

Abstract
Human RAD51 promotes accurate DNA repair by homologous recombination and is involved in protection and repair of damaged DNA replication forks. The active species of RAD51 and related recombinases in all organisms is a nucleoprotein filament assembled on single-stranded DNA (ssDNA). The formation of a nucleoprotein filament competent for the recombination reaction, or for DNA replication support, is a delicate and strictly regulated process, which occurs through filament nucleation followed by filament extension. The rates of these two phases of filament formation define the capacity of RAD51 to compete with the ssDNA-binding protein RPA, as well as the lengths of the resulting filament segments. Single-molecule approaches can provide a wealth of quantitative information on the kinetics of RAD51 nucleoprotein filament assembly, internal dynamics, and disassembly. In this chapter, we describe how to setup a single-molecule total internal reflection fluorescence microscopy (TIRFM) experiment to monitor the initial steps of RAD51 nucleoprotein filament formation in real time and at single-monomer resolution. This approach is based on the unique, stretched-ssDNA conformation within the recombinase nucleoprotein filament and follows the efficiency of Förster resonance energy transfer (EFRET) between two DNA-conjugated fluorophores. We will discuss the practical aspects of the experimental setup, extraction of the FRET trajectories, and how to analyze and interpret the data to obtain information on RAD51 nucleation kinetics, the mechanism of nucleation, and the oligomeric species involved in filament formation.
Keywords: RAD51 DNA strand exchange protein, protein nucleation dynamics, total internal reflection fluorescence microscopy (TIRFM), Forster resonance energy transfer (FRET), single-molecule kinetics, Bayesian statistics, hidden Markov model (HMM)
1. Introduction
The RAD51 DNA strand-exchange protein (recombinase) is critical for the stability of the human genome. It is a key player in homologous recombination, which provides the most accurate means to repair such deleterious DNA lesions as double-strand DNA breaks (DSBs), interstrand DNA crosslinks (ICLs), and collapsed replication forks (Li and Heyer, 2008, Moynahan and Jasin, 2010, Ameziane et al., 2015, Michl et al., 2016). RAD51 also helps to maintain the integrity of damaged replication forks (Schlacher et al., 2012, Schlacher et al., 2011, Kolinjivadi et al., 2017). It is important for the cell, however, to maintain an exact balance in the amount and engagement of RAD51, as both its dearth and overabundance can have deleterious consequences. RAD51 overexpression and misregulation, for example, promote genome instability and allow cancerous cells to develop resistance to radiation and DNA-damaging drugs used in chemotherapy (reviewed in (Klein, 2008) and (Budke et al., 2016)). Both, the recombination- and replication-associated functions of RAD51 depend on the RAD51 nucleoprotein filament assembled on single-stranded DNA (ssDNA). In recombination, the RAD51 nucleoprotein filament catalyzes the reciprocal exchange of DNA sequences between the damaged and template molecules. In DNA replication, RAD51 protects stalled replication forks from excessive nucleolytic degradation by the MRE11 nuclease via a mechanism that may or may not involve the exchange of DNA strands (Schlacher et al., 2012, Schlacher et al., 2011, Kolinjivadi et al., 2017). The ability to selectively manipulate the two functions of the RAD51 nucleoprotein filament can have significant biomedical implications, but it requires knowledge regarding how RAD51 nucleoprotein filaments assemble on the recombination and replication intermediates, and whether the two types of RAD51 nucleoprotein filaments have distinct mechanistic or structural features that can be selectively manipulated. Such information can be obtained through the quantitative analysis, at the single-molecule level, of the kinetics of the RAD51 nucleoprotein filament assembly.
Human RAD51 is a member of the highly conserved family of ATP-dependent recombinases. Recombinases from all species, including bacteriophage UvsX, bacterial RecA, archaeal RadA, yeast Rad51 and human RAD51 share significant structural and functional similarities. A conserved feature of these RecA-like recombinases is that assembly of the active, ATP-bound form of the recombinase on ssDNA forms a right-handed, helical nucleoprotein filament (i.e., a presynaptic filament). The formation of a nucleoprotein competent for DNA strand-exchange reaction requires a bound nucleotide cofactor, either ATP or a non-hydrolysable ATP analog. Each recombinase monomer within the filament occludes 3 nucleotides of ssDNA, and extends the ssDNA within the active form of the filament 1.6-fold beyond its B-form (Xu et al., 2016, Short et al., 2016, Ristic et al., 2005, Galkin et al., 2005, Benson et al., 1994, Qiu et al., 2013, Subramanyam et al., 2013). Crystallographic studies of bacterial RecA (Chen et al., 2008) and high-resolution electron microscopy (EM) studies of human RAD51 (Xu et al., 2016, Short et al., 2016) reveled the remarkably conserved and very unusual structure of the recombinase-extended ssDNA within the presynaptic filament. In each triplet base, the ssDNA extension is not uniform, with the inner triplet bases maintaining stacking interactions and a near-B-DNA conformation, while the backbone between the adjacent triplet bases exhibits significant local stretching (~8Å). The inactive (e.g., ADP-bound) form of the RecA or RAD51 nucleoprotein filament is shorter and more compressed compared to the active, ATP-bound state (Yu et al., 2001). Universal organization of the extended DNA structure within the active recombinase filament is likely to be important for the processes of homology search and DNA strand exchange. Indeed, recent single-molecule studies from the Greene lab (Qi et al., 2015) revealed that homology sampling, at least during the very initial steps in the DNA strand-exchange reaction, proceeds in 3-nucleotide steps, and the fidelity of the recombinase-mediated DNA strand-exchange reaction being defined by the subsequent stabilization of DNA triplet bases.
While the overall conservation of the nucleoprotein filament is striking, the idiosyncratic features of the recombinases from different species are manifold. These include the mechanism of nucleoprotein filament assembly. All recombinases are believed to form the presynaptic filament by a nucleation event that is followed by filament growth (Chabbert et al., 1987) (Figure 1A). The rates, cooperativity, and mechanisms of the nucleation and growth phases define the segments’ lengths within the nucleoprotein filament, which may in turn have significant influence on the mechanism of homology search and the DNA strand exchange reaction, as well as on the requirement for, and sensitivity to, different recombination mediators, various auxiliary proteins, and anti-recombinases. Scanning force microscopy studies revealed a diversity of shapes, lengths, and regularities in the organization of human RAD51 nucleoprotein filaments assembled in the presence of different nucleotide cofactors, with more regular filaments corresponding to the conditions that promote the DNA strand-exchange reaction (Ristic et al., 2005).
Figure 1. Dynamics of human RAD51 nucleation.
A. Reversible nucleation of RAD51 onto ssDNA. Human RAD51 binds ssDNA in an ATP-dependent manner (Tombline et al., 2002). The ATP bound RAD51 nucleoprotein filament is stably nucleated such that each RAD51 monomer binds three nucleotides which facilitates further polymerization of the nucleoprotein filament forming the active pairing unit in strand exchange reactions. Hydrolysis of ATP lowers the affinity of RAD51 for ssDNA and leads to turnover or disassembly of the nucleoprotein filament. B. To form nucleoprotein filaments and perform strand invasion and recombinase activities within the cell, human RAD51 has to compete with Replication Protein A (RPA), the ssDNA binding protein. The stability of the RAD51 nucleus and its extension define the capacity of RAD51 to favorably compete with RPA. C. Strand invasion and homology search activity of the RAD51 nucleoprotein filament, allows it to invade homologous duplex DNA, resulting in a displacement loop structure that can be used as a primer for synthesis of DNA using the intact duplex as a template, resulting in accurate repair of damaged DNA.
Several single-molecule approaches have been developed for the analysis of recombinase nucleoprotein filament formation, dynamics, and disassembly (see (Bell and Kowalczykowski, 2016, Candelli et al., 2013) for relatively recent comprehensive reviews). Each of these approaches provides uniquely valuable information on different aspects of this process. The optical (Amitani et al., 2010, Bell and Kowalczykowski, 2016) or magnetic tweezers (Ristic et al., 2005, van der Heijden et al., 2007, Arata et al., 2009) experiments that monitor RecA or RAD51 nucleoprotein formation by following the changes in the mechanical properties of the DNA molecule have sufficiently high resolution to detect binding or dissociation of individual recombinase molecules. These analyses, however, cannot unambiguously separate ssDNA extension arising from multiple nucleation sites. Additionally, tension exerted on the DNA may also affect nucleoprotein filament formation and dynamics. Visualization of fluorophore-labeled RecA (Forget and Kowalczykowski, 2012) and RAD51 (Forget and Kowalczykowski, 2010, Candelli et al., 2014) forming nucleoprotein filaments on DNA molecules extended by hydrodynamic flow or optical tweezers provided information on the kinetics of filament nucleation and growth originating from distinct nuclei. The drawback of these experiments is that they require “dipping” of the DNA molecule into the solution of fluorescently-labeled recombinase and, therefore, only provide discrete snapshots of the reaction. Power analysis of the time dependence of filament length is used to deduce the oligomeric species involved in the nucleation and growth phases. The two phases can be distinguished by performing several cycles of “dipping” and observation. Candelli and colleagues (Candelli et al., 2014), for example, showed that the RAD51 nucleoprotein filaments grow on ssDNA from heterogeneous nuclei ranging in size from oligomers to dimers and even monomers. This assay, however, is blind to the dynamics of the nucleation step per se. These initial steps of recombinase filament formation can instead be observed and analyzed using a Förster resonance energy transfer (FRET)-based single-molecule assay enabled by total internal reflection fluorescence microscopy (TIRFM). Here, a FRET donor (typically Cy3) and a FRET acceptor (typically Cy5) are incorporated into the DNA substrate, which is then immobilized on the surface of a TIRFM observation flowcell. Binding and dissociation of the recombinase results in increases and decreases to the length of the DNA, respectively, which in turn causes decreases and increases in the FRET efficiency (EFRET) signal (a measure of non-radiative energy transfer from the FRET donor to the FRET acceptor), respectively. This approach has been applied to study RecA (Joo et al., 2006), yeast Rad51 (Qiu et al., 2013), and human RAD51 (Subramanyam et al., 2016) nucleation on ssDNA. These studies showed that while nucleation by RecA and yeast Rad51 occurs mainly via addition of RecA (Joo et al., 2006) or Rad51 (Qiu et al., 2013) monomers, human RAD51 nucleates on the ssDNA primarily via addition of RAD51 dimers (Subramanyam et al., 2016). In this chapter, we provide a protocol for the observation, analysis, and interpretation of human RAD51 filament formation by single-molecule FRET.
