Solis M, Gonzalez‐Lahoz JM. Ciguatera: Eight imported cases. Rev Clin Esp. 2005;205:47–50. [DOI] [PubMed] [Google Scholar]
- 189. Roose SP, Glassman AH, Siris SG, et al. Comparison of imipramine‐ and nortriptyline‐induced orthostatic hypotension: A meaningful difference. J Clin Psychopharmacol. 1981;1:316–319. [DOI] [PubMed] [Google Scholar]
- 190. Thanacoody HK, Thomas SH. Tricyclic antidepressant poisoning: Cardiovascular toxicity. Toxicol Rev. 2005;24:205–214. [DOI] [PubMed] [Google Scholar]
- 191. Woolf AD, Erdman AR, Nelson LS, et al. Tricyclic antidepressant poisoning: An evidence‐based consensus guideline for out‐of‐hospital management. Clin Toxicol. 2007;45:203–233. [DOI] [PubMed] [Google Scholar]
- 192. Feinstein RE, Khawaja IS, Nurenberg JR, Frishman WH. Cardiovascular effects of psychotropic drugs. Curr Probl Cardiol. 2002;27:190–240. [DOI] [PubMed] [Google Scholar]
- 193. Gronbaek M, Deis A, Sorensen TI, et al. Influence of sex, age, body mass index, and smoking on alcohol intake and mortality. BMJ. 1994;308:302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Chagas P, Mazocco L, Piccoli JD, et al. Association of alcohol consumption with coronary artery disease severity. Clin Nutr. 2017;36(4):1036–1039. [DOI] [PubMed] [Google Scholar]
- 195. Renaud S, de Lorgeril M. Wine, alcohol, platelets, and the French paradox for coronary heart disease. Lancet. 1992;339:1523–1526. [DOI] [PubMed] [Google Scholar]
- 196. Zhou Y, Zheng J, Li S, et al. Alcoholic beverage consumption and chronic diseases. Int J Environ Res Public Health. 2016;13(6):522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Leong DP, Smyth A, Teo KK, et al. Patterns of alcohol consumption and myocardial infarction risk: Observations from 52 countries in the INTERHEART case‐control study. Circulation. 2014;130:390–398. [DOI] [PubMed] [Google Scholar]
- 198. Grant BF, Goldstein RB, Saha TD, et al. Epidemiology of DSM‐5 alcohol use disorder results from the National Epidemiologic Survey on Alcohol and Related Conditions III. JAMA Psychiatry. 2015;72:757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Vancampfort D, Hallgren M, Mugisha J, et al. The prevalence of metabolic syndrome in alcohol use disorders: A systematic review and meta‐analysis. Alcohol Alcohol. 2016;51(5):515–521. [DOI] [PubMed] [Google Scholar]
- 200. Saremi A, Hanson RL, Tulloch‐Reid M, et al. Alcohol consumption predicts hypertension but not diabetes. J Stud Alcohol. 2004;65:184–190. [DOI] [PubMed] [Google Scholar]
- 201. Mukamal KJ, Tolstrup JS, Friberg J, Jensen G, Gronbaek M. Alcohol consumption and risk of atrial fibrillation in men and women: The Copenhagen City Heart Study. Circulation. 2005;112:1736–1742. [DOI] [PubMed] [Google Scholar]
- 202. Rosenqvist M. Alcohol and cardiac arrhythmias. Alcohol Clin Exp Res. 1998;22:318S–322S. [DOI] [PubMed] [Google Scholar]
- 203. Guzzo‐Merello G, Cobo‐Marcos M, Gallego‐Delgado M, Garcia‐Pavia P. Alcoholic cardiomyopathy. World J Cardiol. 2014;6:771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Piano MR, Phillips SA. Alcoholic cardiomyopathy: Pathophysiologic insights. Cardiovasc Toxicol. 2014;14:291–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Darke S, Torok M, Duflou J. Sudden or unnatural deaths involving anabolic‐androgenic steroids. J Forensic Sci. 2014;59:1025–1028. [DOI] [PubMed] [Google Scholar]
- 206. Liu PY, Death AK, Handelsman DJ. Androgens and cardiovascular disease. Endocr Rev. 2003;24:313–340. [DOI] [PubMed] [Google Scholar]
- 207. Huang CK, Lee SO, Chang E, Pang H, Chang C. Androgen receptor (AR) in cardiovascular diseases. J Endocrinol. 2016;229:R1–R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Nieminen MS, Ramo MP, Viitasalo M, et al. Serious cardiovascular side effects of large doses of anabolic steroids in weight lifters. Eur Heart J. 1996;17:1576–1583. [DOI] [PubMed] [Google Scholar]
- 209. Urhausen A, Albers T, Kindermann W. Are the cardiac effects of anabolic steroid abuse in strength athletes reversible? Heart. 2004;90:496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Varro A, Baczko I. Possible mechanisms of sudden cardiac death in top athletes: A basic cardiac electrophysiological point of view. Pflugers Arch. 2010;460:31–40. [DOI] [PubMed] [Google Scholar]
- 211. Frati P, Busardo FP, Cipolloni L, Dominicis ED, Fineschi V. Anabolic androgenic steroid (AAS) related deaths: Autoptic, histopathological and toxicological findings. Curr Neuropharmacol. 2015;13:146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Stergiopoulos K, Brennan JJ, Mathews R, Setaro JF, Kort S. Anabolic steroids, acute myocardial infarction and polycythemia: A case report and review of the literature. Vasc Health Risk Manag. 2008;4:1475–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Basaria S. Androgen abuse in athletes: Detection and consequences. J Clin Endocrinol Metab. 2010;95:1533–1543. [DOI] [PubMed] [Google Scholar]
- 214. Ajayi AA, Mathur R, Halushka PV. Testosterone increases human platelet thromboxane A2 receptor density and aggregation responses. Circulation. 1995;91:2742–2747. [DOI] [PubMed] [Google Scholar]
- 215. Rothman RD, Weiner RB, Pope H, et al. Anabolic androgenic steroid induced myocardial toxicity: An evolving problem in an ageing population. BMJ Case Rep. 2011;2011 https://doi.org/10.1136/bcr.05.2011.4280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37:2129–2200. [DOI] [PubMed] [Google Scholar]
- 217. Love JN, Litovitz TL, Howell JM, Clancy C. Characterization of fatal beta blocker ingestion: A review of the American Association of Poison Control Centers data from 1985 to 1995. J Toxicol Clin Toxicol. 1997;35:353–359. [DOI] [PubMed] [Google Scholar]
- 218. Love JN, Howell JM, Litovitz TL, Klein‐Schwartz W. Acute beta blocker overdose: Factors associated with the development of cardiovascular morbidity. J Toxicol Clin Toxicol. 2000;38:275–281. [DOI] [PubMed] [Google Scholar]
- 219. Chan TY. Aconitum alkaloid content and the high toxicity of aconite tincture. Forensic Sci Int. 2012;222:1–3. [DOI] [PubMed] [Google Scholar]
- 220. Chan TY. Incidence and causes of aconitum alkaloid poisoning in Hong Kong from 1989 to 2010. Phytother Res. 2015;29:1107–1111. [DOI] [PubMed] [Google Scholar]
- 221. Chan TY. Aconite poisoning. Clin Toxicol (Phila). 2009;47:279–285. [DOI] [PubMed] [Google Scholar]
- 222. Bosmans F, Maertens C, Verdonck F, Tytgat J. The poison Dart frog's batrachotoxin modulates Na(v)1.8. FEBS Lett. 2004;577:245–248. [DOI] [PubMed] [Google Scholar]
- 223. Ulbricht W. Effects of veratridine on sodium currents and fluxes. Rev Physiol Biochem Pharmacol. 1998;133:1–54. [DOI] [PubMed] [Google Scholar]
- 224. Wang SY, Wang GK. Voltage‐gated sodium channels as primary targets of diverse lipid‐soluble neurotoxins. Cell Signal. 2003;15:151–159. [DOI] [PubMed] [Google Scholar]
- 225. Grobosch T, Binscheck T, Martens F, Lampe D. Accidental intoxication with Veratrum album . J Anal Toxicol. 2008;32:768–773. [DOI] [PubMed] [Google Scholar]
- 226. Eroglu SE, Urgan O, Onur OE, Denizbasi A, Akoglu H. Grayanotoxin (mad honey)—Ongoing consumption after poisoning. Balkan Med J. 2013;30:293–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Jansen SA, Kleerekooper I, Hofman ZL, et al. Grayanotoxin poisoning: “Mad honey disease” and beyond. Cardiovasc Toxicol. 2012;12:208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Silici S, Atayoglu AT. Mad honey intoxication: A systematic review on the 1199 cases. Food Chem Toxicol. 2015;86:282–290. [DOI] [PubMed] [Google Scholar]
- 229. Effect of the antiarrhythmic agent moricizine on survival after myocardial infarction . The cardiac arrhythmia suppression trial ii investigators. N Engl J Med. 1992;327:227–233. [DOI] [PubMed] [Google Scholar]
- 230. Preliminary report: Effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction . The cardiac arrhythmia suppression trial (CAST) investigators. N Engl J Med. 1989;321:406–412. [DOI] [PubMed] [Google Scholar]
- 231. Pratt CM, Moye LA. The cardiac arrhythmia suppression trial. Casting suppression in a different light. Circulation. 1995;91:245–247. [DOI] [PubMed] [Google Scholar]
- 232. Rosen MR. The sicilian gambit—A new approach to the classification of antiarrhythmic drugs based on their actions on arrhythmogenic mechanisms. Circulation. 1991;84:1831–1851. [DOI] [PubMed] [Google Scholar]
- 233. Vaughan Williams EM. Classification of antidysrhythmic drugs. Pharmacol Ther Pt B. 1975;1:115–138. [DOI] [PubMed] [Google Scholar]
- 234. Kirchhof P, Benussi S, Kotecha D, et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J. 2016;37:2893–2962. [DOI] [PubMed] [Google Scholar]
- 235. McLure HA, Rubin AP. Review of local anaesthetic agents. Minerva Anestesiol. 2005;71:59–74. [PubMed] [Google Scholar]
- 236. Bourne E, Wright C, Royse C. A review of local anesthetic cardiotoxicity and treatment with lipid emulsion. Local Reg Anesth. 2010;3:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Wolfe JW, Butterworth JF. Local anesthetic systemic toxicity: Update on mechanisms and treatment. Curr Opin Anaesthesiol. 2011;24:561–566. [DOI] [PubMed] [Google Scholar]
- 238. Di Gregorio G, Neal JM, Rosenquist RW, Weinberg GL. Clinical presentation of local anesthetic systemic toxicity: A review of published cases, 1979 to 2009. Reg Anesth Pain Med. 2010;35:181–187. [DOI] [PubMed] [Google Scholar]
- 239. Dickerson DM, Apfelbaum JL. Local anesthetic systemic toxicity. Aesthet Surg J. 2014;34:1111–1119. [DOI] [PubMed] [Google Scholar]
- 240. Harvey M, Cave G. Lipid emulsion in local anesthetic toxicity. Curr Opin Anaesthesiol. 2017;30:632–638. [DOI] [PubMed] [Google Scholar]
- 241. Gosselin S, Morris M, Miller‐Nesbitt A, et al. Methodology for AACT evidence‐based recommendations on the use of intravenous lipid emulsion therapy in poisoning. Clin Toxicol (Phila). 2015;53:557–564. [DOI] [PubMed] [Google Scholar]
- 242. Barrington MJ, Weinberg GL, Neal JM. A call to all readers: Educating all surgeons on preventing and treatment of local anaesthetic systemic toxicity. ANZ J Surg. 2016;86:636–637. [DOI] [PubMed] [Google Scholar]
- 243. Sadowski ZP, Alexander JH, Skrabucha B, et al. Multicenter randomized trial and a systematic overview of lidocaine in acute myocardial infarction. Am Heart J. 1999;137:792–798. [DOI] [PubMed] [Google Scholar]
- 244. Podrid P, Ganz L. Prophylaxis against ventricular arrhythmias during and after acute myocardial infarction. In: Up to Date. https://www.uptodate.com/contents/prophylaxis-against-ventricular-arrhythmias-during-and-after-acute-myocardial-infarction, accessed: Mar 3, 2017.
