

Abstract
Santalales is a large order, with over 2200 species, most of which are root or aerial (stem) hemiparasites. In this study, we report the newly assembled chloroplast genome of Dendrotrophe varians (140,666 bp) in the family Amphorogynaceae and the cp genomes of Helixanthera parasitica (124,881 bp) and Macrosolen cochinchinensis (122,986 bp), both in the family Loranthaceae. We compared the cp genomes of 11 Santalales including eight currently available cp genomes. Santalales cp genomes are slightly or not reduced in size (119–147 kb), similar to other hemiparasitic species, when compared with typical angiosperm cp genomes (120–170 kb). In a phylogeny examining gene content, the NADH dehydrogenase gene group is the only one among eight functional gene groups that lost complete functionally in all examined Santalales. This supports the idea that the functional loss of ndh genes is the initial stage in the evolution of the plastome of parasitic plants, but the loss has occurred independently multiple times in angiosperms, while they are not found in some parasites. This suggests that the functional loss of ndh genes is not essential for the transition from autotroph to parasite. We additionally examined the correlation between gene content and type of parasitism (obligate/facultative and stem/root parasites) of all hemiparasitic species in which cp genomes have been reported to date. Correlation was not found in any types of parasitism.
Introduction
Parasitism in plants is the result of the transition from autotrophy to heterotrophy, involving a decrease or loss of the ability to photosynthesize, and absorption of the host’s water and nutrients through a haustorium, a specialized structure used to invade hosts [1]. Parasitism in angiosperms is considered to have at least 12 independent origins, and most species are included in the family Orobanchaceae and the order Santalales. Among the 12 recognized lineages of parasitic plants, ca. 4260 species belong to one of two lineages (Orobanchaceae and Santalales), and ca. 220 other species belong to one of 10 lineages (Apodanthaceae, Cassytha, Cuscuta, Cynomoriaceae, Cytinaceae, Hydnoraceae, Krameriaceae, Lennoaceae, Mitrastemonaceae, and Rafflesiaceae) [2–4]. Depending on photosynthetic ability, parasitic plants are divided into two types: photosynthetic parasites (hemiparasites) and nonphotosynthetic parasites (holoparasites). Hemiparasitic plants possess varying rates of photosynthetic ability, while holoparasitic plants obtain all nutrients from their host and show complete loss of photosynthetic ability [5]. Among the 12 lineages of parasitic plants, hemiparasites are found in Cassytha, Cuscuta, Krameriaceae, Orobanchaceae, and Santalales. Hemiparasitic Santalales is composed of obligate and facultative parasites that are distinguished by the essential or nonessential role of a host in the parasite’s full life cycle [2, 3].
The transition from autotrophy to heterotrophy in plants leads to pseudogenization and loss of plastid genes associated with photosynthesis. It also leads to reduction and change in the highly conserved plastome, which consists of two copies of large inverted repeats (IRs) that separate the large and small single copy regions (LSC and SSC) [2, 6–12]. The rate of chloroplast genome degradation in parasitic plants varies with the level of photosynthesis [13]. The complete cp genomes of hemiparasitic plants deposited in GenBank to date (https://www.ncbi.nlm.nih.gov/genome, last accessed May 28, 2017) are slightly or not reduced in size compared to typical cp genomes in angiosperms (120–170 kb) [14–16]. In holoparasitic plants, reductions in cp genome size are typically observed [7, 9, 16–21], and in several species, dramatically reduced cp genome sizes are found [22–24]. Even the possible loss of the entire cp genome has been reported in Rafflesia [25]. Along with a reduction in cp genome size, the conserved quadripartite genome structure has been perturbed in some holoparasitic species [16, 22, 23]. Among the 13 completed cp genomes of hemiparasitic plants, two species, Cassytha filiformis and Striga hermonthica, have lost the typical quadripartite structure [9, 26]. Among the 120–130 genes that are common in typical angiosperm cp genomes [14], pseudogenization and loss of ndh genes are considered the first steps of gene content change in the evolution of heterotrophic plants [13, 16, 27]. Ndh-specific degradations are commonly found in the currently completed cp genomes of hemiparasitic plants, except for Triphysaria versicolor, which is a facultative parasite in Orobanchaceae [9, 15, 16, 26, 28–30]. Unlike hemiparasitic plants, the cp genomes of holoparasitic plants cover the full spectrum of degradation in gene content [23]. In a recent study describing a mechanistic model of plastome degradation and accelerated evolutionary rates, Wicke et al. (2016) suggest that the transition from autotrophy to obligate parasitism relaxes functional constraints on plastid genes in a stepwise manner.
Santalales, which is one of the largest orders in angiosperms, consists of 20 families, ca. 179 genera, and 2460 species [3, 4]. It consists of a relatively small number of autotrophic species and a large number of parasitic species, mostly hemiparasites, which are root or aerial (stem) parasites [31, 32]. The species of Santalales are distributed worldwide, but are found mainly in tropical and subtropical regions. Recent studies of the Santalales have mainly focused on phylogenetic relationships in angiosperms [33–35] and on the phylogenetic classification of Santalales [4, 31, 32, 36–38]. To date, the complete cp genomes of eight Santalales species (Champereia manillana, Osyris alba, Schoepfia jasminodora, Taxillus chinensis, Taxillus sutchuenensis, Viscum album, Viscum crassulae, and Viscum minimum) belonging to five families (Loranthaceae, Opiliaceae, Santalaceae, Schoepfiaceae, and Viscaceae) have been sequenced [15, 29, 30, 39]. These eight cp genomes have a typical structure, which is highly conserved in most angiosperms and is composed of a single circular DNA molecule with a quadripartite structure including an LSC, SSC, and two IRs. These cp genome sizes range from ca. 119 (Schoepfia jasminodora) to 147 (Champereia manillana) kb.
In this study, we first report the complete cp genome of Dendrotrophe varians in the family Amphorogynaceae. Additionally, from the family Loranthaceae, we report the complete cp genome of Helixanthera parasitica of the subtribe Dendrophthoinae and the cp genome of Macrosolen cochinchinensis of the tribe Elytrantheae [38]. We describe general characteristics of the three cp genomes based on their structural features such as genome size, gene content, and gene order. To better understand plastome evolution in hemiparasitic Santalales, we compare these three cp genomes with eight other Santalales cp genomes reported and reconfirm their phylogenetic positions. Finally, we investigate pseudogenization and gene loss in parasitic plant cp genomes, with particular focus on hemiparasitic plants.