2. Observing RAD51 nucleation on ssDNA using total internal reflection microscopy
2.1. Total internal reflection microscope
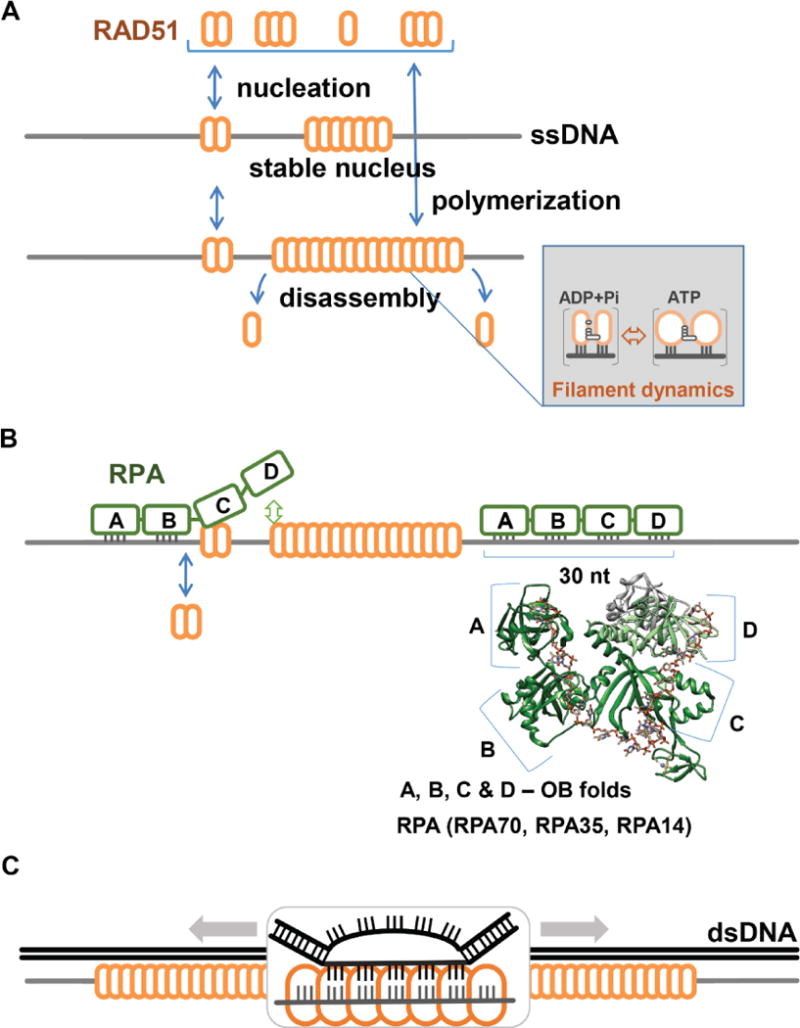
TIRF microscopy provides a convenient system to observe fluorophore-labeled molecules tethered to the surface of a passivated TIRF observation flowcell. The basic TIRF microscope comes in three main types; prism-based, objective-based, and a micro mirror-based (see (Axelrod, 2008, Larson et al., 2014) for detailed descriptions of these types of microscopes). The single-molecule RAD51 filament formation assay can be performed using either of the three microscope configurations. For FRET-based measurements, a single excitation source is sufficient, but the emission signal needs to be recorded in a dual-view system, which separates the emission of the FRET donor and FRET acceptor. Our TIRF-FRET setup uses Cy3 as a donor fluorophore (excited by a 532 nm, DPSS Laser, Coherent Inc.) and Cy5 as an acceptor fluorophore. An evanescent wave generated by total internal reflection of the laser source, specifically illuminates molecules that are tethered to the surface of the observation flowcell (<100nm) (Axelrod, 2008, Bain et al., 2016). The scattered light is removed using a Cy3/Cy5 dual band-pass filter (Semrock, FF01-577/690) in the emission optical path. Images are chromatically separated into Cy3 image (using a Chroma ET605/70m filter) and Cy5 image (using a Chroma ET700/75m filter) using a 630-nm dichroic mirror inside the dual-view system (DV2; Photometrics) (Figure 2A). An evanescent wave generated by total internal reflection of the laser source, specifically illuminates molecules that are tethered to the surface of the slide (<100nm) (Axelrod, 2008, Bain et al., 2016) (Figure 2B).
Figure 2. Observing the RAD51 nucleoprotein filament formation by TIRFM.
A. Typical layout of a prism-type TIRF microscope. An evanescent wave generated using a 532 nm laser illuminates individual fluorescence molecules tethered on the surface of the microscope flow cell. Fluorescence emission from the Cy3 and Cy5 fluorophores is separated in the dual view, and the split image of the flow cell surface is directed to the CCD camera. B. A Poly dT(60) ssDNA substrate is tethered to a biotinylated quartz slide using the biotin-Neutravidin interaction. The evanescent wave produced by the total internal reflection illumination excites the Cy3 fluorophore on the surface-tethered DNA substrate. Both Cy3 and Cy5 emission is observed in separate channels. The dark spots in the microscope field of view below, shows the distribution of fluorescent molecules on the slide. The black line separates the Cy3 and Cy5 channels. Intensities of the Cy3 and Cy5 dyes incorporated into the individual DNA molecules are tracked over time by recording movies over the course of the experiment.
2.2. Preparation of fluorophore-labeled DNA substrates
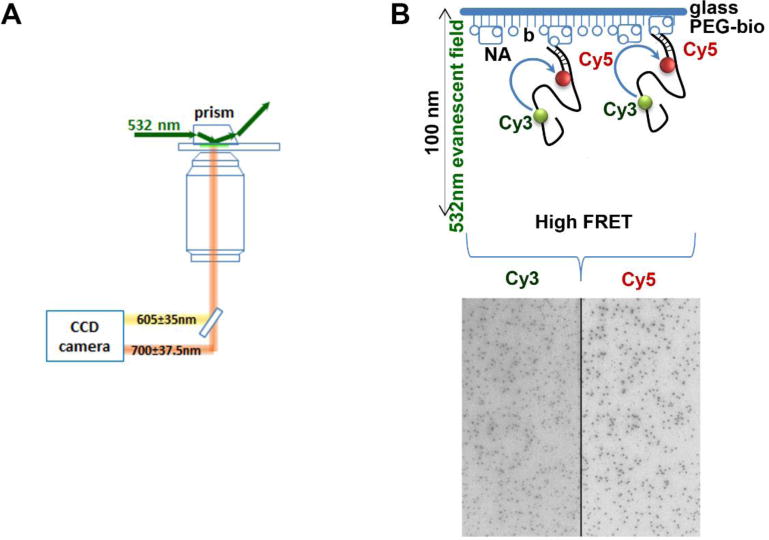
DNA oligonucleotides can be site-specifically labeled with a wide range of fluorophores compatible with most single-molecule TIRFM setups and are available for purchase through commercial companies like IDT, MWG, Operon, etc. In order to visualize the RAD51 nucleoprotein filaments using our FRET-based assay and TIRF microscope, a DNA substrate labeled with a Cy3 donor fluorophore and a Cy5 acceptor fluorophore is prepared and immobilized on the surface of the observation flowcell. A biotin-Neutravidin interaction is used for tethering the substrate to the surface. Therefore, the substrate has to contain a biotin moiety in addition to two fluorophores. The Cy3 and Cy5 fluorophores are positioned such that there will be a sufficient, appreciable change in the EFRET signal upon binding of RAD51 to the substrate. We use a biotinylated oligonucleotide substrate that is composed of two ssDNA oligonucleotides. The first is a short anchor oligo containing the Cy5 dye at the 5’ and the biotin tag at the 3’ end (5Cy5-Bot18-3Bio 5’Cy5-GCCTCGCTGCCGTCGCCA-3’Bio) is used to tether the DNA to the surface of the observation flowcell. The second oligonucleotide contains a complementary sequence to the anchor oligo followed by a poly(dT) sequence containing the Cy3 donor fluorophore. Constructing the substrate from two oligonucleotides is advantageous as they are more inexpensive to synthesize compared to a single oligonucleotide containing the donor, the acceptor, and the biotin. Another advantage of the two-oligonucleotide design is that the length of the poly(dT) sequence can be varied, along with the location of the Cy3 donor (Joo et al., 2006), simply by synthesizing different complementary oligonucleotides with the desired changes. In our assay, we use a poly(dT60) with the Cy3 fluorophore internally positioned 21 bases from the end of the complementary sequence. The resulting oligonucleotide is referred to as Top18-(T21)Cy3(T39) (TGGCGACGGCAGCGAGGCTTTTTTTTTTTTTTTTTTTTT/iCy3/TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT). The two oligonucleotides are annealed in a 1:1 molar ratio (500 nM each) in annealing buffer (10 mM Tris, pH 8.0, 100 mM NaCl, 0.1 mM EDTA) by heating the solution to 95 °C in a heat block for 5 minutes and allowing it to cool down to room temperature in a dark chamber (Figure 3A). The annealed oligos can be analyzed on an 8% TAE-PAGE gel (Figure 3B), aliquoted as required for a single experiment, and stored at –80°C.
Figure 3. Preparation and tethering of fluorescent substrates.
