

Abstract
Pyruvate dehydrogenase complex (PDC) is a large multienzyme complex that catalyzes the irreversible conversion of pyruvate to acetyl-coenzyme A with reduction of NAD+. Distinctive from PDCs in lower forms of life, in mammalian PDC, dihydrolipoyl acetyltransferase (E2; E2p in PDC) and dihydrolipoamide dehydrogenase binding protein (E3BP) combine to form a complex that plays a central role in the organization, regulation, and integration of catalytic reactions of PDC. However, the atomic structure and organization of the mammalian E2p/E3BP heterocomplex are unknown. Here, we report the structure of the recombinant dodecahedral core formed by the C-terminal inner-core/catalytic (IC) domain of human E2p determined at 3.1 Å resolution by cryo electron microscopy (cryoEM). The structure of the N-terminal fragment and four other surface areas of the human E2p IC domain exhibit significant differences from those of the other E2 crystal structures, which may have implications for the integration of E3BP in mammals. This structure also allowed us to obtain a homology model for the highly homologous IC domain of E3BP. Analysis of the interactions of human E2p or E3BP with their adjacent IC domains in the dodecahedron provides new insights into the organization of the E2p/E3BP heterocomplex and suggests a potential contribution by E3BP to catalysis in mammalian PDC.
Graphical Abstract
INTRODUCTION
The mammalian pyruvate dehydrogenase complex (PDC) is a large and highly organized multienzyme system found in both prokaryotes and eukaryotes. PDC, as well as 2-oxoglutarate dehydrogenase complex (OGDC) and branched-chain α-keto acid dehydrogenase complex (BCDC), belongs to the α-keto acid dehydrogenase complex family for which complexes range from 4 to 10 million Da in total molecular weight and share common features in the organization of subunits.1–3 These complexes catalyze the oxidative decarboxylation of α-keto acids to yield acyl-CoA and NADH. In particular, the reaction that PDC catalyzes links glycolysis to the citric acid cycle and to fatty acid biosynthesis. Diminished PDC activity due to defects in PDC components causes metabolic acidosis, serious neurologic defects, and other genetic and physiological disorders.4 PDC is inactivated by a set of pyruvate dehydrogenase kinase (PDK) isoforms and activated by pyruvate dehydrogenase phosphatase (PDP) isoforms.5–8 Decreased PDC activity due to defects in regulatory signaling that alter PDK expression and effector control occurs in many disease states.8–10 Activation of PDC by treatment with PDK inhibitors prevents glucose depletion in insulin-resistant diabetes and damage due to lactic acid in ischemic heart and allows killing of cancer cells undergoing Warburg metabolism.8–14 Consequently, PDC activation is a pivotal target for therapeutic interventions.
PDC has three catalytic components: pyruvate dehydrogenase (E1p), dihydrolipoyl acetyltransferase (E2p), and dihydrolipoyl dehydrogenase (E3).1,2 E1p and E2p are unique to PDC, whereas a common E3 supports all three α-keto acid dehydrogenase complexes in most organisms. E2p forms the core structure of PDC and invariably plays a central role in the organization and the integration of the chemical reactions catalyzed by PDC.2 Multiple copies of E1p and E3 are normally bound to the E2p core. However, the core structure of most eukaryotic PDCs include an additional putatively noncatalytic subunit, E3-binding protein (E3BP, initially protein X),15,16 that specifically binds E3,17–21 whereas prokaryotic and plant PDC and other classes of α-keto acid dehydrogenase complex lack E3BP.22,23 Each mammalian E2p subunit consists of two consecutive N-terminal lipoyl domains (L1 and L2), a small E1p-binding domain (E1pBD), and a C-terminal inner core/catalytic (IC) domain.24 Similarly, mammalian E3BP consists of an N-terminal lipoyl domain (L3), an E3-binding domain (E3BD), and an apparently noncatalytic inner domain.21 The lipoyl domains and the subunit binding domains are connected by extended, mobile linker regions.1,21,22
The core structure of PDC from eukaryotes, Gram-positive bacteria, and Gram-negative Alphaproteobacteria (except out-group) show a dodecahedral organization, whereas most other known Gram-negative bacteria and all OGDCs and BCDCs show 24-meric cubic organization.2,3,22,23,25–33 Atomic-resolution crystal structures of the cubic inner cores from different members of the α-keto acid dehydrogenase complex family, including A. vinelandii PDC, E. coli OGDC, and bovine BCDC, have been reported.27–29 The crystals of the dodecahedral inner cores of E2p from B. stearothermophilus and E. faecalis diffracted to 4.4 Å,32 and a homology model was built based on the high-resolution structure of A. vinelandii E2p.27 These structures show that the IC domains, in different types of α-keto acid dehydrogenase complexes from various prokaryotic and eukaryotic species, have a conserved 3-D fold and trimer-based substructure. This is consistent with their amino acid sequence similarity and the vital function of the α-keto acid dehydrogenase complexes in most species. However, despite rigid conservation of the PDC function, E2p IC domain structures have evolved, as indicated above, with significant diversity in subunit composition, for example, PDC in plants lacks E3BPs and trimer organization (cubic or dodecahedral). Two “substitution” models have been proposed for the E2p/E3BP inner core of mammalian PDC: 48 E2p + 12 E3BP and 40 E2p + 20 E3BP with 12 or 20 copies of E3BP replacing an equivalent number of E2p proteins in the 60-meric inner core.34–37 This highly varied pattern with the eukaryotic E2p IC domains is further complicated by the lack of any high-resolution structures.
In the present study, we used cryo electron microscopy (cryoEM) and single-particle reconstruction to determine a 3.1 Å resolution structure of the pentagonal dodecahedron assemblage of 60 IC domains of recombinant human E2p. The atomic model of the human E2p IC domain shows an appreciable similarity with A. vinelandii PDC, E. coli OGDC, and bovine BCDC structures; however, it also displays marked structural differences primarily on the 3- and 2-fold interfaces. We develop a homology model of the IC domain of human E3BP that shows a conserved structure similar to human E2p but differs in the interface regions between domains, either 3-or 2-fold related. Analysis of possible associations of E2p, E3BP, or mixed subunits of these two suggests a model for the E2p/E3BP core in which E3BP substitutes no more than one E2p within each trimer. While E3BP lacks the His residue to support catalysis by one domain in an active site,21 our homology model for the E3BP IC domain strongly supports that it can perform the other domain role since it contains conserved residues in positions to carry out the primary role in substrate binding within an active site.