Materials and methods
Plant DNA extraction and cp genome sequencing
Dendrotrophe varians was collected from Phnum Bokor National Park, Kampot prov. in Cambodia. Helixanthera parasitica and Macrosolen cochinchinensis were collected from Phuoc Hoa comm., Bac Ai distr., Ninh Thuan prov. and Sa Son comm., Sa Thay distr., Kon Tum prov. in Vietnam, respectively. A voucher specimen of Dendrotrophe varians (CB-2630) was deposited in the Herbarium of Hallym University (HHU) and voucher specimens of Helixanthera parasitica (VK4005) and Macrosolen cochinchinensis (VK2608) were deposited in the Herbarium of the Institute of Ecology and Biological resources, Hanoi, Vietnam (HN). All three species are not endangered or protected species. Whole genomic DNA was isolated from fresh leaf tissue using the CTAB method [40] or a DNeasy Plant Mini Kit (Qiagen, USA) following the manufacture’s protocols. Extracted DNA was quantified with a NanoDrop spectrophotometer. Raw sequence reads were generated using the Illumina MiSeq Sequencing System (http://www.illumina.com). To extract chloroplast DNA contigs, the raw data were filtered by BLAST searches (National Center for Biotechnology Information, http://blast.ncbi.nlm.nih.gov/Blast.cgi) and assembled with the complete cp genome sequences of Santalales species (Champereia manillana [NC_034931], Osyris alba [NC_027960], Schoepfia jasminodora [NC_034228], Taxillus chinensis [KY996492], T. sutchuenensis [KY996493], Viscum album [NC_029039], Viscum crassulae [NC_027959], and Viscum minimum [NC_027829]), which are deposited in GenBank (https://www.ncbi.nlm.nih.gov/genome, last accessed Nov. 09, 2017), as references using Geneious version 9.1.5 (Biomatters, https://www.geneious.com/). The filtered contigs were assembled de novo, and assembled contigs were used as references. To fill gaps between contigs and to confirm the raw data, direct sequencing of polymerase chain reaction (PCR) products was performed using primers designed from contig ends. To confirm SSC/IR and LSC/IR junctions, repetitive sequences were analyzed using REPuter [41], and a PCR-based survey was also conducted at breakpoints. Additionally, several regions with more than 20 nucleotides of poly A and T were further amplified with PCR to check assembly error of raw contigs from MiSeq sequencing. The PCR products were purified using an AccuPrep PCR Purification Kit (BIONEER) following the manufacturer’s protocols. The PCR products were then sequenced in both directions using the same primers as those used for PCR amplification. Primer information is presented in S1 Table. DNA fragment assembly and sequence editing were performed with Geneious version 9.1.5 (Biomatters, https://www.geneious.com/). All sequences were deposited in GenBank (http://www.ncbi.nlm.nih.gov/genbank/).
Gene annotation and sequence analysis
BLAST and DOGMA [42] were used to annotate coding genes, and additional annotation was performed in Geneious using reference sequences of Santalales species. The exact positions of the start and stop codons were confirmed, and translation was generated by BioEdit 7.2.5 [43]. The locations of RNAs were predicted by tRNAscan-SE 2.0 [44]. After gene annotation was complete, circular and linear gene maps were drawn by OGDraw 1.2 [45, 46]. Mauve 2.3.1 [47] and Geneious version 9.1.5 (Biomatters, https://www.geneious.com/) were used to compare structural differences among multiple Santalales cp genomes.
Phylogenetic analysis
To confirm the exact topology among Santalales species, we performed phylogenetic analysis using eight complete cp genome sequences of Santalales available in GenBank, with the three complete cp genome sequences of Cornus controbersa [NC_030260], Diospyros lotus [NC_030786], and Fagopyrum esculentum subsp. ancestrale [NC_010776] as outgroup taxa. Fifty-eight protein-coding sequences that are shared among the 14 representative taxa were used for the analysis. Each gene was aligned using MUSCLE 3.5 [48], and then the aligned genes were concatenated into a dataset. Maximum likelihood (ML) analysis was used to construct a phylogenetic tree as performed in RAxML 8.2.7 [49], and it was implemented in Geneious version 9.1.5. The analysis was performed using the GTR+GAMMA+I model generated by Modeltest [50], and clade support was evaluated by 1000 bootstrap replications. Bayesian phylogenetic analysis was performed in MrBayes 3.2.6 [51]. In the Bayesian phylogenetic analysis, the Markov Chain Monte Carlo (MCMC) algorithm approach was used under the parameter settings of chain length = 1,100,000, heated chains = 4, heated chain temperature = 0.2, subsampling frequency = 200, burn in length = 100,000, and random seed = 18,770.
Results
The cp genome structure of three newly sequenced species
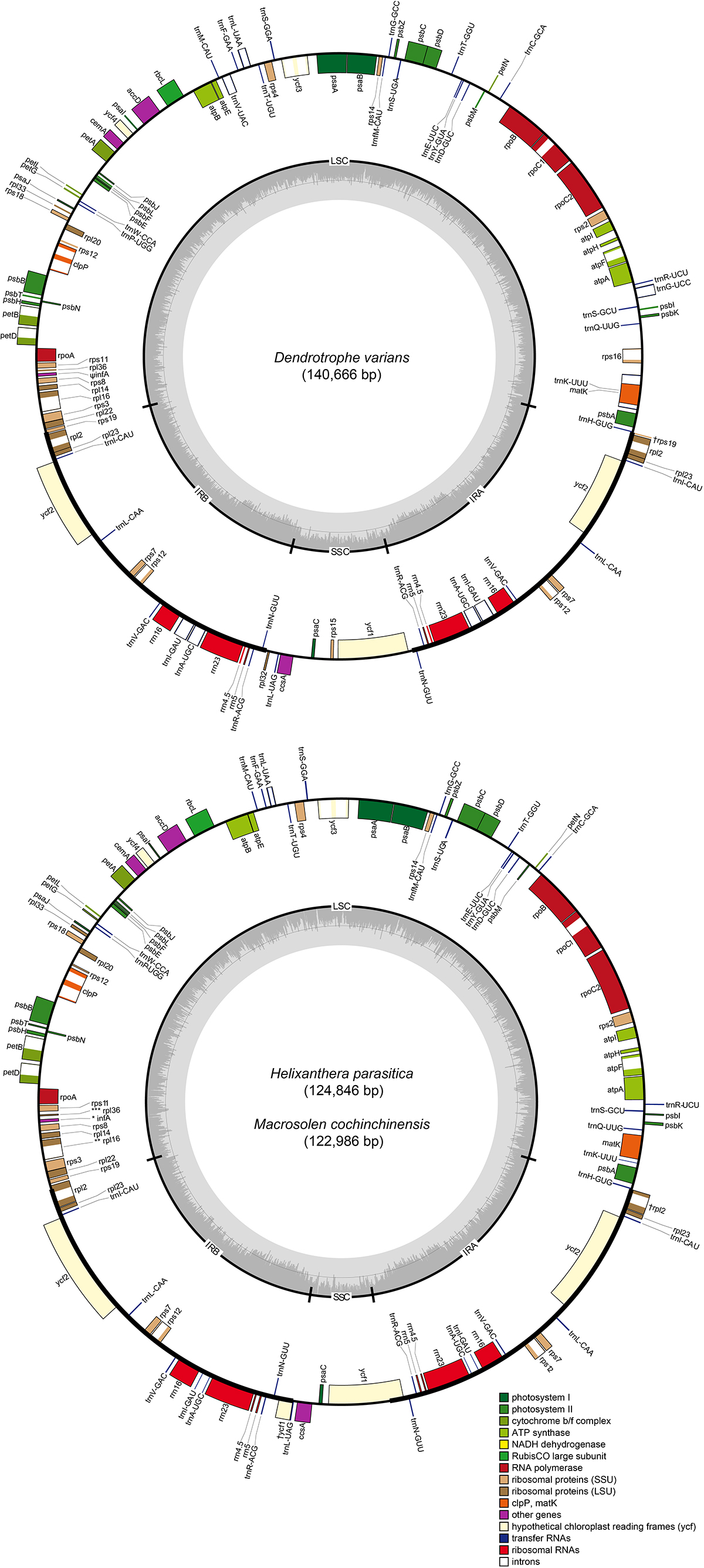
