

Abstract
Protein–protein interactions (PPI) offer unexploited opportunities for CNS drug discovery and neurochemical probe development. Here, we present ZL181, a novel peptidomimetic targeting the PPI interface of the voltage-gated Na+ channel Nav1.6 and its regulatory protein fibroblast growth factor 14 (FGF14). ZL181 binds to FGF14 and inhibits its interaction with the Nav1.6 channel C-tail. In HEK-Nav1.6 expressing cells, ZL181 acts synergistically with FGF14 to suppress Nav1.6 current density and to slow kinetics of fast inactivation, but antagonizes FGF14 modulation of steady-state inactivation that is regulated by the N-terminal tail of the protein. In medium spiny neurons in the nucleus accumbens, ZL181 suppresses excitability by a mechanism that is dependent upon expression of FGF14 and is consistent with a state-dependent inhibition of FGF14. Overall, ZL181 and derivatives could lay the ground for developing allosteric modulators of Nav channels that are of interest for a broad range of CNS disorders.
Keywords: Fibroblast growth factor 14 (FGF14), protein:protein interaction (PPI), voltage-gated sodium channels (Nav1.6), peptidomimetics, minimal functional domains, neurochemical probes, CNS drug discovery
Graphical Abstract
INTRODUCTION
The voltage-gated sodium (Nav) channel is a large trans-membrane protein composed of one of nine pore-forming α-subunits (Nav1.1–Nav1.9), a varied auxiliary β-subunit, and many accessory regulatory proteins that mediate influx of Na+ through rapid voltage-dependent cycling between closed, open, and inactivated states.1–3 In neurons, Nav channels are enriched within the axonal initial segment (AIS), where they contribute to action potential initiation, forward and back-propagation, and repetitive firing.4,5 Mutations (including SNPs) in specific Nav isoforms are associated with numerous motor and cognitive disorders, including epilepsy,6–9 ataxia,10 and autism spectrum disorders.11,12 Yet, the lack of appropriate molecular probes has greatly limited the study of Nav channel function in the normal and diseased brain.
Emerging molecular insights on Nav channels reveal that they operate within a macromolecular complex of regulatory proteins through a matrix of protein–protein interaction (PPI) surfaces that form minimal functional domains (MFD) relevant for function and disease.13–16 Therefore, molecular probes against these MFD may enable the interrogation of Nav channel function with unprecedented precision.17
Previous studies provide evidence that intracellular fibroblast growth factor 14 (FGF14) is a functionally relevant component of the Nav channel macromolecular complex.18,19 FGF14 controls channel gating and axonal targeting in neurons through isoform-specific interactions with the intracellular C-terminal tail of Nav1.6 and other Nav isoforms,18–24 affecting excitability, synaptic transmission, plasticity, and neurogenesis in the cortico-mesolimbic circuit with cognitive and affective behavioral outcomes.18,25–30 Recent translational studies have linked FGF14 to cognitive diseases, neurodegeneration,31 depression, anxiety, addictive behaviors,32–36 and schizophrenia.28,30 Based on this strong premise, we selected the PPI interface between FGF14 and the Nav1.6 channel, a relevant regulator of intrinsic excitability and repetitive firing in the cortico-mesolimbic circuit, as a novel target for probe development.
Here, we identify ZL181, a novel PPI-based peptidomimetic that suppresses binding of FGF14 to Nav1.6 and acts as an FGF14-dependent modulator of Nav1.6 currents and intrinsic excitability of medium spiny neurons (MSNs) of the nucleus accumbens (NAc). Our MFD approach is new in CNS drug discovery and neurochemical probe development, and might open new horizons in therapeutics against brain disorders.
RESULTS AND DISCUSSION
Design and Synthesis of Novel FGF14-Based Modulators of Nav1.6 Channel
To design new PPI probes targeting Nav1.6, we generated a series of analogues derived from Ac-FLPK-CONH2, a previously identified peptide fragment that aligns with the FGF14:Nav1.6 channel complex interface.37 FLPK is a homology model-based peptide previously mapped to the FGF14 surface37 that aligns with V160 in the β-12 sheet of the FGF14 core domain. Substitution of V160 with alanine abolishes FGF14 interaction with Nav1.6, and disrupts functional modulation of Nav1.6 currents without affecting other binding partners of FGF14 (such as the FGF14 monomer in the FGF14:FGF14 dimer complex). Thus, V160 within the β8-β9/β12 pocket is a hot-spot residue that confers structure–function specificity to the FGF14:Nav1.6 complex surface22 and, as such, was the primary determinant of the domain chosen for probe development. The derived peptidomimetics were synthesized either by truncation modifications such as ZL141 (Cbz-FLP-CONH2) and ZL148 (Ac-FLK(Boc)-CONH2) or addition of hydrophobic functional groups such as ZL170 (Ac-Leu-Pro-Lys(Fmoc)-COOMe), ZL171 H-Leu-Pro-Lys(Fmoc)-COOMe, ZL173 (Boc-Phe-Leu-Pro-Lys(Fmoc)-COOMe), ZL175 (H-Phe-Leu-Pro-Lys(Fmoc)-COOMe), ZL176, (Cbz-Phe-Leu-Pro-COOMe), ZL181 (Cbz-FLPK(Boc)-CONH2), ZL182 (Phe-Leu-Pro-Lys (Fmoc)–OH), ZL183 (Ac-Phe-Leu-Pro-Lys-(Boc)-CO- Morpholine), ZL184 (Ac-Phe-Leu-Pro-Lys(Boc)-CO-benzene), ZL185 (Ac-Phe-Leu-Pro-Lys(Boc)-CO- Thiazolidine), ZL186 (H-Phe-Leu-Pro-Lys(Boc)-CONH2), ZL188–02 (Cbz-Phe-Leu-Pro-Lys(Boc)-CONH2), and ZL192 Ac-Phe-Leu-Pro-Lys-CO-Aniline) (Figure 1 or chemical synthesis in Supporting Information Schemes 1–4) through the synthetic approach described in the Supporting Information. Introduction of hydrophobic moieties such as (carboxybenzyl (Cbz) and tert-butyloxycarbonyl (Boc)) were used to improve solubility and cell permeability, which impeded full evaluation of FLPK in cellular assays.37
Figure 1.

Chemical structures of four selected newly synthesized peptidomimetics ZL181, ZL141, ZL148, and ZL182.