MATERIALS AND METHODS
Protein Purification
The sample of the human E2p IC domain (also called human truncated E2p or tE2) was prepared from scE2, which contains a PreScission site in the third linker region, as described previously.33,34 E. coli cells with scE2 plasmid were grown at 37 °C to mid log phase and induced expression by adding 0.5 mM IPTG at 25 °C for 18 h. Harvested cells were disrupted by ultrasonication, and crude extract was fractionated by polyethylene glycol (PEG) 8000 (50 wt %/vol) and polyethylenimine. The active fraction was applied to gel filtration using a Sephacryl S-400 HR column. After assaying for activity and analyzing with SDS-PAGE, those containing high activity was pelleted by ultracentrifugation in a Beckman type 50.1 Ti angle rotor at 35,000 rpm for 4 h at 4 °C. The resuspended pellets were further clarified by centrifugation, and the resulting supernatant was subsequently applied to a Sephacryl S-400 HR column. Fractions (1.1 mL/tube) were monitored according to absorbance at 260 and 280 nm, activity, as well as SDS-PAGE profile. Treatment of scE2 with the PreScission protease (Amersham Biosciences) removed the N-terminal 319 amino acids. The resulting E2p IC domain was purified by gel filtration with a Sephacryl S-300 HR column. The assembly of the recombinant molecules into fully functional, pentagonal, dodecahedral cores was confirmed by a previously described method and by analytical ultra-centrifugation.34,38
CryoEM Sample Preparation and Data Collection
To prepare cryoEM grids, 2.5 μL of sample was applied to a glow-discharged Quantifoil R2/1 grid. The grid was then blotted with filter paper (Whatman #1) to remove excess sample and flash-frozen in liquid ethane with a homemade plunger. The grid was loaded into an FEI Titan Krios electron microscope operated at 300 kV for automated image acquisition with the Leginon software.39 One set of images was recorded on Kodak SO-163 films at a dosage of ~25 e−/Å2 and 59,000× nominal magnification with defocus values ranging from −1.8 to −3.9 μm. The films were digitized with a Nikon Super CoolScan 9000 ED scanner at a step size of 6.35 μm/pixel. The final pixel size was calibrated to 1.14 Å on the sample level using catalase 2D crystals as a standard. A second set of images was recorded with a Gatan K2 Summit direct electron detection camera using the counting mode at 29,000× nominal magnification (calibrated pixel size of 1.02 Å on the sample level) and defocus values ranging from −0.7 to −2.7 μm. The dose rate on the camera was ~8 e− pixel−1 s−1. The total exposure time was 5 s and was fractionated into 25 frames of images with 0.2 s exposure time for each frame. The frame images were aligned and averaged using the GPU-accelerated motion correction program Motioncor.40
Image Processing
For the data acquired with films, a total of 409,191 particles (320 × 320 pixels) were picked from 1,369 micrographs using a combination of the batchboxer in EMAN and the DoGpicker.41,42 The defocus values were determined by CTFFIND,43 and the micrographs were corrected for contrast transfer function (CTF) by phase-flipping with the corresponding defocus and astigmatism values using Bsof t.44 The particles were initially processed using EMAN for an initial 3D reconstruction and refinement enforced with the icosahedral symmetry followed by 2D and 3D classification using RELION.45 The preliminary 3D map generated using EMAN was low-pass filtered to 60 Å to serve as a starting model for the 3D classification of RELION. The 2D class averages and 3D class reconstructions generated by RELION were visually inspected to remove those without high-resolution features. The best 47,768 particles selected from the “good” classes were then sent to 3D autorefinement using RELION. The icosahedral symmetry was applied in the 3D classification and autorefinement of RELION. The final resolution of 4.1 Å was estimated with two independently refined maps from the halves of the data set with the gold-standard FSC at the 0.143 criterion using the relion_postprocess program in RELION.
For the data acquired with the K2 camera, the average images of all frames from each exposure were used for particle picking, defocus determination, and 2D and 3D classification using procedures similar to those for the film data described above. Briefly, a total of 61,418 particles (320 × 320 pixels) manually picked from 1,072 micrographs were subjected to 2D and 3D classification. The “bad” classes that did not have high-resolution features were discarded, and 49,374 particles were combined from the “good” classes for the following 3D autorefinement. For maximizing usable signals from the frame images acquired with the K2 camera, the particles were “polished” following the previously described procedures.46,47 Briefly, 49,374 particles averaged from all frames with whole-image drift correction were first sent to a preliminary 3D autorefinement. Particle images from individual frames were then used to calculate translational alignments for the particle-based drift correction using RELION following the suggested protocol of RELION. Last, particle images from frame 2 to frame 20 (~30 e−/Å2 total dose on sample) were translated using the above optimal alignment and weighted with different B-factors as estimated from the single-frame reconstructions to generate optimal “shiny” average images. The 3D autorefinement enforced with the icosahedral symmetry using these “shiny” particles generated a map at 3.1 Å resolution estimated with two independently refined maps from the halves of the data set with the gold-standard FSC at the 0.143 criterion using the relion_postprocess program in RELION. The local resolution was estimated by ResMap using the two cryoEM maps independently refined from the halves of the data.48
Before visualization and atomic modeling, the cryoEM maps were sharpened by B-factor and low-pass filtered to the stated resolution using the relion_postprocess program.
Atomic Model Building
De novo atomic model building was carried out on the 3.1 Å resolution map using Coot.49 First, we traced an initial backbone using the “C-alpha Baton Mode” tool. Second, we observed bulky side chains in the EM map to assist the amino acid registration. Third, we fitted the side chains into the density using the “Real Space Refine Zone” tool to generate a full atomic model. The torsion, planar, and Ramachandran restraints were enabled to ensure that the structure is in an energetically favorable conformation.