- 245. Lago J, Rodriguez LP, Blanco L, Vieites JM, Cabado AG. Tetrodotoxin, an extremely potent marine neurotoxin: distribution, toxicity, origin and therapeutical uses. Mar Drugs. 2015;13:6384–6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Zimmer T. Effects of tetrodotoxin on the mammalian cardiovascular system. Mar Drugs. 2010;8:741–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Thottumkara AP, Parsons WH, Du Bois J. Saxitoxin. Angew Chem Int Ed Engl. 2014;53:5760–5784. [DOI] [PubMed] [Google Scholar]
- 248. Prabhu MA, Agustinus R, Shenthar J. Suicidal zinc phosphide poisoning unmasking Brugada syndrome and triggering near fatal ventricular arrhythmia. Pacing Clin Electrophysiol. 2016;39:198–201. [DOI] [PubMed] [Google Scholar]
- 249. Srivastava A, Peshin SS, Kaleekal T, Gupta SK. An epidemiological study of poisoning cases reported to the National Poisons Information Centre, All India Institute of Medical Sciences, New Delhi. Hum Exp Toxicol. 2005;24:279–285. [DOI] [PubMed] [Google Scholar]
- 250. Mizusawa Y, Wilde AA. Brugada syndrome. Circ Arrhythm Electrophysiol. 2012;5:606–616. [DOI] [PubMed] [Google Scholar]
- 251. Fuchs J, Rauber‐Luthy C, Kupferschmidt H, et al. Acute plant poisoning: Analysis of clinical features and circumstances of exposure. Clin Toxicol (Phila). 2011;49:671–680. [DOI] [PubMed] [Google Scholar]
- 252. Tranca S, Petrisor CL. A fatal case of Taxus poisoning. Clujul Med. 2013;86:279–281. [PMC free article] [PubMed] [Google Scholar]
- 253. Willaert W, Claessens P, Vankelecom B, Vanderheyden M. Intoxication with taxus baccata: Cardiac arrhythmias following yew leaves ingestion. Pacing Clin Electrophysiol. 2002;25:511–512. [DOI] [PubMed] [Google Scholar]
- 254. Kaplan WD, Trout WE, 3rd . The behavior of four neurological mutants of Drosophila. Genetics. 1969;61:399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Vandenberg JI, Perry MD, Perrin MJ, et al. hERG K(+) channels: Structure, function, and clinical significance. Physiol Rev. 2012;92:1393–1478. [DOI] [PubMed] [Google Scholar]
- 256. Sanguinetti MC, Tristani‐Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. [DOI] [PubMed] [Google Scholar]
- 257. van der Heyden MA, Smits ME, Vos MA. Drugs and trafficking of ion channels: A new pro‐arrhythmic threat on the horizon? Br J Pharmacol. 2008;153:406–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Wible BA, Hawryluk P, Ficker E, et al. HERG‐Lite: A novel comprehensive high‐throughput screen for drug‐induced hERG risk. J Pharmacol Toxicol Methods. 2005;52:136–145. [DOI] [PubMed] [Google Scholar]
- 259. Nogawa H, Kawai T. hERG trafficking inhibition in drug‐induced lethal cardiac arrhythmia. Eur J Pharmacol. 2014;741:336–339. [DOI] [PubMed] [Google Scholar]
- 260. Shah RR. Drug‐induced QT interval prolongation—Regulatory guidance and perspectives on hERG channel studies. Novartis Found Symp. 2005;266:251–280. [PubMed] [Google Scholar]
- 261. Hoffmann P, Warner B. Are hERG channel inhibition and QT interval prolongation all there is in drug‐induced torsadogenesis? A review of emerging trends. J Pharmacol Toxicol Methods. 2006;53:87–105. [DOI] [PubMed] [Google Scholar]
- 262. Roden DM. Drug‐induced prolongation of the QT interval. N Engl J Med. 2004;350:1013–1022. [DOI] [PubMed] [Google Scholar]
- 263. Shah RR. Drug‐induced QT interval prolongation: Regulatory perspectives and drug development. Ann Med. 2004;36(Suppl 1):47–52. [DOI] [PubMed] [Google Scholar]
- 264. Darpö B. Spectrum of drugs prolonging QT interval and the incidence of torsade de pointes. Eur Heart J Suppl. 2001;3:K70–80. [Google Scholar]
- 265. Olasinska‐Wisniewska A, Olasinski J, Grajek S. Cardiovascular safety of antihistamines. Postepy Dermatol Alergol. 2014;31:182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Yap YG, Camm AJ. Arrhythmogenic mechanisms of non‐sedating antihistamines. Clin Exp Allergy. 1999;29(Suppl 3):174–181. [DOI] [PubMed] [Google Scholar]
- 267. Woosley RL. Cardiac actions of antihistamines. Annu Rev Pharmacol Toxicol. 1996;36:233–252. [DOI] [PubMed] [Google Scholar]
- 268. Taglialatela M, Castaldo P, Pannaccione A, et al. Cardiac ion channels and antihistamines: Possible mechanisms of cardiotoxicity. Clin Exp Allergy. 1999;29(Suppl 3):182–189. [DOI] [PubMed] [Google Scholar]
- 269. Hondeghem LM. Drug‐Induced QT prolongation and torsades de pointes: An all‐exclusive relationship or time for an amicable separation? Drug Saf. 2017. https://doi.org/10.1007/s40264-017-0584-4. [DOI] [PubMed] [Google Scholar]
- 270. Isbister GK. Risk assessment of drug‐induced QT prolongation. Aust Prescr. 2015;38:20–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Thomas SH, Behr ER. Pharmacological treatment of acquired QT prolongation and torsades de pointes. Br J Clin Pharmacol. 2016;81:420–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Hondeghem LM, Carlsson L, Duker G. Instability and triangulation of the action potential predict serious proarrhythmia, but action potential duration prolongation is antiarrhythmic. Circulation. 2001;103:2004–2013. [DOI] [PubMed] [Google Scholar]
- 273. Yap YG, Camm AJ. Drug induced QT prolongation and torsades de pointes. Heart. 2003;89:1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Chan A, Isbister GK, Kirkpatrick CM, Dufful SB. Drug‐induced QT prolongation and torsades de pointes: Evaluation of a QT nomogram. QJM. 2007;100:609–615. [DOI] [PubMed] [Google Scholar]
- 275. Astrom‐Lilja C, Odeberg JM, Ekman E, Hagg S. Drug‐induced torsades de pointes: A review of the Swedish pharmacovigilance database. Pharmacoepidemiol Drug Saf. 2008;17:587–592. [DOI] [PubMed] [Google Scholar]
- 276. Arizona Center for Education and Research on Therapeutics (AZCERT) . QTdrug List. https://www.crediblemeds.org/, accessed October 20, 2017.
