

Abstract
The RAD51 DNA strand exchange protein plays an important role in maintaining the integrity of the human genome. It promotes homology-directed DNA repair by exchanging strands between the damaged and the intact DNA molecules. It also plays an important role in stabilizing distressed DNA replication forks. When overexpressed or misregulated, however, RAD51 contributes to “rogue”, genome destabilizing events that can lead to cancer, cell death and to acquisition of chemotherapy resistance by cancerous cells. Human RAD51 is, therefore, an important and highly coveted anticancer drug target. Biochemical, biophysical and structural studies of the human RAD51, and establishment of its structure-activity relationship requires purification of large quantities of protein. In this chapter we describe a robust method for expression and purification of human RAD51 and the methods for assessing its activity based on the ssDNA binding stoichiometry and its capacity to carry out the DNA strand-exchange reaction.
Keywords: RAD51 recombinase, RAD51 protein purification, RAD51-ssDNA binding stoichiometry, DNA strand exchange, homologous recombination
1. Introduction
The Human RAD51 recombinase catalyzes the central step in Homologous Recombination (HR), which is an exchange of the DNA strands between two complementary DNA molecules (Baumann and West, 1998, Morrical, 2015). As result, RAD51 is important for the accurate repair of DNA double strand breaks (DSB’s), Single Strand DNA (ssDNA) gaps and stalled replication forks (Moynahan and Jasin, 2010, Li and Heyer, 2008). In addition to its HR function, RAD51 also protects damaged or stalled DNA replication forks from excessive nucleolytic degradation by the MRN complex (Schlacher et al., 2012, Schlacher et al., 2011, Kolinjivadi et al., 2017). Finally, as an integral member of the Fanconi Anemia pathway, RAD51 (FANCR) also plays an important in role in repair of the DNA inter-strand DNA crosslinks (ICLs) (Wang et al., 2015, Ameziane et al., 2015, Michl et al., 2016).
Despite numerous studies, significant gaps remain in our understanding of the assembly, dynamics and regulation of the RAD51 nucleoprotein filament and mechanism of the DNA strand exchange reaction. Because of its important role in HR and because RAD51 overexpression contributes to radiation and chemotherapy resistance in a range of cancers, RAD51 has become an attractive drug target (see (Hengel et al., 2017, Budke et al., 2016) for recent reviews). Biochemical and biophysical analysis of RAD51, as well as the drug discovery campaigns, require robust methods for purification of the large quantities of this protein. Several methods have been developed for the purification and analysis of RAD51 (Baumann et al., 1997, Benson et al., 1994, Sigurdsson et al., 2001). These methods for protein purification are generally complex, and involve several supporting plasmids during expression. This is because the native RAD51 gene sequence contains multiple rare codons and the expression of soluble RAD51 also requires the presence of a chaperone. Additionally, biochemical studies of the RAD51 protein that require protein modifications, such as for example incorporation of artificial amino acids using amber suppressor systems to mimic tyrosine phosphorylation (Xie et al., 2007, Subramanyam et al., 2016), or site specific protein labeling using genetically encoded aldehyde tags (Carrico et al., 2007, Rabuka et al., 2012), are not compatible with these protocols. Here, we describe a modified and highly optimized method for expression and purification of RAD51 that consistently yields large quantities of pure, functional protein using an E. coli expression system while allowing the use of additional plasmids enabling incorporation of non-natural amino acids or enzymatic modifications.
2. System for the bacterial expression of human RAD51 protein
The RAD51 Complementarity Determining Sequence (CDS) used here for RAD51 expression, is optimized for E. coli codon usage is derived from the human RAD51, transcript variant 1 (NCBI RefSeq: NM_002875). This sequence has been cloned into the first multiple cloning site (MCS) of a pCOLA-Duet1 (Millipore Sigma) expression vector. The E. coli GroE operon has been cloned into the second MCS. The resulting plasmid is hereafter referred to as pCH1/RAD51o (Figure 1A). The pCH1/RAD51o plasmid along with sequence information is available upon request from the Spies lab or from Addgene (#102562). Modifications of the pCH1 system include RAD51 mutants, RAD51 containing a C-terminal 6xHis tag and RAD51 containing a recognition sequence for the formylglycine generating enzyme (FGE) also at the C-terminus. The proteins are then expressed from the pCH1 vector in the Acella™ competent cells [F− ompT hsdSB(rB− mB−) gal dcm (DE3) ΔendA ΔrecA] (EdgeBio Catalog#36795) which have the RecA protein completely deleted from their genome (Figure 1).
Figure 1. E. coli Optimized expression system for Human RAD51 protein.
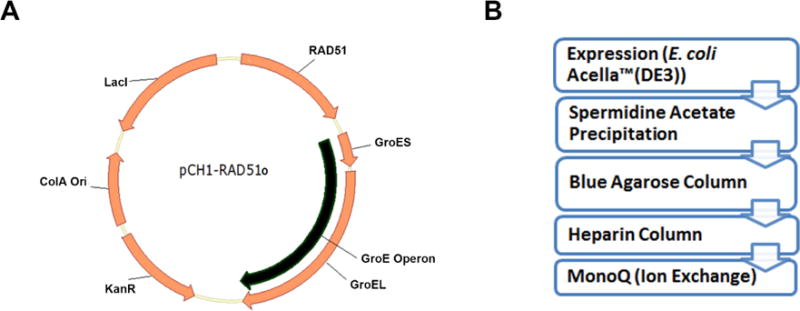
A. The pCH1/RAD51o vector consists of an E. coli codon optimized CDS for the Human RAD51 protein alongwith the E.coli GroE operon expressed under the control of the T7 promoter and a ColA bacterial origin of replication. B. Purification scheme for producing RAD51 protein involves expression of the protein in E. coli Acella (DE3) cells. After expression, the RAD51 protein is precipitated from the clarified lysate using spermidine acetate. The resuspended precipitate is then purified through affinity chromatography (blue agarose and heparin) followed by anion exchange.