The coarse model from Coot was then refined using PHENIX in a pseudocrystallographic manner.50 Note this procedure only improved the atomic model and did not modify the cryoEM map. Briefly, the cryoEM map was put into an artificial crystal lattice to calculate its structure factor using the em_map_to_h-kl. inp utility program in CNS.51 The amplitudes and phases of the structural factor were used as pseudoexperimental diffraction data for model refinement in PHENIX. The restraints of Ramachandran, secondary structure, and non-crystallographic symmetry were used in the refinement.
The cryoEM maps and atomic model were visualized using UCSF Chimera52 and DeepView/Swiss pdp viewer.53
RESULTS
CryoEM Reconstruction of the Dodecahedral Core of Human E2p
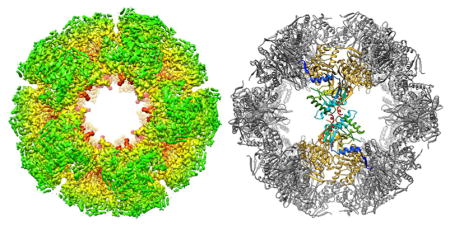
Initially, we acquired cryoEM images of the dodecahedral core of human E2p using photographic films and obtained a 3D reconstruction at 4.1 Å resolution using the best 47,768 particles of a data set of over 400,000 particles (Figure S1). We subsequently reimaged the same batch cryoEM grids with the Gatan K2 Summit direct electron detection camera (hereafter mentioned as K2 camera) using the counting mode. A data set of 61,418 particles was collected using the K2 camera, from which 49,374 were used to obtain a 3D reconstruction at 3.1 Å resolution (Figure 1A, Figure S1, and Table S1).
Figure 1.

Structure of the octahedral core of the human E2p IC domain solved by cryoEM single particle reconstruction. (A) CryoEM 3D reconstruction of the dodecahedral core of human E2p IC domain. Radially colored surface view is shown along the 5-, 3-, and 2-fold axes from left to right. The 2-fold view (right) is cut through to show the interior. (B) Rainbow-colored ribbon of the atomic model of the human E2p IC domain (left) and superimposition of the IC domains (right) of human E2p, A. vinelandii E2p (PDB ID: 1EAA), E. coli E2o (PDB ID: 1E2O), and bovine E2b (PDB ID: 2IHW). The left and right panels are in the same angle of view. The regions with high variability among these structures are highlighted with dashed boxes in the superimposition. The resolvable N-terminal residues are indicated by diamond (human E2p, E. coli E2o, and bovine E2b) or asterisk (A. vinelandii E2p). The inset in the middle shows the extended “tip” in the human E2p IC trimers formed by the loop connecting βI2 and βJ. (C) Structure of the human E2p IC trimer with the subunits colored in different shades of blue. (D) Close-up views of the interface between two subunits within a trimer of the IC domain. The subunits are colored in different shades of blue as in (C), and the prime symbol is used for the 3-fold related subunit on the clockwise position.
The 3D reconstruction at 3.1 Å resolution shows a structure with the IC domains organized as a pentagonal dodecahedron (Figure 1A) with structural features similar to those of B. stearothermophilus or E. faecalis E2p.32 The dodecahedron of the human E2p IC domain has a maximum outer diameter of ~240 Å and a central hollow cavity with a diameter of ~120 Å. Three IC domains closely associate into a trimer located at each of the 20 3-fold axes of the dodecahedron. Through trimer–trimer interactions along the 2-fold axes in a double-handed manner, 20 trimers assemble into a dodecahedral shell with large openings (~70 Å in diameter) along the 5-fold axes. By contrast, the shell of the cubic core of A. vinelandii E2p has an outer diameter of 160 Å and openings of ~30 Å in diameter along the 4-fold axes.27
Atomic Model of the Human E2p IC Domain
A de novo atomic model of the human E2p IC domain was built from the 3.1 Å resolution EM map. The backbone of residues 417–647 is fully traceable except for residues 519 and 520, which lack densities. The structure of the human E2p IC domain contains six α-helices (H1–6), a short C-terminal 310-helix (H7), and ten β-strands (A–J) (Figure 1B). The identification scheme for secondary structure elements described here is the same as that for E. coli E2o (OGDC E2) and bovine E2b (BCDC E2)28,29 but different from that for A. vinelandii E2p in which the short H5 is not assigned.27 The βI1 and βI2 regions exist in the E2’s IC domains of all of these four different species. The assembled trimer has a diameter of 80 Å and height of 70 Å (Figure 1C) and incorporates 51 interdomain hydrogen bonds and total buried areas of 4,352 Å2. The interactions between two IC domains (the one with the prime symbol is on the clockwise position of the exterior view) within a trimer occurs at four different regions (Figure 1D), generally similar to the intratrimer interactions in the cubic cores of A. vinelandii E2p, E. coli E2o, and bovine E2b.27–29 First, βH interacts with βB′ to join the β-sheet as an additional antiparallel strand. Second, βA interacts with βD′, forming a peripheral sheet with βC′. Third, H1 interacts with several residues from the clockwise adjacent domain. Lastly, βI2, βJ, and the loop connecting them interact with their 3-fold related counterparts.
Superposition of the IC domain of human E2p with those of A. vinelandii E2p, E. coli E2o, and bovine E2b shows their high structural similarity in the fold (Figure 1B), which is consistent with their sequence similarity. In the 229-residue structure, the r.m.s.d. of 185 pairs of matching Cα-atoms between the IC domains of human E2p and A. vinelandii E2p is 1.0 Å. Structural comparisons of these three proteins reveal several variable regions, including H2, H7, the β-hairpin turn connecting βC and βD, and the loop connecting βI2 and βJ (Figure 1B, boxed elements). This divergence is in contrast to the general conservation of the overall fold and secondary structure elements mentioned above. Interestingly, H2 and H7 are directly involved in the interaction between two 2-fold related trimers and along with H4 determine the geometry of the 2-fold interface and consequently the symmetric assembly of cubic or dodecahedral cores as discussed below. Both human and A. vinelandii E2p proteins have the same helix length and similar structures for H7, whereas E. coli E2o and bovine E2b have two and three additional residues at the C-termini, respectively. Compared to the hairpin structures connecting βI2 and βJ in A. vinelandii E2p, E. coli E2o, and bovine E2b, human E2p has two extra residues (E603 and K604) in this loop. Three of these loops associate in a trimer to form a tip (Figure 1B) about the 3-fold axis that extends inside the cavity of the dodecahedral core.