From genomic DNAs, 7,245,848 raw reads of Dendrotrophe varians, 8,931,796 raw reads of Helixanthera parasitica, and 9,108,812 raw reads of Macrosolen cochinchinensis were produced. The complete cp genomes of Dendrotrophe varians (140,666 bp), Helixanthera parasitica (124,881 bp), and Macrosolen cochinchinensis (122,986 bp) were determined (S1 Fig). The cp genome sizes are in the range of the previously reported Santalales cp genomes. In Loranthaceae, the cp genomes of Helixanthera parasitica and Macrosolen cochinchinensis are similar in size to the previously reported cp genomes of Loranthaceae species, Taxillus chinensis (121,363 bp) and Taxillus sutchuenensis (122,562 bp). The eight publicly available cp genomes of hemiparasitic Santalales have a typical organization, divided into four parts: two IRs, an LSC, and an SSC. The cp genomes of the three newly sequenced species are collinear to those of most Santalales species except Osyris alba and Viscum minimum, which have large inversions. In the cp genome of Dendrotrophe varians, there are a total of 101 genes, including 67 protein-coding genes, 4 ribosomal RNA (rRNA) genes, and 30 transfer RNA (tRNA) genes. Sixteen genes (5 protein-coding genes, 4 rRNA genes, and 7 tRNA genes) have been duplicated in IRs. One pseudogene (ψinfA) was detected and has a premature stop codon. Two partial sequences of ndhB genes were detected in IRs. One truncated gene of rps19 is located at the LSC-IRA junction. The Helixanthera parasitica and Macrosolen cochinchinensis cp genomes have an identical number of genes. In each genome, there are a total of 92 genes including 63 protein-coding genes, 4 rRNA genes, and 25 tRNA genes. Thirteen genes (4 protein-coding genes, 4 rRNA genes, and 5 tRNA genes) were duplicated in IRs. The infA gene is lost in the Helixanthera parasitica cp genome, and the rpl36 gene is lost in the Macrosolen cochinchinensis cp genome. Two pseudogenes, ψinfA gene in the Helixanthera parasitica cp genome and ψrpl16 gene in the Macrosolen cochinchinensis cp genome, were detected. Both pseudogenes have premature stop codons. The two cp genomes have the same gene segments in IRs and truncated genes at the IR junctions. The partial sequences of ndhB, trnA-UGC, and trnI-GAU genes were detected in IRs. Two truncated genes of rpl2 and ycf1 are located at the LSC-IRA and SSC-IRB junctions, respectively.
General features of Santalales cp genomes
We compared general features of 11 Santalales cp genomes including three newly sequenced cp genomes (Table 1). The largest cp genome is Champereia manillana (147,461 bp) and the smallest cp genome is Schoepfia jasminodora (118,743 bp) which is reduced by 28,718 bp (19.5%) compared to the Champereia manillana cp genome. The reduction is due to the contraction of protein-coding regions. Among the LSC, SSC, and IR regions, many contractions in the IR regions are responsible for the reduction of the Schoepfia jasminodora cp genome. We newly annotated eight cp genomes, which were previously deposited in GenBank, and compared all 11 cp genomes with one another to get the most accurate genome annotation. Inaccurate annotation of genes in the previously deposited genomes was due to typing errors in tRNA genes, incorrect prediction of gene location, and miscounted genes because of gene duplication. The Santalales cp genomes have a total of 91–101 genes including duplicated genes in the IRs. These genes consist of 63–69 protein-coding genes, 23–30 tRNA genes, and 4 rRNA genes. All annotated genes are listed in S2 Table. Among the three major regions (LSC, SSC, and IRs), the IRs have the highest GC content, with average of 43.3%, and the SSC has the lowest GC content, with an average of 26.8%. The GC content of the cp genome of Schoepfia jasminodora is the highest, and that of Viscum minimum is the lowest of all the cp genomes. The Viscum crassulae cp genome has the most compact cp genome with a total of 62.1% coding regions.
Table 1. Comparison of cp DNA features among 11 Santalales species.
| Features | Viscum minimum | Viscum crassulae |
Viscum album |
Dendrotrophe varians |
Osyris alba |
Champereia manillana | Taxillus sutchuenensis | Taxillus chinensis | Helixanthera parasitica | Macrosolen cochinchinensis | Schoepfia jasminodora | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Size (bp) | ||||||||||||
| Total | 131,016 | 126,064 | 128,921 | 140,666 | 147,253 | 147,461 | 122,562 | 121,363 | 124,881 | 122,986 | 118,743 | |
| Protein-coding genes | 66,691 | 66,562 | 64,231 | 64,624 | 66,500 | 66,254 | 63,593 | 63,722 | 64,010 | 64,100 | 58,362 | |
| tRNA genes | 2,735 | 2,662 | 2,736 | 2,792 | 2,656 | 2,792 | 2,139 | 2,141 | 2,287 | 2,287 | 2,559 | |
| rRNA genes | 9,034 | 9,044 | 9,050 | 9,068 | 9,044 | 9,049 | 9,072 | 9,072 | 9,062 | 9,052 | 9,050 | |
| introns | 15,314 | 15,278 | 17,161 | 16,434 | 16,648 | 10,052 | 6,991 | 6,858 | 9,091 | 8,694 | 13,971 | |
| IGSs | 37,242 | 32,518 | 35,743 | 47,748 | 52,405 | 59,314 | 40,767 | 39,570 | 40,431 | 38,853 | 34,801 | |
| LSC | 75,814 | 73,226 | 73,893 | 81,684 | 84,601 | 83,505 | 70,630 | 70,357 | 73,043 | 69,018 | 84,168 | |
| SSC | 9,014 | 8,628 | 8,632 | 10,870 | 13,972 | 7,806 | 6,102 | 6,082 | 6,334 | 6,144 | 9,763 | |
| one IR | 23,094 | 22,105 | 23,198 | 24,056 | 24,340 | 28,075 | 22,915 | 22,462 | 22,752 | 23,912 | 12,406 | |
| Number of genes | ||||||||||||
| Total | 99 | 98 | 96 | 101 | 101 | 101 | 91 | 91 | 92 | 92 | 101 | |
| Protein-coding genes | 66 | 66 | 64 | 67 | 67 | 67 | 64 | 64 | 63 | 63 | 69 | |
| tRNA genes | 29 | 28 | 28 | 30 | 30 | 30 | 23 | 23 | 25 | 25 | 28 | |
| rRNA genes | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | |
| Genes with introns | 15 | 14 | 14 | 16 | 16 | 16 | 8 | 8 | 10 | 10 | 14 | |
| % GC content | ||||||||||||
| Total | 36.2 | 36.4 | 36.4 | 37.8 | 37.7 | 37.4 | 37.3 | 37.3 | 36.5 | 36.6 | 38.1 | |
| LSC | 33.3 | 33.6 | 33.5 | 35.5 | 35.6 | 35.3 | 34.7 | 34.7 | 33.8 | 33.6 | 36.1 | |
| SSC | 24.2 | 24.0 | 24.8 | 29.7 | 31.2 | 27.9 | 26.2 | 26.2 | 25.5 | 24.4 | 30.7 | |
| IRs | 43.2 | 43.4 | 43.2 | 43.7 | 43.1 | 41.9 | 42.8 | 43.0 | 42.3 | 42.2 | 47.9 | |