In-Cell and in Vitro Validation of Peptidomimetics
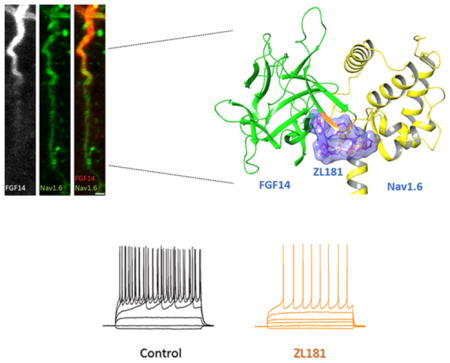
To examine the activity of these compounds, we reconstituted the FGF14:Nav1.6 complex using the split-luciferase complementation assay (LCA), which shows binding of CLuc-FGF14 to CD4-Nav1.6-NLuc at physiological conditions.21,22,38–40 All compounds were dissolved in DMSO and initially tested at 50 μM (Figure S1) and a few selected for further analysis. Compared to DMSO control (0.5%) significant changes in luminescence were observed for ZL141 (132 ± 9%, n = 9, p = 0.0007), ZL181 (75 ± 6%, n = 9, p = 0.0007), and ZL182 (129 ± 13%, n = 9, p = 0.0244), but not for other compounds such as ZL141 (103 ± 14%, n = 5, p = 0.6984, Figure 2a,b). These changes were specific as none of the compounds interfered with luminescence produced by the native luciferase enzyme (Figure 2c). Next, we ranked compounds (exhibiting statistically significant effects on the FGF14:Nav1.6 complex) based on dose–response curves. ZL181 displayed a sigmoidal dose–response inhibition curve (Figure 2d), with an apparent IC50 = 63 μM and a favorable clogP = 3.7,37,41,42 had no effect on LCA reporters expression (Figure 2c) and on other iFGF:Nav1.6 pairs (Figure S3).43 Using surface plasmon resonance (SPR), we calculated a Kd value of 89.6 μM for ZL181 binding to FGF14 (Figure 2e–f). We also used dynamic light scattering to rule out self-aggregation of ZL181,44,45 and we show that the compound does not self-aggregate (Figure S4). Given that the FLPK parent compound aligns with the V160 residue located at the FGF14:Nav1.6 interface,22 we tested ZL181 binding affinity for the FGF14V160A mutant, positing that this mutation would weaken ZL181 binding. Indeed, ZL181 had a weaker binding affinity for FGF14V160A (Kd = 181.5 μM, Figure S5), and we infer that V160 might be a potential binding site for ZL181. Complementary in silico studies performed on the FGF14:Nav1.6 homology model showed ZL181 docking at the previously identified druggable pocket37 including N157, E152, Y158, Y159, V160, and P203 residues of FGF14 (Figure 2g–i). Within this pocket that corresponds to the β12 strand and the β8-β9 NYYV loop of FGF14,37 ZL181 binds to V160 through hydrophobic interaction and forms hydrogen bonds with residues N157, E152, and P203. ZL181 also engages with the side chain of the two respective Y residues46 through a face-to-face π- stacking with Y158 and an edge-to-face π-stacking with Y159 (Figure 2g). We were unable to accurately determine a Kd value of ZL181 for the Nav1.6 C-tail using SPR as binding did not reach steady-state (data not shown). Nevertheless, our in silico model predicted interaction sites of ZL181 with the Nav1.6-C tail through hydrogen bonds, π–π, and hydrophobic interactions at D1833, R1892, and I1886 (Figure S6). These interactions might represent low-affinity binding sites of the compound to the channel C-tail.
Figure 2.

In-cell and in vitro validation of ZL181. (a) Luminescence values from HEK293 cells expressing CLuc-FGF14 and CD4-Nav1.6-NLuc constructs treated with ZL141 (gray), ZL148 (green), ZL181 (orange), ZL182 (blue) at 50 μM or 0.5% DMSO (black). (b) Bar chart with data points overlap represents percent maximal luminescence of ZL141, ZL148, ZL181, and ZL182 normalized to DMSO. (c) Bar chart with data points overlap represents percent maximal luminescence from HEK293 cells expressing the full-length luciferase treated with ZL141, ZL148, ZL181, and ZL182 and normalized to DMSO. (d) LCA-based dose–responses of ZL148, ZL181, and ZL182 against CLuc-FGF14 and CD4-Nav1.6-NLuc. (e) The representative SPR sensorgram of ZL181 binding to FGF14WT and (f) saturation binding curves (n = 3). (g) Ribbon presentation of ZL181 (purple) docking on FGF14 (green) homology model. FLPK and NYYV sequences are highlighted in orange. Hydrogen bonds are shown as purple dotted lines, and π −π interaction as blue dotted line. (h) ZL181 docking at the PPI of the FGF14 (green) and Nav1.6 (yellow) complex. The binding surface of ZL181 (purple) is shown in blue. (i) Zoomed view of ZL181 (purple) docking pose overlay at the PPI interface of the FGF14:Nav1.6 complex. Data represented as mean ± SEM, *p < 0.05; **p < 0.01.
ZL181 Modulates Nav1.6 Channel Activity
To test whether ZL181 affected Nav1.6 currents and their well-described regulation by FGF14, we used patch-clamp electrophysiology of HEK cells stably expressing human Nav1.6 (HEK-Nav1.6) that were transiently transfected with GFP (HEK-Nav1.6-GFP) or FGF14-GFP (HEK-Nav1.6-FGF14-GFP) and treated with ZL181 (20 μM) or DMSO (0.2%) (Figure 3a). In HEK-Nav1.6-GFP cells pretreated with ZL181, Nav1.6 peak current density derived from transient Na+ currents (INa+) was significantly lower (−20.9 ± 3.4 pA/pF) compared to DMSO treatment (−73.8 ± 13.6 pA/pF, n = 12, p = 0.04; Figure 3b,c). In line with previous studies,20,22,40 we found that expression of FGF14-GFP suppresses Nav1.6 peak current density (−18.1 ± 3.8 pA/pF, n = 20 versus −73.8 ± 13.6 pA/pF, n = 12; p = 0.001, ANOVA, posthoc Bonferroni), a phenotype that was enhanced by ZL181 (−7.4 ± 4.4 pA/pF, n = 19 compared to DMSO treatment, p = 0.0039 Figure 3b,c). Western blots ruled out changes in Nav1.6 and FGF14 total protein expression levels in the presence of ZL181 (Figure S2). Additional analysis revealed effects of ZL181 on tau (τ) of fast inactivation that were evident only in the presence of FGF14 (Figure 3d,e). In HEK-Nav1.6-FGF14-GFP cells, ZL181 slowed the transition of the channel from the open to the inactivated state (2.15 ± 0.4 ms, n = 8 compared to GFP DMSO control group (1.0 ± 0.8 ms, n = 10, p = 0.0032, posthoc Dunn test) to a degree that exceeded previously reported effects of FGF14 on this channel property (1.5 ± 0.1 ms, n = 12, p = 0.0218, posthoc Dunn test). In the absence of FGF14, ZL181 had no significant effect on V1/2 of activation, and did not interfere with FGF14 modulation of the channel activation (Figure 3f,g). In contrast, ZL181 competed with FGF14 inducing a 7 mV hyperpolarizing shift of V1/2 of steady-state inactivation (p = 0.037; Figure 3h,i) that rescued channel availability back to the GFP DMSO control group. We next determined if ZL181 had any effect on other FGF14:Nav isoform complexes. We showed that ZL181 had no significant effects on Nav1.1- and Nav1.2-mediated currents in the presence of FGF14 (Figure S7). Detailed analysis of all data presented in Figures 3 and S7 is available in Tables S1 and S3.