- 277. Cantani A, Mocini V. Antihistamines and the torsade de point in children with allergic rhinitis. Eur Rev Med Pharmacol Sci. 2001;5:139–142. [PubMed] [Google Scholar]
- 278. Poluzzi E, Raschi E, Godman B, et al. Pro‐arrhythmic potential of oral antihistamines (H1): Combining adverse event reports with drug utilization data across Europe. PLoS One. 2015;10:e0119551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Briasoulis A, Agarwal V, Pierce WJ. QT prolongation and torsade de pointes induced by fluoroquinolones: Infrequent side effects from commonly used medications. Cardiology. 2011;120:103–110. [DOI] [PubMed] [Google Scholar]
- 280. Rossi M, Giorgi G. Domperidone and long QT syndrome. Curr Drug Saf. 2010;5:257–262. [DOI] [PubMed] [Google Scholar]
- 281. Preston CL. Stockley´s Drug Interactions. 11th ed London: Pharmaceutical Press; 2016. [Google Scholar]
- 282. Alinejad S, Kazemi T, Zamani N, Hoffman RS, Mehrpour O. A systematic review of the cardiotoxicity of methadone. EXCLI J. 2015;14:577–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Raffa RB, Burmeister JJ, Yuvasheva E, Pergolizzi JV, Jr . QTc interval prolongation by d‐propoxyphene: What about other analgesics? Expert Opin Pharmacother. 2012;13:1397–1409. [DOI] [PubMed] [Google Scholar]
- 284. Harel Y, Scott WA, Szeinberg A, Barzilay Z. Pentamidine‐induced torsades de pointes. Pediatr Infect Dis J. 1993;12:692–694. [DOI] [PubMed] [Google Scholar]
- 285. Le Blaye I, Donatini B, Hall M, Krupp P. Acute overdosage with thioridazine: A review of the available clinical exposure. Vet Hum Toxicol. 1993;35:147–150. [PubMed] [Google Scholar]
- 286. Isbister GK, Balit CR, Macleod D, Duffull SB. Amisulpride overdose is frequently associated with QT prolongation and torsades de pointes. J Clin Psychopharmacol. 2010;30:391–395. [DOI] [PubMed] [Google Scholar]
- 287. Hassaballa HA, Balk RA. Torsade de pointes associated with the administration of intravenous haloperidol. Am J Ther. 2003;10:58–60. [DOI] [PubMed] [Google Scholar]
- 288. Wenzel‐Seifert K, Wittmann M, Haen E. QTc prolongation by psychotropic drugs and the risk of torsade de pointes. Dtsch Arztebl Int. 2011;108:687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Tampi RR, Balderas M, Carter KV, et al. Citalopram, QTc prolongation, and torsades de pointes. Psychosomatics. 2015;56:36–43. [DOI] [PubMed] [Google Scholar]
- 290. Loffi M, Toffetti L, Gianni C, Lombardi F. Self‐terminating ventricular fibrillation in vandetanib‐induced torsades de pointes. J Cardiovasc Electrophysiol. 2015;26:811–813. [DOI] [PubMed] [Google Scholar]
- 291. Gao H, Zehl M, Leitner A, et al. Comparison of toad venoms from different Bufo species by HPLC and LC‐DAD‐MS/MS. J Ethnopharmacol. 2010;131:368–376. [DOI] [PubMed] [Google Scholar]
- 292. Yancy CW, Jessup M, Bozkurt B, et al. 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: An update of the 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines and the Heart Failure Society of America. J Am Coll Cardiol. 2016;68(13):1476–1488. [DOI] [PubMed] [Google Scholar]
- 293. Kirilmaz B, Saygi S, Gungor H, et al. Digoxin intoxication: An old enemy in modern era. J Geriatr Cardiol. 2012;9:237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Stucky MA, Goldberger ZD. Digoxin: Its role in contemporary medicine. Postgrad Med J. 2015;91:514–518. [DOI] [PubMed] [Google Scholar]
- 295. Ziff OJ, Kotecha D. Digoxin: The good and the bad. Trends Cardiovasc Med. 2016;26:585–595. [DOI] [PubMed] [Google Scholar]
- 296. Bandara V, Weinstein SA, White J, Eddleston M. A review of the natural history, toxinology, diagnosis and clinical management of Nerium oleander (common oleander) and Thevetia peruviana (yellow oleander) poisoning. Toxicon. 2010;56:273–281. [DOI] [PubMed] [Google Scholar]
- 297. Pirasath S, Arulnithy K. Yellow oleander poisoning in eastern province: An analysis of admission and outcome. Indian J Med Sci. 2013;67:178–183. [PubMed] [Google Scholar]
- 298. Puvaneswaralingam S. Yellow oleander poisoning and suicide in Sri Lanka. Scottish Univ Med J. 2012;21(6):293–295. [Google Scholar]
- 299. Rathore SS, Curtis JP, Wang Y, Bristow MR, Krumholz HM. Association of serum digoxin concentration and outcomes in patients with heart failure. JAMA. 2003;289:871–878. [DOI] [PubMed] [Google Scholar]
- 300. Dvela M, Rosen H, Feldmann T, Nesher M, Lichtstein D. Diverse biological responses to different cardiotonic steroids. Pathophysiology. 2007;14:159–166. [DOI] [PubMed] [Google Scholar]
- 301. Fuerstenwerth H. On the differences between ouabain and digitalis glycosides. Am J Ther. 2014;21:35–42. [DOI] [PubMed] [Google Scholar]
- 302. Clausen T. Acute stimulation of Na/K pump by cardiac glycosides in the nanomolar range. J Gen Physiol. 2002;119:295–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Rang HP, Dale MM, Ritter JM, Flower RJ. Rang and Dale's Pharmacology. 6th ed London: Churchill Livingstone; 2007. [Google Scholar]
- 304. Pincus M. Management of digoxin toxicity. Aust Prescr. 2016;39:18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Roberts DM, Gallapatthy G, Dunuwille A, Chan BS. Pharmacological treatment of cardiac glycoside poisoning. Br J Clin Pharmacol. 2016;81:488–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Chan BS, Buckley NA. Digoxin‐specific antibody fragments in the treatment of digoxin toxicity. Clin Toxicol (Phila). 2014;52:824–836. [DOI] [PubMed] [Google Scholar]
- 307. Dasgupta A, Szelei‐Stevens KA. Neutralization of free digoxin‐like immunoreactive components of oriental medicines Dan Shen and Lu‐Shen‐Wan by the Fab fragment of antidigoxin antibody (Digibind). Am J Clin Pathol. 2004;121:276–281. [DOI] [PubMed] [Google Scholar]
- 308. Dasgupta A, Emerson L. Neutralization of cardiac toxins oleandrin, oleandrigenin, bufalin, and cinobufotalin by digibind: Monitoring the effect by measuring free digitoxin concentrations. Life Sci. 1998;63:781–788. [DOI] [PubMed] [Google Scholar]
- 309. Dasgupta A, Lopez AE, Wells A, Olsen M, Actor J. The Fab fragment of anti‐digoxin antibody (digibind) binds digitoxin‐like immunoreactive components of Chinese medicine Chan Su: Monitoring the effect by measuring free digitoxin. Clin Chim Acta. 2001;309:91–95. [DOI] [PubMed] [Google Scholar]
- 310. Eddleston M, Persson H. Acute plant poisoning and antitoxin antibodies. J Toxicol Clin Toxicol. 2003;41:309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Sabouraud A, Urtizberea M, Cano N, Garnier R, Scherrmann JM. Specific anti‐digoxin Fab fragments: An available antidote for proscillaridin and scilliroside poisoning? Hum Exp Toxicol. 1990;9:191–193. [DOI] [PubMed] [Google Scholar]
- 312. Bania T, Hoffman RS, Howland MA, Goldfrank LR. Accidental Indian hemp (Apocyneacea cannabinum) cardiac glycoside toxicity. Vet Hum Toxicol. 1993;35:328. [Google Scholar]
- 313. Tatlısu MA, Çekirdekçi Eİ, Akyüz Ş, Nurkalem Z. A case of Mobitz type II atrioventricular block due to Nerium oleander poisoning successfully managed with digoxin‐specific Fab antibody fragments. Turk Kardiyol Dern Ars. 2015;43:648–650. [DOI] [PubMed] [Google Scholar]
- 314. Camphausen C, Haas NA, Mattke AC. Successful treatment of oleander intoxication (cardiac glycosides) with digoxin‐specific Fab antibody fragments in a 7‐year‐old child: Case report and review of literature. Z Kardiol. 2005;94:817–823. [DOI] [PubMed] [Google Scholar]
- 315. Brubacher JR, Ravikumar PR, Bania T, Heller MB, Hoffman RS. Treatment of toad venom poisoning with digoxin‐specific Fab fragments. Chest. 1996;110:1282–1288. [DOI] [PubMed] [Google Scholar]
- 316. Eddleston M, Rajapakse S, Rajakanthan, et al. Anti‐digoxin Fab fragments in cardiotoxicity induced by ingestion of yellow oleander: A randomised controlled trial. Lancet. 2000;355:967–972. [DOI] [PubMed] [Google Scholar]
- 317. Fink SL, Robey TE, Tarabar AF, Hodsdon ME. Rapid detection of convallatoxin using five digoxin immunoassays. Clin Toxicol. 2014;52:659–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Rhodes L, Munday R, Briggs L. Ostreopsis siamensis and palytoxin‐related compounds in New Zealand: A risk to human health? In: Proceedings of the 12th International Conference on Harmful Algae, Copenhagen, Denmark. ISSHA and Intergovernmental Oceanographic Commision of UNESCO 2008;326–329.