The RAD51 protein, expressed in the Acella™ competent cells is precipitated using spermidine acetate (Baumann et al., 1997) and then purified using chromatography based on Cibacron™ F3G-A (Blue) affinity followed by a Heparin column which removes any RAD51 bound DNA. The protein prep is then polished and concentrated using an anion exchange column (Figure 1B). The detailed protocol is described below.
3. Equipment, Materials & Reagents
3.1. Equipment
50mL conical tubes.
0.45μM syringe filters
0.2μM bottle top vacuum filters
Large agarose gel running apparatus
Akta (GE) or BioLogic (BioRad) chromatography system
ChemiDoc XRS (BioRad)
Dialysis beakers
Dialysis Tubing (10000MWCO)
HiTrap™ Blue HP chromatography column (GE Life Sciences #17-0412-01)
HiTrap™ Heparin HP chromatography column (GE Life Sciences #17-0406-01)
MonoQ (5/50) Ion Exchange Column (GE Life Sciences #17-5166-01)
Refrigerated floor standing centrifuge
Oak Ridge Centrifuge tubes.
Shaker incubator
Sonicator
Temperature block/bath
Micropipettors
Micropipettor tips
Fluorescence spectrophotometer equipped with a temperature controller and capable of simultaneously detecting fluorescence in two channels: We used Cary Eclipse (Agilent). Alternatively, a spectrofluorimeter from ISS or Shimadzu can be used. The assays described below also can be adapted for a multi-well plate format for use with a plate reader.
UV-Vis spectrophotometer
Quartz or optical glass cuvettes for binding titrations (Starna)
3.2. Materials Required
Antifoam 204 (Sigma# A8311)
Ammonium Sodium Phosphate Dibasic Tetrahydrate (NaNH4PO4) (Sigma # 04266)
ApaLI (NEB# R0507)
Bacto™ Tryptone (BD Biosciences)
Bacto™ Yeast Extract (BD Biosciences)
Chicken egg white lysozyme (Sigma# L7651)
Dithiothreitol (DTT) (Sigma# D0632)
Ethylenediaminetetraacetic acid, EDTA (Sigma# E9884)
Glacial Acetic Acid
Glycerol
HEPES (Sigma# H4034)
Hydrochloric acid
IPTG (EMD Millipore# 420322)
Kanamycin sulfate from Streptomyces kanamyceticus (Sigma# K1377)
Potassium Chloride (Sigma# P5405)
Potassium Phosphate Monobasic (Sigma# P0662)
Potassium Phosphate Dibasic (Sigma# P3786)
Protease inhibitors (Roche# 11836170001)
Proteinase K Solution (20 mg/ml) (Invitrogen/Life Technologies# AM2546)
Sodium Chloride (NaCl)
Sodium Dodecyl Sulphate (SDS)
Sodium Hydroxide (Sigma# S5881)
Sodium Thiocyanate (NaSCN) (Sigma# 251410)
Spermidine (Sigma# S0266)
SYBR® Gold Nucleic Acid Gel Stain (10,000X Concentrate in DMSO) (Invitrogen/Life Technologies# S-11494)
Trizma® Base (Sigma# T6066)
Triton™ X-100
UltraPure™ Agarose (Invitrogen/Life Technologies# 16500-100)
3.3. Oligonucleotides and DNA Substrates
The synthetic oligonucleotide used in the FRET DNA extension assay can be commercially synthesized. We use PAGE purified oligonucleotides synthesized by IDT. The dT60 oligo we use has the Cy3 and Cy5 dyes separated by 25 nucleotides (5`T5-Cy5-T25-Cy3- T30). The oligonucleotides are resuspended in 10mM Tris (pH8.0) and stored at −20°C.
ϕX174 Virion ssDNA (NEB# N3023)
ϕX174 RFI dsDNA (NEB# N3021)
3.4. Buffers and Solutions
Lysis Buffer
100mM Tris-OAc (pH 7.5 at 23°C), 2mM EDTA, 10% Glycerol (V/v), 1mM DTT, Lysozyme (0.5mg/ml), Protease Inhibitors, 0.1% Triton X-100.
Spermidine Acetate Buffer
20mM Tris-OAc (pH 7.5 at 23°C), 7mM Spermidine Acetate (pH 7.5), 5% Glycerol, 0.1mM Dithiothreitol.
T-150 Buffer
50mM Tris-HCl (pH 7.5 at 23°C), 10% Glycerol, 150mM NaCl, 1mM Dithiothreitol.
T-250 Buffer
50mM Tris-HCl (pH 7.5 at 23°C), 10% Glycerol, 250mM NaCl, 1mM Dithiothreitol.