| % Coding regions | ||||||||||||
| Total | 59.8 | 62.1 | 59.1 | 54.4 | 53.5 | 53.0 | 61.1 | 61.8 | 60.3 | 61.3 | 59.0 | |
| Protein-coding genes | 50.9 | 52.8 | 49.8 | 45.9 | 45.2 | 44.9 | 51.9 | 52.5 | 51.3 | 52.1 | 49.2 | |
| tRNA genes | 2.1 | 2.1 | 2.1 | 2.0 | 1.8 | 1.9 | 1.7 | 1.8 | 1.8 | 1.9 | 2.2 | |
| rRNA genes | 6.9 | 7.2 | 7.0 | 6.4 | 6.1 | 6.1 | 7.4 | 7.5 | 7.3 | 7.4 | 7.6 | |
Loss of genes throughout Santalales phylogeny
To confirm the phylogenetic relationships among Santalales species, a phylogenetic tree was constructed using 58 concatenated protein-coding genes common to all taxa (Fig 1). As in previous studies of global angiosperm phylogeny [34, 35], monophyly of the Santalales was well supported, with a bootstrap value of 100%. All branches were supported by 100% bootstrap values, except for the Loranthaceae clade, which had a 99% bootstrap value. Dendrotrophe varians of the Amphorogynaceae was sister to the Viscaceae, and Schoepfia jasminodora of the Schoepfiaceae was sister to the Loranthaceae. A clade made up of Loranthaceae and Schoepfiaceae was sister to a clade consisting of Viscaceae, Amphorogynaceae, Santalaceae, and Opiliaceae. The functional loss of all ndh genes occurred in all 11 Santalales species. Depending on the species, the ndh genes were completely lost, or several of the ndh genes have recognized sequences as pseudogenes or partial sequence of genes. Six genes have been lost (rpl32, rps15, rps16, trnA-UGC, trnI-GAU, and trnK-UUU) in the Loranthaceae. Parallel gene loss or pseudogenization of infA was detected in some branches. The gene trnG-UCC was lost independently in Viscum crassulae, Viscum album, and the clade of Loranthaceae and Schoepfiaceae. Ten pseudogenes were detected in Santalales cp genomes. The ψccsA and ψmatK genes are in the Viscum album cp genome. The ψinfA genes are in the Dendrotrophe varians, Macrosolen cochinchinensis, and Osyris alba cp genomes. The ψndhB, ψndhC, ψndhD, ψndhE, and ψndhK genes are in Osyris alba cp genomes. The ψrpl16 genes are in two Taxillus species and Helixanthera parasitica cp genomes. The ψtrnL-CAA is in the Schoepfia jasminodora cp genome.
Fig 1. Maximum likelihood tree resulting from the 58 concatenated protein-coding genes common to 14 representative taxa.

The BI tree had identical topology with the ML tree. The GTR+I+G model was selected based on Modeltest. The bootstrap support values of the ML tree and the posterior probabilities of the BI tree at all nodes are 100% and 1.00, respectively, except for one node marked by an asterisk, which has a value of 99% and probability of 1.00. ψ, pseudogene; †, partial gene sequence.
Evolutionary dynamics of inverted repeat regions
We compared IR/LSC and IR/SSC junctions in 11 Santalales cp genomes (Fig 2). Santalales species show various sizes of IRs, caused by the evolutionary dynamics of expansion and contraction. They commonly have six duplicated genes (rrn16, rrn23, rrm4.5, rrn5, rps7, and trnV-GAC) and two exons of rps12 in their IRs. All cp genomes except Dendrotrophe varians and Osyris alba have truncated ycf1 genes, which are produced by the presence of IR/SSC junctions within the ycf1 genes. The gene number and order of the IRs in Loranthaceae cp genomes are identical. The IR/LSC junctions of all cp genomes of Loranthaceae species are in exon 2 of rpl2 genes. In the Viscaceae, three Viscum have similar gene numbers and order of genes. The Viscum album cp genome has the most expanded IRs toward the LSC. After expanding from the IRB to the LSC up to the point between the rps3 and rpl22 genes, the IRA extends up to the trnH-GUG gene. The IRs of the cp genomes of Viscum crassulae and Viscum minimum do not include the trnH-GUG genes. The Champereia manillana cp genome, which has the largest IRs (28,075 bp), also has the largest truncated ycf1 gene (2,777 bp) at the junction of IRB/SSC. The Schoepfia jasminodora cp genome has the most highly contracted IRs (12,406 bp) and is the only cp genome in which the IRs do not include duplicated ycf2 genes.
Fig 2. Comparison of the SSC-IR and LSC-IR boundary regions across 11 Santalales species.

Gene content of hemiparasitic plants
Gene content of complete cp genomes of all hemiparasitic plants published to date, including one autotrophic plant Arabidopsis thaliana and two Cuscuta species that are considered ‘photosynthetic holoparasites’ or hemiparasites, are summarized in Fig 3. Among all hemiparasitic plants deposited in GenBank, the cp genomes of Bartsia inaequalis and Pedicularis ishidoyana were excluded due to being incomplete sequences. All genes were categorized into 8 different functional groups. Among the eight functional gene groups, the NADH dehydrogenase gene group is highly variable, with intactness, pseudogenization, or complete loss of genes, while the rRNA gene and ATP synthase groups are intact in all species. All hemiparasitic species except the species of Orobanchaceae share the functional loss of whole ndh genes. In the Santalales, the cp genome of Osyris alba has several ndh pseudogenes (ndhB, ndhC, ndhD, ndhE, and ndhK) [28, 29], but the cp genomes of other species lost all ndh genes completely or have only a partial sequence of ndhA or ndhB gene. The Dendrotrophe varians cp genome has only the exon 2 fragment of the ndhB gene, but the Helixanthera parasitica and Macrosolen cochinchinensis cp genomes have only the exon 1 fragment. Three Viscum cp genomes are composed of exon 1 and exon 2 fragments of ndhB gene.
