

Abstract
Over 300 chiral drug substances lack official United States Pharmacopeia (USP) methods for the enantiomeric purity determination. Because enantiomeric analysis typically requires specialized methods for each drug compound, developing protocols for each of these 300+ substances would be an expensive and laborious endeavor. Alternatively, if a detector capable of determining the enantiomeric composition without chiral separation could be used with certain drug compounds, this could be implemented relatively rapidly into official testing monographs. Circular dichroism (CD) detection following HPLC (HPLC-CD) has been proposed for this purpose but studies performed thus far have not prioritized its compatibility with validated regulatory methods. In this study, HPLC-CD was evaluated for enantiomeric purity determinations of 13 drug substances using HPLC methods consistent with assay protocols described in United States Pharmacopeia (USP) monographs. Of these selected substances, three (sitagliptin, timolol, and levalbuterol) showed no CD activity and one other (levofloxacin) could not be analyzed due to incompatibility of the mobile phase with the CD detector. For the remaining 9 substances, method validation was performed to determine the linearity, accuracy, precision and limits of quantitation of enantiomer impurities, which was compared to limits established by USP. It was found that enantiomeric impurities for four substances (pramipexole, levocetirizine, (S)-citalopram, and tolterodine) could be quantitatively determined at levels suitable to USP specifications. This analysis demonstrated that HPLC-CD does provide an effective enantiomeric characterization strategy for compatible chiral compounds, and can be implemented quickly and economically compared to traditional column-dependent chiral separation or derivatization methods.
Keywords: Circular dichroism, HPLC, Chiral, Enantiomer, USP
1. Introduction
Single enantiomer drugs make up a large and growing portion of over-the-counter and prescription drug products, and in some cases offer improved efficacy compared to racemic alternatives [1–6]. Unfortunately, traditional analysis methods for determining drug substance impurities, such as high-performance liquid chromatography (HPLC), are not suitable for chiral analysis without specialization because opposite enantiomers typically require a specific chiral stationary phase or mobile phase additive to separate enantiomers prior to detection [7,8]. As a result, unique methods must be developed to determine the enantiomeric purity for different compounds. To address this, an alternative strategy has been described involving the use of a circular-dichroism (CD) detector, which can distinguish between enantiomers for a large variety of chemical species [9–19]. Rather than separating enantiomers, the CD detector is positioned following achiral (i.e., not chiral specific) separations, and is used to determine the chiral purity of eluting substances [20]. In this way, enantiomeric analysis can be carried out simultaneously with the assay determination. Further, HPLC-compatible CD detectors are commercially available, and could be incorporated relatively easily into existing analysis assay methods, reducing new method development costs and shortening analysis times associated with chiral drugs.
CD spectroscopy is performed by recording the difference between left- and right-handed circularly polarized light absorbed by the sample (ΔA). This value is referred to as the ellipticity, and it is often expressed in units of millidegrees (mdeg) to represent the extent to which polarized light is rotated after passing through the sample. To determine the enantiomeric composition of a chiral species, the ellipticity is first normalized by the sample quantity by dividing ΔA by the absorption of unpolarized light (A). This value is called the g-factor . The g-factor is different for each chiral compound and varies with the enantiomer composition, as well as environmental factors such as the solvent, temperature, and pH [21]. Under constant conditions, a deviation in the g-factor for a sample with respect to the value for a pure enantiomer indicates the presence of a chiral impurity.
In this study, the ability of HPLC-CD to quantitate enantiomer impurity levels of chiral drug substances was analyzed. Unlike most HPLC-CD analyses which seek to optimize experimental conditions for CD sensitivity, this study restricted parameters to those currently validated and consistent with United States Pharmacopeia (USP) specifications. The USP is a non-profit pharmacopeia whose official methods are generally recognized by the U.S. Food and Drug Administration as a minimum quality standard for drug substances and products. The USP publishes monographs, or lists of analysis methods for specific drug substances, which contain instructions for determining assay and impurity values and also reports currently accepted limits for impurities allowed in each substance. By utilizing these methods without modification, this study examined the potential regulatory impact that HPLC-CD could provide through its incorporation into currently validated assay methods.