- 319. Patocka J, Gupta RC, Wu QH, Kuca K. Toxic potential of palytoxin. J Huazhong Univ Sci Technolog Med Sci. 2015;35:773–780. [DOI] [PubMed] [Google Scholar]
- 320. Deeds JR, Schwartz MD. Human risk associated with palytoxin exposure. Toxicon. 2010;56:150–162. [DOI] [PubMed] [Google Scholar]
- 321. Moore RE, Scheuer PJ. Palytoxin: A new marine toxin from a coelenterate. Science. 1971;172:495–498. [DOI] [PubMed] [Google Scholar]
- 322. Alcala AC, Alcala LC, Garth JS, Yasumura D, Yasumoto T. Human fatality due to ingestion of the crab demania‐reynaudii that contained a palytoxin‐like toxin. Toxicon. 1988;26:105–107. [DOI] [PubMed] [Google Scholar]
- 323. Fukui M, Murata M, Inoue A, Gawel M, Yasumoto T. Occurrence of palytoxin in the trigger fish Melichtys vidua . Toxicon. 1987;25:1121–1124. [DOI] [PubMed] [Google Scholar]
- 324. Granéli E FC, Yasumoto T, et al. Sea urchins poisoning by the benthic dinoflagellate Ostreopsis ovata on the Brazilian coast In: Book of Abstracts of Xth International Conference on Harmful Algae. St. Pete Beach, FL: 2002. [Google Scholar]
- 325. Okano H, Masuoka H, Kamei S, et al. Rhabdomyolysis and myocardial damage induced by palytoxin, a toxin of blue humphead parrotfish. Intern Med. 1998;37:330–333. [DOI] [PubMed] [Google Scholar]
- 326. Cha JK, Christ WJ, Finan JM, et al. Stereochemistry of palytoxin .4. Complete structure. J Am Chem Soc. 1982;104:7369–7371. [Google Scholar]
- 327. Moore RE, Bartolini G, Barchi J, et al. Absolute stereochemistry of palytoxin. J Am Chem Soc. 1982;104:3776–3779. [Google Scholar]
- 328. Nordt SP, Wu J, Zahller S, Clark RF, Cantrell FL. Palytoxin poisoning after dermal contact with zoanthid coral. J Emerg Med. 2011;40:397–399. [DOI] [PubMed] [Google Scholar]
- 329. Ramos V, Vasconcelos V. Palytoxin and analogs: Biological and ecological effects. Marine Drugs. 2010;8:2021–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Tubaro A, Sosa S, Hungerford J. Toxicology and diversity of marine toxins Veterinary Toxicology: Basic and Clinical Principles. Amsterdam: Academic Press/Elsevier; 2012. [Google Scholar]
- 331. Proudfoot A. The early toxicology of physostigmine: A tale of beans, great men and egos. Toxicol Rev. 2006;25:99–138. [DOI] [PubMed] [Google Scholar]
- 332. Coman G, Farcas A, Matei AV, Florian C.. Pesticides mechanisms of action in living organisms In: Simeonov L. MF, Simeonova B, eds. Environmental Security Assessment and Management of obsolete Pesticides in Southeast Europe. NATO Science for Peace and Security Series C: Environmental Security. Dordrecht: Springer; 2013:201–214. [Google Scholar]
- 333. Dawson AH, Eddleston M, Senarathna L, et al. Acute human lethal toxicity of agricultural pesticides: A prospective cohort study. PLoS Med. 2010;7:e1000357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Mikaszewska‐Sokolewicz MA, Pankowska S, Janiak M, et al. Coma in the course of severe poisoning after consumption of red fly agaric (Amanita muscaria). Acta Biochim Pol. 2016;63:1170. [DOI] [PubMed] [Google Scholar]
- 335. Michelot D, Melendez‐Howell LM. Amanita muscaria: Chemistry, biology, toxicology, and ethnomycology. Mycol Res. 2003;107:131–146. [DOI] [PubMed] [Google Scholar]
- 336. Hendrickson RG, Morocco AP, Greenberg MI. Pilocarpine toxicity and the treatment of xerostomia. J Emerg Med. 2004;26:429–432. [DOI] [PubMed] [Google Scholar]
- 337. Pfliegler GP, Palatka K. Attempted suicide with pilocarpine eyedrops. Am J Ophthalmol. 1995;120:399–400. [DOI] [PubMed] [Google Scholar]
- 338. Berdai MA, Labib S, Chetouani K, Harandou M. Atropa belladonna intoxication: A case report. Pan Afr Med J. 2012;11:72. [PMC free article] [PubMed] [Google Scholar]
- 339. Caksen H, Odabas D, Akbayram S, et al. Deadly nightshade (Atropa belladonna) intoxication: An analysis of 49 children. Hum Exp Toxicol. 2003;22:665–668. [DOI] [PubMed] [Google Scholar]
- 340. Mateo Montoya A, Mavrakanas N, Schutz JS. Acute anticholinergic syndrome from Atropa belladonna mistaken for blueberries. Eur J Ophthalmol. 2009;19:170–172. [DOI] [PubMed] [Google Scholar]
- 341. Adegoke SA, Alo LA. Datura stramonium poisoning in children. Niger J Clin Pract. 2013;16:116–118. [DOI] [PubMed] [Google Scholar]
- 342. Boumba VA, Mitselou A, Vougiouklakis T. Fatal poisoning from ingestion of Datura stramonium seeds. Vet Hum Toxicol. 2004;46:81–82. [PubMed] [Google Scholar]
- 343. Higbee MD, Kumar M, Galant SP. Stimulation of endogenous catecholamine release by theophylline: A proposed additional mechanism of action for theophylline effects. J Allergy Clin Immunol. 1982;70:377–382. [DOI] [PubMed] [Google Scholar]
- 344. Fredholm BB. On the mechanism of action of theophylline and caffeine. Acta Med Scand. 1985;217:149–153. [DOI] [PubMed] [Google Scholar]
- 345. Lemery R, Pecarskie A, Bernick J, Williams K, Wells GA. A prospective placebo controlled randomized study of caffeine in patients with supraventricular tachycardia undergoing electrophysiologic testing. J Cardiovasc Electrophysiol. 2015;26:1–6. [DOI] [PubMed] [Google Scholar]
- 346. Voldsgaard A, Koch‐Henriksen N, Magyari M, et al. Early safety and efficacy of fingolimod treatment in Denmark. Acta Neurol Scand. 2017;135:129–133. [DOI] [PubMed] [Google Scholar]
- 347. Linker RA, Wendt G. Cardiac safety profile of first dose of fingolimod for relapsing‐remitting multiple sclerosis in real‐world settings: data from a German Prospective Multi‐Center Observational Study. Neurol Ther. 2016;5:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Baghbanian SM. Follow‐up of hypertension in patients with multiple sclerosis. Iran J Neurol. 2016;15:180–181. [PMC free article] [PubMed] [Google Scholar]
- 349. Steckler TL. Lithium‐ and carbamazepine‐associated sinus node dysfunction: Nine‐year experience in a psychiatric hospital. J Clin Psychopharmacol. 1994;14:336–339. [PubMed] [Google Scholar]
- 350. Tocchetti CG, Cadeddu C, Di Lisi D, et al. From molecular mechanisms to clinical management of antineoplastic drug‐induced cardiovascular toxicity: A translational overview. Antioxid Redox Signal. 2017;27(18):1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Zamorano JL, Lancellotti P, Rodriguez Munoz D, et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur Heart J. 2017;19:9–42. [DOI] [PubMed] [Google Scholar]
- 352. Ewer MS, Ewer SM. Cardiotoxicity of anticancer treatments. Nat Rev Cardiol. 2015;12:547–558. [DOI] [PubMed] [Google Scholar]
- 353. Yan T, Deng S, Metzger A, et al. Topoisomerase II{alpha}‐dependent and ‐independent apoptotic effects of dexrazoxane and doxorubicin. Mol Cancer Ther. 2009;8:1075–1085. [DOI] [PubMed] [Google Scholar]
- 354. Suter TM, Ewer MS. Cancer drugs and the heart: Importance and management. Eur Heart J. 2013;34:1102–1111. [DOI] [PubMed] [Google Scholar]
- 355. Sterba M, Popelova O, Vavrova A, et al. Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid Redox Signal. 2013;18:899–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Zhang S, Liu X, Bawa‐Khalfe T, et al. Identification of the molecular basis of doxorubicin‐induced cardiotoxicity. Nat Med. 2012;18:1639–1642. [DOI] [PubMed] [Google Scholar]
- 357. Lencova‐Popelova O, Jirkovsky E, Jansova H, et al. Cardioprotective effects of inorganic nitrate/nitrite in chronic anthracycline cardiotoxicity: Comparison with dexrazoxane. J Mol Cell Cardiol. 2016;91:92–103. [DOI] [PubMed] [Google Scholar]
- 358. Schwartz CL, Wexler LH, Krailo MD, et al. Intensified chemotherapy with dexrazoxane cardioprotection in newly diagnosed nonmetastatic osteosarcoma: A report from the children's oncology group. Pediatr Blood Cancer. 2016;63:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Wu V. Dexrazoxane: A cardioprotectant for pediatric cancer patients receiving anthracyclines. J Pediatr Oncol Nurs. 2015;32:178–184. [DOI] [PubMed] [Google Scholar]
- 360. van Dalen EC, Caron HN, Dickinson HO, Kremer LC. Cardioprotective interventions for cancer patients receiving anthracyclines. Cochrane Database Syst Rev. 2011;(6):CD003917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Pun SC, Neilan TG. Cardioprotective interventions: Where are we? Am Coll Cardiol. 2016. http://www.acc.org/latest-in-cardiology/articles/2016/07/21/07/28/cardioprotective-interventions. [Google Scholar]
- 362. Oliveira PJ, Bjork JA, Santos MS, et al. Carvedilol‐mediated antioxidant protection against doxorubicin‐induced cardiac mitochondrial toxicity. Toxicol Appl Pharmacol. 2004;200:159–168. [DOI] [PubMed] [Google Scholar]
- 363. van der Meer P, Gietema JA, Suter TM, van Veldhuisen DJ. Cardiotoxicity of breast cancer treatment: No easy solution for an important long‐term problem. Eur Heart J. 2016;37:1681–1683. [DOI] [PubMed] [Google Scholar]
- 364. Chintalgattu V, Rees ML, Culver JC, et al. Coronary microvascular pericytes are the cellular target of sunitinib malate‐induced cardiotoxicity. Sci Transl Med. 2013;5:187ra169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Koulaouzidis G, Lyon AR. Proteasome inhibitors as a potential cause of heart failure. Heart Fail Clin. 2017;13(2):289–295. [DOI] [PubMed] [Google Scholar]
- 366. Mackin P. Cardiac side effects of psychiatric drugs. Hum Psychopharmacol. 2008;23(Suppl 1):3–14. [DOI] [PubMed] [Google Scholar]