Fig 3. Comparison of the cp genome gene content of 16 hemiparasitic plants published to date with one autotrophic plant and two Cuscuta species, based on Funk, H. T., et al. (2007), McNeal, J. R., et al. (2007), Wicke, S., et al. (2013), Petersen, G., et al. (2015), Rossetto, M., et al. (2015), Fan, W., et al. (2016), Su, H. -J. and J. -M. Hu (2016), Wicke, S., et al. (2016), Li, Y., et al. (2017) and Yang, G. -S., et al. (2017).

In the case of Striga hemonthica, pseudogene presence was not confirmed in a previous paper. Black/gray/white boxes indicate each gene present and are considered functional/pseudogene or fragment of gene present/gene absent. AU, autotroph; HE, hemiparasite; HO, holoparasite; O, obligate parasite; F, facultative parasite; S, stem parasite; R, root parasite.
We categorized each species as autotroph/hemiparasite/holoparasite, obligate/facultative parasite, and stem/root parasite according to ecological and morphological characteristics. The one facultative root parasite, Triphysaria versicolor of the Orobanchaceae, has all ndh genes intact, while the other facultative and root parasites, Champereia manillana and Osyris alba of the Santalales, have no intact ndh genes. The obligate and root parasites functionally lost all ndh genes or have several intact ndh genes. All obligate and stem parasites show the functional loss of all ndh genes, but they are different in the degree of gene degradation. In the case of other genes, there is no correlation of parasitic types (obligate/facultative parasite and stem/root parasite) and degree of gene degradation.
Discussion
In the order Santalales, which is a large order including most hemiparasites, more studies on its molecular phylogeny and genomics are needed [31, 32, 38]. The cp genomes of hemiparasitic plants have smaller genetic changes than those of holoparasitic plants, but studies of hemiparasites are important for understanding the evolutionary transition from autotrophs to parasites. In previous molecular phylogenetic studies, the evolutionary relationships of Santalales species have been confirmed by plastid genes (accD, matK, rbcL, and trnL-F), nuclear genes (SSU rDNA, LSU rDNA, and RPB2), and a mitochondrial gene (matR) [4, 31, 32, 36]. In this study, most protein-coding genes in the cp genomes of Santalales species were used to confirm the phylogeny of 11 Santalales species belonging to six families. The topologies of the six families are the same as the topologies in the recently revised classification system based on molecular and morphological data [38]. We confirmed that Dendrotrophe varians of Amphorogynaceae is sister to the three Viscum species of Viscaceae, and that Helixanthera parasitica and Macrosolen cochinchinensis form a clade with two Taxillus species to make up the family Loranthaceae (Fig 1).
The 11 Santalales species have a slightly reduced number of genes in their cp genomes compared to typical angiosperm cp genomes (Table 1) [14]. Common losses of some genes are related to phylogenetic relationships. Some losses of genes in each clade, such as the common losses of the rpl32, rps15, rps16, A-UGC, I-GAU, and K-UUU genes in the Loranthaceae, are considered important characteristics of families, genera, or species (Figs 1 and 3). However, the genes lost in Santalales cp genomes are also lost in other angiosperm lineages and represent phylogenetically independent losses [52]. The parallel loss and pseudogenization of ndh genes and various fragments that are composed of exon 1, exon 2, or both exons of the ndhB gene were detected independently in the phylogeny of Santalales. The rpl32, trnG-UCC, and infA genes were also independently lost or pseudogenized. The gene losses in hemiparasitic Santalales cp genomes are not as extreme as those in holoparasitic plant cp genomes, which have few or no genes [22, 23, 25]. These gradual losses enhanced the understanding about evolutionary history in hemiparasitic Santalales cp genomes. The families of Santalales have a confusing taxonomic history [38, 53–59]. To further clarify phylogenetic relationships among Santalales families, genera, and species, more morphological and molecular data of taxa belonging to all Santalales families are required. Additionally, the cp genomes of autotrophic Santalales species are needed for more in-depth analysis of the evolutionary transition from autotrophs to parasites.
Generally, closely related species tend to have similar IR boundaries with few changes [60–62]. The cp genomes of Santalales species have similar IR boundaries in that the IRB/LSC junctions are near the rpl2, rps19, and rpl22 genes except for the Viscum album and Schoepfia jasminodora cp genomes (Table 1 and Fig 2). Expansions from the IRB to the LSC up to rpl2 are common in angiosperms, and expansions up to rps19 or rpl22 are often found in vascular plants [60]. The IR/LSC and IR/SSC junctions of all four Loranthaceae species are located at the same positions, which are within exon2 of rpl2 and ycf1. Most angiosperms have 20–22 kb of one IR [63, 64]. The size of one IR in four species of Loranthaceae and Dendrotrophe varians of the Amphorogynaceae is slightly bigger than that of most angiosperms. In the Santalales, the cp genome of Schoepfia jasminodora has highly contracted IRs (12,406 bp), which are reduced until the IRs do not include the ycf2 gene (Fig 2). Goulding et al. (1996) suggested two IR expansion models. The first model is a single-strand break and gene conversion leading to a small IR expansion, and the second model is a double-strand break resulting in a larger IR expansion [62]. Among 11 Santalales species, the cp genomes of Osyris alba and Viscum minimum have large inversions and a general structure consisting of two IRs (IRA and IRB) and two single copy regions (LSC and SSC) [29]. These may be caused by mechanisms such as mediation of duplicated tRNA genes [65, 66], mediation of dispersed short repeats [67–70], and HR (homologous recombination) between more than 200 bp inverted repeats [11]. The two IRs of land plant plastomes typically contain four rRNA genes (rrn16, rrn23, rrn4.5 and rrn5) and five tRNA genes (trnA-UGC, trnI-GAU, trnN-GUU, trnR-ACG, and trnV-GAC). In the Loranthaceae, the IRs do not include the intron-containing tRNA genes trnA-UGC and trnI-GAU. Large structural changes in the plastome are evolutionarily and phylogenetically important characteristics in land plants [71]. The Santalales cp genomes also show various large structural changes such as gene deletion, IR expansion, and large inversions. Future phylogenetic studies in the Santalales should analyze additional closely related species and their structural genomic changes.
The ndh complex is the only group (among eight functional groups) that has functionally lost all genes in Santalales. Like the cp genomes of previously reported Santalaceae species, Dendrotrophe varians, Helixanthera parasitica, and Macrosolen cochinchinensis cp genomes have functionally lost all ndh genes. This supports the idea that the functional loss of ndh genes is the initial stage in the transition from autotrophs to parasites [16, 27, 28]. The loss of ndh genes has occurred independently multiple times in angiosperms [52]. It is not only found in various parasitic plants with 12 independent origins, but also in several non-parasitic lineages: the Pinaceae, Gnetales, Orchidaceae, etc. The ndh complex is considered inessential to the plant cp genome, based on previous studies examining varying degrees of loss and pseudogenization in the ndh complex [16, 52, 72].
