

Abstract
Alzheimer's disease (AD) is one of the most common neurodegenerative disorders and causes cognitive impairment and memory deficits of the patients. The mechanism of AD is not well known, due to lack of human brain models. Recently, mini-brain tissues called organoids have been derived from human induced pluripotent stem cells (hiPSCs) for modeling human brain development and neurological diseases. Thus, the objective of this research is to model and characterize neural degeneration microenvironment using three-dimensional (3D) forebrain cortical organoids derived from hiPSCs and study the response to the drug treatment. It is hypothesized that the 3D forebrain organoids derived from hiPSCs with AD-associated genetic background may partially recapitulate the extracellular microenvironment in neural degeneration. To test this hypothesis, AD-patient derived hiPSCs with presenilin-1 mutation were used for cortical organoid generation. AD-related inflammatory responses, matrix remodeling and the responses to DAPT, heparin (completes with heparan sulfate proteoglycans [HSPGs] to bind Aβ42), and heparinase (digests HSPGs) treatments were investigated. The results indicate that the cortical organoids derived from AD-associated hiPSCs exhibit a high level of Aβ42 comparing with healthy control. In addition, the AD-derived organoids result in an elevated gene expression of proinflammatory cytokines interleukin-6 and tumor necrosis factor-α, upregulate syndecan-3, and alter matrix remodeling protein expression. Our study demonstrates the capacity of hiPSC-derived organoids for modeling the changes of extracellular microenvironment and provides a potential approach for AD-related drug screening.
Keywords: : human induced pluripotent stem cells, three-dimensional, organoids, neural differentiation, degeneration
Introduction
Restriction of the access to human brain tissues requires alternative approach to study human brain. Human pluripotent stem cells (hPSCs), including human embryonic stem cells (hESCs) and human induced PSCs (hiPSCs), emerge as a promising source to construct human brain tissue in vitro to study neurological diseases.1–3 For example, the forebrain-like cortical tissues, spheroids, and organoids derived from hPSCs have been investigated recently in three-dimensional (3D) cultures.3–8 Compared to 2D cultures, 3D neural cultures promote neuronal maturation and regional specification, better mimicking human brain structures.9–12 In particular, several studies have utilized 3D neural cultures to investigate Alzheimer's disease (AD) and other neurological disorders.13–16 For example, a human neural stem cell-derived 3D culture was evaluated as a potential in vitro AD model.9 Upon overexpression of familial AD mutations in amyloid precursor protein and presenilin-1 (PS1), this 3D culture showed amyloid β (Aβ) aggregation and recapitulated several Aβ and tau-induced pathology. Moreover, 3D neurospheres were formed in concave microwells using prenatal rat cortical neurons and exhibited Aβ-induced toxicity.17 Using hiPSC-derived neural progenitors, the 3D neural cultures in hydrogels showed higher levels of co-localization of debrin and F-actin than 2D cultures, better recapitulating AD pathology.10 However, this 3D model lacks complex human cortical structures and cannot show differential cellular responses of various neuronal subtypes.
To understand the influence of genetic background of patients in AD progression, AD patient-specific hiPSCs have been recently derived to model neural degeneration16,18–20 In one such study, hiPSCs were derived from two familial and two sporadic AD patients.19 Neurons from AD patients (two familial and one sporadic) had the elevated levels of Aβ40 and phosphor-tau. In another study, extracellular Aβ42 increased along with the decrease in intracellular Aβ42 in familial AD-derived cells compared with the control neural cells.16 While these studies demonstrate the potential of AD patient-specific hiPSCs in understanding neural degeneration mechanism in 2D cultures, the investigations of 3D cortical organoids from AD patient-specific hiPSCs to recapitulate the specific brain regions has been under explored.
While previous investigations demonstrate the potential of hiPSCs in modeling AD,16,19 the 2D cultures used in these studies cannot fully recapitulate AD-associated neuropathology (e.g., demonstration of Aβ aggregation and deposition). On the other hand, modeling brain tissues with cortical layers was performed using hiPSCs2,3,5 and the 3D structures that contain polarized glia progenitors and layer-specific cortical neurons were generated.2,3 While these studies demonstrate the feasibility of using hiPSCs to create 3D cortical tissues, the use of these 3D models to probe AD-associated pathology has not been well demonstrated. This study fills this knowledge gap by using 3D cortical organoids derived from hiPSCs to recapitulate AD-associated changes in extracellular microenvironment.
The objective of this study is to model and characterize neural degeneration microenvironment using 3D cortical organoids derived from patient-specific hiPSCs with PS1 M146 V mutations.21 The organoids were characterized for cortical layer phenotypes, neuronal markers, cytotoxicity, and neural degeneration-associated phenotype (Aβ42 secretion, tau etc.). Gene expression of interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α), matrix protein syndecan-3, and matrix remodeling proteins were also characterized. In addition, the cellular responses to a γ-secretase inhibitor DAPT, heparin (to complete with heparan sulfate proteoglycan [HSPG]-Aβ42 binding),22,23 and heparinase III (to digest HSPG)22 were investigated. Different from the reported studies,11,14 this study investigated the changes of extracellular matrix (ECM) and remodeling in addition to cellular responses to the molecules targeted on ECMs. This study indicates that the 3D cortical organoids derived from hiPSCs of AD patients retain their genetic backgrounds and have applications as patient-specific in vitro models for studying neural degeneration and identifying therapeutic target(s).
Materials and Methods
Undifferentiated hiPSC culture
Human iPSK3 cells were derived from human foreskin fibroblasts transfected with plasmid DNA encoding reprogramming factors OCT4, NANOG, SOX2, and LIN28 (kindly provided by Dr. Stephen Duncan, Medical College of Wisconsin, and Dr. David Gilbert, Department of Biological Sciences of Florida State University).24,25 Human iPSK3 cells were maintained in mTeSR serum-free medium (StemCell Technologies, Inc., Vancouver, Canada) on six-well plates coated with growth factor reduced Geltrex (Life Technologies, Carlsbad, CA). The cells were passaged by Accutase dissociation every 5–6 days and seeded at 1 × 106 cells per well of six-well plates in the presence of 10 μM Y27632 (Sigma) for the first 24 h.26,27
Patient-specific hiPSC with PS1 mutation, the SY-UBH cell line, was derived from the fibroblasts of an early-onset AD individual with PS1 M146 V mutation (kindly provided by Dr. T.K. and Dr. G.B., at Alzheimer's Disease Research Center, Mayo Clinic, Jacksonville, FL). The SY-UBH cells were maintained in mTeSR serum-free medium on Geltrex-coated surface. The cells were passaged by dispase and mechanical scraping every 7 days at 1:3 split ratio in the presence of Y27632 (10 μM) for the first 24 h. Adaptation of this culture to Accutase passaging (same as IPSK3 cells) was attempted. But this culture did not replate well with Accutase passaging. For experimental set up, additional wells were harvested and dissociated by 0.05% Trypsin/ethylenediaminetetraacetic acid for cell counting with hemacytometer. To compare the passaging of different cell lines, SY-UBH cells were also passaged by Accutase and seeded at 3.0–3.5 × 105 cells per well of 24-well plates in 1 mL of mTeSR medium.