- 367. Abdel‐Wahab BA, Metwally ME. Clozapine‐induced cardiotoxicity: Role of oxidative stress, tumour necrosis factor alpha and NF‐kappabeta. Cardiovasc Toxicol. 2015;15:355–365. [DOI] [PubMed] [Google Scholar]
- 368. Cosyns B, Droogmans S, Rosenhek R, Lancellotti P. Drug‐induced valvular heart disease. Heart. 2013;99:7–12. [DOI] [PubMed] [Google Scholar]
- 369. Elangbam CS. Drug‐induced valvulopathy: An update. Toxicol Pathol. 2010;38:837–848. [DOI] [PubMed] [Google Scholar]
- 370. Huang XP, Setola V, Yadav PN, et al. Parallel functional activity profiling reveals valvulopathogens are potent 5‐hydroxytryptamine(2B) receptor agonists: Implications for drug safety assessment. Mol Pharmacol. 2009;76:710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Seferovic PM, Ristic AD, Maksimovic R, et al. Pericardial syndromes: An update after the ESC guidelines 2004. Heart Fail Rev. 2013;18:255–266. [DOI] [PubMed] [Google Scholar]
- 372. Caldeira D, Barra M, Goncalves N, et al. Pericardial bleeding risk with non‐vitamin K oral anticoagulants: A meta‐analysis. Int J Cardiol. 2015;182:187–188. [DOI] [PubMed] [Google Scholar]
- 373. Cornily JC, Pennec PY, Castellant P, et al. Cardiac tamponade in medical patients: A 10‐year follow‐up survey. Cardiology. 2008;111:197–201. [DOI] [PubMed] [Google Scholar]
- 374. Devasahayam J, Pillai U, Lacasse A. A rare case of pericarditis, complication of infliximab treatment for Crohn's disease. J Crohns Colitis. 2012;6:730–731. [DOI] [PubMed] [Google Scholar]
- 375. Cohen M, Rocher F, Brunschwig C, Lebrun C. Recurrent pericarditis due to natalizumab treatment. Neurology. 2009;72:1616–1617. [DOI] [PubMed] [Google Scholar]
- 376. Sonu I, Wong R, Rothenberg ME. 5‐ASA induced recurrent myopericarditis and cardiac tamponade in a patient with ulcerative colitis. Dig Dis Sci. 2013;58:2148–2150. [DOI] [PubMed] [Google Scholar]
- 377. Coman RM, Glover SC, Gjymishka A. Febrile pleuropericarditis, a potentially life‐threatening adverse event of balsalazide—Case report and literature review of the side effects of 5‐aminosalicylates. Expert Rev Clin Immunol. 2014;10:667–675. [DOI] [PubMed] [Google Scholar]
- 378. Park EH, Kim BJ, Huh JK, et al. Recurrent mesalazine‐induced myopericarditis in a patient with ulcerative colitis. J Cardiovasc Ultrasound. 2012;20:154–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Kiyomatsu H, Kawai K, Tanaka T, et al. Mesalazine‐induced pleuropericarditis in a patient with Crohn's disease. Intern Med. 2015;54:1605–1608. [DOI] [PubMed] [Google Scholar]
- 380. Siddiqui MA, Khan IA. Isoniazid‐induced lupus erythematosus presenting with cardiac tamponade. Am J Ther. 2002;9:163–165. [DOI] [PubMed] [Google Scholar]
- 381. Harnett DT, Chandra‐Sekhar HB, Hamilton SF. Drug‐induced lupus erythematosus presenting with cardiac tamponade: A case report and literature review. Can J Cardiol. 2014;30(247):e211–242. [DOI] [PubMed] [Google Scholar]
- 382. Guberman BA, Fowler NO, Engel PJ, Gueron M, Allen JM. Cardiac‐tamponade in medical patients. Circulation. 1981;64:633–640. [DOI] [PubMed] [Google Scholar]
- 383. Xu B, MacIsaac A. Life‐threatening haemorrhagic pericarditis associated with rivaroxaban. Int J Cardiol. 2014;174:E75–E76. [DOI] [PubMed] [Google Scholar]
- 384. Bock J, Doenitz A, Andreesen R, Reichle A, Hennemann B. Pericarditis after high‐dose chemotherapy: More frequent than expected? Onkologie. 2006;29:321–324. [DOI] [PubMed] [Google Scholar]
- 385. Gahler A, Hitz F, Hess U, Cerny T. Acute pericarditis and pleural effusion complicating cytarabine chemotherapy. Onkologie. 2003;26:348–350. [DOI] [PubMed] [Google Scholar]
- 386. Chen CC, Gau JP, You JY, et al. Decitabine‐induced effusions. Leuk Res. 2009;33:e150–151. [DOI] [PubMed] [Google Scholar]
- 387. Kelly K, Swords R, Mahalingam D, Padmanabhan S, Giles FJ. Serosal inflammation (pleural and pericardial effusions) related to tyrosine kinase inhibitors. Target Oncol. 2009;4:99–105. [DOI] [PubMed] [Google Scholar]
- 388. Dadfarmay S, Dixon J. A case of acute cardiomyopathy and pericarditis associated with methylphenidate. Cardiovasc Toxicol. 2009;9:49–52. [DOI] [PubMed] [Google Scholar]
- 389. Balachandran KP, Stewart D, Berg GA, Oldroyd KG. Chronic pericardial constriction linked to the antiparkinsonian dopamine agonist pergolide. Postgrad Med J. 2002;78:49–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390. Champagne S, Coste E, Peyriere H, et al. Chronic constrictive pericarditis induced by long‐term bromocriptine therapy: Report of two cases. Ann Pharmacother. 1999;33:1050–1054. [DOI] [PubMed] [Google Scholar]
- 391. Townsend M, MacIver DH. Constrictive pericarditis and pleuropulmonary fibrosis secondary to cabergoline treatment for Parkinson's disease. Heart. 2004;90:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Meeran MK, Ahmed AH, Parsons FM, Anderson CK. Constrictive pericarditis due to methysergide therapy. S Afr Med J. 1976;50:1595–1597. [PubMed] [Google Scholar]
- 393. Schifferdecker B, Spodick DH. Nonsteroidal anti‐inflammatory drugs in the treatment of pericarditis. Cardiol Rev. 2003;11:211–217. [DOI] [PubMed] [Google Scholar]
- 394. Krehlik JM, Hindson DA, Crowley JJ, Jr , Knight LL. Minoxidil‐associated pericarditis and fatal cardiac tamponade. West J Med. 1985;143:527–529. [PMC free article] [PubMed] [Google Scholar]
- 395. Krauth MT, Herndlhofer S, Schmook MT, et al. Extensive pleural and pericardial effusion in chronic myeloid leukemia during treatment with dasatinib at 100 mg or 50 mg daily. Haematologica. 2011;96:163–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396. Newman M, Malla M, Gojo I. Azacitidine‐induced pericarditis: A case series. Pharmacotherapy. 2016;36:443–448. [DOI] [PubMed] [Google Scholar]
- 397. Mohyuddin T, Elyan M, Kushner I. Pericarditis: A rare complication of methotrexate therapy. Clin Rheumatol. 2007;26:2157–2158. [DOI] [PubMed] [Google Scholar]
- 398. Davey P, Lalloo DG. Drug induced chest pain‐rare but important. Postgrad Med J. 2000;76:420–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399. Benjamini O, Kimhi O, Lishner M. Severe pleuropericarditis and cardiomyopathy induced by high dose interferon alpha‐2b. Israel Med Assoc J. 2007;9:486–487. [PubMed] [Google Scholar]
- 400. Krantz MJ, Garcia JA, Mehler PS. Tramadol‐associated pericarditis. Int J Cardiol. 2005;99:497–498. [DOI] [PubMed] [Google Scholar]
- 401. Hermans C, Straetmans N, Michaux JL, Ferrant A. Pericarditis induced by high‐dose cytosine arabinoside chemotherapy. Ann Hematol. 1997;75:55–57. [DOI] [PubMed] [Google Scholar]
- 402. Gottdiener JS, Appelbaum FR, Ferrans VJ, Deisseroth A, Ziegler J. Cardiotoxicity associated with high‐dose cyclophosphamide therapy. Arch Intern Med. 1981;141:758–763. [PubMed] [Google Scholar]
- 403. Schoenwetter AH, Siber EN. Penicillin hypersensitivity, acute pericarditis, and eosinophilia. JAMA. 1965;191:672–673. [DOI] [PubMed] [Google Scholar]
- 404. Felman RH, Sutherland DB, Conklin JL, Mitros FA. Eosinophilic cholecystitis, appendiceal inflammation, pericarditis, and cephalosporin‐associated eosinophilia. Dig Dis Sci. 1994;39:418–422. [DOI] [PubMed] [Google Scholar]
- 405. Coxib, Traditional NTC , Bhala N, et al. Vascular and upper gastrointestinal effects of non‐steroidal anti‐inflammatory drugs: meta‐analyses of individual participant data from randomised trials. Lancet. 2013;382:769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406. Patrono C, Baigent C. Nonsteroidal anti‐inflammatory drugs and the heart. Circulation. 2014;129:907–916. [DOI] [PubMed] [Google Scholar]
- 407. Howes LG. Selective COX‐2 inhibitors, NSAIDs and cardiovascular events—Is celecoxib the safest choice? Ther Clin Risk Manag. 2007;3:831–845. [PMC free article] [PubMed] [Google Scholar]
- 408. Seta F, Chung AD, Turner PV, et al. Renal and cardiovascular characterization of COX‐2 knockdown mice. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1751–1760. [DOI] [PubMed] [Google Scholar]
- 409. Jia Z, Zhang Y, Ding G, et al. Role of COX‐2/mPGES‐1/prostaglandin E2 cascade in kidney injury. Mediators Inflamm. 2015;2015:147894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Therland KL, Stubbe J, Thiesson HC, et al. Cycloxygenase‐2 is expressed in vasculature of normal and ischemic adult human kidney and is colocalized with vascular prostaglandin E‐2 EP4 receptors. J Am Soc Nephrol. 2004;15:1189–1198. [DOI] [PubMed] [Google Scholar]
- 411. Catella‐Lawson F, Reilly MP, Kapoor SC, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001;345:1809–1817. [DOI] [PubMed] [Google Scholar]
- 412. Ungprasert P, Srivali N, Thongprayoon C. Nonsteroidal anti‐inflammatory drugs and risk of incident heart failure: A systematic review and meta‐analysis of observational studies. Clin Cardiol. 2016;39:111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Aw TJ, Haas SJ, Liew D, Krum H. Meta‐analysis of cyclooxygenase‐2 inhibitors and their effects on blood pressure. Arch Intern Med. 2005;165:490–496. [DOI] [PubMed] [Google Scholar]
- 414. Khatchadourian ZD, Moreno‐Hay I, de Leeuw R. Nonsteroidal anti‐inflammatory drugs and antihypertensives: How do they relate? Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;117:697–703. [DOI] [PubMed] [Google Scholar]