Figure 3.

Complex functional regulation of the Nav1.6-mediated currents by ZL181. (a) Representative traces of Na+ transient currents (INa+) recorded from HEK-Nav1.6 cells transiently expressing GFP or FGF14-GFP constructs in response to depolarizing voltage steps (inset). GFP-expressing cells were treated with 0.2% DMSO (black traces) or with 20 μM ZL181 (orange traces). FGF14-GFP expressing cells were treated with either 0.2% DMSO (blue traces) or 20 μM ZL181 (gray traces). (b) Current–voltage relationships of INa from the experimental groups described in panel (a). (c) Bar chart with data points overlap represents peak current densities derived from panel (a). (d) Representative traces of experimental groups described in panel (a) to illustrate tau (τ) of INa+. (e) Summary bar chart with data points overlap of τ from the indicated experimental groups. Voltage dependence of INa+ activation (f) and steady-state inactivation (h) are plotted as a function of the membrane potential (mV). Bar chart with data points overlap summary of V1/2 of activation (g) and steady-state inactivation (i) in the indicated experimental groups. Data are mean ± SEM; *p < 0.05; **p < 0.01. The fitted parameters are provided in Supporting Information Table S1.
In the absence of FGF14, ZL181 mimics FGF14-induced suppression of Nav1.6 currents. However, none of the other Nav1.6 parameters modulated by FGF14 (i.e., V1/2 of activation and/or steady-state inactivation and tau of fast inactivation)20,22 were affected by ZL181 alone. Thus, in the absence of FGF14, ZL181 might retain a weak FGF14 agonist activity. That might be explained by interactions of the compound with the low affinity sites on the channel C-tail (that might mimic FGF14 interaction) or with yet undetermined sites on the channel that differ from the known FGF14 interacting domain on the C-tail.
In the presence of FGF14, ZL181 exhibits a complex mechanism of action. During the closed-to-open state transition and the onset of fast inactivation, ZL181 acts as an agonist that potentiates FGF14 function, suppressing Nav1.6 currents and slowing tau of fast inactivation to an extent that exceeds the effect of FGF14 alone. This phenotype might find a structural correlate with ZL181 docking to the FGF14 β12/β8-β9 pocket and then recruiting potential low-affinity sites on the Nav1.6 C-tail, stabilizing the FGF14:Nav1.6 complex and enhancing FGF14 functions. At steady-state inactivation, ZL181 acts as an antagonist competing with FGF14 and decreasing channel availability (preventing FGF14-induced hyperpolarizing shift of V1/2 of steady-state inactivation). This switch from an agonist to antagonist might occur in response to conformational changes at the FGF14:Nav1.6 interface during channel inactivation.
ZL181 Functional Properties Are Affected by the FGF14 N-Terminal Domain
Previous work has shown that the N-terminus of FGF14 dictates the magnitude and direction of phenotypes of Nav1.6 currents. In heterologous cells, Nav1.6 peak current density is potentiated by FGF14 constructs lacking the N-terminal tail.20 This phenotype is opposite to the suppression induced by full-length FGF14 (Figure 3a–c), but is closer to the predicted mechanism of action of native FGF14 in neurons.18,27 We posited that ZL181 could provide novel insights into such an apparent discrepancy revealing important information on the FGF14 mechanism of action. We tested this hypothesis using the same experimental design of Figure 3 except that HEK-Nav1.6 cells were transiently transfected with an FGF14 mutant lacking the N-terminal domain (HEK-Nav1.6 and FGF14-ΔNT-GFP). In agreement with previous studies, Nav1.6 peak current amplitude in HEK-Nav1.6-FGF14-ΔNT-GFP cells was potentiated (−99.8 ± 13.9 pA/pF, n = 18) compared to HEK-Nav1.6-GFP cells (−59.0 ± 8.9 pA/pF, n = 13; p < 0.05, posthoc Holm-Sidak; Figure 4a–c). ZL181 competed with FGF14-ΔNT-GFP inducing a suppression of Nav1.6 currents (−25.4 ± 6.9 pA/pF, n = 10) that was significantly different from the corresponding DMSO group in HEK-Nav1.6-FGF14-ΔNT-GFP cells (one-way ANOVA, post-hoc Holm-Sidak, p < 0.0001; Figure 4a–c); neither FGF14-ΔNT-GFP nor ZL181 affected tau of fast inactivation (Figure 4d,e). Importantly, in the presence of FGF14-ΔNT-GFP, ZL181 caused a significant 7 mV depolarizing shift of V1/2 activation compared to DMSO treatment in HEK-Nav1.6-FGF14-ΔNT-GFP cells (one-way ANOVA, posthoc Tukey test, p < 0.01, Figure 4f,g), but had no effects on V 1/2 of steady-state inactivation (Figure 4h,i). Detailed analysis of all data presented in Figure 4 is available in Table S2. Previous studies, confirmed here, demonstrate that in heterologous cells full length FGF14 suppresses Nav1.6 currents, slows tau of fast inactivation, and shifts V1/2 of steady-state inactivation.22 In contrast, FGF14-ΔNT potentiates Nav1.6 currents and shifts V1/2 of activation, but is ineffective on inactivation (both fast and steady-state). Therefore, we concluded that in heterologous cells the N-terminal tail of FGF14 blocks (or significantly alters) the core domain activity as the channel progresses from the open to inactivated state and becomes the dominant structural determinant when the channel is fully inactivated.
Figure 4.

ZL181 functional regulation of Nav1.6-mediated currents is influenced by the FGF14 N-terminal tail. (a) Representative traces of Na+ transient currents (INa+) recorded from HEK-Nav1.6 cells transiently expressing the indicated constructs in response to depolarizing voltage steps (inset); GFP-expressing cells were treated with 0.2% DMSO (black traces) or 20 μM ZL181 (orange traces). FGF14-ΔNT-GFP expressing cells were treated with either 0.2% DMSO (blue traces) or 20 μM ZL181 (gray traces). (b) Current–voltage relationships of INa from the experimental groups described in (a). (c) Bar chart with data points overlap representing peak current densities derived from panel (a). (d) Representative traces of experimental groups described in panel a to illustrate τ of INa+. (e) Summary bar chart with data points overlap of τ from the indicated experimental groups. Voltage dependence of INa+ activation (f) and steady-state inactivation (h) are plotted as a function of the membrane potential (mV). Bar chart with data points overlap summary of V1/2 of activation (g) and steady-state inactivation) (i) in the indicated experimental groups. Data are mean ± SEM; *p < 0.05; **p < 0.01. The fitted parameters are provided in Supporting Information Table S2.