The construct of the human E2p IC domain used in the present study contains part of the flexible linker (residues 408–416) that connects the E1BD to the IC domain, but this linker region is invisible in the EM density map due to its high flexibility. The structure of the N-terminus of the human E2p IC domain is similar to those from the crystal structures of E. coli E2o and bovine E2b with the termini placed above the 2-fold interface of the clockwise neighboring domain within a trimer (Figure 2A). In contrast, the crystal structure of A. vinelandii E2p inner core resolves a few more N-terminal residues; this extended strand makes a sharp turn near the equivalent 2-fold interface and ends close to the 3-fold axis.27 In the EM density map of the human E2p IC domain, there is no density corresponding to these residues of A. vinelandii E2p (Figure 2B). Therefore, it is unlikely that the flexible linker connecting the IC domain and the E1BD of human E2p is organized similarly to that of A. vinelandii E2p. It is more plausible that E1BD and the linker are localized near the 2-fold interface of the core complex, as previously suggested.33
Figure 2.

Position of the N-terminus of the human E2p IC domain. (A) Space-filling models of the IC trimers of human E2p and A. vinelandii E2p, showing how the N-terminal arm of each domain of a trimer extends along an outer edge of a trimer as seen in the 3-fold exterior view. (B) Residues 408–416 of human E2p, corresponding to the region of A. vinelandii E2p structure extending to the 3-fold axis, are not visible in the EM map (transparent gray surface). In contrast to the structure of A. vinelandii E2p, the resolvable N-terminal residues of human E2p, E. coli E2o, and bovine E2b are in similar positions close to the 2-fold axis.
The structure of the E2 active site is highly conserved in α-keto acid dehydrogenase complexes. In human E2p, S566 from one IC domain and H620′ from the clockwise, 3-fold related IC domain of a trimer are two residues directly involved in catalysis (Figure 3A). Using the crystal structures of A. vinelandii E2p bound with the dihydrolipoyl group (DHL) or CoA (PDB ID: 1EAE and 1EAD),54 we modeled DHL and CoA into the structure of the human E2p IC domain (Figure 3A). The DHL, which carries an 8-S-acetyl group for the forward reaction, gains access to the active site from the outside of the dodecahedral core via a narrow passage next to H1. The acetyl-accepting CoA enters the other end of the long active site channel via an entry within the inner core after first passing through a pentagonal opening, and the product acetyl-CoA exits reversely. The exterior of the dodecahedron has a surplus of positively charged residues, which may aid in attracting both the negatively charged lipoyl domains and CoA; in contrast, there is a balance of negative and positive charges in the passage through the open face followed by an interior surface enriched in negatively charged residues, which may help avoid nonspecific binding of the CoA substrate and product (Figure S2). The density of two residues, A519 and G520, which form part of the tight β-turn connecting βE and βF, is missing in the EM density map (Figure 3B), suggesting a high mobility for these two residues. The corresponding β-turn in bovine E2b is proposed to carry out a synchronized substrate-gating mechanism.29 The mobility of this β-turn in human E2p suggests that it may have a similar gating function in substrate binding.
Figure 3.

Interdomain active site of the human E2p IC trimers. (A) One of three active sites in a trimeric unit of human E2p (left) and a close-up view of the active site (right). Three inner domains are shown as ribbons and colored in different shades of blue. The catalytic residues S566 and H620′ from two adjacent IC domains, respectively, are displayed as red spheres. The putative CoA and dihydrolipoamide (DHLA) positions are adopted from A. vinelandii E2p structures (PDB ID: 1EAD and 1EAE). (B) The densities of A519 and G520 are not visible in the EM map (chicken wire) due to their flexibility. The density of the side chain of L521 (colored in purple), whose corresponding residue (L293) in bovine E2b is a gatekeeper residue for binding of the lipoyl group, is well-defined. Only the backbone of the atomic model is shown except for the side chains of L521 and the catalytic residues S566 and H620′.
Two-Fold Related Interface and the Dodecahedral Core of Human E2p
The crystal structures of the cubic cores of A. vinelandii E2p, E. coli E2o, and bovine E2b all reveal a similar interaction between trimers. The C-terminal residues of each domain form a hydrophobic “knob” that is then buried into a hydrophobic “hole” of its 2-fold related domain to form a double “knob–hole” interaction along each 2-fold axis.27–29 Because all E2 proteins in the α-keto acid dehydrogenase complex family maintain hydrophobicity in the C-terminal residue, it was suggested that this common arrangement is maintained whether they form a cubic or dodecahedral core.27 Indeed, the arrangement of a double “knob” and “hole” is similar in the dodecahedral core of human E2p (Figure 4A). Each “knob” formed by residues of H7 sits in the “hole” formed by the hydrophobic residues of H7′, H2′, and the N-terminal end of H4′ in the 2-fold related domain.
Figure 4.

Two-fold interface between trimers determines the dodecahedral assembly of human E2p. (A) Two-fold related dimer unit of the human E2p inner core. The amino acid residues are colored by their hydropathy index (brown, hydrophobic; purple, hydrophilic; white, neutral) to show the double-handed hydrophobic “knob-and-hole” interaction between the 2-fold related subunits. One of the two 2-fold related subunits is marked with the prime symbol. (B) Comparison of the helix H2 of the human E2p IC domain (blue) with that of A. vinelandii E2p IC domain (green). The H2 of A. vinelandii E2p is bent and separated into two fragments. However, the H2 of human E2p IC domain is straight and tilted toward the H6′ and H7′ of the 2-fold related domain. (C) Side view of the 2-fold interface of the human E2p inner core. Two IC domains are colored in different shades of blue. The side chains of the residues (E463, L464, I467, K641′, I643′, I531, and I531′) involved in maintaining the geometry of the 2-fold interaction are displayed. (D) Space-filling models of the dodecahedral human E2p inner core and the cubic A. vinelandii E2p inner core. The included angle between two 2-fold related trimers is 138.2° for the dodecahedron and 109.5° for the cube.