- 415. Garcia Rodriguez LA, Varas C, Patrono C. Differential effects of aspirin and non‐aspirin nonsteroidal antiinflammatory drugs in the primary prevention of myocardial infarction in postmenopausal women. Epidemiology. 2000;11:382–387. [DOI] [PubMed] [Google Scholar]
- 416. Oikonomou E, Vogiatzi G, Papamikroulis GA, et al. Antiplatelet therapy in acute coronary syndromes. Evidence based medicine. Curr Pharm Des. 2016;22(29):4519–4536. [DOI] [PubMed] [Google Scholar]
- 417. Sutcliffe P, Connock M, Gurung T, et al. Aspirin for prophylactic use in the primary prevention of cardiovascular disease and cancer: A systematic review and overview of reviews. Health Technol Assess. 2013;17(43):1–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418. Sondergaard KB, Weeke P, Wissenberg M, et al. Non‐steroidal anti‐inflammatory drug use is associated with increased risk of out‐of‐hospital cardiac arrest: A nationwide case‐time‐control study. Eur Heart J Cardiovasc Pharmacother. 2017;3:100–107. [DOI] [PubMed] [Google Scholar]
- 419. Reilly IA, FitzGerald GA. Inhibition of thromboxane formation in vivo and ex vivo: Implications for therapy with platelet inhibitory drugs. Blood. 1987;69:180–186. [PubMed] [Google Scholar]
- 420. Santilli F, Rocca B, De Cristofaro R, et al. Platelet cyclooxygenase inhibition by low‐dose aspirin is not reflected consistently by platelet function assays: Implications for aspirin “resistance.” J Am Coll Cardiol. 2009;53:667–677. [DOI] [PubMed] [Google Scholar]
- 421. Capone ML, Sciulli MG, Tacconelli S, et al. Pharmacodynamic interaction of naproxen with low‐dose aspirin in healthy subjects. J Am Coll Cardiol. 2005;45:1295–1301. [DOI] [PubMed] [Google Scholar]
- 422. Pasvolsky O, Leader A, Iakobishvili Z, et al. Tyrosine kinase inhibitor associated vascular toxicity in chronic myeloid leukemia. Cardio‐Oncology. 2015;1(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423. Katsi V, Zerdes I, Manolakou S, et al. Anti‐VEGF anticancer drugs: Mind the hypertension. Recent Adv Cardiovasc Drug Discov. 2014;9:63–72. [DOI] [PubMed] [Google Scholar]
- 424. Patel JN, Jiang C, Hertz DL, et al. Bevacizumab and the risk of arterial and venous thromboembolism in patients with metastatic, castration‐resistant prostate cancer treated on Cancer and Leukemia Group B (CALGB) 90401 (Alliance). Cancer. 2015;121:1025–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425. Li Y, Li S, Zhu Y, et al. Incidence and risk of sorafenib‐induced hypertension: A systematic review and meta‐analysis. J Clin Hypertens (Greenwich). 2014;16:177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. Kruzliak P, Novak J, Novak M. Vascular endothelial growth factor inhibitor‐induced hypertension: From pathophysiology to prevention and treatment based on long‐acting nitric oxide donors. Am J Hypertens. 2014;27:3–13. [DOI] [PubMed] [Google Scholar]
- 427. Nalluri SR, Chu D, Keresztes R, Zhu X, Wu S. Risk of venous thromboembolism with the angiogenesis inhibitor bevacizumab in cancer patients: A meta‐analysis. JAMA. 2008;300:2277–2285. [DOI] [PubMed] [Google Scholar]
- 428. Hurwitz HI, Saltz LB, Van Cutsem E, et al. Venous thromboembolic events with chemotherapy plus bevacizumab: A pooled analysis of patients in randomized phase II and III studies. J Clin Oncol. 2011;29:1757–1764. [DOI] [PubMed] [Google Scholar]
- 429. Kappers MHW, de Beer VJ, Zhou ZC, et al. Sunitinib‐induced systemic vasoconstriction in swine is endothelin mediated and does not involve nitric oxide or oxidative stress. Hypertension. 2012;59:151–157. [DOI] [PubMed] [Google Scholar]
- 430. Ikeda M, Okusaka T, Mitsunaga S, et al. Safety and pharmacokinetics of lenvatinib in patients with advanced hepatocellular carcinoma. Clin Cancer Res. 2016;22:1385–1394. [DOI] [PubMed] [Google Scholar]
- 431. Robinson ES, Matulonis UA, Ivy P, et al. Rapid development of hypertension and proteinuria with cediranib, an oral vascular endothelial growth factor receptor inhibitor. Clin J Am Soc Nephrol. 2010;5:477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432. Abdel‐Rahman O, ElHalawani H, Ahmed H. Doublet BRAF/MEK inhibition versus single‐agent BRAF inhibition in the management of BRAF‐mutant advanced melanoma, biological rationale and meta‐analysis of published data. Clin Transl Oncol. 2016;18:848–858. [DOI] [PubMed] [Google Scholar]
- 433. Rao KN, Binbrek AS, Sobel BE. Heart disease and erythropoietin. Future Cardiol. 2008;4:57–64. [DOI] [PubMed] [Google Scholar]
- 434. Malyszko J, Suchowierska E, Malyszko JS, Mysliwiec M. A comprehensive study on hemostasis in CAPD patients treated with erythropoietin. Periton Dialysis Int. 2002;22:582–592. [PubMed] [Google Scholar]
- 435. Fishbane S, Besarab A. Mechanism of increased mortality risk with erythropoietin treatment to higher hemoglobin targets. Clin J Am Soc Nephrol. 2007;2:1274–1282. [DOI] [PubMed] [Google Scholar]
- 436. Tobu M, Iqbal O, Fareed D, et al. Erythropoietin‐induced thrombosis as a result of increased inflammation and thrombin activatable fibrinolytic inhibitor. Clin Appl Thromb Hemost. 2004;10:225–232. [DOI] [PubMed] [Google Scholar]
- 437. Kurtul A, Duran M, Uysal OK, Ornek E. Acute coronary syndrome with intraventricular thrombus after using erythropoietin. Anadolu Kardiyol Der. 2013;13:278–279. [DOI] [PubMed] [Google Scholar]
- 438. Cariou A, Deye N, Vivien B, et al. Early high‐dose erythropoietin therapy after out‐of‐hospital cardiac arrest: A multicenter, randomized controlled trial. J Am Coll Cardiol. 2016;68:40–49. [DOI] [PubMed] [Google Scholar]
- 439. Stavert B, McGuinness MB, Harper CA, Guymer RH, Finger RP. Cardiovascular adverse effects of phenylephrine eyedrops: A systematic review and meta‐analysis. JAMA Ophthalmol. 2015;133:647–652. [DOI] [PubMed] [Google Scholar]
- 440. Hengstmann JH, Goronzy J. Pharmacokinetics of 3H‐phenylephrine in man. Eur J Clin Pharmacol. 1982;21:335–341. [DOI] [PubMed] [Google Scholar]
- 441. Kanfer I, Dowse R, Vuma V. Pharmacokinetics of oral decongestants. Pharmacotherapy. 1993;13:116S–128S; discussion 143S‐146S. [PubMed] [Google Scholar]
- 442. Hatton RC, Winterstein AG, McKelvey RP, Shuster J, Hendeles L. Efficacy and safety of oral phenylephrine: Systematic review and meta‐analysis. Ann Pharmacother. 2007;41:381–390. [DOI] [PubMed] [Google Scholar]
- 443. Tark BE, Messe SR, Balucani C, Levine SR. Intracerebral hemorrhage associated with oral phenylephrine use: A case report and review of the literature. J Stroke Cerebrovasc. 2014;23:2296–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444. Atkinson HC, Potts AL, Anderson BJ. Potential cardiovascular adverse events when phenylephrine is combined with paracetamol: Simulation and narrative review. Eur J Clin Pharmacol. 2015;71:931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445. Atkinson HC, Stanescu I, Salem, II , Potts AL, Anderson BJ. Increased bioavailability of phenylephrine by co‐administration of acetaminophen: Results of four open‐label, crossover pharmacokinetic trials in healthy volunteers. Eur J Clin Pharmacol. 2015;71:151–158. [DOI] [PubMed] [Google Scholar]
- 446. Costantino G, Ceriani E, Sandrone G, Montano N. Ischemic stroke in a man with naphazoline abuse history. Am J Emerg Med. 2007;25:983 e981–982. [DOI] [PubMed] [Google Scholar]
- 447. Leupold D, Wartenberg KE. Xylometazoline abuse induced ischemic stroke in a young adult. Neurologist. 2011;17:41–43. [DOI] [PubMed] [Google Scholar]
- 448. Wenzel S, Sagowski C, Laux G, Kehrl W, Metternich FU. Course and therapy of intoxication with imidazoline derivate naphazoline. Int J Pediatr Otorhinolaryngol. 2004;68:979–983. [DOI] [PubMed] [Google Scholar]
- 449. Alvarez‐Pitti J, Rodriguez‐Varela A, Morales‐Carpi C, Lurbe E, Estan L. Naphazoline intoxication in children. Eur J Pediatr. 2006;165:815–816. [DOI] [PubMed] [Google Scholar]
- 450. Rossato LG, Costa VM, Limberger RP, Bastos Mde L, Remiao F. Synephrine: From trace concentrations to massive consumption in weight‐loss. Food Chem Toxicol. 2011;49:8–16. [DOI] [PubMed] [Google Scholar]
- 451. Bui LT, Nguyen DT, Ambrose PJ. Blood pressure and heart rate effects following a single dose of bitter orange. Ann Pharmacother. 2006;40:53–57. [DOI] [PubMed] [Google Scholar]
- 452. Min B, Cios D, Kluger J, White CM. Absence of QTc‐interval‐prolonging or hemodynamic effects of a single dose of bitter‐orange extract in healthy subjects. Pharmacotherapy. 2005;25:1719–1724. [DOI] [PubMed] [Google Scholar]
- 453. Stohs SJ, Preuss HG, Shara M. A review of the receptor‐binding properties of p‐synephrine as related to its pharmacological effects. Oxid Med Cell Longev. 2011;2011:482973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454. Marcus DM, Grollman AP. Ephedra‐free is not danger‐free. Science. 2003;301:1669–1671; author reply 1669–1671. [DOI] [PubMed] [Google Scholar]
- 455. Etxabe J, Vazquez JA. Morbidity and mortality in Cushing's disease: An epidemiological approach. Clin Endocrinol (Oxf). 1994;40:479–484. [DOI] [PubMed] [Google Scholar]
- 456. Whitworth JA. Mechanisms of glucocorticoid–induced hypertension. Kidney Int. 1987;31:1213–1224. [DOI] [PubMed] [Google Scholar]