USP monographs for over 300 chiral drug substances were examined to determine the current state of available methods to characterize enantiomeric composition. To our best estimate, less than 30 of these monographs contained methods to quantify enantiomeric impurities, indicating the urgent need to implement enantiomeric characterization protocols to ensure drug quality and safety. To determine whether CD could be used for this purpose, multiple chiral drug substances were selected and analyzed by HPLC-CD using USP assay methods. For each substance, method validation was performed to assess the linearity, accuracy, precision, and the limit of quantitation of enantiomeric impurities. Results were compared with specification limits of the impurity set by USP, which allowed an assessment for the immediate impact that HPLC-CD could provide if incorporated into existing monographs.
2. Experimental
2.1. Materials and methods
2.1.1. Reagents
Mobile phases for each drug substance analysis were prepared in a manner consistent with the assay method of USP monographs (supplemental information) [22–34]. All solvents were filtered using a 0.45 μm HVLP filter (Millipore, Billerica, MA) prior to use. Standard impurity solutions were prepared in triplicate from individual enantiomers, or from a pure enantiomer and a racemic mixture (supplemental information). For each substance, the combined concentration of enantiomers was constant while the impurity enantiomer varied from 0 to 5%. At least six impurity levels were prepared for each substance (supplemental information).
2.1.2. Liquid chromatography with circular dichroism detection
HPLC was performed using a Jasco LC-4000 Series (Jasco Inc., Maryland, USA) equipped with an inline CD detector (CD-4095, Jasco Inc.). A photodiode array detector preceded the CD detector and was used to record UV chromatograms. The CD detector recorded ellipticity and g-factor chromatograms, and was allowed to equilibrate for 6 h prior to analysis to ensure stabilization [35]. HPLC methods and parameters were consistent with the assay procedure described in USP drug substance monographs (supplemental information). Each standard solution was analyzed once by HPLC-CD, and the magnitude of the g-factor signal was recorded at the retention time of the drug substance.
2.1.3. Circular dichroism spectroscopy
A Jasco J-815 CD Spectrometer (Jasco, Inc.) was used with Spectra Manager software (Jasco Inc.) to record CD spectra. Recordings were made for each substance in the HPLC assay mobile phase outlined in USP monographs. When possible, solutions were prepared at concentrations equal to that of standard solutions detailed in the monographs, but some solutions required slight dilution to obtain spectra. All spectra were recorded between 200 and 400 nm at a rate of 200 nm/min, and were averaged over 10 accumulations. A 10 mm path length cuvette was used to hold solutions at room temperature, and all spectra were background subtracted from those of the mobile phases.
3. Results and discussion
3.1. Determining compatibility of drug substances with HPLC-CD
All drug substances selected for enantiomeric analysis by HPLC-CD are listed in Table 1, and were chosen based upon several qualifying criteria. First, the USP monograph for each drug substance included a chiral-specific HPLC method for enantiomeric analysis, along with a specification limit for the enantiomeric impurity permitted in the drug substance. Recall that most chiral substances do not currently have a USP method for the enantiomeric impurity determination, so this was the greatest limiting factor in the search for HPLC-CD drug candidates. Second, all drugs contained an achiral HPLC assay method for concentration determination. Since one of the benefits of HPLC-CD includes compatibility with existing methods, the HPLC assay method would be used for analysis while an inline CD detector determined the enantiomeric purity. Lastly, all drugs chosen contained one chiral center, simplifying analysis by limiting drugs to one impurity per substance. Importantly, this does not indicate that HPLC-CD is incompatible with drugs possessing multiple chiral centers. Analysis may be performed on such samples in an identical manner as the current approach by utilizing CD wavelengths specifically tuned to one center, or, as some have demonstrated, by building a mathematical model to determine contributions from each isomer [13]. In all, roughly 20 substances were identified which met all criteria. From these, those with the highest tolerances for the enantiomeric impurity limit (>0.5%) and two others that are widely used (dextromethorphan, esomeprazole) were selected for HPLC-CD analysis.
Table 1.