ZL181 Activity Impairs Intrinsic Excitability of MSNs
Next, we examined the effect of ZL181 on firing properties of medium spiny neurons (MSNs) in the nucleus accumbens (NAc), known to express Nav1.6 channels28 and to exhibit a firing pattern consistent with Nav1.6 activity.47 Using confocal microscopy, we showed that native FGF14 and Nav1.6 are abundantly expressed in the NAc (Figure 5a), especially in small-size neurons consistent with previously characterized MSNs. In these cells, FGF14 and Nav1.6 colocalized within the AIS, the site of initiation of action potential (Figure 5a, and b).
Figure 5.

ZL181 suppresses firing of MSNs in the NAc. (a) Representative confocal images showing colocalization of Nav1.6 (green) and FGF14 (red) in the NAc. Note that Nav1.6 and FGF14 immunoreactivities were intense in the proximal region of the AIS. (b) Representative examples of Nav1.6 channels immunofluorescence intensity line scans along the AIS regions in individual neurons expressing FGF14. Nav1.6 (black), FGF14 (red). (c) Representative traces of action potentials (AP) evoked by multiple current steps (−20, 0, 50, 80, and 110 pA current steps of 800 ms duration) in MSNs from Fgf14+/+ mice treated with either 0.05% DMSO (black) or 50 μM ZL181 (orange); (d) single AP traces from the two corresponding experimental groups shown in c. Representative Input–output curves of (e) number of spikes, and (f) average instantaneous firing frequency (IFF) at varying injected current stimuli recorded in MSN from Fgf14+/+ mice in response to 0.05% DMSO (black) or 50 μM ZL181 (orange) treatment. Bar graph with data points overlap represents (g) voltage threshold and (h) current threshold in MSN from Fgf14+/+ mice in response to 0.05% DMSO (black) and 50 μM ZL181 (orange) treatment. Representative traces of AP in MSN from Fgf14−/− after (i) 0.05% DMSO (black), and 50 μM ZL181 (pink) treatment, (j) single AP traces of 0.05% DMSO (black) or 50 μM ZL181 (pink). Representative input–output curves of (k) number of spikes and (l) average IFF at varying injected current stimuli recorded in MSN from Fgf14−/− mice in response to 0.05% DMSO (black) and 50 μM ZL181 (pink) treatment. Bar chart with data points overlap represents (m) voltage threshold and (n) current threshold in MSN from Fgf14−/− mice in response to 0.05% DMSO (black) and 50 μM ZL181 (pink) treatment. Data are mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.005.
Using whole-cell patch clamp electrophysiology of acute NAc slices, we showed that ZL181 significantly reduced the number of evoked action potentials in MSNs compared to the DMSO control group (Figure 5c), a phenotype that persisted across a range of injected currents (Figure 5e). For example, at a current step of 150 pA, the number of spikes was 6.9 ± 2.3, n = 10 for the ZL181 group versus 18.4 ± 1.3, n = 8 for DMSO (p = 0.0011; Figure 5c,e). ZL181 was also effective in decreasing instantaneous firing frequency (IFF) across all tested current steps (i.e., at a current step of 150 pA, it was 9.3 ± 3.5 Hz, n = 8 for ZL181 versus 26.3 ± 1.5 Hz, n = 8 for DMSO, p = 0.0005); Figure 5f) and in increasing both the action potential voltage threshold (−35.9 ± 2.3 mV for DMSO treated MSNs, n = 8 versus −27.8 ± 2 mV for ZL181 treated MSNs, n = 10; p < 0.05 with Student’s t-test, p = 0.0156; Figure 5g) and current threshold (72.5 ± 11.5 pA, for DMSO treated MSNs, n = 8 versus 135.8 ± 12.5 pA for ZL181 treated MSNs, n = 10; p < 0.01; Student’s t-test; p = 0.0024; Figure 5h, Table S4). None of these phenotypes could be attributed to changes in other active or passive properties (Table S4), but are consistent with a reduction in Nav channel function.
To determine whether the mechanism of action of ZL181 required binding to FGF14 as expected from in vitro studies (Figure 2e,f), patch-clamp experiments were conducted in MSNs derived from Fgf14−/− mice. We found that in the knockout mouse model ZL181 did not significantly alter maximal and instantaneous firing frequency of MSNs compared to the DMSO control group (Figure 5i–l). Furthermore, no changes in the action potential voltage and current threshold were found in the ZL181 group compared to DMSO (Figure 5m,n and Table S5). These results confirmed our expectation that the mechanism of action of ZL181 in the native system depends upon FGF14.
Radial plots depict a comparison of ZL181 activity in HEK-Nav1.6 cells and MSNs (Figure 6). In HEK-Nav1.6 cells ZL181 acts as an antagonist of FGF14-ΔNT inhibiting Na+ peak current density and shifting Nav1.6 voltage sensitivity to a more depolarized level (Figure 6a, green line). In HEK-Nav1.6 cells expressing full length FGF14, ZL181 appears to be state-dependent. At the open channel state (transient peak current), ZL181 cooperates with FGF14 further suppressing Na+ currents and delaying Nav1.6 fast inactivation (Figure 6a, red line); at steady state inactivation ZL181 inhibits FGF14 function decreasing the fraction of available channels (Figure 6a, red line; Figure 4i gray vs black bar), a phenotype driven by the N-terminal tail of the protein.20
Figure 6.

Radial plots summarizing the mechanism of action of ZL181 in heterologous cells and in MSNs. (a) Peak current density (top), V1/2 of activation (right, defined as “voltage-sensitivity”), tau of fast inactivation (bottom) and available fraction of channels calculated from V1/2 of steady-state inactivation (left) in HEK-Nav1.6 cells treated with ZL181 and expressing either full-length FGF14-GFP (red) or FGF14-ΔNT-GFP (green); data sets are normalized to respective controls treated with DMSO (either full-length FGF14-GFP or FGF14-ΔNT-GFP; these two control data sets are displayed in blue in Figures 3 and 4, respectively). The data points inside the gray box represent suppression (green line) or increase (red line) of Nav1.6 channel function in the presence of ZL181 compared to respective DMSO control. (b) Mean firing frequency (top), action potential voltage threshold (right), instantaneous firing frequency (bottom), and action potential current threshold (left) in Fgf14+/+ wild type MSNs exposed to ZL181 (yellow line) compared to DMSO controls (dotted line); data are normalized to their respective controls treated with DMSO (displayed in black in Figure 5a–h). The action potential current threshold value is absolute as it refers to absolute Vm values (i.e., a Vthr of –40 mV is referred to as “40 mV”). The data points inside the gray box represent suppression (yellow line) of intrinsic firing in the presence of ZL181 compared to respective DMSO control.
In MSNs ZL181 reduces maximal and instantaneous firing frequencies and drives neurons to a less excitable state by shifting action potential current and voltage thresholds to a more depolarized level (Figure 6b, yellow line). All these phenotypes are consistent with reduced Na+ currents and channel availability. Extrapolating from the HEK cells results, we concluded that in MSNs ZL181 acts as a state-dependent antagonist inhibiting the FGF14 core domain when the Nav channel is in the open state and competing with the FGF14 N-terminal tail when the channel enters steady state inactivation.48,49 Because none of the effects of ZL181 in MSNs could be reconciled with a mechanism of cooperativity of the compound with FGF14 (as observed in HEK-Nav1.6 cells expressing full length FGF14), we concluded that in the native system the N-terminal tail of FGF14 might be tethered to other proteins (i.e., the β4 subunit)50 and remain functionally silent until the Nav channel enters steady-state inactivation. In summary, ZL181 inhibition of neuronal excitability may reffect state-dependent intramolecular changes at the FGF14:Nav channel interface that enable a switch of structural determinants to occur during the channel cycle.