T-300 Buffer
50mM Tris-HCl (pH 7.5 at 23°C), 10% Glycerol, 300mM NaCl, 1mM Dithiothreitol
T-500 Buffer
50mM Tris-HCl (pH 7.5 at 23°C), 10% Glycerol, 500mM NaCl, 1mM Dithiothreitol
T-600 Buffer
50mM Tris-HCl (pH 7.5 at 23°C), 10% Glycerol, 600mM NaCl, 1mM Dithiothreitol
Blue-A Buffer
100mM Potassium Phosphate (KPi) (pH 7.0 at 23°C), 5% Glycerol, 300mM NaCl, 1 mM EDTA, 1mM Dithiothreitol
Blue-B Buffer
100mM Potassium Phosphate (KPi) (pH 7.0 at 23°C), 5% Glycerol, 2M NaSCN, 1mM EDTA, 1 mM Dithiothreitol
Buffer HA
20mM HEPES-NaOH (pH 7.5 at 23°C), 5% Glycerol, 150mM NaCl, 1mM EDTA, 1mM Dithiothreitol
Buffer HB
20mM HEPES-NaOH (pH 7.5 at 23°C), 5% Glycerol, 2M NaCl, 1mM EDTA, 1mM Dithiothreitol
FRET Buffer (Ca2+-ATP)
20mM HEPES-KOH (pH 7.5 at 23°C), 5mM CaCl2, 5mM MgCl2, 150mM KCl, 1mM ATP, 1mM Dithiothreitol
FRET Buffer (Mg2+-ATP)
20mM HEPES-NaOH (pH 7.5 at 23°C), 150mM NaNH4PO4, 2mM MgCl2, 1mM ATP, 1mM Dithiothreitol
0.5M NaNH4PO4 (this reagent tends to precipitate at room temperature during prolonged storage and is made fresh before use)
10% Sodium Dodecyl Sulphate (SDS)
Tris-Acetate-EDTA Buffer (TAE)
40mM Trizma Base, 20mM acetic acid, 1 mM EDTA.
UltraPure™ Phenol:Chloroform:Isoamyl Alcohol (25:24:1, v/v) (Invitrogen/Life Technologies# 15593-031)
RPA Protein purified as described earlier (Henricksen et al., 1994).
4. Expression of RAD51 Wild type protein
4.1
Inoculate two 10mL LB cultures supplemented with 40μg/mL Kanamycin with E. coli Acella™ (DE3)/pCH1-RAD51o. Incubate the cultures at 37°C overnight (~14 hours) at 230 rpm.
4.2
Inoculate 8mL of the overnight culture into 1 liter of LB Broth supplemented with 40μg/mL Kanamycin and 300μL 5% Antifoam 204. You will need two 1 liter cultures. Incubate the inoculated cultures at 37°C at 230 rpm until the OD600 reaches 0.6-0.7 (~2.5 hours). Induce the RAD51 expression with 0.1mM IPTG (100μl of 1M Stock) followed by incubation at 37°C for 4 hours. Higher IPTG concentrations result in an insoluble protein. The cells in the culture can be pelleted and stored at −80°C until further use. Typical yield is 2g of cells per 1 liter of culture.
5. Purification of RAD51
All purification steps described below should be carried out on ice or at 4°C. All buffers should be pre-chilled and degassed (if air bubbles are observed).
5.1
Day1
Take 4g of the Acella™ (DE3)/pCH1-RAD51o cell pellet (from two 1 liter cultures) from the −80°C and thaw on ice for 1 hour. Resuspend the pellet in Lysis Buffer (5mL/g of cells) for 30 minutes at 4°C and lyse by sonication for about 4 minutes (Amplitude 40, 2 second pulse, 2 minutes’ timer repeated twice). Spin the sonicated cells in the centrifuge at ~38759 rcf for 1 hour and 20 minutes at 4°C. Collect the supernatant and transfer to a dialysis bag.
5.2
Dialyze the supernatant at 4°C in 500ml of pre-chilled Spermidine Acetate Buffer (see buffers and solutions) which needs to be replaced with fresh buffer after three hours and then again after another three hours. The final buffer change is kept overnight. While changing buffers, make sure to mix the precipitate by flipping the dialysis bag several times to distribute the forming precipitate throughout the buffer volume. This ensures uniform and efficient precipitation.
5.3
Day 2
Separate the precipitated proteins by centrifugation at ~38759 rcf for 20 minutes at 4°C.
5.4
Remove the supernatant and set aside. Resuspend the pellet in 10ml T-150 Buffer (see buffers and solutions) using gentle pipetting on ice. Note that some fraction of the pellet will remain insoluble. Spin the pellet at ~38759 rcf for 20 minutes at 4°C.
5.5
Remove the supernatant from the T-150 wash and set aside. Resuspend the pellet in 10mL T-250 Buffer and spin at ~38759 rcf for 20 minutes at 4°C. It is important to note that these first two pellets be only partially solubilized. The RAD51 protein, if completely solubilized in these early fractions contains more impurities that are difficult to separate later. Repeat this step with 10mL of T-300 and 5mL of T-500 and T-600 Buffers. Analyze samples from all preserved supernatants by electrophoresis on a 12% SDS-PAGE to determine which fractions contain RAD51. Wild type RAD51 is generally observed in T300, T500 & T600 fractions (Figure 2A). Different mutants and modified forms of RAD51, however, may behave differently.
Figure 2. Step-by-step purification of RAD51 protein.