Summary of g-factors determined for chiral drug substances.
| Drug Substance | Solventa,b | Elution Method | Wavelength (nm) | g-Factors (x1e4)
|
|
|---|---|---|---|---|---|
| Spectrum | HPLC-CD | ||||
| Dexchlorpheniramine | ACN-potassium phosphate (40 mM) (pH* 3.0) | Gradient | 245 | 5.89 | 6.50 |
| Dextromethorphan | ACN-ammonium nitrate (23 mM), docusate sodium (23 mM) (7:3) (pH*3.4) | Isocratic | 280 | 6.27 | 7.18 |
| (S)-citalopram | ACN-potassium phosphate (25 mM) (pH* 3.0) | Gradient | 242 | 4.87 | 5.44 |
| Esomeprazole | ACN-sodium phosphate (9 mM) (pH* 7.6) (7:3) | Isocratic | 230 | 12.30 | 12.78 |
| Levalbuterol | ACN-methanol-phosphoric acid-water | Gradient | – | – | – |
| Levetiracetam | ACN-potassium phosphate (20 mM) (pH* 5.5) | Gradient | 230 | −124.82 | −167.05 |
| Levocetirizine | ACN-water-sulfuric acid (4 mM) (930:66:4) | Isocratic | 230 | 3.64 | 3.92 |
| Levofloxacin | Methanol-ammonium acetate (11 mM), cupric sulfate (5 mM), L-isoleucine (10 mM) (3:7) | Isocratic | 230 | −5.31 | – |
| Pramipexole | ACN-potassium phosphate (67 mM), sodium 1-octanesulfonate (23 mM) (pH* 3.0) | Gradient | 240 | −3.11 | −2.90 |
| Sitagliptin | ACN-potassium phosphate (10 mM) (pH* 2.0) (3:17) | Isocratic | – | – | – |
| Timolol | ACN-water (0.05% trifluoroacidic acid) | Gradient | – | – | – |
| Tolterodine | ACN-water (0.01% phosphoric acid) (33:67) | Isocratic | 231 | 16.65 | 14.87 |
| Valsartan | ACN-water (0.01% acetic acid) (1:1) | Isocratic | 233 | −3.79 | −4.43 |
ACN refers to acetonitrile.
All isocratic conditions are reported as (v:v). Gradient details are provided in supplemental information.
CD spectra of selected drug substances were recorded to determine which were suitable for analysis by HPLC-CD. To obtain spectra, recordings were made using a standalone CD spectrometer. While the HPLC inline CD spectrometer could be used for this purpose, the standalone instrument was chosen for this step due to its increased performance characteristics. To mimic the solvent environment that would be present during HPLC-CD, all recordings were made in the HPLC mobile phases described in USP assay methods (Table 1, supplemental information). Fig. 1 displays the CD spectrum for (S)-citalopram, which shows bands of positive and negative activity from 200 to 290 nm, indicating it was amenable to HPLC-CD. Of the 13 candidates considered, 3 (sitagliptin, timolol, and levalbuterol) showed no CD activity, and were eliminated from subsequent analyses.
Fig. 1.
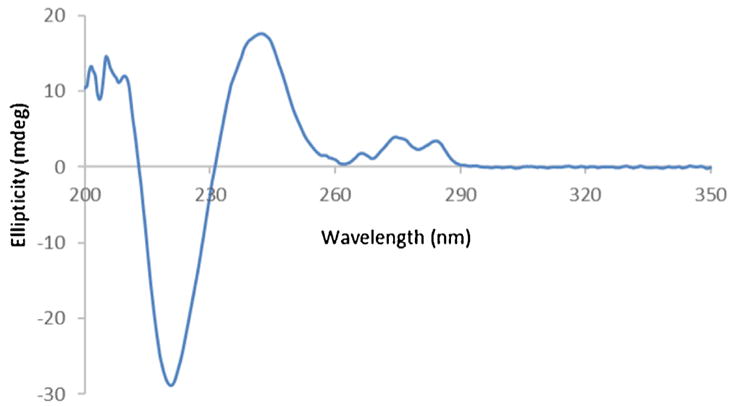
CD spectrum of 0.133 mg/mL (S)-citalopram oxalate in acetonitrile and potassium phosphate (pH = 3.0) (65:35, v:v).