We categorized the hemiparasitic species as obligate/facultative and stem/root parasites to examine the correlation between parasitic types and gene content of hemiparasitic plants. Unlike an obligate parasite, a facultative parasite can survive without a host, like an autotroph, and is an opportunistic parasite [73]. Its cp genome may be expected to be similar in size and gene number to that of autotrophs. Four facultative hemiparasites, Castilleja paramensis, Champereia manillana, Osyris alba, and Triphysaria versicolor, have different degrees of ndh gene loss, ranging from containing all genes (Triphysaria versicolor) to none (Champereia manillana and Osyris alba). Additionally, the Champereia manillana cp genome lost infA and rpl32 genes, and the infA gene of the Osyris alba cp genome was pseudogenized. The degrees of gene loss and pseudogenization in obligate hemiparasites also vary. The range of cp genome size of four facultative hemiparasites is from 147,253 to 152,448 bp, which is included in the range for obligate hemiparasites (from 121,363 to 160,910 bp). There is no tendency for gene content or cp genome size to be related with parasitic type of obligate or facultative parasitism. A common change in gene content in stem or root parasites was also not detected. From this, it may be concluded that the degrees of change in cp genome size and gene content of hemiparasitic plants are not correlated with parasitic type.
Supporting information
Genes drawn inside the circle are transcribed clockwise, while those drawn outside the circle are transcribed counterclockwise. Dark and light gray bars of the inner circle are graphs of GC and AT content, respectively. ψ, pseudogene; †, truncated gene at the LSC/IR or SSC/IR junction. The infA gene marked by an asterisk is a pseudogene in the M. cochinchinensis cp genome. The rpl16 gene marked by two asterisks is a pseudogene in the H. parasitica cp genome. The rpl36 gene marked by three asterisks is lost in the M. cochinchinensis cp genome.
(JPG)
{kind=link}
(PDF)
+, gene present and considered functional; -, gene not present or not functional. ψ, pseudogene; †, partial gene sequence including truncation at the LSC/IR or SSC/IR junction. Gray shaded boxes indicate genes in IRs.
(XLSX)
Acknowledgments
We would like to thank Mr. Chhang Phourin (Forestry Administration of Cambodia) and Dr. Seong-Hyun Cho (International Biological Material Research Center, Korea Research Institute of Bioscience and Biotechnology) for plant material of Dendrotrophe varians, and we thank Dr. Tran The Bach (Institute of Ecology and Biological Resources) for plant material of Helixanthera parasitica and Macrosolen cochinchinensis. We also thank Prof. Richard Whitkus (Sonoma State University) and Dr. Elizabeth Hassell Kern (Ewha Womans University) for proof-reading the manuscript.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This research was supported by the BK21 Plus Program (Creative Academy of Eco Science, 31Z20130012990) funded by the Ministry of Education and National Research Foundation of Korea. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Kuijt J. Biology of parasitic flowering plants University of California Press: Berkeley and Los Angeles; 1969. [Google Scholar]
- 2.Westwood JH, Yoder JI, Timko MP, dePamphilis CW. The evolution of parasitism in plants. Trends in Plant Sci. 2010;15(4): 227–235. doi: 10.1016/j.tplants.2010.01.004 [DOI] [PubMed] [Google Scholar]
- 3.Nickrent DL. The parasitic plant connection 1997. [cited 09 Nobemver 2015]. Southern Illinois University; Available from: http://parasiticplants.siu.edu/ [Google Scholar]
- 4.Su H-J, Hu J-M, Anderson FE, Der JP, Nickrent DL. Phylogenetic relationships of Santalales with insights into the origins of holoparasitic Balanophoraceae. Taxon. 2015;64(3): 491–506. [Google Scholar]
- 5.Irving LJ, Cameron DD. Chapter 3 You are what you eat: interactions between root parasitic plants and their hosts. Adv Bot Res. Volume 50: Academic Press. 2009. pp 87–138. [Google Scholar]
- 6.dePamphilis CW, Palmer JD. Loss of photosynthetic and chlororespiratory genes from the plastid genome of a parasitic flowering plant. Nature. 1990;348: 337–339. doi: 10.1038/348337a0 [DOI] [PubMed] [Google Scholar]
- 7.McNeal JR, Kuehl JV, Boore JL, dePamphilis CW. Complete plastid genome sequences suggest strong selection for retention of photosynthetic genes in the parasitic plant genus Cuscuta. BMC Plant Biol. 2007;7(1): 57 doi: 10.1186/1471-2229-7-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krause K. From chloroplasts to “cryptic” plastids: evolution of plastid genomes in parasitic plants. Curr Genet. 2008;54(3): 111 doi: 10.1007/s00294-008-0208-8 [DOI] [PubMed] [Google Scholar]
- 9.Wicke S, Müller KF, dePamphilis CW, Quandt D, Bellot S, Schneeweiss GM. Mechanistic model of evolutionary rate variation en route to a nonphotosynthetic lifestyle in plants. Proc Natl Acad Sci U S A. 2016;113(32): 9045–9050. doi: 10.1073/pnas.1607576113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shinozaki K, Ohme M, Tanaka M, Wakasugi T, Hayashida N, Matsubayashi T, et al. The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J. 1986;5(9): 2043–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palmer JD. Plastid chromosomes: structure and evolution. The Molecular Biology of Plastids. 1991;7: 5–53. [Google Scholar]
- 12.Jansen RK, Cai Z, Raubeson LA, Daniell H, Leebens-Mack J, Müller KF, et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci U S A. 2007;104(49): 19369–19374. doi: 10.1073/pnas.0709121104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barrett CF, Freudenstein JV, Li J, Mayfield-Jones DR, Perez L, Pires JC, et al. Investigating the path of plastid genome degradation in an early-transitional clade of heterotrophic orchids, and implications for heterotrophic angiosperms. Mol Biol Evol. 2014;31(12): 3095–3112. doi: 10.1093/molbev/msu252 [DOI] [PubMed] [Google Scholar]
- 14.Ruhlman TA, Jansen RK. The plastid genomes of flowering plants. Chloroplast Biotechnology: Methods and Protocols. 2014. pp. 3–38. [DOI] [PubMed] [Google Scholar]
- 15.Su H-J, Hu J-M. The complete chloroplast genome of hemiparasitic flowering plant Schoepfia jasminodora. Mitochondrial DNA B Resour. 2016;1(1): 767–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wicke S, Müller KF, dePamphilis CW, Quandt D, Wickett NJ, Zhang Y, et al. Mechanisms of functional and physical genome reduction in photosynthetic and nonphotosynthetic parasitic plants of the Broomrape family. Plant Cell. 2013;25(10): 3711–3725. doi: 10.1105/tpc.113.113373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bellot S, Cusimano N, Luo S, Sun G, Zarre S, Gröger A, et al. Assembled plastid and mitochondrial genomes, as well as nuclear genes, place the parasite family Cynomoriaceae in the Saxifragales. Genome Biol Evol. 2016;8(7): 2214–2230. doi: 10.1093/gbe/evw147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X, Zhang T-C, Qiao Q, Ren Z, Zhao J, Yonezawa T, et al. Complete chloroplast genome sequence of holoparasite Cistanche deserticola (Orobanchaceae) reveals gene loss and horizontal gene transfer from its host Haloxylon ammodendron (Chenopodiaceae). PLoS One. 2013;8(3): e58747 doi: 10.1371/journal.pone.0058747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Funk HT, Berg S, Krupinska K, Maier UG, Krause K. Complete DNA sequences of the plastid genomes of two parasitic flowering plant species, Cuscuta reflexa and Cuscuta gronovii. BMC Plant Biol. 2007;7(1): 45 doi: 10.1186/1471-2229-7-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cusimano N, Wicke S. Massive intracellular gene transfer during plastid genome reduction in nongreen Orobanchaceae. New Phytol. 2016;210(2): 680–93. doi: 10.1111/nph.13784 [DOI] [PubMed] [Google Scholar]