CONCLUSION
In summary, we report the discovery and characterization of ZL181, a novel peptidomimetic based on FGF14. ZL181 is the first PPI-based modulator of Nav channels that was developed with an MFD approach based on the FGF14 interaction interface with Nav1.6. ZL181 modulates Nav1.6 currents in heterologous cells in a mode that depends on the channel state and the N-terminal tail of FGF14. In MSNs, where FGF14 and Nav1.6 are abundantly expressed, ZL181 suppresses neuronal excitability by a mechanism consistent with a state-dependent inhibition of native FGF14. The structure–function specificity conferred by the MFD approach and the translational value of FGF14 for neuropsychiatric disorders provide a strong premise for selecting ZL181 and other probes targeting this PPI complex as targets for future drug development.28,30–36
METHODS
Plasmids
CLuc-FGF14 (human), CD4-Nav1.6-NLuc (human), Fgf14–6xmyc (human) (Figure 2S) and the construct expressing full-length Firefly (Photinus pyralis) luciferase were engineered and characterized as previously described.19,20,39,51 FGF14-GFP (human) and FGF14-ΔNT-GFP (human) were cloned into the GFP plasmid (pQBI-fC2; Quantum Biotechnology Inc., Montreal, Canada) as previously described.19,40 The CLuc-FGF13 (human) construct was generated by replacing FGF14 with the FGF13 (1a isoform) coding sequence in the CLuc-FKBP fusion vector.52
Cell Culture and Transient Transfections
HEK-Nav1.6 cells were gifts from Enzo Wanke and Cinzia Lecchi, Università degli Studi di Milano-Bicocca, Milano, Italy. HEK-Nav1.6 cells were maintained in growth medium composed of equal volumes of DMEM and F-12 (Invitrogen, Waltham, MA) supplemented with 0.05% glucose, 0.5 mm pyruvate, 10% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin incubated at 37 °C with 5% CO2. And 80 or 500 μg/mL G418 was used to maintain Nav1.1/Nav1.6 or Nav1.2 stable expression in cells, respectively (Invitrogen, Waltham, MA). Cells were transfected according to manufacturer’s instructions at 90–100% confluency with equal amount of plasmid pairs using Lipofectamine 2000 (Invitrogen, Waltham, MA).39,40,51
Chemical Synthesis of Peptidomimetics
Available in the Supporting Information.
Split-Luciferase Complementation Assay (LCA)
Assays were performed as previously described.21,22,31,37,39,40 Briefly, HEK-293 cells (~4.5 × 105 per 24-well plates-mm dish) were transiently cotransfected with pairs of plasmids or single plasmids using Lipofectamine 2000 (Invitrogen). At 24 h after transfection with CLuc-FGF14 or CD4-Nav1.6-NLuc (or other indicated plasmids), cells were detached from the 24-well plate using a 0.04% trypsin:EDTA mixture dissolved in PBS. Cell suspension was centrifuged for 5 min at 6000 rmp and seeded in white, clear-bottom 96-well tissue culture plates (Greiner Bio-One) in 200 μL of medium. The cells were incubated for another 24 h, and then the growth medium was replaced with 100 μL of serum-free, phenol red-free DMEM/F-12 medium (Invitrogen). Peptidomimetics were dissolved in DMSO, and were added to a final concentration of 1–500 μM in the serum-free medium and cells were incubated with peptidomimetics for 1 h. The final concentration of DMSO was maintained at 0.5%. The bioluminescence reaction was initiated by automatic injection of 100 μL of d-luciferin substrate (1.5 mg/mL dissolved in PBS) using a Synergy H4 multimode microplate reader (BioTek, Winooski, VT). Luminescence readings were initiated after 3 s of mild plate shaking and performed at 2 min intervals for 20 min with integration times of 0.5 s. Raw signal intensity was computed by averaging peak luminescence plus two adjacent time points. Normalized signal intensity was expressed as percentage of mean signal intensity relative to control treated with 0.5% DMSO. Full-length Photinus luciferase activity was determined by transfecting HEK293 cells, as above, with pGL3 firefly luciferase plasmid, as described previously.39 Cells were maintained at 37 °C throughout the measurements. Additional information for LCA can be found in a dedicated book chapter.51
Immunoblot
HEK-Nav1.6 cells were transfected with FGF14-6xmyc, and incubated with either DMSO (0.5%) or ZL181 (50 μM). Cells were then washed with cold PBS. Subsequently, 50 μL of lysis buffer (20 mM Tris-HCl, 150 mM NaCl, 1% NP-40) and 1 μL Protease inhibitor cocktail (set #3, Calbiochem, Billerica, MA) were added. Cell extracts were collected, sonicated for 20 s, and centrifuged at 4 °C, 13,000 g for 15 min, and 2× sample buffer containing 50 mM tris (2-carboxyethyl) phosphine (TCEP) was added. Mixtures were heated for 10 min at 55 °C and resolved on 4–15% polyacrylamide gels (BioRad, Hercules, CA). Resolved proteins were transferred to PVDF membranes (Millipore, Bedford, MA) for 2 h at 4 °C and blocked in tris-buffered saline (TBS) with 3% nonfat dry milk and 0.1% Tween-20. Membranes were then incubated in blocking buffer containing mouse monoclonal anti-Myc (1:1000; 9E10 clone Santa Cruz Biotechnology) or mouse monoclonal anti-PanNav channel (1:1000; Sigma) antibody overnight. Washed membranes were incubated with goat anti-mouse HRP antibody (1:5000–10,000; Vector Laboratories, Burlingame, CA) and detected with ECL Advance Western blotting Detection kit (GE Healthcare). Subsequently, protein bands were visualized using FluorChem HD2 System and analyzed with AlphaView 3.1 software (ProteinSimple, Santa Clara, CA).