3.2. Optimizing CD detection wavelength
Spectra for the 10 remaining substances were used to select the detection wavelength for the inline CD detector. Since the inline detector measures the g-factor using a constant wavelength, selection of wavelengths corresponding to large CD signals was important to maximize sensitivity. However, the selection of the detection wavelength was constrained by several factors. First, wavelengths less than 10 nm from polarity changes in spectra were avoided. Since the inline CD detector utilized a mandatory bandwidth of 20 nm, signals of different polarity would cancel out and decrease sensitivity. For example, a detection wavelength of 235 nm for (S)-citalopram would not be optimal due to positive (231–245 nm) and negative (225–230 nm) contributions. In addition, the g-factor at these wavelengths would be more susceptible to slight differences in mobile phase composition, which would decrease precision. Next, wavelengths below 230 nm were eliminated due to excessive CD noise observed in the region. This noise is due to the absorbance of organic solvents such as acetonitrile and methanol, which are common additions to HPLC mobile phases.
The above procedure was used to narrow down detection wavelengths to a small window for each substance. For example, the CD spectrum of (S)-citalopram indicated the best wavelength fell between 240 and 255 nm. For further optimization, standard preparations of pure drug enantiomers were analyzed by HPLC-CD using conditions consistent with assay methods prescribed in USP monographs. To illustrate, chromatograms for the HPLC-CD analysis of (S)-citalopram are shown in Fig. 2. The upper chromatogram displays the UV response, which was recorded by a photodiode array detector. This detector was positioned before the CD detector, showing how conventional assay determinations may be performed concurrently with enantiomeric analysis. The middle and lower chromatograms show the ellipticity and g-factor responses, respectively, which were both recorded using the inline CD detector. As forecast by the CD spectrum, the ellipticity response showed a large positive peak during the elution of (R)-citalopram (37.5 min). Unlike traditional peak shapes, the g-factor response was rectangular. This is because the g-factor is determined by the enantiomer composition, which does not change during elution. Large spikes are typically observed at the beginning and end of the g-factor response due to errors associated with the lower elution concentrations present during these times. To obtain the g-factor for each sample, the magnitude of the signal at the retention time was recorded (dashed lines).
Fig. 2.

Chromatograms for the HPLC-CD analysis of (S)-citalopram. A UPDA detector (upper) recorded absorbance at 237 nm while a CD detector recorded the ellipticity (middle) and g-factor (bottom) at 242 nm. Insets display (S)-citalopram elution at 37.5 min.
Injections for each substance were performed while the CD detector wavelength was altered within the detection window determined from the CD spectra. The detection wavelength which corresponded to the greatest absolute value of the g-factor was chosen for each substance. A complete summary of chosen detection wavelengths is presented in Table 1, alongside the g-factors determined from spectra and from HPLC-CD. Note the slight discrepancies between g-factors recorded from the different sources, which is attributable to two factors. First, several methods utilized gradient mobile phases, meaning that the mobile phase composition was time-dependent. Because the exact mobile phase composition during substance elution is difficult to determine precisely for recording spectra, spectral g-factors will inevitably vary from those recorded during HPLC-CD. Although attempts were made to replicate the precise mobile phase composition for spectral recordings, these were nevertheless best approximations. The second reason g-factors varied slightly was because spectra were recorded using a 1 nm bandwidth, whereas detection following HPLC utilized a 20 nm bandwidth. A smaller bandwidth can be used to record spectra because the signal is not time dependent, so the detector can accumulate data over a longer period. For HPLC detection, a larger bandwidth is necessary to improve the sensitivity during the elution period. Thus, even g-factors for substances utilizing isocratic elution (i.e. constant mobile phase composition) were slightly different from their spectral values.
Not all drug substances which showed CD spectral activity were compatible with HPLC-CD. The g-factor for levofloxacin could not be determined by the inline CD detector due to excessive noise, which was due to the presence of copper (II) sulfate in the mobile phase. This mobile phase component absorbed strongly at the wavelengths for which levofloxacin displayed CD signals, obscuring any measurement of the g-factor. The reason the g-factor was determinable from the spectrum was because this was recorded using a separate CD spectrometer, whose more intense light source was better suited to analyze strongly absorbing samples. Thus even when compounds show CD activity, parameters such as the mobile phase composition in USP monograph methods may still limit the effectiveness of HPLC-CD.
3.3. Method validation
Once detection parameters were established, the accuracy, precision, linearity, and LOQ for the enantiomer impurities were determined. This was done through HPLC-CD analysis of standard solutions containing the assay concentration of the drug substance, which was spiked with a small amount (≤5%) of the chiral impurity. Each analytical characteristic was determined using criteria detailed below.