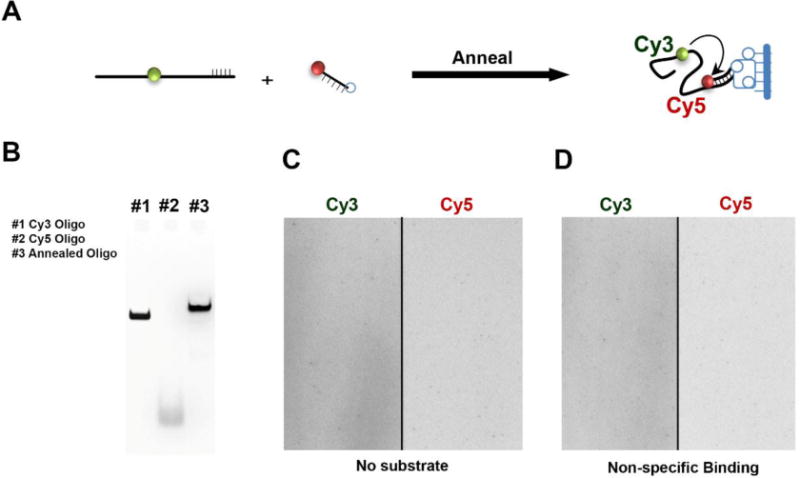
A. Scheme for annealing of the Cy3- and Cy5-labelled oligos to generate the DNA substrate used for the single-molecule RAD51 binding experiment. B. PAGE gel used to verify the annealing of the two ssDNA substrates. C & D. Image of a flow cell illuminated by evanescent wave produced by total internal reflection of the 532 nm, DPSS Laser source (Cy3 excitation). The absence of appreciable fluorescent spots in in both the Cy3 (left) and Cy5 (right) channels confirms that there is no non-specific binding on introduction of the annealed substrate.
2.3. Preparation of reagents for single-molecule experiments
The following reagents need to be prepared for all single-molecule experiments. Attention to detail while preparing these solutions is required, as they are critical to the proper immobilization of substrates and the photostability of the organic fluorophores while performing experiments.
2.3.1. 0.2 mg/mL Neutravidin
Mix 2 mg of Neutravidin (Thermo Fisher catalog # 31000) with 10 mL of Phosphate Buffered Saline (Thermo Fisher catalog # 10010023). Do not vortex. Aliquot the solution and store the aliquots at 4 °C, where they will be stable for use for up to a month.
2.3.2. 24 mM Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid)
Add 60 mg Trolox powder (Sigma catalog #391913) directly to 10 mL Milli-Q water in 15-ml sterilized tube. Before Trolox dissolves, add 60 μl of 2 M NaOH (~12 mM). Shake the solution few times by hand and incubate at 25 °C for approximately 60 hours on a rotary shaker under a fluorescent lamp. Monitor Trolox solubilization and the color of solution. The solution should turn cloudy after incubation, can be filtered using a 0.2 μM syringe filter, and can be stored at 4° C and used for up to 1 month. NaOH is added because the Trolox solubilization results in a drop in pH (pH 3 – 4), and higher solubility can be achieved by dissolving Trolox in a solution with neutral pH, rather than in acidic medium. A good preparation of Trolox has a yellow color and an OD400 of > 0.12. The higher peak at 400 nm corresponds to the ability of Trolox to more effectively quench excited triplet states of the fluorophores that can lead to photoblinking and photobleaching, thus resulting in extended visualization times with minimal photophysical side effects (Ha and Tinnefeld, 2012).
2.3.3. 1X Bovine Serum Albumin (BSA)
Mix 10 mg of BSA (Sigma catalog #A7030) in 1 mL of T50 buffer (10 mM Tris pH 8.0, 50 mM NaCl). Mix and centrifuge the solution for 1 min at 4 °C at 1000xg. The solution can be stored at 4 °C for a month.
2.3.4. 100X Gloxy (glucose oxidase-catalase)
Prepare 40mg/mL catalase (Sigma catalog #E3289) solution in T50 buffer. Weigh 10mg glucose oxidase (Sigma catalog #G2133) and add 90μl of T50 buffer. Add 10μl of the catalase solution. Mix the two by tapping; do not vortex. Centrifuge the solution for 1 minute at 1000 × g and collect the supernatant. The solution can be stored at 4 °C for a month.
2.4. Oxygen scavenging system
Employing an enzymatic oxygen-scavenging system such as the glucose oxidase-catalase (gloxy) system, effectively reduces photobleaching. In addition, using Trolox, a vitamin E analogue, further eliminates photoblinking and reduces photobleaching. One of the most popular oxygen scavenging-systems in use today employs glucose oxidase and catalase to remove molecular oxygen by oxidizing glucose (Shi et al., 2010) (See reaction scheme below).
2.5. Surface passivation and tethering of the DNA substrates
The poly(dT) partial overhang substrate is annealed to the anchor oligomer in Annealing Buffer as described above. Surface-passivated and functionalized TIRFM observation flowcells are prepared by chemically treating the quartz slide and borosilicate coverslip components of the flowcells with detergents and strong alkali (potassium hydroxide) followed by aminosilanation (uniformly coating the slide with negative charge) and coating the reaction surface with a mixture of mPEG (polyethyleneglycol) and biotinylated-PEG. The coated slide and coverslips can be stored in nitrogen-dried, vacuum-sealed, conical tubes at –20 °C. Before use, the tubes are equilibrated to room temperature for about 20 minutes in a dark chamber to prevent condensation onto the treated surface. The passivated and functionalized quartz slides and borosilicate glass coverslips are then assembled in the observation flowcells using double-sided tape and epoxy (detailed procedures for cleaning, passivating, functionalizing, and assembling the flowcells can be found in (Joo and Ha, 2012a, Bain et al., 2016)) and depicted later in (Figure 4). This procedure reduces the non-specific binding of proteins and nanoscopic impurities in buffers that might auto-fluoresce. In the absence of any DNA substrate, no more than four to five fluorescent spots should be observed in the field of view (57 μm × 150 μm/256 × 512 pixels) (Figure 3C). Higher abundance of non-specific fluorescent spots may stem from imperfections in the quartz or borosilicate glass, contaminants in the solutions, or from air pollution. Inject 50 pM of the annealed DNA substrate in one 100 μL of T50 buffer (Tris pH 8.0, 50 mM NaCl) into the flowcell to test the quality of the surface passivation and functionalization. Since the flowcell has not yet been treated with Neutravidin, addition of the fluorophore-labeled DNA should not, at this step, change the number of fluorescent spots, thereby indicating the absence of non-specific adsorption of the DNA to the surface of the flowcell (Figure 3D). One hundred microliters (reaction volume) of 0.2 mg/ml Neutravidin is then added to the flowcell. In the case of flow experiments, we use 300 μl of 0.2 mg/ml Neutravidin. After 3-minute incubation at room temperature, excess Neutravidin is washed out of the flowcell using 300 μl T50 buffer. Fifty pM annealed DNA substrate is added to the Imaging Buffer (24 mM Trolox, 20 mM HEPES pH 7.5, 2 mM MgCl2, 0.8% Glucose, 150 mM NaNH4PO4, 1 mM ATP, 1 mM DTT, 0.1 mg/ml BSA, 0.04 mg/ml Catalase and 1 mg/ml Glucose Oxidase (1X gloxy)) and flowed into the reaction chamber. Excess DNA substrate is washed out by flowing in 300 μL of fresh imaging buffer.
Figure 4. Assembly of flow cells for TIRFM Imaging.
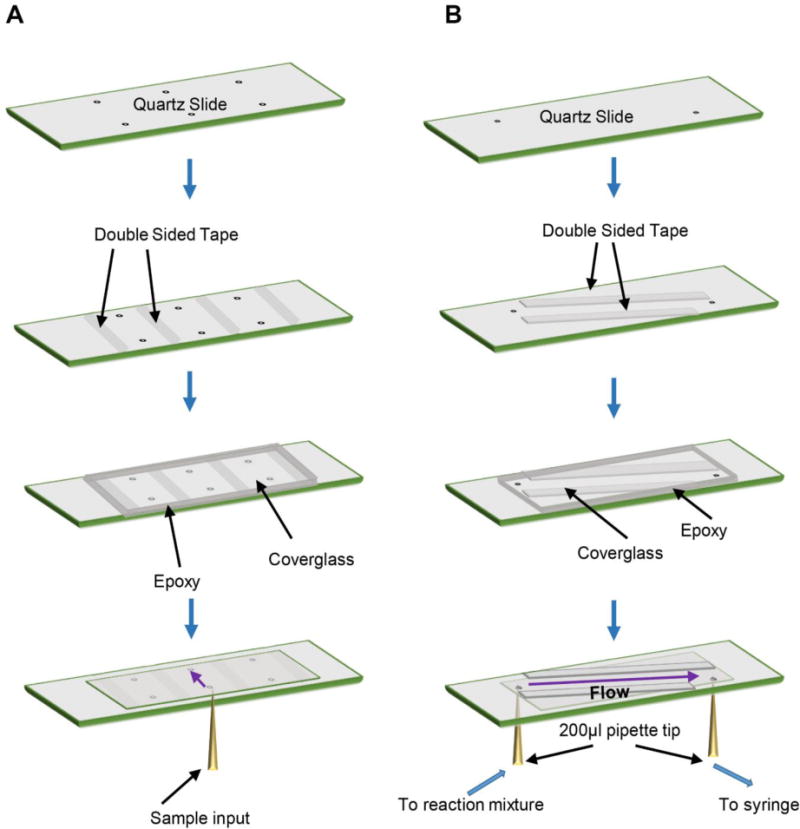
A. To prepare flowcells for equilibrium TIRFM experiments, a quartz slide is drilled, passivated and PEG-treated as described in section 2.5. Double-sided tape is applied to the slide to mark out three flow cells per slide. A passivated and surface treated cover glass is then applied onto the slide covering the pre-drilled holes, and the edges of the cover glass are then sealed with epoxy. Once dry, excess epoxy is removed from the slide to make sure it lies flat on a table with the cover glass facing down. Samples can be pipetted into the flowcell from one direction using a 200 μL pipette while the excess sample flows out of the pre-drilled hole at the other end and absorbed by a clean absorbent tissue. Note that the slide is placed on prism type TIRF microscopes with the cover glass facing down while the sample is introduced from the top, while the figure shows an inverted scheme with introduction of the sample from the bottom for clarity in assembly of the flowcell. B. Preparation of flowcells for pre-equilibrium experiments is similar as described above with the following differences. Holes in the quartz slide are pre-drilled in a diagonal direction and the double-sided tape is placed such that a long flowcell is created along the diagonal of the slide. The cover glass is placed in a similar manner and sealed with epoxy. 200 μL pipette tips, attached to flow tubing are glued to the ports. One end of the tube is inserted into the reaction mixture and is drawn out the other end, through the flowcell using a syringe. The solution can be exchanged by pausing flow and placing the tubing into another sample tube before re-establishing flow.