Neural differentiation and cortical organoid formation from hiPSCs
Briefly, human iPSK3 cells or SY-UBH cells were seeded into Ultra-Low Attachment (ULA) 24-well plates (Corning, Inc., Corning, NY) at 3.0–3.5 × 105 cells per well in 1 mL of mTeSR medium and grown for 2 days. Y27632 (10 μM) was added during the seeding and removed after 24 h. Then, the culture was switched to neural differentiation medium composed of Dulbecco's modified Eagle's medium/nutrient mixture F-12 (DMEM/F12) plus 2% B27 serum-free supplement (Life Technologies).28–30 At day 1 in neural medium, the cells were treated with dual SMAD signaling inhibitors: 10 μM SB431542 (Sigma) and 100 nM LDN193189 (Sigma). After 7 days, the cells were incubated with cyclopamine (1 μM) and fibroblast growth factor (FGF)-2 (10 ng/mL; Life Technologies) for another 8 days. The suspension cultures were maintained in FGF-2 until day 22 and matured for up to 71 days in FGF-2 and 5 ng/mL brain-derived neurotrophic factor (BDNF) (R&D Systems). Supernatants were collected for quantitative Aβ42 ELISA and lactate dehydrogenase (LDH) activity assay. For immunocytochemistry characterizations, the aggregates were replated onto Geltrex-coated surface for 5–7 days. To compare aggregate formation, both iPSK3 and SY-UBH cells were seeded in ULA 96-well plates at 1.0 × 104 cells per well in 200 μL of mTeSR medium for 2 days and then were induced for neural differentiation. The aggregate size of both cell lines was evaluated at different days.
Aggregate size characterization
The images of aggregates (both iPSK3 cells and SY-UBH cells) were captured over the culture time by a phase contrast microscopy. The captured images were converted to binary images using ImageJ software (http://rsb.info.nih.gov/ij) and analyzed with the “particle analysis tool.” Through particle analysis in ImageJ software, the Feret's diameter of each aggregate in the images can be calculated and the average of aggregate diameter was presented.31,32
Biochemical assays: Live/Dead, MTT, and LDH assay
Live/Dead® staining kit (Molecular Probes) was used to assess cell viability. Immediately after harvesting, the cells were incubated in DMEM/F12 containing 1 μM calcein AM and 2 μM ethidium homodimer I for 30 min. The samples were then washed and imaged under a fluorescent microscope (Olympus IX70, Melville, NY). Using ImageJ software, the staining intensity (red for dead cells and green for live cells) was measured and each value was subtracted from the background intensity. The viability was calculated as the percentage of green intensity over total intensity. For MTT assay, the spheroids at day 9 were incubated with 5 mg/mL 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma) solution. The absorbance of the samples was measured at 500 nm using a microplate reader (Bio-Rad, Richmond, CA).
The cytotoxicity was assessed using LDH activity assay kit (Sigma; MAK066). Briefly, a total volume of 100 μL of spent medium and LDH reaction mixture was mixed well and the initial absorbance at 450 nm was measured using a microplate reader (Bio-Rad iMark™). The mixture was incubated at 37°C and taken measurement every 5 min. The LDH activity was calculated through the subtraction of final and initial measurements in comparison to the standard curve.
ELISA of Aβ42
The secreted Aβ42 was quantified through Human Aβ42 Quantikine® ELISA kit (R&D; DAB142) according to the manufacturer's instructions. Briefly, 100 μL per well of samples was added to the provided microplate and incubated for 2 h at 4°C. The samples were removed and the wells were washed after incubation. Then, 200 μL of human Aβ42 conjugate buffer was added to each well and incubated for another 2 h at 4°C. After the washing, 200 μL of substrate solution was added to each well and incubated for 30 min at room temperature. And, 50 μL of stop solution was then added to each well. The absorbance was measured at 450 nm using a microplate reader with wavelength correction at 540 or 570 nm.
Immunocytochemistry
Briefly, the samples were fixed with 4% paraformaldehyde (PFA) and permeabilized with 0.2–0.5% Triton X-100 for intracellular markers. The samples were then blocked and incubated with various mouse or rabbit primary antibodies (Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/tea). After washing, the cells were incubated with the corresponding secondary antibody: Alexa Fluor® 488 goat anti-Mouse IgG or Alexa Fluor® 594 goat anti-Rabbit IgG (Life Technologies). The samples were stained with Hoechst 33342 and visualized using a fluorescent microscope (Olympus IX70) or a confocal microscope (Zeiss LSM 880). The proportion of positive cells was calculated based on the area of marker of interest normalized to the nuclei using ImageJ analysis, indicating the relative expression among different conditions.
Flow cytometry
To quantify the levels of various marker expression, the cells were harvested by trypsinization and analyzed by flow cytometry. Briefly, 1 × 106 cells per sample were fixed with 4% PFA and washed with staining buffer (2% fetal bovine serum in phosphate-buffered saline). The cells were permeabilized with 100% cold methanol for intracellular markers, blocked, and then incubated with primary antibodies against Aβ42, total tau, or SATB2 followed by the corresponding secondary antibody (Supplementary Table S1). The cells were acquired with BD FACSCanto™ II flow cytometer (Becton Dickinson) and analyzed against isotype controls using FlowJo software.
Reverse transcription–polymerase chain reaction
Total RNA was isolated from neural cell samples using the RNeasy Mini kit (Qiagen, Valencia, CA) according to the manufacturer's protocol followed by the treatment of DNA-Free RNA kit (Zymo, Irvine, CA). Reverse transcription was carried out using 2 μg of total RNA, anchored oligo-dT primers (Operon, Huntsville, AL), and Superscript III (Invitrogen, Carlsbad, CA) (according to the protocol of the manufacturer). Primers specific for target genes were designed using the software Oligo Explorer 1.2 (Genelink, Hawthorne, NY; Supplementary Table S2). The gene β-actin was used as an endogenous control for normalization of expression levels. Real-time reverse transcription–polymerase chain reactions (RT-PCRs) were performed on an ABI7500 instrument (Applied Biosystems, Foster City, CA), using SYBR1 Green PCR Master Mix (Applied Biosystems). The amplification reactions were performed as follows: 2 min at 50°C, 10 min at 95°C, and 40 cycles of 95°C for 15 s and 55°C for 30 s, and 68°C for 30 s. Fold variation in gene expression was quantified by means of the comparative Ct method: 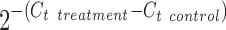 , which is based on the comparison of expression of the target gene (normalized to the endogenous control β-actin) between the test samples and the day 17 iPSK3 cell sample.