In comparison with the above-mentioned crystal structures, the IC domain of human E2p differs markedly from those of the other E2 proteins in its straight H2, which is bent in A. vinelandii E2p, E. coli E2o, and bovine E2b (Figure 4B). The straight H2 of human E2p is tilted toward the 2-fold related domain and requires repositioning of H7′ of the 2-fold related IC domain in forming the “knob–hole” interaction. The included angle between the 2-fold related trimers is 109.5° in the cubic cores of A. vinelandii E2p (Figure 4D), E. coli E2o, and bovine E2b. However, in human E2p, avoidance steric strain in forming the two H2–H7′ interactions between each pair of 2-fold related domains fosters a larger included angle of 138.2° to achieve the dodecahedral symmetry (Figure 4C and D). In addition, two hydrophobic residues, I531 and its 2-fold related counterpart I531′, contact each other opposite the H2–H7′ interaction (Figure 4C), stabilizing the 2-fold related interface. The two “knob–holes” merge via L646–L646′ interaction in the center of the 2-fold axis. Despite the conservation of the “knob–hole” and secondary structure elements, sequence alignment does not show conservation of the residues involved in the 2-fold interactions in human E2p, yeast E2p, B. stearothermophilus E2p, and E. faecalis E2p, which all form dodecahedral cores. This variation suggests that the residues stabilizing the 2-fold interface have evolved differently to meet the requirements for the organization, dynamic structure, and activity regulation in different species.
Homology Model of the IC Domain of Human E3BP
E3BP is an essential component of human PDC, and its absence results in greatly diminished overall PDC activity.21 The structure of the IC domain of E3BP is necessary for understanding the organization and function of the E2p/E3BP core complex, but there is no high-resolution structure available yet. Thanks to the very high level of sequence identity (49% identity and 69% similarity) between the IC domains of human E2p and E3BP (Figure 5A), we were able to build a homology model of the IC domain of human E3BP with SWISS-MODEL55 using the structure of the human E2p IC domain as a template (Figure 5B). The homology model of the human E3BP IC domain is highly similar to the structure of the human E2p IC domain (Figure S3A) consistent with their sequence identity. Sequence alignment also shows notable differences between the IC domains of E2p and E3BP in several regions (Figure 5). Interestingly, these regions are located in βB, H2, βD, βI2, βJ, H6, H7, and the loop connecting βF and H4, all of which are involved in intra- or intertrimer interfaces (Figure 5B). It is worth noting that the putative H7 of E3BP is both one residue shorter (Figure 5A) and less stabilized than its equivalent in E2p and is consequently unable to form a typical “knob” and “hole” interaction between 2-fold related domains. Three residues of H2, which participate in the 2-fold interaction in the dodecahedral core of E2p, are also absent in E3BP (Figure 5A, Figure S3A). These differences might compromise the stability of the 2-fold interface, as it was reported that purified E3BP is unstable and does not form dodecahedral complexes but may form dimers instead.35 In contrast to the absent residues, the loop connecting βI and βJ in E3BP has three additional residues (Figure 5A, Figure S3A). The function of this longer loop is unknown; however, it may contribute to the selective binding of E3BP to E2p to form a heterotrimer.
Figure 5.

Sequence and structure conservation of the IC domains between E2p and E3BP in humans. (A) Sequence alignment of the IC domains of E2p and E3BP in humans. The regions in E3BP with low identity are colored in red. (B) Homology model of the E3BP IC domain (ribbon) and its interactions with the 3- and 2-fold related E2p IC domains (transparent space-filling models). The regions in E3BP with low identity are colored in red as in (A).
DISCUSSION
The following have been experimentally observed: first, each bovine E2p/E3BP dodecahedron has ~12 copies of E3BP in vivo,20 and each human E2p/E3BP dodecahedron has at most 20 copies of E3BP when E2p and E3BP are overexpressed.35,37 Second, ~12 copies of E3BP were estimated in the E2p/E3BP complex when overexpression of E3BP did not occur.34 Third, following subunit dissociation of the E2p/E3BP complex, reversal of dissociating conditions by rapid dialysis produced E2p 60-mers with little, if any, E3BP.18,56 Fourth, trimers formed first and then reassembled into dodecahedrons as dissociating agents were further lowered.56 Fifth, E3BP was only incorporated into reconstituted products when the lowering of the concentration of the dissociating agent was carried out very slowly.57 Finally, biophysical studies have eliminated the possibility that E3BP adds to E2p 60-mer, strongly suggesting that E3BP substitutes for E2p within trimers of the dodecahedron.34–37 These observations favor the incorporation of one E3BP into a trimer with two E2ps but did not completely eliminate production of trimers formed by three E3BP.
Beyond E2p trimers, we have used the homology model of the E3BP IC domain to assess whether E3BP can form stable trimers by itself or with two E2p IC domains. As a first indication of stability, we used PDBePISA58 to calculate the solvation free energy (ΔiG) change upon formation of different trimer combinations (Figure S3B). It is worth noting that the above calculation of solvation free energy was based on the homology model of the E3BP IC domain, and no conformational changes were considered during oligomerization. The similarly favorable ΔiG with incorporation of one E3BP IC domain but not with a homotrimer of E3BP further supports the heterotrimer structure with one E3BP and two copies of E2p and is consistent with models for the structure of the inner core based on experimental approaches.34,35,37 As noted above, the major differences in sequence in E3BP reside in the regions involved in intra- or intertrimer interactions. The three extra residues in the loop connecting βI2 with βIJ in E3BP may form a special structure that selectively favors binding to two E2ps while repelling insertion of another E3BP inner domain to thereby confer strict maintenance of the 2 E2p + 1 E3BP stoichiometry in heterotrimers.