For experiments performed under flow, we use a similar experimental protocol, except 75 pM of the DNA substrate is used instead of 50 pM. The concentration of the annealed substrate is calibrated such that there is sufficient spacing between the observed fluorescent spots and no overlap between the diffraction-limited fluorescent spots corresponding to the individual molecules. Too high a density of surface-tethered molecules may lead to errors during image analysis, or may preclude the extraction of the trajectories (see below). The optimal density corresponds to approximately three to five hundred molecules in the field of view (Figure 2B).
2.6. Acquisition of single molecule data for equilibrium binding experiments
The construction of the imaging chambers for the flow experiments is depicted in (Figure 4B). To initiate the RAD51 filament formation, RAD51 (in imaging buffer) is added at various concentrations into the flowcell containing the immobilized DNA substrates. For equilibrium experiments, the sample is equilibrated for ~5 minutes prior to imaging and the recording is started when the system has fully equilibrated.
Single-molecule data is acquired by the Andor iXon EMCCD camera, using software written in Visual C++ (available at https://cplc.illinois.edu/software/). Movies are recorded in the *.pma format which is analyzed by the IDL suite (ITT Visual Information Solutions) (see below), using customized scripts (available upon request from the Spies lab). For equilibrium experiments, forty movies, each of fifteen-second duration (100 ms time resolution, 400 background value and 1600 data scalar) are recorded using a gain of 230 at 45.6 mW green laser intensity. These short movies are used to measure the equilibrium state of the entire system. The recorded movies contain Cy3 and Cy5 fluorescence intensities for all of the molecules in the field-of-view over each frame recorded. These time based-changes in the fluorescence intensities of the donor and acceptor fluorophores at a specified location are referred to as fluorescence trajectories. These fluorescence trajectories correspond to single fluorescent molecules and are extracted from the movies using IDL scripts. If the surface-tethered DNA substrates are separated further than the diffraction limit (ensured by the low ratio of biotinylated to non-biotinylated PEG molecules and the concentrations of the Neutravidin and biotinylated molecules during surface immobilization reaction) and in the absence of non-specific binding, each fluorescence trajectory can be attributed to the binding of RAD51 (or the absence of binding of RAD51) to a single, surface-tethered DNA molecule.
2.7. Acquisition of single molecule kinetic data for pre-equilibrium experiments
For measuring pre-equilibrium dynamics of RAD51 nucleation events, three minute movies are recorded to capture all events of RAD51 nucleation on ssDNA. The main differences between these pre-equilibrium experiments and the equilibrium experiments described in the previous section are that the RAD51 protein sample is flowed into a modified flowcell (Figure 4B). We begin recording the movie with the DNA tethered to the surface, but no RAD51 present. After 10 seconds form the beginning of the movie we flow the RAD51 protein sample into the flowcell. Otherwise we use the same recording conditions as described for the equilibrium experiments.
2.8. Extracting and organizing single-molecule fluorescence trajectories
To extract single-molecule fluorescence trajectories and to correlate signals from the donor and acceptor channels, we use an smFRET data acquisition and analysis package which can be downloaded from the University of Illinois Center for the Physics of Living Cells website (https://cplc.illinois.edu/software/). Note that this package requires the IDL suite, movie files with *.pma extension, 512 by 512 pixels frames (containing the donor and the acceptor channels, 256 by 512 pixels each), and a mapping file. The mapping file, which is essential to accurately align the donor and acceptor channels, should be recorded within a week of the experimental data recording using a flowcell that is sparsely populated with beads that fluoresce in both donor and acceptor channels. The IDL script, uses the mapping file to correlate fluorescent spots in the donor channel with the corresponding fluorescent spots in the acceptor channel and then uses this alignment to identify the locations in the field-of-view that contain individual, surface-tethered DNA molecules labeled with both Cy3 and Cy5 fluorophores. For the FRET-based experiments with surface-tethered DNA molecules, only the first ten frames are usually used to map the location of the molecules. It is expected that each acceptable molecule will generate a circular, diffraction-limited fluorescent spot whose fluorescence intensity has a Gaussian distribution and is present in both channels. The images are first analyzed in the Cy3 channel, and the location of the individual molecules are identified as two-dimensional Gaussian peaks of fluorescence intensity by comparing the fluorescence around the bright spot in two circles, with the diameter of 6 and 8 pixels. Then, the corresponding location in the Cy5 channel is checked for the presence of an acceptable Gaussian peak. Peaks that are deformed or present only in one channel are ignored, as these either stem from imperfections in the quartz coverslip or the borosilicate glass or are due to two molecules located too close to one another. The selected peaks are marked for trajectory extraction. Fluorescence trajectories represent the time-based changes in the Cy3 and Cy5 fluorescence intensity of each individual molecule over time.
Several hundred individual fluorescence trajectories are extracted from each recorded video and visualized. Fluorescence trajectories can be visualized using customized MATLAB (The MathWorks, Inc.), scripts (available upon request from the Spies lab) and can be used to generate the corresponding EFRET trajectories using the following equation to calculate FRET-
where and are the sensitized emission intensity of the donor and acceptor, respectively (Ha, 2001). The measured raw intensities of the donor and acceptor channels must be corrected by measuring the percentage of fluorescence leakage from the donor channel to the acceptor channel (denoted further as β). The donor leakage is then subtracted from the acceptor channel intensities and added back to the donor intensity.
The donor leakage intensity depends (β) on the optical setup of the TIRF microscope and can be determined by immobilizing an oligonucleotide substrate labeled only with the fluorescent donor (in our experiments, this is Cy3). Movies are recorded until all the Cy3 molecules are completely photobleached, i.e. there are no spots visible in either the donor or acceptor channels. The fluorescence trajectories are extracted from the movies as described above. Residual background intensities after the photobleaching step are subtracted from the fluorescence intensity values for both the donor and acceptor fluorescence trajectories (Figure 5). Using the background-corrected trajectories, average values of the Cy3 intensity before and after Cy3 photobleaching are picked out for each fluorescence trajectory. Similar values are obtained for Cy5 after Cy3 photobleaching. Change in background corrected donor intensity (ΔID) corresponds to the difference between the Cy3 intensities before and after photobleaching. The change in background corrected acceptor intensity (ΔIDA) corresponds to the difference in Cy5 intensities before and after Cy3 (donor) photobleaching. The ratios are calculated (the percentage of donor intensity that leaks into the acceptor channel upon donor excitation (Ha, 2001)) to get donor leakage for that trace (i.e., β = (ΔIDA/ΔID)). Averaging the donor leakage values for several such fluorescence trajectories gives us the donor leakage intensity for the optical setup. For our experimental system, the donor leakage correction was measured to be 7%. Applying this correction, we get the equation for EFRET (FRET Efficiency) given below –
Where, and are the background-corrected acceptor and donor intensities. respectively. , is a parameter representing relative detection efficiencies and quantum yields of the two dyes, and can be determined from photobleaching events and is calculated as the ratio of change in the acceptor intensity, ΔICy5 to change in the donor intensity, ΔICy3 upon acceptor photobleaching (γ = ΔICy5/ΔICy3) (Roy et al., 2008, Joo and Ha, 2012b).
Figure 5. Background correction and measurement of donor leakage.
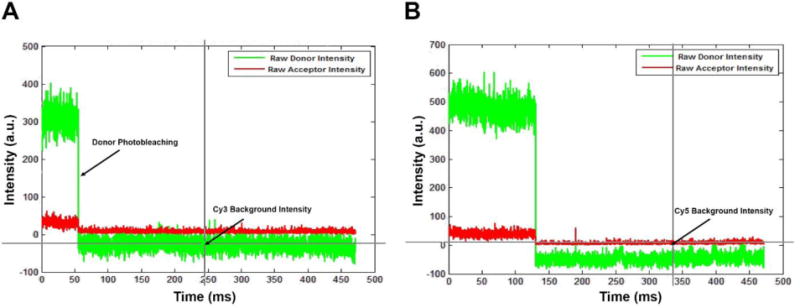
A&B. Fluorescence time trajectories for the Cy3 and Cy5 channels of surface-tethered Cy3 labeled ssDNA molecules showing single step photobleaching events. Donor and acceptor background intensities are measured by recording the values of Cy3 and Cy5 intensities after the Cy3 molecule has photobleached (Shown in crosshairs). The background intensity values are averaged for several molecules in a movie to account for small fluorescence changes in the local environment of the molecules. The drop in the intensity of the Cy5 trajectory as the Cy3 fluorophore undergoes photobleaching corresponds to the amount of donor leakage from the Cy3 channel to the Cy5 channel. This is measured as a fraction and defined as the ratio of change in background corrected acceptor intensity to the change in background corrected donor intensity (β = (ΔIDA/ΔID)).
3. Ensemble analysis of single-molecule data
Ensemble analysis of RAD51 filament formation is a useful method to quickly determine many of the physico-chemical properties of the equilibrium-bound state of the RAD51 filament. For example, it can be used to identify the number of equilibrium states, stoichiometric information, and relative affinities. For the RAD51 wild-type protein, two major states are observed (Figure 6A), one corresponding to free DNA and the other representing the fully extended nucleoprotein filament (Figure 6A). Intermediate states might be observed in mutants harboring deficiencies in the binding of short oligonucleotides, or those that have nucleation defects. These generally manifest in broadening of the peaks, or the appearance of new, intermediate peaks, in a FRET histogram.
Figure 6. Single-molecule experiments to visualize equilibrium distributions of the RAD51-DNA FRET states.