Protein Expression and Purification
cDNAs encoding human FGF14WT (accession number NP_787125; aa 64–252) or the C-terminal domain of human Nav1.6 (accession number #NP_001171455; aa 1756-1939) were subcloned into suitable pET bacterial expression vectors (pET28a-FGF14; pET30a-Nav1.6) with a 6× His-tag at the N-terminal site; these plasmids were a gift of Drs. Regina Goetz and Moosa Mohammadi.52 The mutation coding for FGF14V160A was generated by site-directed mutagenesis and PCR using FGF14 as a template. Upon transformation with corresponding cDNA clones, recombinant proteins FGF14WT and FGF14V160A were expressed in the bacterial strain E. coli BL21 (DE3) pLys (Invitrogen) after induction with 0.1 mM isopropyl thio-β-D-galacto-pyranoside (IPTG) for 24 h at 16°C. After induction with IPTG, bacterial cells were harvested and lysed by sonication at 4 °C in lysis/binding buffer containing sodium phosphate 10 mM (prepared from 1 M of Na2HPO4 V160A and NaH2PO4)+ CHAPS 0.1% pH 7.0 (for FGF14/FGF14 proteins), HEPES 25 mM + NaCl 150 mM + glycerol 10% (Nav1.6) pH 7.5 containing 0.1 mM phenyl methyl sulphonyl fluoride (PMSF). The respective proteins were centrifuged at 18000 g for 30 min at 4 °C. For purification of FGF14WT, and FGF14V160A, the supernatant was applied to pre-equilibrated heparin and the proteins were then eluted with NaCl 0.2–2.0 M in the elution (sodium phosphate 10 mM+ NaCl 0.2–2.0 M pH 7.0) buffer. For purification of the Nav1.6 C-tail, the supernatant was applied first to Ni2+ NTA column and eluted with imidazole (200 mM). Finally, all concentrated proteins were purified on an AKTA purifier using Superdex 200 Hiload 16 × 60 columns (both products from GE Healthcare Bio-Sciences, Pittsburgh, PA) and equilibrated in Tris-HCl 50 mM + NaCl 150 mM, pH 7.5.
Surface Plasmon Resonance Spectroscopy
According to previous publication,52 we have performed the surface plasmon resonance spectroscopy (SPR) experiment. Six independent SPR experiments were performed on a Biacore T100 instrument (Biacore GE) and the interaction of FGF14 or FGF14V160A with ZL181 (0–400 μM) was studied at 25 °C. The proteins were immobilized on a CM5 sensor chip using acetate 5.5 with amine coupling kit (GE Healthcare, Pittsburgh, PA). No protein was coupled to the control flow channel of the chip. ZL181 (0–400 μM) dissolved in HBS-P+ (50 μL/min) buffer (100 mM HEPES, 150 mM NaCl, 0.005% (v/v) P20), pH7.4 were injected over the chip for 180 s. Next, HBS-P+ buffer without protein was passed over the chip for 180 s to monitor dissociation, and the chip surface was then regenerated with NaCl (200 mM). For each injection of ZL181 binding to the FGF14WT or FGF14V160A nonspecific responses were subtracted from the responses obtained for control prior to data analysis. Maximal equilibrium responses were plotted against the concentrations of ZL181, and the equilibrium dissociation constant (kd) was calculated from the fitted saturation binding curve using the Biacore software. Binding curves were plotted in Origin 8.6 (Origin Lab Corporation).
Dynamic Light Scattering (DLS)
Samples of the compound, ZL181, were prepared by first dissolving powdered sample in DMSO (50 mM) and serial dilution was done to achieve intermediate stocks of 25 mM, 10 mM, 5 mM, and 500 μM stock concentrations in DMSO. These stocks were then diluted with the same volume of HEPES buffer (pH 7.4) to get 100, 50, 20, 10, and 1 μM working concentrations, respectively. The final concentration of DMSO used was 0.2% in each sample. For each sample concentration, the radius of gyration (Rg), of the major component by mass, was determined using 4 μL aliquots pipetted into a quartz sample cuvette placed in a Malvern Zetasizer μV dynamic light scattering (DLS) instrument (Malvern Instruments Ltd., Malvern, UK) at 25 °C.45,53 The major component was always near 100% of the mass. In the absence of aggregation, the instrument’s automatic intensity optimization routine always used the same settings: 100%. As recommended by the manufacturer, the autocorrelation curves were examined to ensure that scattering from low sample concentrations were not too small to distinguish from buffer.54
In Silico Docking
The docking study was performed with the Schrödinger Small-Molecule Drug Discovery Suite55 using the FGF14:Nav1.6 homology model.22 The chain C of FGF14 was kept for docking and prepared with Protein Prepared Wizard.55 ZL181 was prepared with LigPrep55 and the initial lowest energy conformations were calculated. The grid center was chosen on selected hotspot residues at the PPI of FGF14:Nav1.6 with the coordination of x = 15.77, y = −9.73, z = −18.99. Grid box size was set to 24 × 24 × 24 Å3, and a finer scaling factor of 0.5 was used. Grid generating and docking were both employed with Glide55 using SP-Peptide protocol.56 Docking poses were incorporated into Schrödinger Maestro55 for a visualization of ligand–receptor interactions. The homology structure of FGF14:Nav1.6 was incorporated into Maestro with the top ranked docking pose of ZL181 for an overlay analysis.
Animals
Fgf14−/− mice were maintained on an inbred C57/BL6J background (greater than ten generations of backcrossing to C57/ BL6J). All genotypes described were confirmed by Charles River Laboratories International, Inc. (Houston, TX). Fgf14+/+ wild-type controls (C57/BL6J) were either Fgf14−/− littermates or were purchased from Jackson Laboratory (Bar Harbor, ME). Mice were housed, n ≤ 5 per cage, with food and water ad libitum. Mice were closely monitored for health and overall well-being daily by veterinary staff and the investigators. Animal maintenance and experiments were performed in accordance with US National Institutes of Health (NIH) guidelines and were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Texas Medical Branch.
Immunocytochemistry
Detailed methods for the colocalization of FGF14 and Nav1.6 in nucleus accumbens in brain tissue slices can be found in previous studies.28 Briefly, mice (C57/BL6J) were bred i n the UTMB animal care facility. Adult mice were deeply anesthetized with 2,2,2-tribromoethanol (250 mg/kg i.p.; Sigma-Aldrich, St. Louis, MO), then perfused intracardially with cold 1× phosphate buffer (PBS, pH = 7.4; flow rate = 8–10 mL/min for 2–5 min), followed by 10 min of cold 1% formaldehyde containing methanol (a dilution of 37% formaldehyde solution in 1X PBS, catalog number fxfor37 gal, American MasterTech Scientific Inc., Lodi CA). Whole brains were then removed and postfixed in 1% formaldehyde solution for 1 h at 4 °C and then cryopreserved in 20–30% sucrose/PBS at 4 °C until use (Brains were allowed to completely sink to the bottom of the container before sectioning). In preparation for sectioning, brains were embedded in OCT compound (Tissue-Tek, Ted Pella, Inc., Redding, CA) and 10–15 μm thick sagittal or coronal sequential brain sections were prepared at −18 to −20 °C using a Leica CM1850 cryostat (Leica Microsystems, Buffalo Grove, IL). Sections were then mounted on glass microscope slides (Fisherbrand Superfrost Plus, Fisher Scientific, Pittsburgh, PA) and stored at −80 °C. Before staining, frozen sections were immersed in acetone for 7–10 min followed by 1X PBS washing and incubating with a blocking buffer containing a mixture of 5% NGS and 3% DS in 1X TBS containing 0.3% Triton X-100 for 1 h, followed by overnight incubation with primary antibodies in 3% bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO) and 1× PBS containing 0.1% Tween-20 at 4 °C. Primary antibodies used in this study were mouse anti-FGF14 (1:300, clone N56/21, UC Davis/NIH NeuroMab Facility cat# 75-096, RRI-D:AB_2104060) and rabbit anti-Nav1.6 (1:300, Alomone Laboratories, Jerusalem, Israel, cat# ASC-009). Confocal images were acquired using a Zeiss LSM-510 META confocal microscope with a Plan-Apochromat (63×/1.46 oil) objective (Zeiss, Oberkochen, Germany). Multitrack acquisition was performed with excitation lines at 488 nm for Alexa 488, and 543 nm for Alexa 568. Z-series stack confocal images were taken at fixed intervals: 0.4 μm for 63× with the same pinhole setting for all channels; frame size was either 1024 × 1024 or 512 × 512 pixels. All confocal images were processed using ImageJ US NIH (http://imagej.nih.gov/ij).