- 21.Wolfe KH, Mordent CW, Ems SC, Palmer JD. Rapid evolution of the plastid translational apparatus in a nonphotosynthetic plant: loss or accelerated sequence evolution of tRNA and ribosomal protein genes. J Mol Evol. 1992;35(4): 304–317. doi: 10.1007/bf00161168 [DOI] [PubMed] [Google Scholar]
- 22.Bellot S, Renner SS. The plastomes of two species in the endoparasite genus Pilostyles (Apodanthaceae) each retain just five or six possibly functional genes. Genome Biol Evol. 2016;8(1): 189–201. doi: 10.1093/gbe/evv251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roquet C, Coissac É, Cruaud C, Boleda M, Boyer F, Alberti A, et al. Understanding the evolution of holoparasitic plants: the complete plastid genome of the holoparasite Cytinus hypocistis (Cytinaceae). Ann Bot. 2016;118(5): 885–96. doi: 10.1093/aob/mcw135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naumann J, Der JP, Wafula EK, Jones SS, Wagner ST, Honaas LA, et al. Detecting and characterizing the highly divergent plastid genome of the nonphotosynthetic parasitic plant Hydnora visseri (Hydnoraceae). Genome Biol Evol. 2016;8(2): 345–363. doi: 10.1093/gbe/evv256 PMC4779604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Molina J, Hazzouri KM, Nickrent D, Geisler M, Meyer RS, Pentony MM, et al. Possible loss of the chloroplast genome in the parasitic flowering plant Rafflesia lagascae (Rafflesiaceae). Mol Biol Evol. 2014;31(4): 793–803. doi: 10.1093/molbev/msu051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu C-S, Wang T-J, Wu C-W, Wang Y-N, Chaw S-M. Plastome evolution in the sole hemiparasitic genus laurel dodder (Cassytha) and insights into the plastid phylogenomics of Lauraceae. Genome Biol Evol. 2017;9(10): 2604–2614. doi: 10.1093/gbe/evx177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barrett CF, Davis JI. The plastid genome of the mycoheterotrophic Corallorhiza striata (Orchidaceae) is in the relatively early stages of degradation. Am J Bot. 2012;99(9): 1513–1523. doi: 10.3732/ajb.1200256 [DOI] [PubMed] [Google Scholar]
- 28.Fan W, Zhu A, Kozaczek M, Shah N, Pabón-Mora N, González F, et al. Limited mitogenomic degradation in response to a parasitic lifestyle in Orobanchaceae. Sci Rep. 2016;6: 36285 doi: 10.1038/srep36285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petersen G, Cuenca A, Seberg O. Plastome evolution in hemiparasitic mistletoes. Genome Biol Evol. 2015;7(9): 2520–2532. doi: 10.1093/gbe/evv165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, Zhou J-g, Chen X-l, Cui Y-x, Xu Z-c, Li Y-h, et al. Gene losses and partial deletion of small single-copy regions of the chloroplast genomes of two hemiparasitic Taxillus species. Sci Rep. 2017;7 doi: 10.1038/s41598-017-13401-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malecot V, Nickrent DL. Molecular phylogenetic relationships of Olacaceae and related Santalales. Syst Bot. 2008;33(1): 97–106. [Google Scholar]
- 32.Der JP, Nickrent DL. A molecular phylogeny of Santalaceae (Santalales). Syst Bot. 2008;33(1): 107–16. [Google Scholar]
- 33.Soltis DE, Soltis PS, Chase MW, Mort ME, Albach DC, Zanis M, et al. Angiosperm phylogeny inferred from 18S rDNA, rbcL, and atpB sequences. Bot J Linn Soc. 2000;133(4): 381–461. [Google Scholar]
- 34.Hilu KW, Borsch T, Müller K, Soltis DE, Soltis PS, Savolainen V, et al. Angiosperm phylogeny based on matK sequence information. Am J Bot. 2003;90(12): 1758–1776. doi: 10.3732/ajb.90.12.1758 [DOI] [PubMed] [Google Scholar]
- 35.Soltis DE, Senters AE, Zanis MJ, Kim S, Thompson JD, Soltis PS, et al. Gunnerales are sister to other core eudicots: implications for the evolution of pentamery. Am J Bot. 2003;90(3): 461–470. doi: 10.3732/ajb.90.3.461 [DOI] [PubMed] [Google Scholar]
- 36.Vidal-Russell R, Nickrent DL. Evolutionary relationships in the showy mistletoe family (Loranthaceae). Am J Bot. 2008;95(8): 1015–1029. doi: 10.3732/ajb.0800085 [DOI] [PubMed] [Google Scholar]
- 37.Vidal-Russell R, Nickrent DL. The first mistletoes: origins of aerial parasitism in Santalales. Mol Phylogenet Evol. 2008;47(2): 523–537. doi: 10.1016/j.ympev.2008.01.016 [DOI] [PubMed] [Google Scholar]
- 38.Nickrent DL, Malécot V, Vidal-Russell R, Der JP. A revised classification of Santalales. Taxon. 2010;59(2): 538–558. [Google Scholar]
- 39.Yang G-S, Wang Y-H, Wang Y-H, Shen S-K. The complete chloroplast genome of a vulnerable species Champereia manillana (Opiliaceae). Conserv Genet Resour. 2017;9(3): 415–418. doi: 10.1007/s12686-017-0697-1 [Google Scholar]
- 40.Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin. 1987;19: 11–15. doi: citeulike-article-id:678648. [Google Scholar]
- 41.Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29(22): 4633–4642. doi: 10.1093/nar/29.22.4633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20(17): 3252–3255. doi: 10.1093/bioinformatics/bth352 [DOI] [PubMed] [Google Scholar]
- 43.Thomas AH. Bioedit: a user-friendly biological sequence alignment editor and analysis. Nucleic Acids Symp Ser. 1999;41: 95–98. [Google Scholar]
- 44.Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5): 955–964. doi: 10.1093/nar/25.5.0955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lohse M, Drechsel O, Bock R. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet. 2007;52(5–6): 267–274. doi: 10.1007/s00294-007-0161-y [DOI] [PubMed] [Google Scholar]
- 46.Lohse M, Drechsel O, Kahlau S, Bock R. OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013;41(W1): W575–W581. doi: 10.1093/nar/gkt289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7): 1394–1403. doi: 10.1101/gr.2289704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5): 1792–1797. doi: 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9): 1312–1313. doi: 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14(9): 817–818. doi: 10.1093/bioinformatics/14.9.817 [DOI] [PubMed] [Google Scholar]
- 51.Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17(8): 754–755. doi: 10.1093/bioinformatics/17.8.754 [DOI] [PubMed] [Google Scholar]
- 52.Wicke S, Schneeweiss GM, dePamphilis CW, Müller KF, Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol. 2011;76(3): 273–297. doi: 10.1007/s11103-011-9762-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuijt J. Mutual affinities of Santalalean families. Brittonia. 1968;20(2): 136–147. [Google Scholar]
- 54.Kuijt J, Hansen B. Flowering plants. Eudicots: Santalales, Balanophorales: Springer; 2014. [Google Scholar]
- 55.Malecot V. Histoire, classification et phylogénie des Olacaceae Brown (Santalales): Paris 6; 2002.