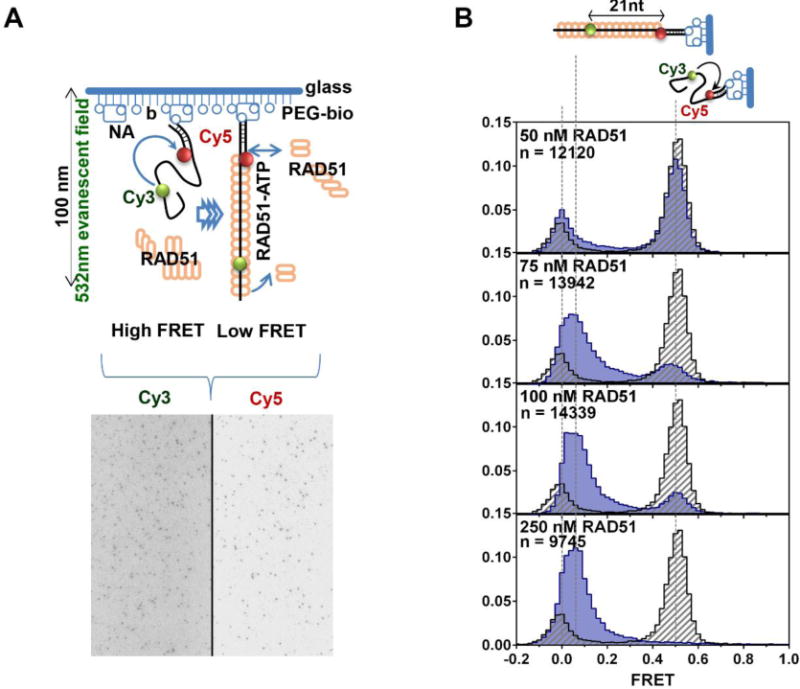
A. Single-molecule TIRFM experiment visualizing binding of RAD51 protein onto ssDNA FRET substrate. In the absence of RAD51 binding, the acceptor Cy5 fluorophore on the substrate DNA is excited via FRET (High EFRET). Upon binding, RAD51 extends the ssDNA substrate leading to a decrease in energy transfer and reduction in Cy5 emission with a corresponding increase in Cy3 intensity (Low EFRET). The dark spots in the microscope field of view below, shows the distribution of fluorescent molecules on the slide. The black line separates the Cy3 and Cy5 channels. Both Cy3 and Cy5 emission can be tracked simultaneously using a dual view system. Changes in Cy3 and Cy5 intensities as RAD51 binds the ssDNA substrate can be tracked over time by recording movies over the course of the experiment. B. Analysis of the equilibrium ssDNA binding using single-molecule TIRFM. Distributions of the FRET states of the DNA substrates in the presence of the indicated concentrations of RAD51 overlaid with the distributions of the FRET states in the absence of protein. Unbound ssDNA (grey) yields a histogram centered on FRET value of (~0.5), while fully extended RAD51 nucleoprotein filament (blue) yields a histogram centered on ~0.1 FRET value. Concentrations of RAD51, as well the number of molecules used to build each histogram are indicated in each panel.
3.1. Preparing FRET histograms
All fluorescence trajectories extracted from the 15-second movies are background corrected such that intensities for photobleached donor and acceptor are always zero. To generate FRET histograms, we use a MATLAB script (available upon request) in which five frames from each movie are included in the calculation of the EFRET values for each molecule and for all molecules in the movie, then bin and plot these values as a FRET histogram normalized for the number of counts. The MATLAB script allows us to set cutoff values for acceptable fluorescence intensities to exclude experimental artifacts like auto-fluorescent aggregates, or light scattering artifacts. The number of bins in the histogram can also be adjusted to the dataset (we have optimized this value to 80 bins, but this can be changed). The program outputs the absolute, as well as normalized, counts per bin, which can be used to create FRET histogram plots in other programs such as GraphPad Prism (GraphPad software). Under our experimental conditions, unbound ssDNA molecules yield a histogram centered on an EFRET value of ~0.5, while fully extended RAD51 nucleoprotein filament yields a histogram centered on ~0.1 EFRET value (Figure 6B). The progression of complete binding and elongation of the ssDNA substrate can be visualized by varying the RAD51 concentration in the reactions. Molecules in which the acceptor (Cy5) undergoes premature photobleaching represent a small fraction of the histogram (<3%) centered on the 0 EFRET value (Figure 6B).
4. Real-time observation and analysis of RAD51 nucleation using ebFRET
4.1. Using Hidden Markov Models to analyze single-molecule data
The data collected in time-dependent, single-molecule experiments are signal versus time trajectories (i.e., the EFRET trajectories in the current case), which are measurements of the signal from an individual molecule (i.e., the ssDNA labeled with Cy3 and Cy5 fluorophores in the current case) or macromolecular complex (i.e., the RAD51 nucleoprotein filament in the current case) taken at consecutive time points. Analysis of these signal trajectories involves correlating the values of the signal at different time points to the underlying conformational dynamics of the individual molecule (Kinz-Thompson et al., 2016). Unfortunately, despite being sensitive enough to report on individual molecules, the signal trajectories collected using most single-molecule techniques are relatively noisy, and this confounds the ability to correlate the signal values to the underlying dynamics. Currently, the most popular approach for inferring the dynamics of the individual molecule from the noisy, observed signal trajectory is to use a hidden Markov model (HMM) (Bishop, 2006). Conceptually, HMMs posit that each data point in a signal trajectory corresponds to an unobserved (i.e., “hidden”) underlying state of the molecule, and that, at each time point, the hidden state has a certain probability of transitioning to a different hidden state for the next time point (c.f., Figure 7A). By determining the HMMs that are consistent with an observed signal trajectory, one can determine the number of underlying hidden states, the signal values corresponding to those states, and the probability of transitioning between those states (i.e., the rate constants for transitions between the states).
Figure 7. Analysis of human RAD51 nucleation using Hidden Markov Models.
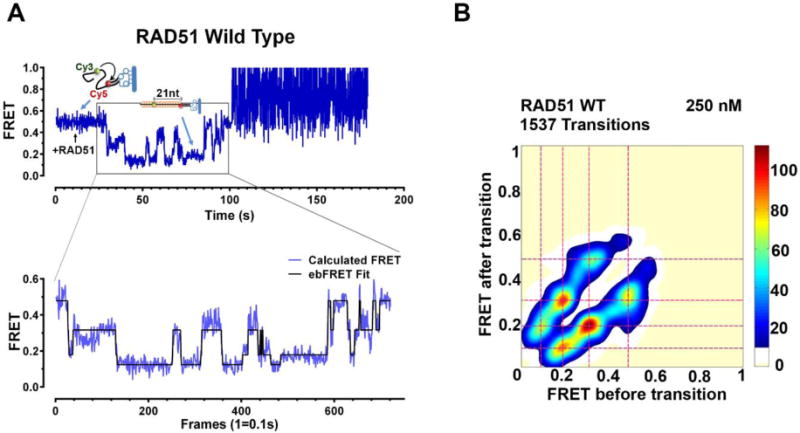
A. Representative EFRET trajectories from pre-equilibrium experiments visualizing step wise RAD51 nucleation onto ssDNA in real time. RAD51 wild type protein is added to the flowcell at t~10 seconds. The region of the trajectory that includes the first observed transition and all subsequent transitions until one of the dyes photo-bleaches (grey box); or the transitions stop occurring (i.e. equilibrium is achieved) or the recording is terminated. The remainder of the trajectory is excluded from empirical Bayesian analysis. The black lines overlaying the FRET data (blue) represent the best HMM fit to the trajectory, which describes the nucleation of the 21-base (nucleotide distance between the Cy3 and Cy5 fluorophores) ssDNA substrate through four distinct EFRET states. B. Transition Density Plot (TDP) showing the transition densities corresponding to each EFRET state for the wild type RAD51 protein nucleation. 1537 transitions were analyzed from 85 molecules at a concentration of 250 nM RAD51. Warmer colors (red) indicate more frequent transitions. The frequency scale is shown on the right of the TDP.
As described in the previous section, the donor and acceptor fluorescence trajectories that are observed in an smFRET experiment are used to calculate EFRET trajectories. Because EFRET is a proxy for the distance between the donor and acceptor fluorophore, the hidden states in an EFRET trajectory correspond to conformational states of the underlying molecule. Consequently, an HMM analysis of an EFRET trajectory provides a structure-based interpretation of the dynamics of the individual molecule corresponding to that EFRET trajectory, which was measured in the smFRET experiment. There are many software packages that are appropriate for the analysis of EFRET trajectories using HMMs (e.g., QuB (Qin et al., 2000), HaMMy (McKinney et al., 2006), vbFRET (Bronson et al., 2009), (Bronson et al., 2010), or ebFRET (Van De Meent et al., 2013), (van de Meent et al., 2014), and these primarily differ in the approach with which they determine which HMM(s) are consistent with the observed EFRET trajectory being analyzed. Many of these packages, including QuB (Qin et al., 2000) and HaMMy (McKinney et al., 2006), utilize the maximum-likelihood approach, which yields the one HMM that best ‘fits’ the EFRET trajectory. This approach ignores the fact that, statistically, multiple HMMs will be consistent with an EFRET trajectory. Perhaps more importantly, because the HMM with the best ‘fit’ (i.e., the largest likelihood value) is the one in which each data point in the EFRET trajectory has been assigned to a unique hidden state, determining an HMM using a maximum-likelihood approach may over-fit the number of hidden states in an EFRET trajectory. To avoid these shortcomings of using maximum-likelihood-based approaches, a Bayesian inference-based approach has been developed that can be used to find the range of HMMs that are consistent with an EFRET trajectory, while simultaneously avoiding over-fitting (Bishop, 2006, Bronson et al., 2009, Bronson et al., 2010, Van De Meent et al., 2013, van de Meent et al., 2014). Both vbFRET and its successor, ebFRET, use this statistically rigorous, Bayesian inference-based approach to analyze EFRET trajectories with HMMs (Bronson et al., 2009, Bronson et al., 2010, Van De Meent et al., 2013, van de Meent et al., 2014). Regardless of the approach that is used, determining the HMMs corresponding to the hundreds to thousands of EFRET trajectories that comprise a single smFRET experiment allows one to gain enough statistical insight so as to infer the kinetic scheme and set of parameters governing the behavior of the population of individual molecules that have been observed in the experiment.