, which is based on the comparison of expression of the target gene (normalized to the endogenous control β-actin) between the test samples and the day 17 iPSK3 cell sample.
Treatment with DAPT, heparin, and heparinase
The day 35 organoids from iPSK3 and SY-UBH groups were seeded into ULA 96-well plates and one organoid with comparable size was seeded in each well. The cultures were treated with different molecules: (1) 5 μM DAPT (a γ-secretase inhibitor) (Sigma; D5942); (2) 100 U/mL heparin (Sigma; H3149), which binds HSPGs to reduce Aβ42 uptake; or (3) 0.05 U/mL heparinase III (Sigma; H8891), which degrades HSPGs, for 72 h, respectively. The culture without treatment was used as the control. The treated samples were evaluated by immunocytochemistry (Aβ42, glutamate, GABA, TBR1, and BRN2) and Live/Dead assay. The culture supernatants were collected for LDH activity assay.
Statistical analysis
To assess the statistical significance of the experimental results, Student's t-test was performed on the measurements from three replicates. The results were expressed as mean ± standard deviation. A p-value <0.05 was considered statistically significant.
Results
Generation and characterization of cortical organoids
The patient-derived hiPSC line with PS1 mutation SY-UBH was first characterized for pluripotency, which exhibited typical undifferentiated PSC colony morphology and the high expression of pluripotent markers Oct-4, Nanog, Sox2, and Rex1 (Supplementary Fig. S1A, S1B).33,34 The cells also demonstrated the ability to differentiate into three-germ layers (Supplementary Fig. S1C). Then, cortical organoids were derived using the protocol as shown in Figure 1A. To better form cell aggregates, the post-passaged cells were initially maintained in mTeSR medium for 2 days, and the aggregates were incubated with neural differentiation medium. For cortical organoid generation, the preformed aggregates were induced by dual SMAD inhibitors and the cortical patterning was tuned by the inhibition of sonic hedgehog signaling. The aggregates were maintained up to 70 days in the differentiation medium in the presence of BDNF and FGF-2. The neural sphere formation of iPSK3 and SY-UBH lines was compared (Supplementary Fig. S2A, S2B). The iPSK3 group showed a better aggregate formation, as the average aggregate size was much larger than the aggregate size of SY-UBH group for each time point (Fig. 1B). For instance, diameter of iPSK3 aggregates was 869 ± 31 μm at day 10, while the aggregate size for SY-UBH cells was 530 ± 28 μm (Fig. 1C). At day 65, the aggregate size increased to 1815 ± 149 μm for iPSK3 group and 1006 ± 251 μm for SY-UBH group. The MTT activity and the DNA content of iPSK3 group were higher than that of the SY-UBH group (Supplementary Fig. S2C). The aggregates of SY-UBH group were harvested and analyzed for neural markers. At day 15, the SY-UBH aggregates showed positive expression of neural markers Nestin and Pax6 (Supplementary Fig. S1D, iPSK3 group neural differentiation can be seen in Refs.26–29). RT-PCR analysis of TBR1 (a cortical deep layer VI marker) and vGluT1 (a glutamatergic neuron marker) showed that the expression was comparable at day 17 for both iPSK3 and SY-UBH groups, while at day 28 iPSK3 group had higher TBR1 expression than SY-UBH group (4.6 ± 0.2 vs. 1.1 ± 0.2) (Fig. 1D). At day 70, the structure of cortical organoids was evaluated by confocal microscopy for the expression of TBR1 and SATB2 (a marker for superficial cortical II–IV layer) (Fig. 1E). A clear layer separation was observed for iPSK3 group and SY-UBH group. These results indicate cortical tissue development in vitro for hiPSCs with PS1 M146 V mutation.
FIG. 1.

Derivation of cortical spheroids and organoids from hiPSCs. (A) Schematic illustration of cortical organoids generation from hiPSCs. (B) The average diameter of spheroids at different time points for iPSK3 cells and SY-UBH cells. *p < 0.05, indicates the statistical difference for average spheroid diameter between the two groups at the same time point. (C) Phase contrast images of organoids at day 12 and 65 for iPSK3 cells and SY-UBH cells. Scale bar: 200 μm. (D) RT-PCR analysis of gene expression of TBR1 and vGlut1 at day 17 and 28. **p < 0.01. (E) Confocal images showed the cortical layer structures for samples of day 70 for iPSK3 cells and SY-UBH cells. The white dotted line indicates cortical layer separation. Red: TBR1, green: SATB2, blue: Hoe (Hoechst 33342). Scale bar: 200 μm. BDNF, brain-derived neurotrophic factor; FGF, fibroblast growth factor; hiPSC, human induced pluripotent stem cell; RT-PCR, reverse transcription–polymerase chain reaction. Color images available online at www.liebertpub.com/tea
The cortical organoids were also characterized at day 28, 42, and 54 for neuronal subtypes and synaptic markers (Fig. 2A, B and Supplementary Figs. S3 and S4). At day 28, the deeper cortical layer VI marker TBR1 and another superficial layer II–IV marker BRN2 (layer III) were observed for both groups. The expression of glutamate (Glut) (a glutamatergic neuron marker) and GABA (a GABAergic neuron marker) indicated the presence of glutamatergic and GABAergic neurons. The presynaptic marker synapsin I and postsynaptic marker PSD95 were observed at day 28. SATB2 expression (developmentally appears later than TBR1) was detected at day 42 (Supplementary Fig. S3). Presence of GFAP expression, an astrocyte marker, was also observed (Supplementary Fig. S3). The quantitative expression for Glut, GABA, TBR1, and BRN2 was comparable for iPSK3 and SY-UBH groups at day 28 (Fig. 2C). When comparing day 28 and 42 samples for SY-UBH group, the expression of neuronal markers increased with culture time (Fig. 2D), showing the development of cortical tissues. At day 54, the presence of TBR1, BRN2, SATB2, Glut, GABA, and MAP2 showed the persistent expression of these markers.
FIG. 2.