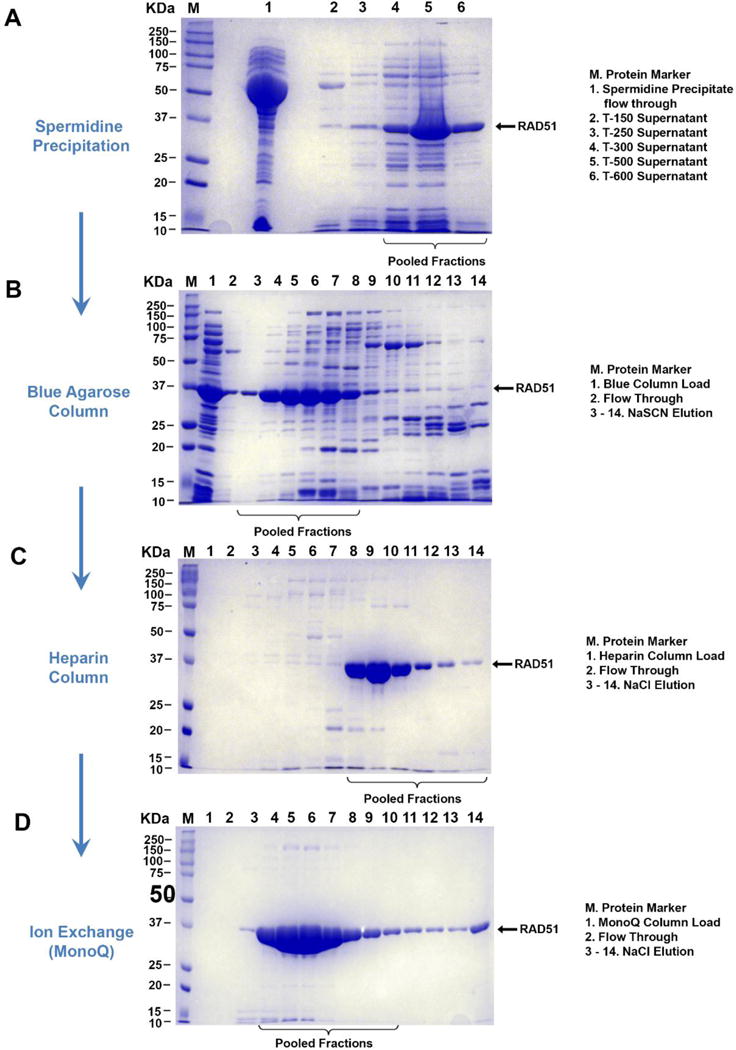
A. The clarified lysate is dialyzed overnight in spermidine acetate buffer. The precipitate is then selectively resuspended in increasing concentrations of sodium chloride. The RAD51 wild type protein is generally soluble in salt concentrations above 300mM. Any RAD51 protein found in fractions having lower salt concentrations, is generally difficult to separate from other proteins in further purification steps. B. Pooled fractions containing RAD51 from the spermidine precipitation are then passed through a Blue Agarose column. The RAD51 protein is eluted from the column using a sodium thiocyanate gradient. C. Fractions containing RAD51 are then further loaded on a Heparin affinity column and RAD51 eluted using a sodium chloride gradient. D. The protein is relatively pure at this step and is polished and concentrated using a MonoQ anion exchange step. Small fractions are collected during elution at this stage to minimized dilution of the RAD51 protein.
5.6
Pool the RAD51-containing fractions together. Combine the T600 fraction with the T300 & T500 fractions and filter the mixture through a 0.45μM syringe filter before loading the sample onto the blue column. This step must be done fast, just before loading onto the next column as pooled fractions tend to precipitate. Another option would be to load the samples individually.
5.7
Load the filtered sample onto three 5mL HiTrap™ Blue Agarose columns (connected in series) cleaned with Blue-B Buffer and equilibrated with Blue-A Buffer (see buffers and solutions).
5.8
Wash the columns with 5 column volumes of Blue-A Buffer or until the absorbance signal stabilizes.
5.9
Elute the protein from the columns using a 60 ml 0 - 2M NaSCN gradient in Blue-B Buffer at 5mL/min. Collect 4 mL fractions. The wild type RAD51 typically elutes before 800mM NaSCN within the first 40% of the NaSCN gradient (Figure 2B). Pool fractions containing RAD51 dialyze in 2 liters of buffer HA overnight at 4°C.
5.10
Day 3
Filter the dialyzed RAD51 from step 2.9 to remove small precipitates (if any) and load onto three 5mL HiTrap™ Heparin FF Columns using Buffer HA (see buffers and solutions) as binding buffer. Wash the column with 5 column volumes of loading buffer or until the absorbance signal has stabilized. Elute RAD51 with an 80mL 150 mM – 2 M NaCl gradient (Buffer HA – Buffer HB) at 5mL/min (Figure 2C). Collect 5 ml fractions.
5.11
Collect the fractions containing RAD51 and pool together to be dialyzed in 2 litres of HA Buffer overnight at 4°C. The protein can be eluted over a longer gradient ~100mL at this scale depending on separation and column performance. It would be ideal to keep dialysis for approximately 12 hours as the protein tends to precipitate when dialyzed longer.
5.12
Day 4
Filter the dialyzed fractions from the Heparin Column and load the protein a 1mL MonoQ (5/50) Ion Exchange Column using Buffer HA as binding buffer followed by a linear NaCl salt gradient to 1.2M over 8 mL to elute RAD51 and a final step up to 2M over 10ml at 1mL/min. It is best to collect small fractions during this step ~0.3μL (Figure 2D). This allows us to retain fractions containing the highest concentrations of RAD51 while simultaneously separating the small impurities.
5.13
Pool the fractions containing Human RAD51 and dialyze the purified protein overnight in HA buffer containing 10% glycerol at 4°C.
5.14
Day 5
The concentration of the protein measured spectrophotometrically at 280 nm using a molar extinction coefficient of 14900M−1cm−1. The extinction coefficient for the mutant or tagged forms of the protein may be different and need to be calculated based on the changes in the sequence.
5.15
The protein can be aliquoted and flash frozen in liquid nitrogen.