The E3BP subunit has one less lipoyl domain and correspondingly one less connecting linker region. Despite the consequently lower mass of a 60-subunit E2p/E3BP complex than that of the E2p 60-mer, the E2/E3BP complex was found by equilibrium centrifugation and small-angle X-ray studies to have a larger exclusion volume than that of the E2p 60-mer.34 One contribution to this might involve weaker connections along the 2-fold interface allowing greater expansion of the inner core of the lighter E2/E3BP complex. At the 2-fold interface, the interaction between the IC domains of E2p and E3BP has a ΔiG less favorable than the ΔiG for two E2ps but much more favorable than two E3BPs (Figure S3C). Therefore, statistically, E2p-E3BP 2-fold connections would be favored. However, much stronger E2p–E2p interactions along the 2-fold axis might by default favor heterotrimers pairing up along the 2-fold axis to form a unique but weaker E3BP–E3BP interaction along the 2-fold axis. This would allow the E2p IC domains to maintain their normal conformation in 2-fold connections and dominate in imposing the correct dodecahedron angles. This hypothesis is similar to the previously proposed 48E2p + 12E3BP model in which 12 copies of E3BP formed trimers with 2 E2p and were placed as six dimers on 2-fold axis in the dodecahedron in a completely equivalent and symmetric fashion.34 The availability of a high-resolution structure for E2p dodecahedron and homology model for E3BP from this study will greatly aid further studies to elucidate the organization and dynamics of the native structure of the E2p/E3BP complex.
An active site in the human E2p trimer is located on the interface between two E2p subunits and is comprised of residues from both subunits that bind, contain, and position the DHL and CoA substrates and facilitate transfer of an acetyl group between these substrates, whereas each IC domain in the trimer participates in two active sites. At the aligned position, E3BP lacks the critical catalytic His residue and, by itself, is inactive in the transacetylation reaction.21 However, we find that on its other active site side (counterclockwise side within the trimer), the E3BP IC domain may carry out the role of substrate binding and confinement. There is high conservation of aligned residues in the E3BP sequence with those of E2p. Six of the aligned E3BP residues implicated in the binding of DHL are identical with their E2p counterpart, and the other two are conservatively substituted (Table S2); all are positioned within standard binding distances of DHL in the active site (Figure S4). Similarly, for the E2p residues gauged by their positions to bind CoA, there are 14 identical aligned residues in E3BP and three conservatively substituted (Table S2) and again, all are near CoA in the homology model (Figure S4). Two residues of E3BP (Arg444, Phe445) are not conservative substitutions; like the aligned E2p residues (Ala592, S593), these are positioned near the adenine ring of CoA but could form even more favorable interactions. Furthermore, in our homology model, E3BP folding in regions contributing substrate-binding residues readily conserves comparable conformations to E2p. Also conserved in E3BP are residues that are positioned at the entry to the lipoyl binding site at the surface of the E2p trimer, where the lipoyl domain that covalently holds dihydrolipoyl on lysyl ε-amino must interact in delivering the dihydrolipoyl group to the active site. Thus, we suggest that the E3BP IC domain in a heterotrimer can participate in the primary role of substrate binding and enclosing one active site and thereby aids catalysis. This would indicate that it is proper to call the E3BP inner domain an IC domain (i.e., both inner and catalytic).
In summary, the 3.1 Å resolution cryoEM structure of the human E2p IC domain presented here is the first high-resolution structure of the inner core dodecahedron of a mammalian dihydrolipoyl acetyltransferase. This structure will open the door for further analyses of catalytic function and inner core dynamics using a variety of experimental and modeling approaches. Indeed, the atomic structure of the human E2p IC domain has enabled us to derive a homology model of the E3BP IC domain and a model for the E2p/E3BP complex in which the IC domains of E2p and E3BP form heterotrimers. Beyond describing how human E3BP IC domains could be integrated with human E2p into the dodecahedron structure, we also provide the first evidence that, like human E2p, human E3BP likely participates in catalysis based on the very similar structure and conservation of residues of E3BP that play a primary substrate binding/enclosing role.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH (R01GM071940, DE025567, and AI094386) and the NSF (DMR-1548924) to Z.H.Z. and American Heart Association Postdoctoral Fellowship (14POST18870059) to J.J. The authors acknowledge the use of instruments at the Electron Imaging Center for NanoMachines supported by the NIH (1S10RR23057 and 1S10OD018111), the NSF (DBI-1338135), and CNSI at UCLA.
Footnotes
Notes
The authors declare no competing financial interest.
Accession Codes
Three-dimensional cryoEM density map of the dodecahedral core of human E2p IC domain has been deposited in the Electron Microscopy Data Bank under accession number EMD-7610. The coordinates of the human E2p IC domain have been deposited in the Protein Data Bank under accession number 6CT0.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.8b00357.
Data collection and structure refinement statistics (Table S1), E2p IC substrate binding residues and aligned residues of E3BP (Table S2), resolution of the cryoEM 3D reconstructions (Figure S1), electrostatic surface of the human E2p IC trimer (Figure S2), homology model of the human E3BP IC domain (Figure S3), and active site substrate binding residues (Figure S4) (PDF)
References
- 1.Patel MS, Roche TE. Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 1990;4:3224–3233. doi: 10.1096/fasebj.4.14.2227213. [DOI] [PubMed] [Google Scholar]