A relatively new, yet powerful, approach to analyzing the hundreds to thousands of EFRET trajectories collected in an smFRET experiment is to simultaneously analyze the entire population of EFRET trajectories corresponding to a single experimental dataset. Conventionally, software programs such as QuB (Qin et al., 2000), HaMMy (McKinney et al., 2006), and vbFRET (Bronson et al., 2009, Bronson et al., 2010) analyze each EFRET trajectory in a dataset separately, with each EFRET trajectory yielding its own HMM. A significant disadvantage of such an approach is that the results from the HMM(s) that are determined from the analysis of one EFRET trajectory in a dataset will not inform on the HMM(s) that will be consistent with another EFRET trajectory within the entire population of EFRET trajectories in the dataset. Nonetheless, the ultimate goal of these analyses is most often to generate a unified description of the entire population of individual molecules. Thus, the ideal approach would be one in which the entire population of EFRET trajectories in a dataset are simultaneously analyzed with a so-called ‘hierarchical’ HMM, which accounts for the different types of EFRET trajectories that are observed across the entire population of EFRET trajectories in a dataset. As such, this approach determines the consensus HMM(s) that are consistent with the dynamics of the entire population of individual molecules (Van De Meent et al., 2013, van de Meent et al., 2014). Such a hierarchical HMM approach enables one to quantitatively characterize the structural and/or dynamic heterogeneity that is frequently present across an entire population of molecules (e.g., the scenario in which an unlabeled ligand binds to and alters the dynamics of the molecules, thereby resulting in two sub-populations of molecules, and therefore EFRET trajectories, which differ in terms of their dynamics). Equally as important, a hierarchical HMM approach also provides the consensus kinetic scheme and set of parameters that governs the behavior of the entire population of molecules that is observed in the single experimental dataset (e.g., even in cases where a rarely sampled conformational state is not observed in every EFRET trajectory, the consensus kinetic scheme and parameter set that are obtained from a hierarchical HMM approach will include this conformational state). In a step that moves the field closer to such a hierarchical HMM approach, the ebFRET software package utilizes the same Bayesian inference-based approach that is used in vbFRET to find the HMMs that are consistent with each EFRET trajectory, but, in a second step, it then uses this information to find the consensus HMM(s) that are consistent with the entire population of EFRET trajectories in the dataset (Van De Meent et al., 2013, van de Meent et al., 2014). In the following sections, we describe how we have used ebFRET to determine the consensus HMMs and corresponding kinetic mechanism that best describes the entire population of EFRET trajectories obtained from smFRET experiments reporting on the nucleation of human RAD51 nucleoprotein filaments.
4.2. Using ebFRET to determine the set of Hidden Markov Models that are consistent with an entire population of EFRET trajectories
In preparation to run ebFRET, download the ebFRET program from http://ebfret.github.io onto your local computer, open MATLAB, select the Set Path button from the main interface, select the Add Folder button, and then select the ebFRET-gui/src folder that was downloaded onto your local computer; note that bolded text denotes MATLAB- and ebFRET-specific terminology. These steps will point MATLAB to the ebFRET code. Although it is not necessary, we suggest using a computer containing multiple processors and running ebFRET on several of these processors to speed up the calculation times; to do so, run the MATLAB command parpool(‘local’,num_proc), where the num_proc variable is the number of processors to be used. The ebFRET interface can then be launched by running the MATLAB command gui = ebFRET(). Donor and acceptor fluorescence trajectories can then be loaded into ebFRET by selecting the Load item from the File menu, and selecting the files containing the donor and acceptor fluorescence trajectories using the Raw donor-acceptor time series (.dat) file option. If desired, donor and acceptor fluorescence trajectories obtained from separate smFRET experiments can be analyzed together by loading the additional donor and acceptor fluorescence trajectory files, and choosing the option Keep in the subsequent dialog box. Although ebFRET loads donor and acceptor fluorescence trajectories, the program will first calculate the EFRET trajectories corresponding to the loaded donor and acceptor fluorescence trajectories and then analyze the subsequent EFRET trajectories. Because the EFRET values that would be calculated for data points corresponding to times when the donor fluorophore is photoblinked or photobleached are undefined (i.e., EFRET = 0/(0+0)), these data points must be removed prior to calculation of the EFRET trajectories. Although this can be accomplished using ebFRET by selecting the Remove Photo-bleaching item from the Analysis menu, in the case of our analysis of RAD51 nucleation dynamics, we used a custom MATLAB script (available upon request from the Spies lab) to trim the beginning of the EFRET trajectories to remove the wait time before binding of the first RAD51, and also to trim the end of the EFRET trajectories to include only those times before donor or acceptor fluorophore photobleaching.
After setting up ebFRET, HMMs can be determined by setting the Min and Max variables in the main ebFRET interface to the minimum and maximum number of states to be considered, respectively, and clicking the Run button in the same interface. An analysis in progress can be interrupted by selecting the Stop button, and the entire ebFRET session can be saved by selecting the Save item in the File menu. Saved sessions can be reloaded by selecting the Load item in the File menu and using the ebFRET saved session (.mat) file type. Running ebFRET entails a search for the parameters that yield the highest probability of the data being generated by a particular HMM. This probability is often referred to as the ‘evidence’ of the model. Notably, however, ebFRET actually uses a lower bound to the evidence, which is often just referred to as the ‘lower bound’. If the parameter search is run long enough, ebFRET is guaranteed to find the parameters that yield the highest lower bound. However, in practice, it is not usually possible to run the parameter search for as long as would be required to achieve this guaranteed outcome. Thus, in practice, the parameter search is typically stopped when the value of the lower bound converges and thus effectively stops increasing. ebFRET determines what this stopping point is by detecting the point in the parameter search at which the relative increase of the lower bound is less than the value that is specified by the Precision variable in main ebFRET interface; for a robust parameter search, a value for the precision of less than 10−6 is recommended, which is even less than the default value. Additionally, the parameter search process is repeated a number of times, specified by the Restarts variable in the main ebFRET interface. Each parameter search runs with different starting parameter values in order to increase the probability of rapidly finding the parameter values that will result in the highest lower bound; for a robust analysis, a value for the restarts of at least 10 is recommended, which is greater than the default value.
When ebFRET is finished running, the results can be obtained by selecting the Export item from the File menu and then selecting the Analysis Summary item. This will export a ‘comma separated value’, .csv, file to your local computer that can be opened using any spreadsheet program. This .csv file contains three sections of results entitled Lower_Bound, Statistics, and Parameters. The Lower_Bound section contains statistics describing the lower bounds on a per trajectory and per data point level. The Statistics section summarizes the data points assigned to each of the different states, including the fraction of data points assigned to each state (Occupancy), the EFRET values of the data points assigned to each state (Observation), and the number and probability of transitioning from each state to each of the other states (Transitions). It is worth noting that, in these calculations, ebFRET inherently accounts for the uncertainty of assigning a data point to each of the different states. The Parameters section provides the values of the variables that describe the consensus HMM(s) that is(are) consistent with the ensemble of EFRET trajectories that were analyzed (i.e., the so-called ‘hyper-parameters’), including the mean of the EFRET distribution corresponding to each state (Center), the width of the EFRET distribution corresponding to each state reported as an inverse variance (Precision), the number of time points spent in each state (Dwell_Time), and the probability of transitioning from each state to each of the other states (Transition_Matrix).
4.3. Selecting the HMM that best describes the entire population of EFRET trajectories and analyzing the corresponding kinetic mechanism
After an ebFRET run is complete, the user must choose between the resulting HMMs in order to determine the corresponding kinetic mechanism (i.e., the number of states and the rates of transitions between those states) that best describes the EFRET trajectories that were analyzed. Because ebFRET is a Bayesian method, the HMM that generates the highest lower bound will be the HMM, and thus kinetic mechanism, that best describes the EFRET trajectories. In order to compare the lower bounds of the various HMMs, we recommend using either the sum of the lower bound of all the trajectories (i.e., Total), or the mean and standard deviation (i.e., Mean and Std) of the lower bounds of all the trajectories as a metric for whether one of the HMMs yields a distinctly higher lower bound. Ideally, a plot of the lower bounds versus the increasing number of states in the HMMs will exhibit a peaked lower bound at some specific complexity (i.e., specific number of states). However, because the amount of information in a population of EFRET trajectories to be analyzed is sometimes not enough to make the distinction between the lower bounds obvious, we find that the lower bounds in such plots sometimes plateau rather than peak. This can make it difficult to select the kinetic mechanism that best describes the EFRET trajectories. In such a case, we advocate for parsimony, such that the HMM that has the lowest complexity (i.e., the lowest number of states) within the plateaued region of the plot is selected as the HMM that best describes the EFRET trajectories; this will avoid overfitting and over-interpretation of the data. In the analysis of RAD51 nucleation dynamics, the lower bounds plateaued starting with the HMM that was composed of four states, so we therefore used this four-state HMM in the subsequent analysis. With four distinct conformational states of the nucleating filament, this suggests that the nucleating species of RAD51 is a dimer (Subramanyam et al., 2016).
Once an HMM has been selected, information about the kinetic mechanism of interest can be obtained by calculating rate constants for transitions from each state to the other states. For the rate constant for the transition from generic states i to j, denoted , this calculation can be performed with the formula , where is the transition probability for the transition from state i to j, which is an element of the transition matrix located in Transition_Matrix entry of the Analysis Summary, and is the time resolution of the dataset in units of seconds (see (Kinz-Thompson et al., 2016) for additional details). Further insights relating to the kinetic mechanism of interest can be obtained by generating a transition density plot (TDP). A TDP is a contour surface plot that reports the relative frequencies of transitions between different states by plotting the EFRET at each time point on the x-axis versus the EFRET at each subsequent time point on the y-axis (Figure 7B). TDPs allow us to better understand the underlying kinetic mechanism by providing a visual representation of which states transition into which other states and the relative frequencies with which these transitions occur. Notably, in smFRET experiments in which the molecules of interest do not undergo frequent conformational changes, the molecules predominantly remain in the same EFRET state during the majority of the EFRET trajectories. Consequently, the relative frequencies of the consecutive time points in which no transitions occur (i.e., the on-diagonal peaks in the TDP) can dominate the TDP and obscure the relative frequencies of the consecutive time points in which transitions do occur (i.e., the off-diagonal peaks in the TDP). However, because the HMM that has been selected as best describing the EFRET trajectories contains all of the information that is required to determine when transitions do occur, we can use the selected HMM to generate a TDP that plots only the EFRET values of those steps in which consecutive time points undergo a transition. This is done by generating a TDP using the ‘idealized’ EFRET trajectories, or ‘Viterbi paths’, which are generated by turning each EFRET value in an EFRET trajectory into the average EFRET value of the corresponding hidden state determined by the HMM (black line in Figure 7A). The idealized EFRET trajectories can be exported as a .dat file from ebFRET by selecting the Export item from the File menu, then selecting the Traces item, defining the filename, toggling off all of the channels except the Viterbi Mean channel, and then setting the Number of States variable such that it corresponds to the HMM that has been selected. Once the idealized EFRET trajectories have been generated, a TDP can be generated by downloading a custom MATLAB script called plot_TDP.m from http://ebfret.github.io onto your local computer, and running the script using the MATLAB command plot_TDP(‘filename.dat’), where filename.dat corresponds to the .dat file containing the idealized EFRET trajectories; further instructions for running this script can be found at http://ebfret.github.io. As was seen in the TDP of the EFRET trajectories of RAD51 nucleation dynamics using an HMM containing four states, high transition frequencies were only observed between states corresponding to adjacent EFRET values (Figure. 7B). Also, we note that this behavior was independent of the number of states that were used to determine the HMM (e.g., eight states), and no substantial differences in the TDP are observed for HMMs with more than four states. Therefore, TDPs are a robust analysis method that are relatively independent of the results of an HMM.