Characterization of cortical organoids for neural marker expression. (A) Representative fluorescent images of neural markers: BRN2, TBR1, glutamate (Glut), β-tubulin III (β-tub), and GABA, and synaptic markers synapsin I (SYN I) and PSD95, for SY-UBH and iPSK3 cells at day 28. Scale bar: 100 μm. (B) Representative fluorescent images of neural markers for SY-UBH cells at day 42. Scale bar: 100 μm. (C) Quantification of neural markers (GABA, glutamate, BRN2, and TBR1) expression for SY-UBH and iPSK3 groups at day 28. p = 0.091 (glutamate) and p = 0.074 (BRN2). (D) Quantification of neural marker expression for SY-UBH samples at day 28 and 42. p = 0.052 (BRN2). *p < 0.05, **p < 0.01. Color images available online at www.liebertpub.com/tea
Characterization of AD-associated phenotype
The AD-associated marker expression was also evaluated for day 28 and 42 samples (Fig. 3). At day 28, the SY-UBH group displayed the expression of tau (a microtubule protein), Aβ42 (amyloid aa1-42) and p-tau (phosphorylated tau protein) (Fig. 3A). However, the expression of Aβ42 and p-tau were lower for iPSK3 group (Fig. 3B). The expression of markers tau, Aβ42, and p-tau were also observed for SY-UBH samples at day 42 (Fig. 3C). From flow cytometry analysis, both SY-UBH and iPSK3 groups showed a similar level for tau expression (65.5% for SY-UBH and 73.3% for iPSK3), while SY-UBH group upregulated the expression of Aβ42 (62.0% vs. 49.4% for SY-UBH vs. iPSK3) (Fig. 3D). For SY-UBH group, the relative expression of tau, Aβ42, and p-tau increased from day 28 to 42 (Fig. 3E).
FIG. 3.

Characterization of Aβ42 and tau expression. (A) Representative fluorescent images of AD-associated markers: tau, Aβ42, and p-tau for SY-UBH and iPSK3 samples at day 28. Scale bar: 100 μm. (B) Fluorescent images of SY-UBH samples at day 42. Scale bar: 100 μm. (C) Expression of Aβ42 and tau by flow cytometry. Black line: negative control; blue line: SY-UBH group; red line: iPSK3 group. (D) Quantification of Aβ42 and tau expression for SY-UBH and iPSK3 samples at day 28. (E) Quantification of Aβ42 and tau expression for SY-UBH samples at day 28 and 42. *p < 0.05, **p < 0.01, ***p < 0.001. AD, Alzheimer's disease. Color images available online at www.liebertpub.com/tea
Live/Dead assay indicates that the day 35 SY-UBH organoids had some cell death, while for the iPSK3 group nearly no cell death was observed (Fig. 4A). The LDH activity of the culture supernatants indicated that the cytotoxicity of SY-UBH group dramatically increased from day 8 to 26, whereas for iPSK3 group, the LDH levels fluctuated in a small range (Fig. 4B). Besides, the cellular expression of Aβ42 for SY-UBH group showed a higher level than the iPSK3 group based on confocal images (Fig. 4C). The extracellular level of Aβ42 measured by ELISA was comparable at day 8 and 25 for SY-UBH and iPSK3 groups, while the Aβ42 secretion was significantly elevated for SY-UBH group at day 48 (Fig. 4D). These data demonstrate that SY-UBH derived cortical organoids could partially recapitulate the AD-related phenotype.
FIG. 4.

Characterization of viability, cytotoxicity, and Aβ42 secretion. (A) Live/Dead assay for SY-UBH and iPSK3 groups at day 35. Scale bar: 200 μm. (B) LDH activity in culture supernatants for SY-UBH and iPSK3 groups at different time points. Both groups were seeded with the same number of cells and fed with the same volume of medium. (C) Confocal images showed intercellular accumulation of Aβ42 at day 70. Scale bar: 200 μm. (D) The secreted Aβ42 levels in in culture supernatants of for SY-UBH and iPSK3 groups determined by ELISA. **p < 0.05. LDH, lactate dehydrogenase. Color images available online at www.liebertpub.com/tea
AD-associated inflammation response and changes in ECM
Another important event that occurred in the pathogenesis of AD is inflammation response, which may cause dysfunction of neurons.35,36 To evaluate the neuro-inflammation process of the derived cortical organoids, the gene expression of IL-6 and TNF-α was analyzed. IL-6 and TNF-α for SY-UBH group were expressed at a higher level than those of iPSK3 group at day 17 (1.6 ± 0.2 vs. 0.9 ± 0.1 for IL-6 and 4.8 ± 0.1 vs. 1.0 ± 0.1 for TNF-α) and day 28 (3.4 ± 0.4 vs. 1.7 ± 0.3 for IL-6 and 5.8 ± 0.5 vs. 1.6 ± 0.2 for TNF-α) (Fig. 5A, B).
FIG. 5.

Gene expression of AD-associated inflammation markers and ECM-associated proteins. RT-PCR analysis of gene expression of TNF-α (A) IL-6 (B) MMP2 and MMP3 (C) Syndecan-3 (D) of SY-UBH and iPSK3 groups. *p < 0.05. ECM, extracellular matrix; IL, interleukin; MMP, matrix metalloproteinase; TNF, tumor necrosis factor.
Next, the expression of ECM protein and remodeling proteins was analyzed, including matrix metalloproteinase (MMP) (e.g., MMP2 and MMP3) and syndecan-3, a transmembrane HSPG that mediates cell spreading and neurite outgrowth.37 The results showed that MMP2 and MMP3 expression was lower for SY-UBH samples than the iPSK3 group at day 17 (0.4 ± 0.03 vs. 1.0 ± 0.1 for MMP2 and 0.6 ± 0.1 vs. 0.9 ± 0.1 for MMP3), day 28 (0.4 ± 0.01 vs. 0.5 ± 0.02 for MMP2 and 0.9 ± 0.3 vs. 1.5 ± 0.04 for MMP3), and day 35 (0.3 ± 0.01 vs. 1.3 ± 0.01 for MMP2 and 0.5 ± 0.2 vs. 2.0 ± 0.04 for MMP3) (Fig. 5C). For syndecan-3 expression, SY-UBH condition resulted in an increased level at day 28 comparing with iPSK3 group (3.5 ± 1.2 vs. 1.6 ± 0.3) (Fig. 5D). These data demonstrate differential expression of MMPs and syndecan-3 in 3D cortical organoids of healthy and AD-specific hiPSCs, suggesting their possible roles in modulating neural degeneration.