- 56.Peng Y, Chen Z, Gong X, Zhong Y, Shi S. Phylogenetic position of Dipentodon sinicus: evidence from DNA sequences of chloroplast rbcL, nuclear ribosomal 18S, and mitochondria matR genes. Botanical Bulletin of Academia Sinica. 2003;44. [Google Scholar]
- 57.Reed CF. The comparative morphology of the Olacaceae, Opiliaceae and Octoknemaceae. Mem Soc Brot. 1955;10: 29–79. [Google Scholar]
- 58.Wurdack KJ, Davis CC. Malpighiales phylogenetics: gaining ground on one of the most recalcitrant clades in the angiosperm tree of life. Am J Bot. 2009;96(8): 1551–1570. doi: 10.3732/ajb.0800207 [DOI] [PubMed] [Google Scholar]
- 59.Xiang QY, Moody ML, Soltis DE, zhu Fan C, Soltis PS. Relationships within Cornales and circumscription of Cornaceae—matK and rbcL sequence data and effects of outgroups and long branches. Mol Phylogenet Evol. 2002;24(1): 35–57. [DOI] [PubMed] [Google Scholar]
- 60.Zhu A, Guo W, Gupta S, Fan W, Mower JP. Evolutionary dynamics of the plastid inverted repeat: the effects of expansion, contraction, and loss on substitution rates. New Phytol. 2016;209(4): 1747–1756. doi: 10.1111/nph.13743 [DOI] [PubMed] [Google Scholar]
- 61.Wang R-J, Cheng C-L, Chang C-C, Wu C-L, Su T-M, Chaw S-M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol Biol. 2008;8(1): 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goulding SE, Wolfe K, Olmstead R, Morden C. Ebb and flow of the chloroplast inverted repeat. Mol Gen Genet. 1996;252(1–2): 195–206. [DOI] [PubMed] [Google Scholar]
- 63.Sugiura M. The chloroplast genome 10 Years Plant Molecular Biology: Springer; 1992. pp. 149–168. [DOI] [PubMed] [Google Scholar]
- 64.Palmer JD. Comparative organization of chloroplast genomes. Ann Rev Genet. 1985;19 doi: 10.1146/annurev.ge.19.120185.001545 [DOI] [PubMed] [Google Scholar]
- 65.Haberle RC, Fourcade HM, Boore JL, Jansen RK. Extensive rearrangements in the chloroplast genome of Trachelium caeruleum are associated with repeats and tRNA genes. J Mol Evol. 2008;66(4): 350–361. doi: 10.1007/s00239-008-9086-4 [DOI] [PubMed] [Google Scholar]
- 66.Hiratsuka J, Shimada H, Whittier R, Ishibashi T, Sakamoto M, Mori M, et al. The complete sequence of the rice (Oryza sativa) chloroplast genome: intermolecular recombination between distinct tRNA genes accounts for a major plastid DNA inversion during the evolution of the cereals. Mol Gen Genet. 1989;217(2): 185–194. [DOI] [PubMed] [Google Scholar]
- 67.Ogihara Y, Terachi T, Sasakuma T. Intramolecular recombination of chloroplast genome mediated by short direct-repeat sequences in wheat species. Proc Natl Acad Sci U S A. 1988;85(22): 8573–8577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Greiner S, Wang X, Rauwolf U, Silber MV, Mayer K, Meurer J, et al. The complete nucleotide sequences of the five genetically distinct plastid genomes of Oenothera, subsection Oenothera: I. sequence evaluation and plastome evolution. Nucleic Acids Res. 2008;36(7): 2366–2378. doi: 10.1093/nar/gkn081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guisinger MM, Kuehl JV, Boore JL, Jansen RK. Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: rearrangements, repeats, and codon usage. Mol Biol Evol. 2010;28(1): 583–600. doi: 10.1093/molbev/msq229 [DOI] [PubMed] [Google Scholar]
- 70.Day A, Madesis P. DNA replication, recombination, and repair in plastids Cell and molecular biology of plastids. Springer; 2007. pp. 65–119. [Google Scholar]
- 71.Raubeson LA, Jansen RK. Chloroplast genomes of plants, plant diversity and evolution: genotypic and phenotypic variation in higher plants CABI Publishing; 2005. doi: 10.1079/9780851999043.0045 [Google Scholar]
- 72.Martín M, Sabater B. Plastid ndh genes in plant evolution. Plant Physiol Biochem. 2010;48(8): 636–645. doi: 10.1016/j.plaphy.2010.04.009 [DOI] [PubMed] [Google Scholar]
- 73.Nickrent DL. Phylogenetic origins of parasitic plants Parasitic plants of the Iberian Peninsula and Balearic islands. Mundi-Prensa Libros, S. A. 2002. pp. 29–56. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genes drawn inside the circle are transcribed clockwise, while those drawn outside the circle are transcribed counterclockwise. Dark and light gray bars of the inner circle are graphs of GC and AT content, respectively. ψ, pseudogene; †, truncated gene at the LSC/IR or SSC/IR junction. The infA gene marked by an asterisk is a pseudogene in the M. cochinchinensis cp genome. The rpl16 gene marked by two asterisks is a pseudogene in the H. parasitica cp genome. The rpl36 gene marked by three asterisks is lost in the M. cochinchinensis cp genome.
(JPG)
(PDF)
+, gene present and considered functional; -, gene not present or not functional. ψ, pseudogene; †, partial gene sequence including truncation at the LSC/IR or SSC/IR junction. Gray shaded boxes indicate genes in IRs.
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.