- 2.Reed LJ. A trail of research from lipoic acid to alpha-keto acid dehydrogenase complexes. J Biol Chem. 2001;276:38329–38336. doi: 10.1074/jbc.R100026200. [DOI] [PubMed] [Google Scholar]
- 3.Roche TE, Cox DJ. Multifunctional 2-Oxo acid dehydrogenase complexes. In: Agius L, Sherratt HSA, editors. Channeling in Intermediary Metabolism. Portland Press Ltd; London: 1996. pp. 115–132. [Google Scholar]
- 4.Patel MS, Naik S, Wexler ID, Kerr DS. Gene regulation and genetic defects in the pyruvate dehydrogenase complex. J Nutr. 1995;125:1753S–1757S. doi: 10.1093/jn/125.suppl_6.1753S. [DOI] [PubMed] [Google Scholar]
- 5.Roche TE, Baker JC, Yan X, Hiromasa Y, Gong X, Peng T, Dong J, Turkan A, Kasten SA. Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Prog Nucleic Acid Res Mol Biol. 2001;70:33–75. doi: 10.1016/s0079-6603(01)70013-x. [DOI] [PubMed] [Google Scholar]
- 6.Roche TE, Hiromasa Y, Turkan A, Gong X, Peng T, Yan X, Kasten SA, Bao H, Dong J. Essential roles of lipoyl domains in the activated function and control of pyruvate dehydrogenase kinases and phosphatase isoform 1. Eur J Biochem. 2003;270:1050–1056. doi: 10.1046/j.1432-1033.2003.03468.x. [DOI] [PubMed] [Google Scholar]
- 7.Patel MS, Korotchkina LG. Regulation of the pyruvate dehydrogenase complex. Biochem Soc Trans. 2006;34:217–222. doi: 10.1042/BST20060217. [DOI] [PubMed] [Google Scholar]
- 8.Roche TE, Hiromasa Y. Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell Mol Life Sci. 2007;64:830–849. doi: 10.1007/s00018-007-6380-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tso SC, Qi X, Gui WJ, Wu CY, Chuang JL, Wernstedt-Asterholm I, Morlock LK, Owens KR, Scherer PE, Williams NS, Tambar UK, Wynn RM, Chuang DT. Structure-guided development of specific pyruvate dehydrogenase kinase inhibitors targeting the ATP-binding pocket. J Biol Chem. 2014;289:4432–4443. doi: 10.1074/jbc.M113.533885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeoung NH, Harris CR, Harris RA. Regulation of pyruvate metabolism in metabolic-related diseases. Rev Endocr Metab Disord. 2014;15:99–110. doi: 10.1007/s11154-013-9284-2. [DOI] [PubMed] [Google Scholar]
- 11.Mayers RM, Leighton B, Kilgour E. PDH kinase inhibitors: a novel therapy for Type II diabetes? Biochem Soc Trans. 2005;33:367–370. doi: 10.1042/BST0330367. [DOI] [PubMed] [Google Scholar]
- 12.Bersin RM, Stacpoole PW. Dichloroacetate as metabolic therapy for myocardial ischemia and failure. Am Heart J. 1997;134:841–855. doi: 10.1016/s0002-8703(97)80007-5. [DOI] [PubMed] [Google Scholar]
- 13.McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, Wu H, Schell MJ, Tsang TM, Teahan O, Zhou S, Califano JA, Jeoung NH, Harris RA, Verma A. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J Biol Chem. 2008;283:22700–22708. doi: 10.1074/jbc.M801765200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sutendra G, Michelakis ED. Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front Oncol. 2013;3:38. doi: 10.3389/fonc.2013.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Marcucci O, Lindsay JG. Component X. An immunologically distinct polypeptide associated with mammalian pyruvate dehydrogenase multi-enzyme complex. Eur J Biochem. 1985;149:641–648. doi: 10.1111/j.1432-1033.1985.tb08972.x. [DOI] [PubMed] [Google Scholar]
- 16.Jilka JM, Rahmatullah M, Kazemi M, Roche TE. Properties of a newly characterized protein of the bovine kidney pyruvate dehydrogenase complex. J Biol Chem. 1986;261:1858–1867. [PubMed] [Google Scholar]
- 17.Rahmatullah M, Gopalakrishnan S, Andrews PC, Chang CL, Radke GA, Roche TE. Subunit associations in the mammalian pyruvate dehydrogenase complex. Structure and role of protein X and the pyruvate dehydrogenase component binding domain of the dihydrolipoyl transacetylase component. J Biol Chem. 1989;264:2221–2227. [PubMed] [Google Scholar]
- 18.Powers-Greenwood SL, Rahmatullah M, Radke GA, Roche TE. Separation of protein X from the dihydrolipoyl transacetylase component of the mammalian pyruvate dehydrogenase complex and function of protein X. J Biol Chem. 1989;264:3655–3657. [PubMed] [Google Scholar]
- 19.Maeng CY, Yazdi MA, Niu XD, Lee HY, Reed LJ. Expression, purification, and characterization of the dihydrolipoamide dehydrogenase-binding protein of the pyruvate dehydrogenase complex from Saccharomyces cerevisiae. Biochemistry. 1994;33:13801–13807. doi: 10.1021/bi00250a034. [DOI] [PubMed] [Google Scholar]
- 20.Sanderson SJ, Miller C, Lindsay JG. Stoichiometry, organisation and catalytic function of protein X of the pyruvate dehydrogenase complex from bovine heart. Eur J Biochem. 1996;236:68–77. doi: 10.1111/j.1432-1033.1996.00068.x. [DOI] [PubMed] [Google Scholar]
- 21.Harris RA, Bowker-Kinley MM, Wu P, Jeng J, Popov KM. Dihydrolipoamide dehydrogenase-binding protein of the human pyruvate dehydrogenase complex. DNA-derived amino acid sequence, expression, and reconstitution of the pyruvate dehydrogenase complex. J Biol Chem. 1997;272:19746–19751. doi: 10.1074/jbc.272.32.19746. [DOI] [PubMed] [Google Scholar]
- 22.Perham RN. Swinging arms and swinging domains in multifunctional enzymes: catalytic machines for multistep reactions. Annu Rev Biochem. 2000;69:961–1004. doi: 10.1146/annurev.biochem.69.1.961. [DOI] [PubMed] [Google Scholar]
- 23.Broz AK, Tovar-Mendez A, Mooney BP, Johnston ML, Miernyk JA, Randall DD. A novel regulatory mechanism based upon a dynamic core structure for the mitochondrial pyruvate dehydrogenase complex? Mitochondrion. 2014;19(Pt B):144–153. doi: 10.1016/j.mito.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 24.Thekkumkara TJ, Ho L, Wexler ID, Pons G, Liu TC, Patel MS. Nucleotide sequence of a cDNA for the dihydrolipoamide acetyltransferase component of human pyruvate dehydrogenase complex. FEBS Lett. 1988;240:45–48. doi: 10.1016/0014-5793(88)80337-5. [DOI] [PubMed] [Google Scholar]
- 25.Reed LJ, Hackert ML. Structure-function relationships in dihydrolipoamide acyltransferases. J Biol Chem. 1990;265:8971–8974. [PubMed] [Google Scholar]
- 26.Wagenknecht T, Grassucci R, Radke GA, Roche TE. Cryoelectron microscopy of mammalian pyruvate dehydrogenase complex. J Biol Chem. 1991;266:24650–24656. [PubMed] [Google Scholar]