6. FRET-Based DNA Binding assay to measure ssDNA binding Activity of the RAD51 recombinase
The purification procedure prescribed above consistently yields fully active RAD51 protein. It is imperative, however, to confirm that each preparation of the protein is indeed 100% active. The percentage of the RAD51 specific activity can be derived from the ssDNA binding stoichiometry. Active form of all RecA-family recombinases, including the human RAD51, is a nucleoprotein filament formed on the ssDNA (Sung and Robberson, 1995). Each ATP-bound RAD51 protomer in the nucleoprotein filament occludes 3 nucleotides of ssDNA and the ssDNA is extended approximately 1.6-fold relative to the B-form (Sung and Robberson, 1995). Both of these properties can be assessed simultaneously as the change in the DNA extension upon the RAD51 binding can followed using Förster resonance energy transfer (FRET) (Clegg, 2002) between the FRET donor (Cy3) and FRET acceptor (Cy5) fluorophores incorporated into the DNA substrate (Subramanyam et al., 2016, Subramanyam et al., 2013, Grimme and Spies, 2011). To determine the RAD51:ssDNA binding stoichiometry we typically use a dT60 ssDNA oligonucleotide internally labelled with Cy3 and Cy5 dyes (dT60-Cy3/Cy5). The two dyes are positioned 5 and 30 nucleotides from the 5′ end of the dT60 oligomer. The 25-nucleotide separation yields a high FRET signal (~0.6) for the protein-free ssDNA in the reaction buffer containing physiological concentrations of monovalent and divalent cations. Extension of the ssDNA to 1.6-fold over the B-form by the fully-formed, ATP-bound RAD51 nucleoprotein results in a transition from a high FRET (~0.6) to a low FRET (~0.2) state (Figure 3). To access the quality of the RAD51 prep, the assay is carried out under the stoichiometric binding conditions (ssDNA is present at 600 nM nucleotides or 10 nM dT60 molecules) where the FRET value increases linearly with each addition of RAD51 and then reaches saturation at 1 active RAD51 monomer per 3 nucleotides of ssDNA.
Figure 3. FRET Based DNA binding and Extension Assay.
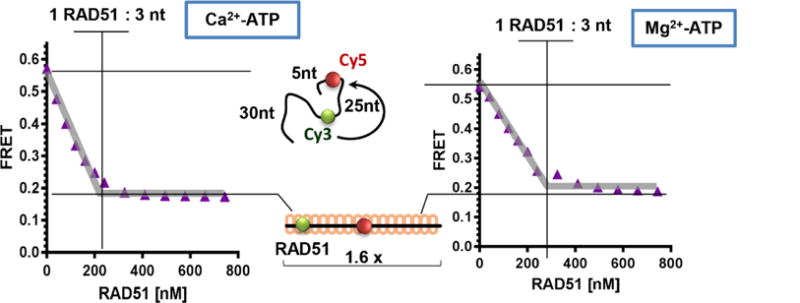
The substrate for this assay is a poly dT60 ssDNA oligomer, which is labelled internally with Cy3 (FRET donor) and Cy5 (FRET acceptor) fluorescent dyes separated by 25 nucleotides. Free ssDNA yields a high FRET due to the proximity of Cy3 and Cy5. Addition of RAD51 into the reaction mixture initiates nucleoprotein filament formation, which straightens and extends the oligomer, which is observed as a decrease in FRET as the two fluorophores move away from one another. The assay can be performed in conditions that permit ATP hydrolysis (Mg2+-ATP) or where ATP hydrolysis is inhibited (Ca2+-ATP). The biochemical characteristics of the wild type RAD51 protein does not vary much between these two conditions but mutants with altered DNA binding properties (Subramanyam et al., 2016) can be separated using this assay.
The FRET-based ssDNA binding assay can be carried out under two experimental regimes, in the buffer containing ammonium sodium phosphate where the RAD51 protein is allowed to hydrolyze ATP (Shim et al., 2006, Subramanyam et al., 2016), or when the ATP hydrolysis (but not the ATP binding) is restricted due to the presence of Ca2+. The wild type RAD51 readily forms active nucleoprotein filaments on the 60-mer ssDNA oligonucleotide under both conditions. For the RAD51 mutant that may display lower binding affinity or a defective ssDNA binding to a relatively short ssDNA, we suggest using a Ca2+ condition, which promotes more regular and stable nucleoprotein filaments (Ristic et al., 2005).
Fluorimeter Settings for FRET-Based Binding Assays
6.1
Cy3 excitation wavelength at 530 nm and emission wavelength at 565 nm.
6.2
Cy5 excitation wavelength at 630 nm and emission wavelength at 660 nm.
6.3
Excitation and emission slit widths set to 10 nm each.
6.4
PMT voltage set, so that the amplitude of the Cy3 signal is below 60% of the available scale.
6.5
The assays are carried out in kinetics mode with the Cy3 and Cy5 signals recorded every second. The kinetic mode allows one to monitor the change in the signal upon each RAD51 addition and to only analyze the data when the signal achieves equilibrium.
FRET DNA Binding and Extension Assay Protocol
6.6
Place fluorescence cuvette containing FRET reaction buffer (See Solutions & Buffers) and allow the sample to equilibrate at 37°C. Zero the reaction. Record the Cy3 and Cy5 baseline values for at least 1 min.
6.7
Add 600nM nucleotides (10nM molecules) of dT60-Cy3/Cy5 oligo into the cuvette, mix by pipetting and measure the Cy3 and Cy5 fluorescence values once the fluorescence has stabilized.
6.8
RAD51 protein is then titrated into the reaction with a proper mixing after each addition. For each addition, continue the recording until the equilibrium is achieved and the Cy3 and the Cy5 fluorescence stabilizes. Record the stable fluorescence signals for at least one minute before making the next RAD51 addition.