3.3.1. Linearity
The g-factors of eluting drug substances were determined by analysis of standard impurity solutions spanning at least 7 different enantiomeric compositions distributed between 0 and 5%. Fig. 3 displays a calibration curve constructed for citalopram, which demonstrates how the g-factor decreased as the concentration of (R)-citalopram, the chiral impurity, increased. A linear least squares regression model was applied to each data set, and the coefficients of determination (r2) are reported in Table 2. Six substances showed strong linearity (r2 coefficients ≥ 0.990), indicating that the g-factor was highly correlated to the impurity concentration. Two substances, dexchlorpheniramine and pramipexole, had r2 values slightly lower (0.978 and 0.988, respectively), but were still great enough to suggest linearity. In contrast, the impurity for just one substance, levetiracetam, was determined to have poor linearity (r2 = 0.866).
Fig. 3.
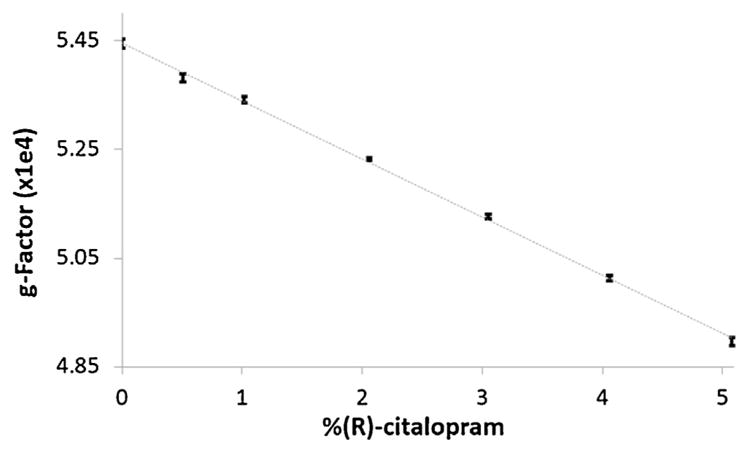
Calibration curve for the g-factor versus (R)-citalopram percentage in standard solutions. Error bars represent the standard deviation of measurements from 3 different solutions.
Table 2.
Linearity, accuracy, precision, and LOQ determinations for impurities of chiral drug substances.
| Drug Substance | r2 | Accuracy (%) | Precision (%) | LOQ (%) | USP Limit (%) |
|---|---|---|---|---|---|
| Dexchlorpheniramine | 0.978 | 1.9 | 2.8 | 2.8 | 2.0 |
| Dextromethorphan | 0.995 | 1.0 | 0.5 | 1.0 | 0.1 |
| (R)-citalopram | 0.999 | 0.5 | 1.0 | 1.0 | 3.0 |
| Esomeprazole | 0.990 | 0.4 | 0.8 | 0.8 | 0.2 |
| Levetiracetam | 0.866 | 4.0 | >4.0 | >4.0 | 0.8 |
| Levocetirizine | 0.999 | 0.6 | 1.9 | 1.9 | 2.0 |
| Pramipexole | 0.988 | 0.6 | 1.0 | 1.0 | 1.0 |
| Tolterodine | 0.995 | 0.3 | 1.0 | 1.0 | 1.0 |
| Valsartan | 0.992 | 1.0 | 2.1 | 2.1 | 1.0 |
3.3.2. Accuracy
Accuracy was determined using the percent recovery of impurities at each concentration level for standard solutions described above. For this study, the lowest impurity level at which the recovery was within 80–120% was used to denote the accuracy limit. As displayed in Table 2, impurities for most substances had an accuracy level at or below 1.0%. To put this in context, the table also reports the maximum enantiomeric impurity quantities allowed in each drug substance per USP, which are similar in magnitude to the accuracy determinations. Drugs with higher limits such as (S)-citalopram and levocetirizine (3.0 and 2.0%, respectively) had accuracy levels well below impurity specification levels. In contrast, impurities for drugs with the lowest specification levels (dextromethorphan and esomeprazole, 0.1 and 0.2%, respectively) could not be determined at levels low enough to meet USP standards. These results suggest that the HPLC-CD sensitivity is limited to the nearest percentage (±1%) for accuracy determinations. This level was suitable to quantify impurities for 6 drug substances at or below the USP limit.