- 457. Goodwin JE, Geller DS. Glucocorticoid‐induced hypertension. Pediatr Nephrol. 2012;27:1059–1066. [DOI] [PubMed] [Google Scholar]
- 458. Saruta T. Mechanism of glucocorticoid‐induced hypertension. Hypertens Res. 1996;19:1–8. [DOI] [PubMed] [Google Scholar]
- 459. Hunter RW, Bailey MA. Glucocorticoids and 11beta‐hydroxysteroid dehydrogenases: Mechanisms for hypertension. Curr Opin Pharmacol. 2015;21:105–114. [DOI] [PubMed] [Google Scholar]
- 460. Grossman A, Messerli FH, Grossman E. Drug induced hypertension—An unappreciated cause of secondary hypertension. Eur J Pharmacol. 2015;763:15–22. [DOI] [PubMed] [Google Scholar]
- 461. Jalili J, Askeroglu U, Alleyne B, Guyuron B. Herbal products that may contribute to hypertension. Plast Reconstr Surg. 2013;131:168–173. [DOI] [PubMed] [Google Scholar]
- 462. Ploeger B, Mensinga T, Sips A, et al. The pharmacokinetics of glycyrrhizic acid evaluated by physiologically based pharmacokinetic modeling. Drug Metab Rev. 2001;33:125–147. [DOI] [PubMed] [Google Scholar]
- 463. Nimkarn S. Apparent mineralocorticoid excess—Update. Adv Exp Med Biol. 2011;707:47–48. [DOI] [PubMed] [Google Scholar]
- 464. Ferrari P. The role of 11beta‐hydroxysteroid dehydrogenase type 2 in human hypertension. Biochim Biophys Acta. 2010;1802:1178–1187. [DOI] [PubMed] [Google Scholar]
- 465. Morales JM, Andres A, Rengel M, Rodicio JL. Influence of cyclosporin, tacrolimus and rapamycin on renal function and arterial hypertension after renal transplantation. Nephrol Dial Transplant. 2001;16(Suppl 1):121–124. [DOI] [PubMed] [Google Scholar]
- 466. Reis F, Parada B, de Lemos ET, et al. Hypertension induced by immunosuppressive drugs: A comparative analysis between sirolimus and cyclosporine. Transpl Proc. 2009;41:868–873. [DOI] [PubMed] [Google Scholar]
- 467. El‐Gowelli HM, El‐Mas MM. Central modulation of cyclosporine‐induced hypertension. Naunyn Schmiedebergs Arch Pharmacol. 2015;388:351–361. [DOI] [PubMed] [Google Scholar]
- 468. Hoorn EJ, Walsh SB, McCormick JA, et al. Pathogenesis of calcineurin inhibitor‐induced hypertension. J Nephrol. 2012;25:269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469. Ho S, Clipstone N, Timmermann L, et al. The mechanism of action of cyclosporin A and FK506. Clin Immunol Immunopathol. 1996;80:S40–45. [DOI] [PubMed] [Google Scholar]
- 470. Seibert F, Behrendt C, Schmidt S, et al. Differential effects of cyclosporine and tacrolimus on arterial function. Transpl Int. 2011;24:708–715. [DOI] [PubMed] [Google Scholar]
- 471. Hannah J, Casian A, D'Cruz D. Tacrolimus use in lupus nephritis: A systematic review and meta‐analysis. Autoimmun Rev. 2016;15:93–101. [DOI] [PubMed] [Google Scholar]
- 472. Grzesk E, Malinowski B, Wicinski M, et al. Cyclosporine‐A, but not tacrolimus significantly increases reactivity of vascular smooth muscle cells. Pharmacol Rep. 2016;68:201–205. [DOI] [PubMed] [Google Scholar]
- 473. Wasilewski G, Przybylowski P, Janik L, et al. Inadequate blood pressure control in orthotopic heart transplant: Is there a role of kidney function and immunosuppressive regimen? Transplant Proc. 2014;46:2830–2834. [DOI] [PubMed] [Google Scholar]
- 474. Rozman B, Praprotnik S, Logar D, et al. Leflunomide and hypertension. Ann Rheum Dis. 2002;61:567–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475. van Riel PL, Smolen JS , Emery P, et al. Leflunomide: A manageable safety profile. J Rheumatol Suppl. 2004;71:21–24. [PubMed] [Google Scholar]
- 476. Miller AE. Teriflunomide: A once‐daily oral medication for the treatment of relapsing forms of multiple sclerosis. Clin Ther. 2015;37:2366–2380. [DOI] [PubMed] [Google Scholar]
- 477. Montani D, Seferian A, Savale L, Simonneau G, Humbert M. Drug‐induced pulmonary arterial hypertension: A recent outbreak. Eur Respir Rev. 2013;22:244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478. Herve P, Launay JM, Scrobohaci ML, et al. Increased plasma serotonin in primary pulmonary hypertension. Am J Med. 1995;99:249–254. [DOI] [PubMed] [Google Scholar]
- 479. Kramer MS, Lane DA. Aminorex, dexfenfluramine, and primary pulmonary hypertension. J Clin Epidemiol. 1998;51:361–364. [DOI] [PubMed] [Google Scholar]
- 480. Montani D, Gunther S, Dorfmuller P, et al. Pulmonary arterial hypertension. Orphanet J Rare Dis. 2013;8:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481. Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France: Results from a national registry. Am J Respir Crit Care Med. 2006;173:1023–1030. [DOI] [PubMed] [Google Scholar]
- 482. Savale L, Chaumais MC, Cottin V, et al. Pulmonary hypertension associated with benfluorex exposure. Eur Respir J. 2012;40:1164–1172. [DOI] [PubMed] [Google Scholar]
- 483. Valent P, Hadzijusufovic E, Schernthaner GH, et al. Vascular safety issues in CML patients treated with BCR/ABL1 kinase inhibitors. Blood. 2015;125:901–906. [DOI] [PubMed] [Google Scholar]
- 484. Kloner RA, Hutter AM, Emmick JT, et al. Time course of the interaction between tadalafil and nitrates—Reply. J Am Coll Cardiol. 2004;43:2150–2151. [DOI] [PubMed] [Google Scholar]
- 485. Riha HM, Summers BB, Rivera JV, Van Berkel MA. Novel therapies for angiotensin‐converting enzyme inhibitor‐induced angioedema: A systematic review of current evidence. J Emerg Med. 2017;53(5):662–679. [DOI] [PubMed] [Google Scholar]
- 486. Brown T, Gonzalez J, Monteleone C. Angiotensin‐converting enzyme inhibitor‐induced angioedema: A review of the literature. J Clin Hypertens (Greenwich). 2017;19(12):1377–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487. Bernstein JA, Cremonesi P, Hoffmann TK, Hollingsworth J. Angioedema in the emergency department: A practical guide to differential diagnosis and management. Int J Emerg Med. 2017;10:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488. Woodard‐Grice AV, Lucisano AC, Byrd JB, et al. Sex‐dependent and race‐dependent association of XPNPEP2 C‐2399A polymorphism with angiotensin‐converting enzyme inhibitor‐associated angioedema. Pharmacogenet Genomics. 2010;20:532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489. Pickering TG. Effects of stress and behavioral interventions in hypertension: The rise and fall of omapatrilat. J Clin Hypertens (Greenwich). 2002;4:371–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490. Bohm M, Young R, Jhund PS, et al. Systolic blood pressure, cardiovascular outcomes and efficacy and safety of sacubitril/valsartan (LCZ696) in patients with chronic heart failure and reduced ejection fraction: Results from PARADIGM‐HF. Eur Heart J. 2017;38:1132–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491. Rotermann M, Dunn S, Black A. Oral contraceptive use among women aged 15 to 49: Results from the Canadian Health Measures Survey. Health Rep. 2015;26:21–28. [PubMed] [Google Scholar]
- 492. Blackburn RD, Cunkelman A, Zlidar VM. Oral contraceptives—An update. Popul Rep A. 2000;9:1–16, 25–32. [PubMed] [Google Scholar]
- 493. Hannaford PC, Selvaraj S, Elliott AM, et al. Cancer risk among users of oral contraceptives: Cohort data from the Royal College of General Practitioner's oral contraception study. BMJ. 2007;335:651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494. Lidegaard O, Lokkegaard E, Jensen A, Skovlund CW, Keiding N. Thrombotic stroke and myocardial infarction with hormonal contraception. N Engl J Med. 2012;366:2257–2266. [DOI] [PubMed] [Google Scholar]
- 495. Shufelt CL, Merz CNB. Contraceptive hormone use and cardiovascular disease. J Am Coll Cardiol. 2009;53:221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496. Heit JA, Spencer FA, White RH. The epidemiology of venous thromboembolism. J Thromb Thrombolysis. 2016;41:3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497. Dong W, Colhoun HM, Poulter NR. Blood pressure in women using oral contraceptives: Results from the health survey for England 1994. J Hypertens. 1997;15:1063–1068. [DOI] [PubMed] [Google Scholar]
- 498. ChasanTaber L, Willett WC, Manson JE, et al. Prospective study of oral contraceptives and hypertension among women in the United States. Circulation. 1996;94:483–489. [DOI] [PubMed] [Google Scholar]
- 499. Margolis KL, Adami HO, Luo J, Ye W, Weiderpass E. A prospective study of oral contraceptive use and risk of myocardial infarction among Swedish women. Fertil Steril. 2007;88:310–316. [DOI] [PubMed] [Google Scholar]
- 500. U.S. Selected Practice Recommendations for Contraceptive Use, 2013: Adapted from the World Health Organization selected practice recommendations for contraceptive use, 2nd edition . Division of reproductive health, national center for chronic disease prevention and health promotion. MMWR Recomm Rep. 2013;21:1–60. [PubMed] [Google Scholar]
- 501. Horton LG, Simmons KB, Curtis KM. Combined hormonal contraceptive use among obese women and risk for cardiovascular events: A systematic review. Contraception. 2016;94(6):590–604. [DOI] [PubMed] [Google Scholar]
- 502. Tepper NK, Curtis KM, Steenland MW, Marchbanks PA. Blood pressure measurement prior to initiating hormonal contraception: A systematic review. Contraception. 2013;87:631–638. [DOI] [PubMed] [Google Scholar]