6.9
Typical protein concentrations range from 40nM to 1000μM. Ideally, the total volume of the titrated protein should not exceed 10% of the reaction volume and the dilution should be factored in the final concentration of the titrations. We try to avoid pipetting extremely small volumes (<0.5μL), to avoid pipetting errors.
6.10
For each RAD51 concentration select the period of at least 1 minute of measurement before the next protein addition and record the average signals for Cy3 and Cy5 fluorescence.
6.11
Subtract the average baseline fluorescence from the average fluorescence recorded at each RAD51 concentration.
6.12
Resulting values for Cy3 and Cy5 fluorescence can be plotted as functions of RAD51 concentration. The FRET values are calculated as a fraction of donor acceptor intensity adjusted by correction factors (Grimme and Spies, 2011). For Cary Eclipse fluorescence spectrophotometer (Agilent) FRET between Cy3 and Cy5 dyes can be calculated using the equation –
6.13
The FRET values should saturate at a RAD51 concentration corresponding to ~1:3 protein:nucleotides (Subramanyam et al., 2016, Subramanyam et al., 2013) (Figure 3).
7. Three-strand DNA Strand Exchange Assay
The ssDNA-binding by RAD51 described above is a prerequisite of all known RAD51 cellular function. The full functionally of the RAD51 protein can be further evaluated in a more delicate namesake (aka recombinase) activity of RAD51. In homologous recombination and homology-directed DNA repair, the RAD51 nucleoprotein filament performs an ATP-dependent homologous DNA pairing and DNA strand exchange reactions (Ciccia and Elledge, 2010, Mehta and Haber, 2014). The RAD51-mediated DNA strand exchange activity can be reconstituted in vitro in the so-called three strand DNA strand exchange reaction (Baumann et al., 1996, Sigurdsson et al., 2001). The active RAD51 nucleoprotein filament is formed on ϕX174 Virion single stranded DNA (ssDNA) with the help of the ssDNA binding protein RPA. The RAD51 nucleoprotein filament then pairs with and invades a linear ϕX174 RFI double stranded (dsDNA) substrate. The strand exchange activity of RAD51 leads to the displacement of the complementary linear ssDNA from the dsDNA, forming a nicked circular product, through a series of joint molecule intermediates, all of which can easily be separated on an agarose gel (Subramanyam et al., 2016, Sigurdsson et al., 2001) (Figure 4A). The reaction depends on the presence of the ssDNA binding protein RPA, which, when added after RAD51, stimulates the DNA strand exchange by destabilizing remaining secondary structure in the ssDNA and by sequesters the displaced DNA strand. Under conditions permitting ATP hydrolysis, RPA can be also added prior to RAD51. The yield of such RPA-first reaction is, however; significantly lower than that of the RAD51-first reaction due to the competition between the two proteins.
Figure 4. Three Strand Exchange Recombinase Assay.
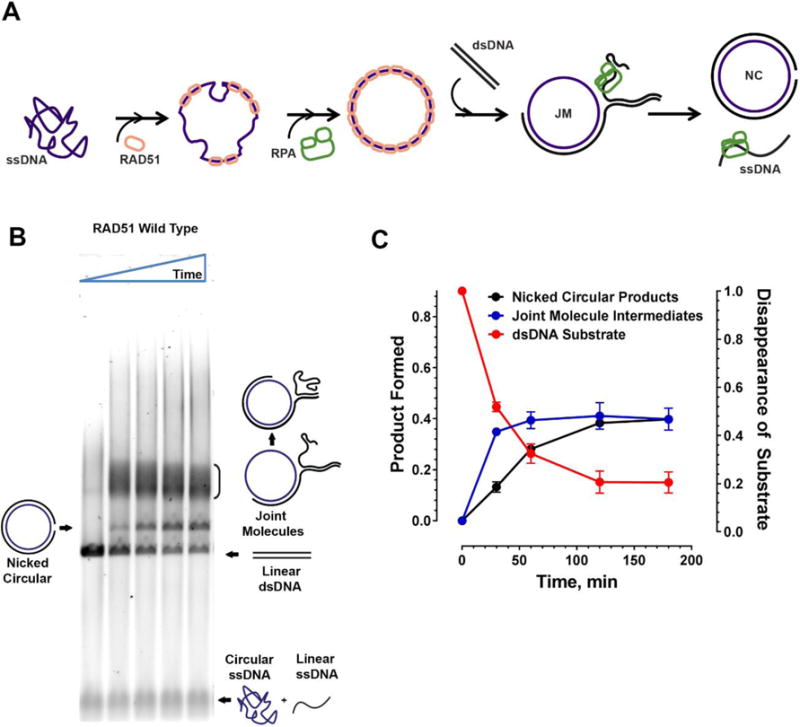
A Active RAD51 nucleoprotein filament is formed on ϕX174 Virion circular ssDNA in reaction conditions permitting ATP hydrolysis followed by addition of RPA which helps remove secondary structures in the ssDNA allowing stable nucleoprotein filament formation. The RAD51 nucleoprotein then invades ϕX174 RFI linear dsDNA to form nicked circular dsDNA products through several joint-molecule intermediates showing various stages of branch migration. The RPA also helps sequester any free displaced ssDNA following strand exchange thereby preventing a reverse reaction. B. Typical agarose gel image from a strand exchange assay used to resolve the substrates, intermediates and the products of the reaction over a time course of 0, 30, 60, 120 and 180 minutes. In reactions containing the wild type protein, nicked circular products can be observed as early as 30 minutes. C. Quantitation of the strand exchange assay gel showing appearance of nicked circular products (black) and joint molecules (blue) on the left Y axis. Disappearance of the linear dsDNA substrate (red) is quantitated on the right Y axis. Time in minutes is indicated on the X axis.