3.3.3. Precision
Precision was assessed using the relative standard deviation (RSD) of recoveries for impurities in standard solutions. An RSD ≤ 10% is generally considered acceptable for impurity determinations, and the lowest impurity level for which this condition was met is reported in Table 2 for each substance. As the table shows, precision levels were slightly worse than accuracy levels, as only four substances demonstrated precision at or below the impurity level specified by USP.
3.3.4. Limits of quantification (LOQs)
The LOQs of enantiomer impurities were determined in a manner consistent with guidelines established by the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use [36]. For this approach, the LOQ is established by demonstrating both accuracy and precision at a particular impurity level. This means that the higher of the two impurity levels determined by accuracy and precision would be the LOQ. Table 2 shows that five substances (dexchlorpheniramine, dextromethorphan, esomeprazole, levetiracetam, and valsartan) had LOQs for their enantiomer impurities above USP impurity limit, while four ((S)-citalopram, pramipexole, levocetirizine, tolterodine) had LOQs at or below the limit. For the latter, these results indicated that HPLC-CD could quantify enantiomer impurities at levels suitable to meet current regulations. The best performing drug substances in this study had LOQs nearly equivalent to those of previous literature reports. Recall that these studies sought to optimize conditions favorable to CD detection rather than focus on compatibility with validated methods, and in doing so, were able to achieve LOQs around 0.5% [10,12,16,19]. The use of organic mobile phase components in the USP methods likely resulted in slightly poorer performance, but nevertheless, most LOQs were near this level.
Unexpectedly, the magnitude of g-factors showed poor correlation to analytical sensitivity. This is demonstrated most strikingly by comparing results for levocetirizine and levetiracetam. Despite having a g-factor 42 times that of levocetirizine, LOQ estimates for levetiracetam were 4 times greater. Indeed, even when levetiracetam was eliminated from the dataset, as it performed significantly worse than all other drug candidates, the correlation coefficient between the magnitude of the g-factor and the LOQ was just −0.39.
4. Conclusions
Of the 13 drug substances selected for HPLC-CD analysis, 4 ((S)-citalopram, levocetirizine, pramipexole, and tolterodine) were identified as compatible with drug substance assay methods and enantiomeric impurity limits established by USP. Additionally, most of the impurities of HPLC-CD compatible drugs were quantitative to sensitivity levels of ±1%. While this compares unfavorably to conventional chiral HPLC separations, which can quantify impurity levels an order of magnitude lower (±0.1%), this means that drugs with higher impurity tolerances would be suitable for HPLC-CD analysis, and could greatly benefit from the incorporation of CD detection. USP monographs for over 300 substances lack procedures to determine enantiomeric purity, and integrating HPLC-CD into even a fraction of these monographs could enhance safety and limit development costs. Additionally, once implemented, CD detection could be performed concurrently with assay methods, providing further benefit over chiral-specific methods.
Supplementary Material
Acknowledgments
Funding
Work was supported by the Research Participation Program at the Center for Drug Evaluation and Research administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. department of Energy and the U.S. Food and Drug Administration.
Abbreviations
- ACN
acetonitrile
- CD
circular dichroism
- HPLC
high-performance liquid chromatography
- HPLC-CD
high-performance liquid chromatography with circular dichroism detection
- USP
United States Pharmacopeia
Footnotes
FDA disclaimer
This article reflects the views of the authors and should not be construed to represent FDA’s views or policies.