Cellular treatment with biomolecules
The day 35 cortical organoids from the two groups were treated with different biomolecules, heparin, heparinase III, and DAPT, for 3 days. Studies have shown that heparin could attenuate the Aβ plaque-induced cytotoxicity and inflammatory response.38 In this study, the treatment of heparin and heparinase III increased cell viability and heparinase treatment reduced LDH activity for SY-UBH group, while there was no change in viability for iPSK3 group (Fig. 6A–C). DAPT is the inhibitor of γ-secretase and could block the amyloid precursor protein cleavage and Aβ42 generation.9,19 In this study, DAPT improved cell survival of SY-UBH, as the cell viability of SY-UBH group was comparable with the viability for iPSK3 group after the treatment of DAPT (Fig. 6A, B). In addition, the LDH activity was decreased after DAPT treatment for SY-UBH group (Fig. 6C). The expression of neuronal and AD-related markers, including Glut, GABA, β-tubulin III, BRN2, TBR1, and Aβ42, was assessed after the treatment of different drugs (Fig. 7A). The level of Aβ42 for SY-UBH group was reduced from 66.2% to 50.7% with the treatment of DAPT (Fig. 7B). The portion of GABA+ cells slightly decreased (glutamate+ cells increased) and TBR1 expression was slightly lower for SY-UBH group with DAPT treatment (Fig. 7C). For heparin, the treatment reduced Aβ42 level and impacted the expression of neuronal markers (i.e., lower GABA, higher Glut, and lower TBR1 with heparin treatment) for SY-UBH group (Fig. 8). The heparinase treatment reduced Aβ42 level, but the expression of neuronal markers was not significantly different from no treatment control. By contrast, for iPSK3 group, heparin and heparinase treatment did not alter Aβ42 and neuronal marker expression (Supplementary Fig. S5). These findings demonstrate that heparin and heparinase III may counteract Aβ42 impairment to neurons possibly through blocking the binding of Aβ42 peptides.
FIG. 6.

The effect of drug treatment of the derived cortical organoids on cell viability and cytotoxicity. The day 35 organoids from SY-UBH and iPSK3 groups were treated with heparin, heparinase III, and DAPT for day 3. (A) Live/Dead assay after the treatment. Green: Live, red: Dead. Scale bar: 200 μm. (B) Cell viability after the treatment. (C) LDH activity after the treatment. *p < 0.05. Color images available online at www.liebertpub.com/tea
FIG. 7.

Treatment of cortical organoids derived from SY-UBH cells with DAPT. (A) Representative fluorescent images for markers Glut (glutamate), GABA, β-tubulin III (βIII-tub), BRN2, TBR1, and Aβ42 after the treatment. Scale bar: 100 μm. (B) Flow cytometry histogram showed the expression of Aβ42 for cells with and without DAPT treatment (3 days). Black line: negative control; blue line: no treatment group; red line: DAPT treatment group. (C) Quantification of GABA and glutamate expression (with or without DAPT treatment). (D) Quantification of Aβ42, BRN2, and TBR1 expression (with or without treatment). *p < 0.05. Color images available online at www.liebertpub.com/tea
FIG. 8.

Treatment of cortical organoids derived from SY-UBH cells with heparin and heparinase III. (A) Representative fluorescent images for markers Glut (glutamate), GABA, β-tubulin III (βIII-tub), BRN2, TBR1, p-tau, and Aβ42 for SY-UBH samples after the treatments (3 days). Scale bar: 100 μm. (B) Quantification of Aβ42, GABA, glutamate, BRN2, and TBR1 expression for samples after the treatments. *p < 0.05. Color images available online at www.liebertpub.com/tea
Discussion
As an age-progressive disease, the mechanism of AD pathology is usually difficult to investigate partially due to the lack of proper brain models. An efficient and robust approach for modeling AD would largely accelerate our steps in uncovering the disease's mechanism. In this study, hiPSC-derived 3D cortical organoids was applied to investigate AD phenotype, ECM remodeling, and cellular responses to a few compounds.
In this study, cortical organoids from AD patient-derived hiPSCs with PS1 M146 V mutation were generated to recapitulate the disease's pathology. Our data demonstrated the differences in cortical organoid formation efficiency and TBR1 gene expression between healthy and AD hiPSCs. Comparing with the healthy organoids, AD-derived cortical organoids may induce imbalanced glutamatergic and GABAergic neurons, the main neuronal subtypes constituting human cortex. The generated cortical organoids from healthy hiPSCs had a different thickness between the TBR1+ and SATB2+ cell layers compared to AD iPSC group. To date, hiPSCs have provided us a useful tool for in vitro neurological disease modeling.1,39,40 Several groups have derived specific neural cells to model some AD-related events using hiPSCs from sporadic or familiar AD patients.9,16,19,41,42 Although these AD models could partially reflect AD-associated pathology, most of the studies utilized simple 2D cultures, which are lack of the interstitial compartments and specific architecture of human brain tissue. The recently developed 3D organoids or spheroids have been used to model various neurological diseases, for example, microcephaly and Zika virus infection,1,39 as organoids contain ordered structure with cell-ECM interactions and represent partial components of human tissues or organs.43 A few studies have shown the feasibility of organoid technology in modeling AD phenotype.11,14 The effects of AD-associated gene mutation on neural patterning from hiPSCs and AD phenotype have not been well investigated. Our results indicate that AD-related gene mutation may impact the composition of cortical organoids and alter neural tissue property.
This work could partially recapitulate some aspects of AD pathology including Aβ42 secretion and neural cell death. A significantly higher level of both cell-bound Aβ42 and cell-secreted Aβ42 from AD organoids was observed compared to healthy control. An increased expression of p-tau through immunostaining was also detected, which was time dependent. The sequential emergence of amyloid aggregation and hyper-phosphorylated tau remains unclear,44,45 and our model did not display a distinct timeline on which events occur. Moreover, our study demonstrated significant cell death and a high level of LDH activity for AD organoid culture, indicating the AD-related cytotoxicity.
AD progression is a complex process involving not only the hallmarks of Aβ aggregation and tau hyperphosphorylation but also other episodes, such as synaptic dysfunction, neuro-inflammation, gliosis, DNA damage, U1 tangles, ECM remodeling, and others.36,38,46 In this study, the synaptic activities were examined using synaptic markers. A further study would employ electrophysiological tools to test the electrical activities and synaptic connections in our models. Also, the gene expression of inflammatory cytokines was evaluated. AD-derived organoids had higher gene expression of IL-6 and TNF-α than the control at day 28. At this early stage, IL-6 and TNF-α secretion was not attempted. The secretion of IL-6 and TNF-α by IL-1β-activated astrocytoma cells and lipopolysaccharide-stimulated astrocytes47–49 could impair microglia and surrounding neural cells, possibly aggravate tau pathology, and finally result in neurodegeneration and neuron loss.36,50 The increased gene expression of IL-6 and TNF-α and the existence of astrocytes in AD organoids may be related to toxicity and neural cell death in our study.