Preparation of DNA substrates
7.1
Linear double stranded DNA is prepared by digesting ϕX174 RFI dsDNA using the ApaLI restriction enzyme. We typically digest 60μg of dsDNA at a time.
7.2
The digested DNA is cleaned using Phenol:Chloroform:Isoamyl Alcohol extraction and precipitated using Isopropanol. Vigorous pipetting and vortexing the mixture should be avoided as it tends to shear the DNA. The DNA is resuspended in nuclease free water and the concentration determined using A260 and is then aliquoted and stored at −80°C. Quality of the purified DNA is paramount as sheared DNA causes smearing in the gels and interferes with the results and quantitation of the strand exchange assay. All reagents used should be checked for the presence of nucleases.
7.3
Similarly, ϕX174 Virion concentration is measured using A260.
DNA Strand Exchange Reaction
7.4
Typical reaction volumes are 90μL which allows us to take 5 time points. All additions should be mixed by gentle pipetting as vigorous mixing or vortexing can shear the DNA substrates.
7.5
Prepare 4X strand exchange Reaction Buffer containing 80mM HEPES-NaOH (pH 7.5 at 23°C), 40% Glycerol, 4mM MgCl2 and 4mM Dithiothreitol.
7.6
DNA strand exchange is initiated by incubating 7.5μM RAD51 with 30μM (nucleotides) ϕX174 ssDNA in 4X Reaction Buffer (See Solutions and Buffers) and 2.5mM ATP for 5 minutes at 37°C.
7.7
150mM of Ammonium Sodium Phosphate (NaNH4PO4) is added along with 2μM RPA and incubated for another 5 minutes.
7.8
15μM (bp) of ApaLI digested Linear dsDNA is added and further incubated at 37°C for the time course of the reaction (typically 180 minutes).
7.9
15μL of the reaction can be drawn at different time points and deproteinized by addition of 0.8% SDS and Proteinase K (800μg/mL) followed by incubation at 37°C for 30 minutes.
7.10
The samples are then loaded on a 0.9% TAE agarose gel and run at 3.5V/cm overnight at room temperature. In our experiments, the quality of Agarose and TAE seems to affect the result of the gels, and hence we always use UltraPure™ Agarose (see materials) and freshly prepared TAE buffer.
Visualization, quantitation and interpretation of DNA Strand Exchange Data
7.11
The agarose gel is then stained with SYBR® Gold for 10 minutes and destained for 20 minutes in MilliQ H2O. The gel can be visualized in a ChemiDoc XRS or equivalent imaging system by exciting the dye-nucleic acid complexes at 495 nm and 300 nm and an emission maximum at 537 nm (Figure 4B).
7.12
The appearance of the nicked circular products can be quantitated using ImageJ software normalized to the amount of initial linear dsDNA substrate. The appearance and processing of joint-molecule intermediates can be estimated by subtracting the normalized intensity of the nicked circular products from the normalized intensity of the dsDNA for that lane (Subramanyam et al., 2016) (Figure 4C). This assay is generally performed in triplicate and the results plotted with standard error mean.
8. Conclusions
The E. coli Acella™ (DE3)/pCH1-RAD51o system can be used to produce large quantities of soluble human RAD51 protein. The purification scheme outlined above allows purification of RAD51 without the use of affinity tags, which may interfere with the recombinase functions or other biochemical assays. The procedure takes five consecutive days, but is robust and straightforward. It can be readily extended to various RAD51 mutants and to the constructs containing tags, as well as recognition and labeling epitopes.
Acknowledgments
The authors’ research is supported by National Institutes of Health (NIH) Grant GM108617 and by University of Iowa Holden Comprehensive Cancer Center Collaborative Pilot Grant NIH P30 CA086862 (to M.S.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank Drs. XXX for critical reading of the manuscript and for valuable discussions.