References
- 1.Murakami H. From racemates to single enantiomers – chiral synthetic drugs over the last 20 years. In: Sakai K, Hirayama N, Tamura R, editors. Novel Optical Resolution Technologies. Springer; Berlin, Heidelberg: 2007. pp. 273–299. [DOI] [PubMed] [Google Scholar]
- 2.Mohan SJ, Mohan EC, Yamsani MR. Chirality and its importance in pharmacetuical field – an overview. Int J Pharm Sci Nanotechnol. 2009;1(4):309–316. [Google Scholar]
- 3.Andersson T. Single-isomer drugs. Clin Pharmacokinet. 2004;43(5):279–285. doi: 10.2165/00003088-200443050-00001. [DOI] [PubMed] [Google Scholar]
- 4.Agranat I, Caner H, Caldwell J. Putting chirality to work: the strategy of chiral switches. Nat Rev Drug Discov. 2002;1(10):753–768. doi: 10.1038/nrd915. [DOI] [PubMed] [Google Scholar]
- 5.Tucker GT. Chiral switches. The Lancet. 9209;355:1085–1087. doi: 10.1016/S0140-6736(00)02047-X. [DOI] [PubMed] [Google Scholar]
- 6.Nguyen LA, He H, Pham-Huy C. Chiral drugs: an overview. Int J Biomed Sci. 2006;2(2):85–100. [PMC free article] [PubMed] [Google Scholar]
- 7.Maier NM, Franco P, Lindner W. Separation of enantiomers: needs, challenges, perspectives. J Chromatogr A. 2001;906(1):3–33. doi: 10.1016/s0021-9673(00)00532-x. [DOI] [PubMed] [Google Scholar]
- 8.Yamamoto C, Okamoto Y. Chiral Analysis. Elsevier; Amsterdam: 2006. CHAPTER 7 – HPLC resolution using polysaccharide derivatives as CSP; pp. 215–239. [Google Scholar]
- 9.Gergely A, Szász G, Szentesi A, Gyimesi-Forrás K, Kökösi J, Szegvári D, Veress G. Evaluation of CD detection in an HPLC system for analysis of DHEA and related steroids. Anal Bioanal Chem. 2006;384(7):1506–1510. doi: 10.1007/s00216-006-0318-4. [DOI] [PubMed] [Google Scholar]
- 10.de Andrés F, Castañeda G, Ríos Á. Achiral liquid chromatography with circular dichroism detection for the determination of carnitine enantiomers in dietary supplements and pharmaceutical formulations. J Pharm Biomed Anal. 2010;51(2):478–483. doi: 10.1016/j.jpba.2009.02.018. [DOI] [PubMed] [Google Scholar]
- 11.Zougagh M, Aranda P, Castañeda G, Ríos Á. Supercritical fluid extraction—achiral liquid chromatography with circular dichroism detection for the determination of menthone enantiomers in natural peppermint oil samples. Talanta. 2009;79(2):284–288. doi: 10.1016/j.talanta.2009.03.047. [DOI] [PubMed] [Google Scholar]
- 12.Lecoeur-Lorin M, Delépée R, Ribet JP, Morin P. Chiral analysis of milnacipran by a nonchiral HPLC −circular dichroism: improvement of the linearity of dichroic response by temperature control. J Sep Sci. 2008;31(16–17):3009–3014. doi: 10.1002/jssc.200800291. [DOI] [PubMed] [Google Scholar]
- 13.Lecoeur-Lorin M, Delépée R, Adamczyk M, Morin P. Simultaneous determination of optical and chemical purities of a drug with two chiral centers by liquid chromatography-circular dichroism detection on a nonchiral stationary phase. J Chromatogr A. 2008;1206(2):123–130. doi: 10.1016/j.chroma.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 14.Bringmann G, Münchbach M, Feineis D, Messer K, Diem S, Herderich M, Clement HW, Stichel-Gunkel C, Kuhn W. Endogenous alkaloids in man: XXXVIII. Chiral and achiral determination of the neurotoxin TaClo (1-trichloromethyl-1,2,3,4-tetrahydro-β-carboline) from blood and urine samples by high-performance liquid chromatography–electrospray ionization tandem mass spectrometry. J Chromatogr B. 2002;767(2):321–332. doi: 10.1016/s1570-0232(01)00589-x. [DOI] [PubMed] [Google Scholar]
- 15.Sánchez FG, Díaz AN, de Vicente ABM. Enantiomeric resolution of bupivacaine by high-performance liquid chromatography and chiroptical detection. J Chromatogr A. 2008;1188(2):314–317. doi: 10.1016/j.chroma.2008.02.070. [DOI] [PubMed] [Google Scholar]