In human central nervous system, ECM constitutes the essential physical structures for neural cells and provides diverse biochemical signals for neurogenesis, cell migration, differentiation and synaptic plasticity.51 The AD-associated activities could modulate the expression of ECM molecules and MMPs, which may result in dysfunction of synapse and neuron loss.52–54 For instance, previous works demonstrated an increased level of some ECM components, for example, collagen IV, perlecan, and fibronectin in brain samples of AD patients.55 However, these events and how ECM being involved in AD pathology were not fully investigated in hPSC-based AD models.
In this study, AD-derived organoids altered the expression of MMP2, MMP3, and syndecan-3, comparing with the control. MMP2 plays an important role in neurogenesis, remodeling of the basal lamina,55 and regeneration of axons,54,56 and MMP3 facilitates synaptic remodeling and degrades Aβ components.57,58 Syndecan-3, a protein of HSPG family that mediates cell spreading, neurite outgrowth, and inflammation process,59–61 has been associated with amyloid plaques formation.62,63 The differences of MMPs and the upregulation of syndecan-3 level between AD organoids and healthy control organoids suggested that AD-induced toxicity may also be related to changes of ECM composition and remodeling.
In AD research, pharmacological testing and candidate drug targets screening usually use primary cultures and animal models and no reliable drugs have been successfully developed, as studies done in these systems could not be easily translated to clinics.64 hiPSC-derived AD models have become a promising approach to evaluate potential drugs.11,65 In our study, the derived cortical organoids were treated with different compounds including DAPT, heparin, and heparinase III. Since PS1 M146 V mutation could cause abnormal translation of γ-secretase and stimulate the generation of amyloid plaques,66–69 modulation of γ-secretase activity is one of the targets. Our results showed that DAPT treatment inhibited endogenous aggregation of Aβ42 and thus led to a lower cytotoxicity and higher cell survival. Since DAPT also inhibited notch signaling,70 additional tests with other γ-secretase inhibitors may be required in future. Another potential therapeutic candidate in AD is heparin, which exhibits anti-inflammatory effects and inhibitory effects on HSPGs assembly and thus may attenuate cytotoxicity of amyloid plaques.38,71,72 The results in this study show that heparin decreased Aβ42 level in cells from AD organoids. Similarly, heparinase III also decreased Aβ42 level. As a proteinase, heparinase III selectively degrades HSPGs from ECM, thereby reduces the binding of proinflammatory factors such as IL-8, and prevents Aβ-HSPGs interactions.73,74 Heparinase III was found to decrease intracellular tau protein aggregation,75 thus it may also prove as a neuroprotective agent. However, heparin and heparinase III treatments resulted in smaller differences in cell viability, which may be attributed to the limited diffusion in 3D organoids as the specific 3D configuration and structure of organoids reduce the exposure of cells to large compounds such as heparin and heparinase III.11
It needs to be noted that this study evaluated the hiPSC line with PS1 M146 V mutations (de-identified) and line-to-line variations maybe exist. While additional AD patient-derived hiPSCs are yet to be evaluated for future study, the elevated cytotoxicity, Aβ42 secretion, and syndecan-3 expression in this study do not appear to be coincidently associated with SY-UBH cells. Generating isogenic iPSC clones (preferably three clones for each group) is another approach to validate the differences between iPSK3 line and SY-UBH line. In addition, the current cortical organoids are still far away from complicated human forebrain. Recent study of fusion or assembly of brain region-specific organoids represents the direction to better mimic human brain development.76,77 Moreover, current cortical organoids lack vascularization structure and microglia components,8 which need to be incorporated in next-generation organoid design.
Conclusions
In this study, cortical organoids were derived from hiPSC with PS1 M146 V mutations and characterized for inflammatory-related gene expression, ECM remodeling and assembly, and the influence of γ-secretase inhibitor and heparin/heparinase to modulate ECM-Aβ interactions. Our study applied organoid methods to model neural degenerative microenvironment. While the organoid model is a useful tool to investigate cell–cell communication and cell–matrix interactions, future work would be required to understand the connections between different cell types such as neurons, microglia, and astrocytes in modeling human forebrain.
Supplementary Material
Acknowledgments
The authors would like to thank Ms. Ruth Didier in the FSU Department of Biomedical Sciences for her help with flow cytometry analysis, Dr. Brian K. Washburn and Kristina Poduch in the FSU Department of Biological Sciences for their help with RT-PCR analysis, Dr. Stephen Duncan at the Medical College of Wisconsin and Dr. David Gilbert in the FSU Department of Biological Sciences for human iPSK3 cells. The authors would also like to thank the Neuroregeneration lab at Mayo Clinic (FL) for technical support on disease-relevant cells. This work is supported by the FSU Bridge Grant and partially the National Science Foundation (NSF CAREER grant No. 1652992 to Y.L). Research reported in this publication was partially supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under Award Number R03NS102640 (to Y.L. and Y.Z.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure Statement
No competing financial interests exist.