- 27.Mattevi A, Obmolova G, Schulze E, Kalk KH, Westphal AH, de Kok A, Hol WG. Atomic structure of the cubic core of the pyruvate dehydrogenase multienzyme complex. Science. 1992;255:1544–1550. doi: 10.1126/science.1549782. [DOI] [PubMed] [Google Scholar]
- 28.Knapp JE, Mitchell DT, Yazdi MA, Ernst SR, Reed LJ, Hackert ML. Crystal structure of the truncated cubic core component of the Escherichia coli 2-oxoglutarate dehydrogenase multienzyme complex. J Mol Biol. 1998;280:655–668. doi: 10.1006/jmbi.1998.1924. [DOI] [PubMed] [Google Scholar]
- 29.Kato M, Wynn RM, Chuang JL, Brautigam CA, Custorio M, Chuang DT. A synchronized substrate-gating mechanism revealed by cubic-core structure of the bovine branched-chain alpha-ketoacid dehydrogenase complex. EMBO J. 2006;25:5983–5994. doi: 10.1038/sj.emboj.7601444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stoops JK, Cheng RH, Yazdi MA, Maeng CY, Schroeter JP, Klueppelberg U, Kolodziej SJ, Baker TS, Reed LJ. On the unique structural organization of the Saccharomyces cerevisiae pyruvate dehydrogenase complex. J Biol Chem. 1997;272:5757–5764. doi: 10.1074/jbc.272.9.5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neveling U, Klasen R, Bringer-Meyer S, Sahm H. Purification of the pyruvate dehydrogenase multienzyme complex of Zymomonas mobilis and identification and sequence analysis of the corresponding genes. J Bacteriol. 1998;180:1540–1548. doi: 10.1128/jb.180.6.1540-1548.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Izard T, Aevarsson A, Allen MD, Westphal AH, Perham RN, de Kok A, Hol WG. Principles of quasi-equivalence and Euclidean geometry govern the assembly of cubic and dodecahedral cores of pyruvate dehydrogenase complexes. Proc Natl Acad Sci U S A. 1999;96:1240–1245. doi: 10.1073/pnas.96.4.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu X, Hiromasa Y, Tsen H, Stoops JK, Roche TE, Zhou ZH. Structures of the human pyruvate dehydrogenase complex cores: a highly conserved catalytic center with flexible N-terminal domains. Structure. 2008;16:104–114. doi: 10.1016/j.str.2007.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hiromasa Y, Fujisawa T, Aso Y, Roche TE. Organization of the cores of the mammalian pyruvate dehydrogenase complex formed by E2 and E2 plus the E3-binding protein and their capacities to bind the E1 and E3 components. J Biol Chem. 2004;279:6921–6933. doi: 10.1074/jbc.M308172200. [DOI] [PubMed] [Google Scholar]
- 35.Brautigam CA, Wynn RM, Chuang JL, Chuang DT. Subunit and catalytic component stoichiometries of an in vitro reconstituted human pyruvate dehydrogenase complex. J Biol Chem. 2009;284:13086–13098. doi: 10.1074/jbc.M806563200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vijayakrishnan S, Kelly SM, Gilbert RJ, Callow P, Bhella D, Forsyth T, Lindsay JG, Byron O. Solution structure and characterisation of the human pyruvate dehydrogenase complex core assembly. J Mol Biol. 2010;399:71–93. doi: 10.1016/j.jmb.2010.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vijayakrishnan S, Callow P, Nutley MA, McGow DP, Gilbert D, Kropholler P, Cooper A, Byron O, Lindsay JG. Variation in the organization and subunit composition of the mammalian pyruvate dehydrogenase complex E2/E3BP core assembly. Biochem J. 2011;437:565–574. doi: 10.1042/BJ20101784. [DOI] [PubMed] [Google Scholar]
- 38.Yang D, Song J, Wagenknecht T, Roche TE. Assembly and full functionality of recombinantly expressed dihydrolipoyl acetyltransferase component of the human pyruvate dehydrogenase complex. J Biol Chem. 1997;272:6361–6369. doi: 10.1074/jbc.272.10.6361. [DOI] [PubMed] [Google Scholar]
- 39.Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. Automated molecular microscopy: the new Leginon system. J Struct Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 40.Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 42.Voss NR, Yoshioka CK, Radermacher M, Potter CS, Carragher B. DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J Struct Biol. 2009;166:205–213. doi: 10.1016/j.jsb.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol. 2003;142:334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 44.Heymann JB, Belnap DM. Bsoft: image processing and molecular modeling for electron microscopy. J Struct Biol. 2007;157:3–18. doi: 10.1016/j.jsb.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 45.Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scheres SH. Beam-induced motion correction for sub-megadalton cryo-EM particles. eLife. 2014;3:e03665. doi: 10.7554/eLife.03665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang J, Pentelute BL, Collier RJ, Zhou ZH. Atomic structure of anthrax protective antigen pore elucidates toxin translocation. Nature. 2015;521:545–549. doi: 10.1038/nature14247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kucukelbir A, Sigworth FJ, Tagare HD. Quantifying the local resolution of cryo-EM density maps. Nat Methods. 2014;11:63–65. doi: 10.1038/nmeth.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 50.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr, Sect D: Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 52.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 53.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 54.Mattevi A, Obmolova G, Kalk KH, Teplyakov A, Hol WG. Crystallographic analysis of substrate binding and catalysis in dihydrolipoyl transacetylase (E2p) Biochemistry. 1993;32:3887–3901. doi: 10.1021/bi00066a007. [DOI] [PubMed] [Google Scholar]
- 55.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 56.Behal RH, DeBuysere MS, Demeler B, Hansen JC, Olson MS. Pyruvate dehydrogenase multienzyme complex. Characterization of assembly intermediates by sedimentation velocity analysis. J Biol Chem. 1994;269:31372–31377. [PubMed] [Google Scholar]
- 57.McCartney RG, Sanderson SJ, Lindsay JG. Refolding and reconstitution studies on the transacetylase-protein X (E2/X) subcomplex of the mammalian pyruvate dehydrogenase complex: evidence for specific binding of the dihydrolipoamide dehydrogenase component to sites on reassembled E2. Biochemistry. 1997;36:6819–6826. doi: 10.1021/bi9630016. [DOI] [PubMed] [Google Scholar]
- 58.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.