References
- AMEZIANE N, MAY P, HAITJEMA A, VAN DE, VRUGT HJ, VAN ROSSUM-FIKKERT SE, RISTIC D, WILLIAMS GJ, BALK J, ROCKX D, LI H, ROOIMANS MA, OOSTRA AB, VELLEUER E, DIETRICH R, BLEIJERVELD OB, MAARTEN ALTELAAR AF, MEIJERS-HEIJBOER H, JOENJE H, GLUSMAN G, ROACH J, HOOD L, GALAS D, WYMAN C, BALLING R, DEN DUNNEN J, DE WINTER JP, KANAAR R, GELINAS R, DORSMAN JC. A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. 2015;6:8829. doi: 10.1038/ncomms9829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAUMANN P, BENSON FE, HAJIBAGHERI N, WEST SC. Purification of human Rad51 protein by selective spermidine precipitation. Mutat Res. 1997;384:65–72. doi: 10.1016/s0921-8777(97)00028-1. [DOI] [PubMed] [Google Scholar]
- BAUMANN P, BENSON FE, WEST SC. Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell. 1996;87:757–66. doi: 10.1016/s0092-8674(00)81394-x. [DOI] [PubMed] [Google Scholar]
- BAUMANN P, WEST SC. Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem Sci. 1998;23:247–51. doi: 10.1016/s0968-0004(98)01232-8. [DOI] [PubMed] [Google Scholar]
- BENSON FE, STASIAK A, WEST SC. Purification and characterization of the human Rad51 protein, an analogue of E. coli RecA. EMBO Journal. 1994;13:5764–71. doi: 10.1002/j.1460-2075.1994.tb06914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUDKE B, LV W, KOZIKOWSKI AP, CONNELL PP. Recent Developments Using Small Molecules to Target RAD51: How to Best Modulate RAD51 for Anticancer Therapy? ChemMedChem. 2016;11:2468–2473. doi: 10.1002/cmdc.201600426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARRICO IS, CARLSON BL, BERTOZZI CR. Introducing genetically encoded aldehydes into proteins. Nat Chem Biol. 2007;3:321–322. doi: 10.1038/nchembio878. [DOI] [PubMed] [Google Scholar]
- CICCIA A, ELLEDGE SJ. The DNA Damage Response: Making It Safe to Play with Knives. Molecular Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLEGG RM. FRET tells us about proximities, distances, orientations and dynamic properties. J Biotechnol. 2002;82:177–9. [PubMed] [Google Scholar]
- GRIMME JM, SPIES M. FRET-based assays to monitor DNA binding and annealing by Rad52 recombination mediator protein. Methods Mol Biol. 2011;745:463–83. doi: 10.1007/978-1-61779-129-1_27. [DOI] [PubMed] [Google Scholar]
- HENGEL SR, SPIES MA, SPIES M. Small-Molecule Inhibitors Targeting DNA Repair and DNA Repair Deficiency in Research and Cancer Therapy. Cell Chemical Biology. 2017;24:1101–1119. doi: 10.1016/j.chembiol.2017.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HENRICKSEN LA, UMBRICHT CB, WOLD MS. Recombinant replication protein A: expression, complex formation, and functional characterization. J Biol Chem. 1994;269:11121–32. [PubMed] [Google Scholar]
- KOLINJIVADI AM, SANNINO V, DE ANTONI A, TECHER H, BALDI G, COSTANZO V. Moonlighting at replication forks - a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. 2017;591:1083–1100. doi: 10.1002/1873-3468.12556. [DOI] [PubMed] [Google Scholar]
- LI X, HEYER WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Research. 2008;18:99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEHTA A, HABER JE. Sources of DNA Double-Strand Breaks and Models of Recombinational DNA Repair. Cold Spring Harbor Perspectives in Biology. 2014;6:a016428. doi: 10.1101/cshperspect.a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MICHL J, ZIMMER J, TARSOUNAS M. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. The EMBO Journal. 2016;35:909–923. doi: 10.15252/embj.201693860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORRICAL SW. DNA-pairing and annealing processes in homologous recombination and homology-directed repair. Cold Spring Harb Perspect Biol. 2015;7:a016444. doi: 10.1101/cshperspect.a016444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOYNAHAN ME, JASIN M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010;11:196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RABUKA D, RUSH JS, DEHART GW, WU P, BERTOZZI CR. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat Protocols. 2012;7:1052–1067. doi: 10.1038/nprot.2012.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RISTIC D, MODESTI M, VAN DER HEIJDEN T, VAN NOORT J, DEKKER C, KANAAR R, WYMAN C. Human Rad51 filaments on double- and single-stranded DNA: correlating regular and irregular forms with recombination function. Nucleic Acids Res. 2005;33:3292–302. doi: 10.1093/nar/gki640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHLACHER K, CHRIST N, SIAUD N, EGASHIRA A, WU H, JASIN M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–42. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHLACHER K, WU H, JASIN M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22:106–16. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIM KS, SCHMUTTE C, YODER K, FISHEL R. Defining the salt effect on human RAD51 activities. DNA Repair (Amst) 2006;5:718–30. doi: 10.1016/j.dnarep.2006.03.006. [DOI] [PubMed] [Google Scholar]
- SIGURDSSON S, TRUJILLO K, SONG B, STRATTON S, SUNG P. Basis for avid homologous DNA strand exchange by human Rad51 and RPA. J Biol Chem. 2001;276:8798–806. doi: 10.1074/jbc.M010011200. [DOI] [PubMed] [Google Scholar]
- SUBRAMANYAM S, ISMAIL M, BHATTACHARYA I, SPIES M. Tyrosine phosphorylation stimulates activity of human RAD51 recombinase through altered nucleoprotein filament dynamics. Proceedings of the National Academy of Sciences. 2016;113:E6045–E6054. doi: 10.1073/pnas.1604807113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUBRAMANYAM S, JONES WT, SPIES M, SPIES MA. Contributions of the RAD51 N-terminal domain to BRCA2-RAD51 interaction. Nucleic Acids Res. 2013;41:9020–32. doi: 10.1093/nar/gkt691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUNG P, ROBBERSON DL. DNA strand exchange mediated by a RAD51-ssDNA nucleoprotein filament with polarity opposite to that of RecA. Cell. 1995;82:453–461. doi: 10.1016/0092-8674(95)90434-4. [DOI] [PubMed] [Google Scholar]
- WANG AT, KIM T, WAGNER JE, CONTI BA, LACH FP, HUANG AL, MOLINA H, SANBORN EM, ZIERHUT H, CORNES BK, ABHYANKAR A, SOUGNEZ C, GABRIEL SB, AUERBACH AD, KOWALCZYKOWSKI SC, SMOGORZEWSKA A. A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Mol Cell. 2015;59:478–90. doi: 10.1016/j.molcel.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIE J, SUPEKOVA L, SCHULTZ PG. A genetically encoded metabolically stable analogue of phosphotyrosine in Escherichia coli. ACS Chem Biol. 2007;2:474–8. doi: 10.1021/cb700083w. [DOI] [PubMed] [Google Scholar]