- 16.Bertucci C, Andrisano V, Cavrini V, Castiglioni E. Reliable assay of extreme enantiomeric purity values by a new circular dichroism based HPLC detection system. Chirality. 2000;12(2):84–92. doi: 10.1002/(SICI)1520-636X(2000)12:2<84::AID-CHIR5>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 17.Meyring M, Mühlbacher J, Messer K, Kastner-Pustet N, Bringmann G, Mannschreck A, Blaschke G. In vitro biotransformation of (R)- and (S)-thalidomide: application of circular dichroism spectroscopy to the stereochemical characterization of the hydroxylated metabolites. Anal Chem. 2002;74(15):3726–3735. doi: 10.1021/ac0203138. [DOI] [PubMed] [Google Scholar]
- 18.Luykx DMAM, Goerdayal SS, Dingemanse PJ, Jiskoot W, Jongen PMJM. HPLC and tandem detection to monitor conformational properties of biopharmaceuticals. J Chromatogr B. 2005;821(1):45–52. doi: 10.1016/j.jchromb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 19.Lorin M, Delepee R, Maurizot JC, Ribet JP, Morin P. Sensitivity improvement of circular dichroism detection in HPLC by using a low-pass electronic noise filter: application to the enantiomeric determination purity of a basic drug. Chirality. 2007;19(2):106–113. doi: 10.1002/chir.20352. [DOI] [PubMed] [Google Scholar]
- 20.Kirkpatrick D, Yang J, Trehy M. Determination of the enantiomeric purity of epinephrine by HPLC with circular dichroism detection. J Liquid Chromatogr Relat Technol. 2017;40(11):556–563. doi: 10.1080/10826076.2017.1333962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yasui SC, Keiderling TA. Vibrational circular dichroism of polypeptides. 8. Poly(lysine) conformations as a function of pH in aqueous solution. J Am Chem Soc. 1986;108(18):5576–5581. [Google Scholar]
- 22.Escitalopram Oxalate Official Monograph. United States Pharmacopeia; Rockville, MD: 2017. pp. 4054–4056. [Google Scholar]
- 23.Dexchlorpheniramine Maleate Official Monograph. United States Pharmacopeia; Rockville, MD: 2017. pp. 3685–3687. [Google Scholar]
- 24.Levalbuterol Hydrochloride Official Monograph. United States Pharmacopeia; Rockville, MD: 2017. pp. 4803–4804. [Google Scholar]
- 25.Tolterodine Tartrate Official Monograph. United States Pharmacopeia; Rockville MD: 2017. pp. 6518–6520. [Google Scholar]
- 26.Valsartan Official Monograph. United States Pharmacopeia; Rockville MD: 2017. pp. 6649–6650. [Google Scholar]
- 27.Levetiracetam Official Monograph. United States Pharmacopeia; Rockville MD: 2017. pp. 4806–4808. [Google Scholar]
- 28.Levocetirizine Dihydrochloride Official Monograph. United States Pharmacopeia; Rockville, MD: 2017. pp. 4825–4826. [Google Scholar]
- 29.Levofloxacin Official Monograph. United States Pharmacopeia; Rockville, MD: 2017. pp. 4831–4833. [Google Scholar]
- 30.Pramipexole Dihydrochloride Official Monograph. United States Pharmacopeia; Rockville, MD: 2017. pp. 5795–5797. [Google Scholar]
- 31.Sitagliptin Phosphate Official Monograph. United States Pharmacopeia; Rockville, MD: 2017. pp. 6171–6172. [Google Scholar]
- 32.Timolol Maleate Official Monograph. United States Pharmacopeia; Rockville, MD: 2017. pp. 8416–8418. [Google Scholar]
- 33.Dextromethorphan Hydrobromide Official Monograph. United States Pharmacopeia; Rockville, MD: 2017. pp. 3705–3707. [Google Scholar]
- 34.Esomeprazol Magnesium Official Monograph. United States Pharmacopeia; Rockville, MD: 2017. pp. 4057–4059. [Google Scholar]
- 35.Bossu’ E, Cotichini V, Gostoli G, Farina A. Determination of optical purity by nonenantioselective LC using CD detection. J Pharm Biomed Anal. 2001;26(5–6):837–848. doi: 10.1016/s0731-7085(01)00483-6. [DOI] [PubMed] [Google Scholar]
- 36.Register F, editor. Validation of Analytical Procedures: Methodology. ICH; 1997, 2018. pp. 27463–27467. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.