References
- 1.Lancaster M.A., Renner M., Martin C.A., et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pasca A.M., Sloan S.A., Clarke L.E., et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods 12, 671, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mariani J., Simonini M.V., Palejev D., et al. Modeling human cortical development in vitro using induced pluripotent stem cells. Proc Natl Acad Sci U S A 109, 12770, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van den Ameele J., Tiberi L., Vanderhaeghen P., and Espuny-Camacho I. Thinking out of the dish: what to learn about cortical development using pluripotent stem cells. Trends Neurosci 37, 334, 2014 [DOI] [PubMed] [Google Scholar]
- 5.Eiraku M., Watanabe K., Matsuo-Takasaki M., et al. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 3, 519, 2008 [DOI] [PubMed] [Google Scholar]
- 6.Marti-Figueroa C.R., and Ashton R.S. The case for applying tissue engineering methodologies to instruct human organoid morphogenesis. Acta Biomater 54, 35, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shah S.B., and Singh A. Cellular self-assembly and biomaterials-based organoid models of development and diseases. Acta Biomater 53, 29, 2017 [DOI] [PubMed] [Google Scholar]
- 8.Di Lullo E., and Kriegstein A.R. The use of brain organoids to investigate neural development and disease. Nat Rev Neurosci 18, 573, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi S.H., Kim Y.H., Hebisch M., et al. A three-dimensional human neural cell culture model of Alzheimer's disease. Nature 515, 274, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang D., Pekkanen-Mattila M., Shahsavani M., Falk A., Teixeira A.I., and Herland A. A 3D Alzheimer's disease culture model and the induction of P21-activated kinase mediated sensing in iPSC derived neurons. Biomaterials 35, 1420, 2014 [DOI] [PubMed] [Google Scholar]
- 11.Lee H.K., Velazquez Sanchez C., Chen M., et al. Three dimensional human neuro-spheroid model of Alzheimer's disease based on differentiated induced pluripotent stem cells. PLoS One 11, e0163072, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y., Muffat J., Omer A., et al. Induction of expansion and folding in human cerebral organoids. Cell Stem Cell 20, 385, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pistollato F., Ohayon E.L., Lam A., et al. Alzheimer disease research in the 21st century: past and current failures, new perspectives and funding priorities. Oncotarget 7, 38999, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raja W.K., Mungenast A.E., Lin Y.T., et al. Self-organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate Alzheimer's disease phenotypes. PLoS One 11, e0161969, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vazin T., Ball K.A., Lu H., et al. Efficient derivation of cortical glutamatergic neurons from human pluripotent stem cells: a model system to study neurotoxicity in Alzheimer's disease. Neurobiol Dis 62, 62, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kondo T., Asai M., Tsukita K., et al. Modeling Alzheimer's disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell 12, 487, 2013 [DOI] [PubMed] [Google Scholar]
- 17.Choi Y.J., Park J., and Lee S.H. Size-controllable networked neurospheres as a 3D neuronal tissue model for Alzheimer's disease studies. Biomaterials 34, 2938, 2013 [DOI] [PubMed] [Google Scholar]
- 18.Nieweg K., Andreyeva A., van Stegen B., Tanriover G., and Gottmann K. Alzheimer's disease-related amyloid-beta induces synaptotoxicity in human iPS cell-derived neurons. Cell Death Dis 6, e1709, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Israel M.A., Yuan S.H., Bardy C., et al. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature 482, 216, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldstein L.S., Reyna S., and Woodruff G. Probing the secrets of Alzheimer's disease using human-induced pluripotent stem cell technology. Neurotherapeutics 12, 121, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang J., Zhao H., Ma Y., et al. Early pathogenic event of Alzheimer's disease documented in iPSCs from patients with PSEN1 mutations. Oncotarget 8, 7900, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanekiyo T., Zhang J., Liu Q., Liu C.C., Zhang L., and Bu G. Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-beta uptake. J Neurosci 31, 1644, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu C.C., Zhao N., Yamaguchi Y., et al. Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-beta clearance and aggregation in Alzheimer's disease. Sci Transl Med 8, 332ra44, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Si-Tayeb K., Noto F.K., Sepac A., et al. Generation of human induced pluripotent stem cells by simple transient transfection of plasmid DNA encoding reprogramming factors. BMC Dev Biol 10, 81, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Si-Tayeb K., Noto F.K., Nagaoka M., et al. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 51, 297, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan Y., Martin L., Bosco D., et al. Differential effects of acellular embryonic matrices on pluripotent stem cell expansion and neural differentiation. Biomaterials 73, 231, 2015 [DOI] [PubMed] [Google Scholar]
- 27.Bejoy J., Song L., Zhou Y., and Li Y. Wnt-YAP interactions during neural tissue patterning of human induced pluripotent stem cells. Tissue Eng Part A 2017. [Epub ahead of print]; DOI: 10.1089/ten.TEA.2017.0153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan Y., Bejoy J., Xia J., Guan J., Zhou Y., and Li Y. Neural patterning of human induced pluripotent stem cells in 3-D cultures for studying biomolecule-directed differential cellular responses. Acta Biomater 42, 114, 2016 [DOI] [PubMed] [Google Scholar]
- 29.Yan Y., Song L., Madinya J., Ma T., and Li Y. Derivation of cortical spheroids from human induced pluripotent stem cells in a suspension bioreactor. Tissue Eng Part A. 2017. [Epub ahead of print]; DOI: 10.1089/ten.TEA.2016.0400 [DOI] [PubMed] [Google Scholar]
- 30.Song L., Tsai A.C., Yuan X., et al. Neural differentiation of spheroids derived from hiPSC-MSC co-culture. Tissue Eng Part A. 2017. [Epub ahead of print]; DOI: 10.1089/ten.TEA.2017.0403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan Y., Li Y., Song L., Zeng C., and Li Y. Pluripotent stem cell expansion and neural differentiation in 3-D scaffolds of tunable Poisson's ratio. Acta Biomater 49, 192, 2017 [DOI] [PubMed] [Google Scholar]
- 32.Sart S., Yan Y., Li Y., et al. Crosslinking of extracellular matrix scaffolds derived from pluripotent stem cell aggregates modulates neural differentiation. Acta Biomater 30, 222, 2016 [DOI] [PubMed] [Google Scholar]
- 33.Wren M.C., Zhao J., Liu C.C., et al. Frontotemporal dementia-associated N279K tau mutant disrupts subcellular vesicle trafficking and induces cellular stress in iPSC-derived neural stem cells. Mol Neurodegener 10, 46, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao J., Davis M.D., Martens Y.A., et al. APOE epsilon4/epsilon4 diminishes neurotrophic function of human iPSC-derived astrocytes. Hum Mol Genet 26, 2690, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marsh S.E., Abud E.M., Lakatos A., et al. The adaptive immune system restrains Alzheimer's disease pathogenesis by modulating microglial function. Proc Natl Acad Sci U S A 113, E1316, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heppner F.L., Ransohoff R.M., and Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 16, 358, 2015 [DOI] [PubMed] [Google Scholar]
- 37.Bespalov M.M., Sidorova Y.A., Tumova S., et al. Heparan sulfate proteoglycan syndecan-3 is a novel receptor for GDNF, neurturin, and artemin. J Cell Biol 192, 153, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bergamaschini L., Donarini C., Rossi E., De Luigi A., Vergani C., and De Simoni M.G. Heparin attenuates cytotoxic and inflammatory activity of Alzheimer amyloid-beta in vitro. Neurobiol Aging 23, 531, 2002 [DOI] [PubMed] [Google Scholar]