5. Conclusions
Nucleation of the RAD51 filament on ssDNA is a critical step in the formation of the active species in homologous recombination and even slight deviations towards either a more or a less stable RAD51 filament, as well as slight differences in the filament nucleation and growth kinetics may cause aberrant recombination or an unstable RAD51 nucleoprotein filament. In this chapter we discussed the analysis of human RAD51 nucleoprotein filament formation using a FRET-based assay enabled by TIRF microscopy. This assay takes advantage of the recombinase-stretched ssDNA within the active form of the RAD51 filament, which allows the experimenter to monitor a dynamic, stepwise formation of a stable RAD51-ssDNA complex by following the ssDNA extension. The experiment is carried out by first imaging a protein-free, surface-tethered DNA decorated with the Cy3 (FRET donor) and Cy5 (FRET acceptor) dyes separated by 21 nucleotides of ssDNA and then flowing in RAD51. FRET-based studies require positioning of the donor and acceptor fluorophores within 25 nucleotides from one another within the substrate that typically does not exceed 100 nucleotides in length. Such substrates allow formation of relatively short nucleoprotein filaments (up to 30 – 35 monomers of RAD51), which coincidently corresponds to an average segment size within discontinuous RAD51 filaments formed under conditions permitting ATP hydrolysis (Holthausen et al., 2011). A flow experiment, like the one described in this chapter, follows the very first steps in the RAD51 nucleoprotein filament assembly, namely, a formation of a stable nucleus from which the filament can then grow. In the case of human RAD51 under conditions permitting ATP hydrolysis, filament nucleation occurs through a dynamic association and dissociation of RAD51 dimers (Subramanyam et al., 2016). Similar approaches have been applied to other RecA-family recombinases (including yeast Rad51 protein (Qiu et al., 2013), and RecA (Joo et al., 2006)), albeit with different kinetics and via different oligomeric species.
The utility of the assay described in this chapter is that it can be readily extended to other experimental conditions. Nucleoprotein filament formation can be monitored, for example, in the presence of Ca2+, where RAD51 can bind ATP and forms an extended active nucleoprotein filament, but cannot hydrolyze ATP (Bugreev and Mazin, 2004) and where it forms longer and less dynamic filaments (Holthausen et al., 2011), or in the presence of different small-molecule modulators of the RAD51-ssDNA interaction (see (Hengel et al., 2017) for review). The partial duplex DNA substrate described here can be substituted with the DNA substrates resembling various DNA repair intermediates and stalled replications forks that are expected to attract RAD51 in the cell. Analysis of RAD51 dynamics on different DNA substrates can help to parse out the differences and similarities between the RAD51 nucleoprotein filaments acting in homology-directed DNA repair and in stabilization of DNA replication forks. Additionally, various post-translationally modified forms of RAD51 can be examined. We have shown recently that RAD51 phosphorylated at Tyr54 has a low affinity for ssDNA, but forms the nucleoprotein filament with higher cooperativity (Subramanyam et al., 2016). RAD51, however, is subject to numerous post-translational modifications, including tyrosine phosphorylation by the c-Abl kinase at Y54 and Y315 residues, tyrosine phosphorylation by the Arg kinase, threonine phosphorylation on T309 by the CHK1 kinase, SUMOylation by UBL1 (Popova et al., 2011), and ubiquitylation mediated by the FBH1 helicase (Chu et al., 2015). How these modifications affect the dynamics of nucleoprotein filament assembly and stability remains to be determined. More importantly, the RAD51-DNA interactions are controlled by the recombination mediator BRCA2, by RAD51 paralogs and Shu complex (Zelensky et al., 2014, Prakash et al., 2015, Godin et al., 2016), by antirecombinogenic DNA helicases (Daley et al., 2014), and by the heteroduplex rejection machinery (Spies and Fishel, 2015). These modulators of RAD51 activity or their individual RAD51-interacting domains can be incorporated into the FRET-based, single-molecule experiments described here. Given the flexibility with which it can be adapted to pursue all of these experiment avenues, the assay described in this chapter should become a powerful tool in the field’s effort to build a comprehensive mechanistic description of RDA51 nucleoprotein filament formation, the most critical step in homologous recombination and an important step in the maintenance of robust and accurate DNA replication. With the increasing availability of single-molecule TIRFM instrumentation and the robust data analysis routine afforded by ebFRET (van de Meent et al., 2014), we expect this FRET-based analysis of the RAD51 nucleoprotein filament formation and stability to be extended to many conditions, RAD51 mutants, and RAD51 nucleoprotein filament modulators.
Acknowledgments
The authors’ research is supported by National Institutes of Health (NIH) Grant GM108617 and by University of Iowa Holden Comprehensive Cancer Center Collaborative Pilot Grant NIH P30 CA086862 to M.S. and NIH Grants GM084288 and GM 119386 to R.L.G. C.D.K. was supported by the Department of Energy Office of Science Graduate Fellowship Program (DOE SCGF), made possible in part by the American Recovery and Reinvestment Act of 2009, administered by ORISE-ORAU under contract number DE-AC05-06OR23100, as well as by Columbia University’s NIH Training Program in Molecular Biophysics (T32-GM008281). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank Colleen Caldwell and Fletcher Bain for critical reading of the manuscript and for valuable discussions.
References
- AMEZIANE N, MAY P, HAITJEMA A, VAN DE, VRUGT HJ, VAN ROSSUM-FIKKERT SE, RISTIC D, WILLIAMS GJ, BALK J, ROCKX D, LI H, ROOIMANS MA, OOSTRA AB, VELLEUER E, DIETRICH R, BLEIJERVELD OB, MAARTEN ALTELAAR AF, MEIJERS-HEIJBOER H, JOENJE H, GLUSMAN G, ROACH J, HOOD L, GALAS D, WYMAN C, BALLING R, DEN DUNNEN J, DE WINTER JP, KANAAR R, GELINAS R, DORSMAN JC. A novel Fanconi anaemia subtype associated with a dominant-negative mutation. RAD51. 2015;6:8829. doi: 10.1038/ncomms9829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AMITANI I, LIU B, DOMBROWSKI CC, BASKIN RJ, KOWALCZYKOWSKI SC. Watching Individual Proteins Acting on Single Molecules of DNA. In: NILS GW, editor. Methods in Enzymology. Academic Press; 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARATA H, DUPONT A, MINÉ-HATTAB J, DISSEAU L, RENODON-CORNIÈRE A, TAKAHASHI M, VIOVY JL, CAPPELLO G. Direct observation of twisting steps during Rad51 polymerization on DNA. Proceedings of the National Academy of Sciences. 2009;106:19239–19244. doi: 10.1073/pnas.0902234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AXELROD D. Methods in Cell Biology. Academic Press; 2008. Chapter 7 Total Internal Reflection Fluorescence Microscopy. [DOI] [PubMed] [Google Scholar]
- BAIN FE, WU CG, SPIES M. Single-molecule sorting of DNA helicases. Methods. 2016;108:14–23. doi: 10.1016/j.ymeth.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BELL JC, KOWALCZYKOWSKI SC. Mechanics and Single-Molecule Interrogation of DNA Recombination. Annu Rev Biochem. 2016;85:193–226. doi: 10.1146/annurev-biochem-060614-034352. [DOI] [PubMed] [Google Scholar]
- BENSON FE, STASIAK A, WEST SC. Purification and characterization of the human Rad51 protein, an analogue of E. coli RecA. EMBO Journal. 1994;13:5764–71. doi: 10.1002/j.1460-2075.1994.tb06914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BISHOP CM. Pattern Recognition and Machine Learning 2006 [Google Scholar]
- BRONSON JE, FEI J, HOFMAN JM, GONZALEZ RL, WIGGINS CH. Learning Rates and States from Biophysical Time Series: A Bayesian Approach to Model Selection and Single-Molecule FRET Data. Biophysical Journal. 2009;97:3196–3205. doi: 10.1016/j.bpj.2009.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRONSON JE, HOFMAN JM, FEI J, GONZALEZ RL, WIGGINS CH. Graphical models for inferring single molecule dynamics. BMC Bioinformatics. 2010;11:S2. doi: 10.1186/1471-2105-11-S8-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUDKE B, LV W, KOZIKOWSKI AP, CONNELL PP. Recent Developments Using Small Molecules to Target RAD51: How to Best Modulate RAD51 for Anticancer Therapy? ChemMedChem. 2016;11:2468–2473. doi: 10.1002/cmdc.201600426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUGREEV DV, MAZIN AV. Ca2+ activates human homologous recombination protein Rad51 by modulating its ATPase activity. Proc Natl Acad Sci U S A. 2004;101:9988–93. doi: 10.1073/pnas.0402105101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CANDELLI A, HOLTHAUSEN JT, DEPKEN M, BROUWER I, FRANKER MA, MARCHETTI M, HELLER I, BERNARD S, GARCIN EB, MODESTI M, WYMAN C, WUITE GJ, PETERMAN EJ. Visualization and quantification of nascent RAD51 filament formation at single-monomer resolution. Proc Natl Acad Sci U S A. 2014;111:15090–5. doi: 10.1073/pnas.1307824111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CANDELLI A, MODESTI M, PETERMAN EJ, WUITE GJ. Single-molecule views on homologous recombination. Q Rev Biophys. 2013;46:323–48. doi: 10.1017/S0033583513000073. [DOI] [PubMed] [Google Scholar]
- CHABBERT M, CAZENAVE C, HELENE C. Kinetic studies of recA protein binding to a fluorescent single-stranded polynucleotide. Biochemistry. 1987;26:2218–25. doi: 10.1021/bi00382a022. [DOI] [PubMed] [Google Scholar]