Electrophysiology Experiments
HEK cells stably expressing the human Nav1.1 (HEK-Nav1.1) or Nav1.2 (HEK-Nav1.2) or Nav1.6 (HEK-Nav1.6) were transfected with GFP or human FGF14-GFP or human FGF14-ΔNT-GFP, plated at low density on glass coverslips for 3–4 h, and subsequently transferred to the recording chamber. Recordings were performed at room temperature (20–22 °C) using a MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA) after incubation of 30–60 min either with 0.2% DMSO or 20 μM ZL181 in extracellular solution. The composition of recording solutions consisted of the following salts: extracellular (mM): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, 10 glucose, pH 7.3; intracellular (mM): 130 CH3O3SCs, 1 EGTA, 10 NaCl, 10 HEPES, pH 7.3. Membrane capacitance and series resistance were estimated by the dial settings on the amplifier and compensated for electronically by 70–80%. Data were acquired at 20 kHz and filtered at 5 kHz prior to digitization and storage. All experimental parameters were controlled by Clampex 9.2 software (Molecular Devices) and interfaced to the electrophysiological equipment using a Digidata 1200 analog–digital interface (Molecular Devices). Voltage-dependent inward currents for HEK-Nav1.6 and HEK-Nav1.1/HEK-Nav1.2 cells were evoked by depolarization test potentials between −100 mV (Nav1.6) or −60 mV (Nav1.1/Nav1.2) and +60 mV from a holding potential of −70 mV. Steady-state (fast) inactivation of Nav channels was measured with a paired-pulse protocol. From the holding potential, cells were stepped to varying test potentials between −120 mv (Nav1.6) or −110 mV (Nav1.1/Nav1.2) and +20 mV (prepulse) prior to a test pulse to −20 mV (Nav1.6) or −10 mV (Nav1.1/Nav1.2).
Current densities were obtained by dividing Na+ current (INa) amplitude by membrane capacitance. Current–voltage relationships were generated by plotting current density as a function of the holding potential. Conductance (GNa) was calculated by the following equation:
where INa is the current amplitude at voltage Vm and Erev is the Na+ reversal potential.
Steady-state activation curves were derived by plotting normalized GNa as a function of test potential and fitted using the Boltzmann equation:
where GNa,max is the maximum conductance, Va is the membrane potential of half-maximal activation, Em is the membrane voltage, and k is the slope factor. For steady-state inactivation, normalized current amplitude (INa/INa,max) at the test potential was plotted as a function of prepulse potential (Vm) and fitted using the Boltzmann equation:
where Vh is the potential of half-maximal inactivation, Em is the membrane voltage, and k is the slope factor.
Transient INa inactivation decay was estimated with the standard exponential equation. Inactivation time constant (tau, τ) was fitted with the following equation:
where A1 and f1 are the amplitude and time constant, respectively. The variable C is a constant offset term along the Y axis. The goodness of fitting was determined by correlation coefficient (R), and the cutoff of R was set at 0.85.
For ex vivo patch-clamp electrophysiology, coronal sections (300 μm thick slices) containing the NAc were prepared using a modified procedure.57,58 Briefly, the animals were anesthetized with 3% isoflurane and decapitated. The brains were rapidly removed and placed in cold (0–4 °C) oxygenated cutting solution containing 72 mM Tris HCL, 18 mM Tris-BASE, 2.5 mM KCl, 1.2 mM NaH2PO4, 25 mM NaHCO3, 20 mM HEPES, 25 mM glucose, 5 mM sodium ascorbate, 3 mM sodium pyruvate, 10 mM MgSO4 and 0.5 mM CaCl2, 20 mM Sucrose, pH 7.35. Slices were cut with a vibratome (VT1200 S, Leica Microsystems, Wetzlar, Germany), incubated in the cutting solution at 32 °C for approximately 30 min and then transferred to regular artificial cerebrospinal fluid (aCSF) consisting of 124 mM NaCl, 3.2 mM KCl, 1 mM NaH2PO4, 26 mM NaHCO3, 1 mM MgCl2, 2 mM CaCl2, 10 mM glucose at room temperature for 1 h. Slices were then transferred to a submerged recording chamber and continuously perfused with aCSF bubbled with a mixture of 95% O2/5% CO2 (pH 7.4). The temperature of aCSF was maintained at 30–32 °C by an inline solution heater and temperature controller (TC-344B, Warner Instruments, Hamden, CT). Whole-cell patch-clamp recordings were performed using Axopatch 200B and Multiclamp 700B amplifiers (Molecular Devices, Sunnyvale, CA). Somatic recording from visually identified MSNs were performed with pipettes (resistance of 3–6 MΩ) filled with internal solution containing 145 mM K-gluconate, 2 mM MgCl2, 0.1 mM EGTA, 2.5 mM MgATP, 0.25 mM NaGTP, 5 mM phosphocreatine, 10 mM HEPES, pH 7.2, osmolarity. Access resistance was monitored throughout the recording and was typically <25 MΩ. Data acquisition and stimulation were performed with a Digidata 1322A Series interface and pClamp 9 software (Molecular Devices, Sunnyvale, CA). Data were filtered at 2 kHz, digitized at 20 kHz, and were analyzed off-line with pClamp 10 software. To measure MSN intrinsic firing 20 μM of NBQX (Tocris Bioscience, Bristol, England), 100 μM of DL-AP5 (Tocris Bioscience, Bristol, England) and 20 μM of bicuculline (Sigma-Aldrich, St. Louis, MO) were added to regular aCSF in order to prevent glutamatergic and GABAergic synaptic transmissions after seal formation and cell membrane rapture MNSs were held in voltage-clamp mode for about 2 min with subsequent switch to current clamp mode to assess neuronal firing. A series of 800 ms current pulses (from −20 to +200 pA, 10 pA increments) was elicited to obtain AP firing trains from MSNs held at −70 mV with constant current injection. Input-output relationships were plotted as number of spikes against given current step. Only spikes with overshoot were taken into analysis. Action potential (AP) current threshold (Ithr) is defined as the current step at which at least one spike was induced while AP voltage threshold (Vthr) is the voltage at which the first-order derivative of the rising phase of the AP exceeded 10 mV/ ms.57,58
Statistical Analysis
Statistical values were calculated as mean and standard error of the mean (mean ± SEM), unless otherwise specified. The statistical significance (*p < 0.05) of different groups was determined by Student’s t test, one-way ANOVA with posthoc Bonferroni’s method or Kruskal–Wallis one-way ANOVA on ranks with posthoc Dunn’s method using Sigma Stat (San Jose, CA) and Graph Prism (La Jolla, CA) software. Dose–response modulation was determined by
where k is the Michaelis constant, n is cooperative sites, x is an independent variable, and y is the dependent variable. Graphs were plotted in Origin 8.6 Software (Origin Lab Corporation, Northampton, MA).