- 39.Qian X., Nguyen H.N., Song M.M., et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165, 1238, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ovchinnikov D.A., and Wolvetang E.J. Opportunities and limitations of modelling Alzheimer's disease with induced pluripotent stem cells. J Clin Med 3, 1357, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yagi T., Ito D., Okada Y., et al. Modeling familial Alzheimer's disease with induced pluripotent stem cells. Hum Mol Genet 20, 4530, 2011 [DOI] [PubMed] [Google Scholar]
- 42.Muratore C.R., Rice H.C., Srikanth P., et al. The familial Alzheimer's disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum Mol Genet 23, 3523, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yin X., Mead B.E., Safaee H., Langer R., Karp J.M., and Levy O. Engineering stem cell organoids. Cell Stem Cell 18, 25, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng W.H., Bastianetto S., Mennicken F., Ma W., and Kar S. Amyloid beta peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience 115, 201, 2002 [DOI] [PubMed] [Google Scholar]
- 45.LaFerla F.M. Pathways linking Abeta and tau pathologies. Biochem Soc Trans 38, 993, 2010 [DOI] [PubMed] [Google Scholar]
- 46.Bai B., Hales C.M., Chen P.C., et al. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer's disease. Proc Natl Acad Sci U S A 110, 16562, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chakrabarty P., Jansen-West K., Beccard A., et al. Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J 24, 548, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heyser C.J., Masliah E., Samimi A., Campbell I.L., and Gold L.H. Progressive decline in avoidance learning paralleled by inflammatory neurodegeneration in transgenic mice expressing interleukin 6 in the brain. Proc Natl Acad Sci U S A 94, 1500, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perry R.T., Collins J.S., Wiener H., Acton R., and Go R.C. The role of TNF and its receptors in Alzheimer's disease. Neurobiol Aging 22, 873, 2001 [DOI] [PubMed] [Google Scholar]
- 50.Prokop S., Miller K.R., and Heppner F.L. Microglia actions in Alzheimer's disease. Acta Neuropathol 126, 461, 2013 [DOI] [PubMed] [Google Scholar]
- 51.Lau L.W., Cua R., Keough M.B., Haylock-Jacobs S., and Yong V.W. Pathophysiology of the brain extracellular matrix: a new target for remyelination. Nat Rev Neurosci 14, 722, 2013 [DOI] [PubMed] [Google Scholar]
- 52.Heneka M.T., Carson M.J., El Khoury J., et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol 14, 388, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heneka M.T., Golenbock D.T., and Latz E. Innate immunity in Alzheimer's disease. Nat Immunol 16, 229, 2015 [DOI] [PubMed] [Google Scholar]
- 54.Brkic M., Balusu S., Libert C., and Vandenbroucke R.E. Friends or foes: matrix metalloproteinases and their multifaceted roles in neurodegenerative diseases. Mediators Inflamm 2015, 620581, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lepelletier F.X., Mann D.M., Robinson A.C., Pinteaux E., and Boutin H. Early changes in extracellular matrix in Alzheimer's disease. Neuropathol Appl Neurobiol 43, 167, 2017 [DOI] [PubMed] [Google Scholar]
- 56.Li W., Poteet E., Xie L., Liu R., Wen Y., and Yang S.H. Regulation of matrix metalloproteinase 2 by oligomeric amyloid beta protein. Brain Res 1387, 141, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meighan S.E., Meighan P.C., Choudhury P., et al. Effects of extracellular matrix-degrading proteases matrix metalloproteinases 3 and 9 on spatial learning and synaptic plasticity. J Neurochem 96, 1227, 2006 [DOI] [PubMed] [Google Scholar]
- 58.Mlekusch R., and Humpel C. Matrix metalloproteinases-2 and −3 are reduced in cerebrospinal fluid with low beta-amyloid1-42 levels. Neurosci Lett 466, 135, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Horssen J., Wesseling P., van den Heuvel L.P., de Waal R.M., and Verbeek M.M. Heparan sulphate proteoglycans in Alzheimer's disease and amyloid-related disorders. Lancet Neurol 2, 482, 2003 [DOI] [PubMed] [Google Scholar]
- 60.Zhang G.L., Zhang X., Wang X.M., and Li J.P. Towards understanding the roles of heparan sulfate proteoglycans in Alzheimer's disease. Biomed Res Int 2014, 516028, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kehoe O., Kalia N., King S., et al. Syndecan-3 is selectively pro-inflammatory in the joint and contributes to antigen-induced arthritis in mice. Arthritis Res Ther 16, R148, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reizes O., Lincecum J., Wang Z., et al. Transgenic expression of syndecan-1 uncovers a physiological control of feeding behavior by syndecan-3. Cell 106, 105, 2001 [DOI] [PubMed] [Google Scholar]
- 63.van Horssen J., Kleinnijenhuis J., Maass C.N., et al. Accumulation of heparan sulfate proteoglycans in cerebellar senile plaques. Neurobiol Aging 23, 537, 2002 [DOI] [PubMed] [Google Scholar]
- 64.Franco R., and Cedazo-Minguez A. Successful therapies for Alzheimer's disease: why so many in animal models and none in humans? Front Pharmacol 5, 146, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yahata N., Asai M., Kitaoka S., et al. Anti-Abeta drug screening platform using human iPS cell-derived neurons for the treatment of Alzheimer's disease. PLoS One 6, e25788, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bekris L.M., Yu C.E., Bird T.D., and Tsuang D.W. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol 23, 213, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ryazantseva M., Skobeleva K., and Kaznacheyeva E. Familial Alzheimer's disease-linked presenilin-1 mutation M146 V affects store-operated calcium entry: does gain look like loss? Biochimie 95, 1506, 2013 [DOI] [PubMed] [Google Scholar]
- 68.Oddo S., Caccamo A., Shepherd J.D., et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409, 2003 [DOI] [PubMed] [Google Scholar]
- 69.Wang R., Wang B., He W., and Zheng H. Wild-type presenilin 1 protects against Alzheimer disease mutation-induced amyloid pathology. J Biol Chem 281, 15330, 2006 [DOI] [PubMed] [Google Scholar]
- 70.Yamamizu K., Iwasaki M., Takakubo H., et al. In vitro modeling of blood-brain barrier with human iPSC-derived endothelial cells, pericytes, neurons, and astrocytes via notch signaling. Stem Cell Reports 8, 634, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 71.Ma Q., Cornelli U., Hanin I., et al. Heparin oligosaccharides as potential therapeutic agents in senile dementia. Curr Pharm Des 13, 1607, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bergamaschini L., Rossi E., Vergani C., and De Simoni M.G. Alzheimer's disease: another target for heparin therapy. ScientificWorldJournal 9, 891, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reynolds M.R., Singh I., Azad T.D., et al. Heparan sulfate proteoglycans mediate Abeta-induced oxidative stress and hypercontractility in cultured vascular smooth muscle cells. Mol Neurodegener 11, 9, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Taylor D.R., Whitehouse I.J., and Hooper N.M. Glypican-1 mediates both prion protein lipid raft association and disease isoform formation. PLoS Pathog 5, e1000666, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Holmes B.B., DeVos S.L., Kfoury N., et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A 110, E3138, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xiang Y., Tanaka Y., Patterson B., et al. Fusion of regionally specified hPSC-derived organoids models human brain development and interneuron migration. Cell Stem Cell 21, 383, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Birey F., Andersen J., Makinson C.D., et al. Assembly of functionally integrated human forebrain spheroids. Nature 545, 54, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.