- 503. Petitti DB. Hormonal contraceptives and arterial thrombosis—Not risk‐free but safe enough. New Engl J Med. 2012;366:2316–2318. [DOI] [PubMed] [Google Scholar]
- 504. Lalude OO. Risk of cardiovascular events with hormonal contraception: Insights from the Danish cohort study. Curr Cardiol Rep. 2013;15:374. [DOI] [PubMed] [Google Scholar]
- 505. Noilhan C, Barigou M, Bieler L, et al. Causes of secondary hypertension in the young population: A monocentric study. Ann Cardiol Angeiol (Paris). 2016;65:159–164. [DOI] [PubMed] [Google Scholar]
- 506. Cagnacci A, Zanin R, Napolitano A, Arangino S, Volpe A. Modification of 24‐h ambulatory blood pressure and heart rate during contraception with the vaginal ring: A prospective study. Contraception. 2013;88:539–543. [DOI] [PubMed] [Google Scholar]
- 507. Kovacs L, Bartfai G, Apro G, et al. The effect of the contraceptive pill on blood pressure: A randomized controlled trial of three progestogen‐oestrogen combinations in Szeged, Hungary. Contraception. 1986;33:69–77. [DOI] [PubMed] [Google Scholar]
- 508. Schindler AE, Campagnoli C, Druckmann R, et al. Classification and pharmacology of progestins. Maturitas. 2003;46(Suppl 1):S7–S16. [DOI] [PubMed] [Google Scholar]
- 509. Sitruk‐Ware R. Pharmacological profile of progestins. Maturitas. 2004;47:277–283. [DOI] [PubMed] [Google Scholar]
- 510. van Rooijen M, von Schoultz B, Silveira A, Hamsten A, Bremme K. Different effects of oral contraceptives containing levonorgestrel or desogestrel on plasma lipoproteins and coagulation factor VII. Am J Obstet Gynecol. 2002;186:44–48. [DOI] [PubMed] [Google Scholar]
- 511. Jick SS, Hernandez RK. Risk of non‐fatal venous thromboembolism in women using oral contraceptives containing drospirenone compared with women using oral contraceptives containing levonorgestrel: Case‐control study using United States claims data. BMJ. 2011;342:d2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512. Oelkers W, Foidart JM, Dombrovicz N, Welter A, Heithecker R. Effects of a new oral contraceptive containing an antimineralocorticoid progestogen, drospirenone, on the renin‐aldosterone system, body weight, blood pressure, glucose tolerance, and lipid metabolism. J Clin Endocrinol Metab. 1995;80:1816–1821. [DOI] [PubMed] [Google Scholar]
- 513. Adams MR, Anthony MS, Manning JM, Golden DL, Parks JS. Low‐dose contraceptive estrogen‐progestin and coronary artery atherosclerosis of monkeys. Obstet Gynecol. 2000;96:250–255. [DOI] [PubMed] [Google Scholar]
- 514. Chen Z, Yuhanna IS, Galcheva‐Gargova Z, et al. Estrogen receptor alpha mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J Clin Invest. 1999;103:401–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515. Dragoman M, Curtis KM, Gaffield ME. Combined hormonal contraceptive use among women with known dyslipidemias: A systematic review of critical safety outcomes. Contraception. 2015;94(3):280–287. [DOI] [PubMed] [Google Scholar]
- 516. Fotherby K. Oral contraceptives and lipids. BMJ. 1989;298:1049–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517. Adams MR, Clarkson TB, Shively CA, Parks JS, Kaplan JR. Oral contraceptives, lipoproteins, and atherosclerosis. Am J Obstet Gynecol. 1990;163:1388–1393. [DOI] [PubMed] [Google Scholar]
- 518. Odutayo A, Cherney D, Miller J, et al. Transdermal contraception and the renin‐angiotensin‐aldosterone system in premenopausal women. Am J Physiol Renal Physiol. 2015;308:F535–540. [DOI] [PubMed] [Google Scholar]
- 519. Fung MM, Poddar S, Bettencourt R, Jassal SK, Barrett‐Connor E. A cross‐sectional and 10‐year prospective study of postmenopausal estrogen therapy and blood pressure, renal function, and albuminuria: The Rancho Bernardo Study. Menopause. 2011;18:629–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520. Seely EW, Walsh BW, Gerhard MD, Williams GH. Estradiol with or without progesterone and ambulatory blood pressure in postmenopausal women. Hypertension. 1999;33:1190–1194. [DOI] [PubMed] [Google Scholar]
- 521. Schunkert H, Danser AH, Hense HW, et al. Effects of estrogen replacement therapy on the renin‐angiotensin system in postmenopausal women. Circulation. 1997;95:39–45. [DOI] [PubMed] [Google Scholar]
- 522. Hulley S, Grady D, Bush T, et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA. 1998;280:605–613. [DOI] [PubMed] [Google Scholar]
- 523. Schierbeck LL, Rejnmark L, Tofteng CL, et al. Effect of hormone replacement therapy on cardiovascular events in recently postmenopausal women: Randomised trial. BMJ. 2012;345:e6409. [DOI] [PubMed] [Google Scholar]
- 524. Dubey RK, Imthurn B, Barton M, Jackson EK. Vascular consequences of menopause and hormone therapy: Importance of timing of treatment and type of estrogen. Cardiovasc Res. 2005;66:295–306. [DOI] [PubMed] [Google Scholar]
- 525. Manson JE, Chlebowski RT, Stefanick ML, et al. Menopausal hormone therapy and health outcomes during the intervention and extended poststopping phases of the women's health initiative randomized trials. J Am Med Assoc. 2013;310:1353–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526. Tuomikoski P, Lyytinen H, Korhonen P, et al. Coronary heart disease mortality and hormone therapy before and after the women's health initiative. Obstet Gynecol. 2014;124:947–953. [DOI] [PubMed] [Google Scholar]
- 527. Rossouw JE, Prentice RL, Manson JE, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA. 2007;297:1465–1477. [DOI] [PubMed] [Google Scholar]
- 528. Hamoda H. Long‐term benefits and risks of HRT (Section 11): Venous thromboembolism. Post Reprod Health. 2016;22:77–79. [DOI] [PubMed] [Google Scholar]
- 529. Braithwaite RS, Chlebowski RT, Lau J, et al. Meta‐analysis of vascular and neoplastic events associated with tamoxifen. J Gen Intern Med. 2003;18:937–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530. Hernandez RK, Sorensen HT, Pedersen L, Jacobsen J, Lash TL. Tamoxifen treatment and risk of deep venous thrombosis and pulmonary embolism: A Danish population‐based cohort study. Cancer. 2009;115:4442–4449. [DOI] [PubMed] [Google Scholar]
- 531. Barrett‐Connor E, Grady D, Sashegyi A, et al. Raloxifene and cardiovascular events in osteoporotic postmenopausal women: Four‐year results from the MORE (Multiple Outcomes of Raloxifene Evaluation) randomized trial. JAMA. 2002;287:847–857. [DOI] [PubMed] [Google Scholar]
- 532. Ettinger B, Black DM, Mitlak BH, et al. Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene: Results from a 3‐year randomized clinical trial. Multiple Outcomes of Raloxifene Evaluation (MORE) Investigators. JAMA. 1999;282:637–645. [DOI] [PubMed] [Google Scholar]
- 533. Biglia N, Maffei S, Lello S, Nappi RE. Tibolone in postmenopausal women: A review based on recent randomised controlled clinical trials. Gynecol Endocrinol. 2010;26:804–814. [DOI] [PubMed] [Google Scholar]
- 534. Lubiniecki GM, Berlin JA, Weinstein RB, Vaughn DJ. Thromboembolic events with estramustine phosphate‐based chemotherapy in patients with hormone‐refractory prostate carcinoma: Results of a meta‐analysis. Cancer. 2004;101:2755–2759. [DOI] [PubMed] [Google Scholar]
- 535. Barbui T, Falanga A. Thalidomide and thrombosis in multiple myeloma. J Thromb Haemost. 2003;1:421–422. [DOI] [PubMed] [Google Scholar]
- 536. Carrier M, Le Gal G, Tay J , Wu C, Lee AY. Rates of venous thromboembolism in multiple myeloma patients undergoing immunomodulatory therapy with thalidomide or lenalidomide: A systematic review and meta‐analysis. J Thromb Haemost. 2011;9:653–663. [DOI] [PubMed] [Google Scholar]
- 537. De Stefano V, Za T , Rossi E. Venous thromboembolism in multiple myeloma. Semin Thromb Hemost. 2014;40:338–347. [DOI] [PubMed] [Google Scholar]
- 538. Minnema MC, Fijnheer R, De Groot PG, Lokhorst HM. Extremely high levels of von Willebrand factor antigen and of procoagulant factor VIII found in multiple myeloma patients are associated with activity status but not with thalidomide treatment. J Thromb Haemost. 2003;1:445–449. [DOI] [PubMed] [Google Scholar]
- 539. Bolland MJ, Grey A. A comparison of adverse event and fracture efficacy data for strontium ranelate in regulatory documents and the publication record. BMJ Open. 2014;4:e005787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540. Osborne V, Layton D, Perrio M, Wilton L, Shakir SA. Incidence of venous thromboembolism in users of strontium ranelate: An analysis of data from a prescription‐event monitoring study in England. Drug Saf. 2010;33:579–591. [DOI] [PubMed] [Google Scholar]
- 541. Ibanez B, James S, Agewall S, et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST‐segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST‐segment elevation of the European Society of Cardiology (ESC). Eur Heart J. 2017. https://doi.org/10.1093/eurheartj/ehx637 [Google Scholar]
- 542. Barrueto F, Jr . Beta blocker poisoning. In: UnToDate. https://www.uptodate.com/contents/beta-blocker-poisoning, accessed: Oct 17, 2017.
- 543. American Society of Regional Anesthesia and Pain Medicine (ASRA) . ASRA Guidelines in local anaesthetics systemic toxicity management. 2010.
- 544. Berul CI, Seslar SP, Zimetbaum PJ. Acquired long QT syndrome. In: UnToDate. https://www.uptodate.com/contents/acquired-long-qt-syndrome, accessed: Oct 17, 2017.