- CHEN Z, YANG H, PAVLETICH NP. Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature. 2008;453:489–4. doi: 10.1038/nature06971. [DOI] [PubMed] [Google Scholar]
- CHU WK, PAYNE MJ, BELI P, HANADA K, CHOUDHARY C, HICKSON ID. FBH1 influences DNA replication fork stability and homologous recombination through ubiquitylation of RAD51. Nat Commun. 2015;6 doi: 10.1038/ncomms6931. [DOI] [PubMed] [Google Scholar]
- DALEY JM, GAINES WA, KWON Y, SUNG P. Regulation of DNA Pairing in Homologous Recombination. Cold Spring Harbor Perspectives in Biology. :2014. doi: 10.1101/cshperspect.a017954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FORGET AL, KOWALCZYKOWSKI SC. Single-molecule imaging brings Rad51 nucleoprotein filaments into focus. Trends Cell Biol. 2010;20:269–76. doi: 10.1016/j.tcb.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FORGET AL, KOWALCZYKOWSKI SC. Single-molecule imaging of DNA pairing by RecA reveals a three-dimensional homology search. Nature. 2012;482:423–7. doi: 10.1038/nature10782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GALKIN VE, ESASHI F, YU X, YANG S, WEST SC, EGELMAN EH. BRCA2 BRC motifs bind RAD51-DNA filaments. Proc Natl Acad Sci U S A. 2005;102:8537–42. doi: 10.1073/pnas.0407266102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GODIN SK, SULLIVAN MR, BERNSTEIN KA. Novel insights into RAD51 activity and regulation during homologous recombination and DNA replication. Biochem Cell Biol. 2016;94:407–418. doi: 10.1139/bcb-2016-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HA T. Single-Molecule Fluorescence Resonance Energy Transfer. Methods. 2001;25:78–86. doi: 10.1006/meth.2001.1217. [DOI] [PubMed] [Google Scholar]
- HA T, TINNEFELD P. Photophysics of Fluorescent Probes for Single-Molecule Biophysics and Super-Resolution Imaging. Annual Review of Physical Chemistry. 2012;63:595–617. doi: 10.1146/annurev-physchem-032210-103340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HENGEL SR, SPIES MA, SPIES M. Small-Molecule Inhibitors Targeting DNA Repair and DNA Repair Deficiency in Research and Cancer Therapy. Cell Chem Biol. 2017;24:1101–1119. doi: 10.1016/j.chembiol.2017.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLTHAUSEN JT, WYMAN C, KANAAR R. Regulation of DNA strand exchange in homologous recombination. DNA Repair. 2011;9:1264–1272. doi: 10.1016/j.dnarep.2010.09.014. [DOI] [PubMed] [Google Scholar]
- JOO C, HA T. Preparing Sample Chambers for Single-Molecule FRET. Cold Spring Harbor Protocols. 2012a;2012 doi: 10.1101/pdb.prot071530. pdb.prot071530. [DOI] [PubMed] [Google Scholar]
- JOO C, HA T. Single-Molecule FRET with Total Internal Reflection Microscopy. Cold Spring Harbor Protocols. 2012b;2012 doi: 10.1101/pdb.top072058. pdb.top072058. [DOI] [PubMed] [Google Scholar]
- JOO C, MCKINNEY SA, NAKAMURA M, RASNIK I, MYONG S, HA T. Real-Time Observation of RecA Filament Dynamics with Single Monomer Resolution. Cell. 2006;126:515–527. doi: 10.1016/j.cell.2006.06.042. [DOI] [PubMed] [Google Scholar]
- KINZ-THOMPSON CD, BAILEY NA, GONZALEZ RL. Precisely and Accurately Inferring Single-Molecule Rate Constants. Methods in enzymology. 2016;581:187–225. doi: 10.1016/bs.mie.2016.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLEIN HL. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair (Amst) 2008;7:686–93. doi: 10.1016/j.dnarep.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOLINJIVADI AM, SANNINO V, DE ANTONI A, TECHER H, BALDI G, COSTANZO V. Moonlighting at replication forks - a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. 2017;591:1083–1100. doi: 10.1002/1873-3468.12556. [DOI] [PubMed] [Google Scholar]
- LARSON J, KIRK M, DRIER EA, O’BRIEN W, MACKAY JF, FRIEDMAN LJ, HOSKINS AA. Design and construction of a multiwavelength, micromirror total internal reflectance fluorescence microscope. Nature Protocols. 2014;9:2317. doi: 10.1038/nprot.2014.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI X, HEYER WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18:99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCKINNEY SA, JOO C, HA T. Analysis of Single-Molecule FRET Trajectories Using Hidden Markov Modeling. Biophysical Journal. 2006;91:1941–1951. doi: 10.1529/biophysj.106.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MICHL J, ZIMMER J, TARSOUNAS M. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. The EMBO Journal. 2016;35:909–923. doi: 10.15252/embj.201693860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOYNAHAN ME, JASIN M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010;11:196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POPOVA M, HENRY S, FLEURY F. Posttranslational Modifications of Rad51 Protein and Its Direct Partners: Role and Effect on Homologous Recombination – Mediated DNA Repair. In: KRUMAN I, editor. DNA Repair. InTech; Rijeka, Croatia: 2011. [Google Scholar]
- PRAKASH R, ZHANG Y, FENG W, JASIN M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harbor Perspectives in Biology. 2015;7 doi: 10.1101/cshperspect.a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QI Z, REDDING S, LEE JAY, GIBB B, KWON Y, NIU H, GAINES WILLIAMA, SUNG P, GREENE ERICC. DNA Sequence Alignment by Microhomology Sampling during Homologous Recombination. Cell. 2015;160:856–69. doi: 10.1016/j.cell.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QIN F, AUERBACH A, SACHS F. A direct optimization approach to hidden Markov modeling for single channel kinetics. Biophysical journal. 2000;79:1915–1927. doi: 10.1016/S0006-3495(00)76441-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QIU Y, ANTONY E, DOGANAY S, RAN KOH H, LOHMAN TM, MYONG S. Srs2 prevents Rad51 filament formation by repetitive motion on DNA. Nat Commun. 2013;4:2281. doi: 10.1038/ncomms3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RISTIC D, MODESTI M, VAN DER HEIJDEN T, VAN NOORT J, DEKKER C, KANAAR R, WYMAN C. Human Rad51 filaments on double- and single-stranded DNA: correlating regular and irregular forms with recombination function. Nucleic Acids Res. 2005;33:3292–302. doi: 10.1093/nar/gki640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROY R, HOHNG S, HA T. A practical guide to single-molecule FRET. Nat Meth. 2008;5:507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHLACHER K, CHRIST N, SIAUD N, EGASHIRA A, WU H, JASIN M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–42. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHLACHER K, WU H, JASIN M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22:106–16. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHI X, LIM J, HA T. Acidification of the Oxygen Scavenging System in Single-Molecule Fluorescence Studies: In Situ Sensing with a Ratiometric Dual-Emission Probe. Analytical Chemistry. 2010;82:6132–6138. doi: 10.1021/ac1008749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHORT JM, LIU Y, CHEN S, SONI N, MADHUSUDHAN MS, SHIVJI MKK, VENKITARAMAN AR. High-resolution structure of the presynaptic RAD51 filament on single-stranded DNA by electron cryo-microscopy. Nucleic Acids Research. 2016;44:9017–9030. doi: 10.1093/nar/gkw783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SPIES M, FISHEL R. Mismatch Repair during Homologous and Homeologous Recombination. Cold Spring Harbor Perspectives in Biology. 2015;7 doi: 10.1101/cshperspect.a022657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUBRAMANYAM S, ISMAIL M, BHATTACHARYA I, SPIES M. Tyrosine phosphorylation stimulates activity of human RAD51 recombinase through altered nucleoprotein filament dynamics. Proc Natl Acad Sci U S A. 2016;113:E6045–E6054. doi: 10.1073/pnas.1604807113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUBRAMANYAM S, JONES WT, SPIES M, SPIES MA. Contributions of the RAD51 N-terminal domain to BRCA2-RAD51 interaction. Nucleic Acids Res. 2013;41:9020–32. doi: 10.1093/nar/gkt691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOMBLINE G, SHIM KS, FISHEL R. Biochemical characterization of the human RAD51 protein. II. Adenosine nucleotide binding and competition. J Biol Chem. 2002;277:14426–33. doi: 10.1074/jbc.M109916200. [DOI] [PubMed] [Google Scholar]
- VAN DE MEENT JW, BRONSON JE, WOOD F, GONZALEZ RL, JR, WIGGINS CH. Hierarchically-coupled hidden Markov models for learning kinetic rates from single-molecule data. Proceedings of the 30th International Conference on Machine Learning. 2013 [PMC free article] [PubMed] [Google Scholar]
- VAN DER HEIJDEN T, SEIDEL R, MODESTI M, KANAAR R, WYMAN C, DEKKER C. Real-time assembly and disassembly of human RAD51 filaments on individual DNA molecules. Nucleic Acids Res. 2007;35:5646–57. doi: 10.1093/nar/gkm629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de MEENT JW, BRONSON JONATHANE, WIGGINS CHRISH, GONZALEZ JR, RUBEN L. Empirical Bayes Methods Enable Advanced Population-Level Analyses of Single-Molecule FRET Experiments. Biophysical Journal. 2014;106:1327–1337. doi: 10.1016/j.bpj.2013.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XU J, ZHAO L, XU Y, ZHAO W, SUNG P, WANG HW. Cryo-EM structures of human RAD51 recombinase filaments during catalysis of DNA-strand exchange. Nat Struct Mol Biol. 2016;24:40. doi: 10.1038/nsmb.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YU X, JACOBS SA, WEST SC, OGAWA T, EGELMAN EH. Domain structure and dynamics in the helical filaments formed by RecA and Rad51 on DNA. Proceedings of the National Academy of Sciences. 2001;98:8419–8424. doi: 10.1073/pnas.111005398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZELENSKY A, KANAAR R, WYMAN C. Mediators of Homologous DNA Pairing. Cold Spring Harbor Perspectives in Biology. 2014;6 doi: 10.1101/cshperspect.a016451. [DOI] [PMC free article] [PubMed] [Google Scholar]