Supplementary Material
Acknowledgments
Funding
This work was supported by NIH Grants R01MH095995 (F.L.), R01MH111107 (F.L., J.Z.), R01DA038446 (J.Z.), P30DA028821 (J.Z.), John Sealy Memorial Endowments Fund (F.L.), NICHD NIH Fellowship Award Number F32HD093292 (S.R.A.), Jeane B. Kempner Postdoctoral Fellowship (O.F.), NIEHS T32 Environmental Toxicology Fellowship Grant No. T32-ES007254 (T.F.J.), and Keck Center for Interdisciplinary Bioscience Training of the Gulf Coast Consortia NIGMS Grant T32 GM089657-04 (S.R.A.).
The authors acknowledge the Sealy Center for Structural Biology and Molecular Biophysics at the University of Texas Medical Branch at Galveston for providing research resources. We also thank Dr. Heather Lander for critical reading of the manuscript.
Footnotes
Author Contributions
S.R.A. and Z.L. contributed equally to this work. S.R.A and F.L. conceived and designed all biophysical and neurobiology experiments, and wrote the vast majority the manuscript. O.F.contributed to the writing, and edited the manuscript. Z.L., H.C., and J.Z. conceived, designed, and chemically synthesized the peptidomimetics. S.R.A. performed LCA, Western blot, and patch clamp experiments in the recombinant cell system. N.I.P.-E. assisted with all cell culture experiments. A.S. performed protein purification and surface plasmon resonance experiments. M.N.N., F.S., and T.F.J. performed patch clamp electrophysiology in brain tissue slices. H.C. performed in silico docking and assisted chemical characterization analysis of the molecules. M.A. performed tissue cryo-sectioning, immunohistochemistry and acquired confocal images. M.A.W. and O.F. performed dynamic light scattering experiments.
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneur-o.7b00399.
Additional methods describing peptidomimetics synthesis; in-cell screening of additional peptidomimetics; total protein expression levels of Nav1.6 and FGF14 in the presence of ZL181; in-cell screening of ZL181 against the FGF13:Nav1.6 complex; DLS data for ZL181; SPR binding of ZL181 to FGF14V160A mutant; ZL181 docking to the Nav1.6 C-tail; functional role of ZL181 on the FGF14:Nav1.1 and FGF14:Nav1.2 complexes; analysis of Nav1.6 channels and FGF14 parameter in the presence of ZL181; analysis of Nav1.6 channels and FGF14-ΔNT parameter in the presence of ZL181; analysis of FGF14:Nav1.1 and FGF14:Nav1.2 channels in the presence of ZL181; active and passive properties of medium spiny neurons from FGF14WT mice in the presence of ZL181; active and passive properties of medium spiny neurons from FGF14KO mice in the presence of ZL181 (PDF)
References
- 1.Catterall WA. Voltage-gated sodium channels at 60: structure, function and pathophysiology. J Physiol. 2012;590:2577–2589. doi: 10.1113/jphysiol.2011.224204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leterrier C, Brachet A, Fache MP, Dargent B. Voltage-gated sodium channel organization in neurons: protein interactions and trafficking pathways. Neurosci Lett. 2010;486:92–100. doi: 10.1016/j.neulet.2010.08.079. [DOI] [PubMed] [Google Scholar]
- 3.Wildburger NC, Ali SR, Hsu WC, Shavkunov AS, Nenov MN, Lichti CF, LeDuc RD, Mostovenko E, Panova-Elektronova NI, Emmett MR, Nilsson CL, Laezza F. Quantitative proteomics reveals protein-protein interactions with fibroblast growth factor 12 as a component of the voltage-gated sodium channel 1.2 (nav1.2) macromolecular complex in Mammalian brain. Mol Cell Proteomics. 2015;14:1288–1300. doi: 10.1074/mcp.M114.040055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fleidervish IA, Lasser-Ross N, Gutnick MJ, Ross WN. Na+ imaging reveals little difference in action potential-evoked Na+ influx between axon and soma. Nat Neurosci. 2010;13:852–860. doi: 10.1038/nn.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu W, Tian C, Li T, Yang M, Hou H, Shu Y. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat Neurosci. 2009;12:996–1002. doi: 10.1038/nn.2359. [DOI] [PubMed] [Google Scholar]
- 6.Catterall WA, Kalume F, Oakley JC. NaV1.1 channels and epilepsy. J Physiol. 2010;588:1849–1859. doi: 10.1113/jphysiol.2010.187484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scheffer IE, Zhang YH, Jansen FE, Dibbens L. Dravet syndrome or genetic (generalized) epilepsy with febrile seizures plus? Brain & development. 2009;31:394–400. doi: 10.1016/j.braindev.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Volkers L, Kahlig KM, Verbeek NE, Das JH, van Kempen MJ, Stroink H, Augustijn P, van Nieuwenhuizen O, Lindhout D, George AL, Jr, Koeleman BP, Rook MB. Nav 1.1 dysfunction in genetic epilepsy with febrile seizures-plus or Dravet syndrome. European journal of neuroscience. 2011;34:1268–1275. doi: 10.1111/j.1460-9568.2011.07826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu XJ, Zhang YH, Sun HH, Liu XY, Wu HS, Wu XR. Phenotype and SCN1A gene mutation screening in 39 families with generalized epilepsy with febrile seizures plus. Zhonghua Er Ke Za Zhi. 2012;50:580–586. [PubMed] [Google Scholar]
- 10.Savio-Galimberti E, Gollob MH, Darbar D. Voltage-gated sodium channels: biophysics, pharmacology, and related channelopathies. Front Pharmacol. 2012;3:124. doi: 10.3389/fphar.2012.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weiss LA, Escayg A, Kearney JA, Trudeau M, MacDonald BT, Mori M, Reichert J, Buxbaum JD, Meisler MH. Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol Psychiatry. 2003;8:186–194. doi: 10.1038/sj.mp.4001241. [DOI] [PubMed] [Google Scholar]
- 12.Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, Song Y, El-Fishawy P, Murtha RC, Choi M, Overton JD, Bjornson RD, Carriero NJ, Meyer KA, Bilguvar K, Mane SM, Sestan N, Lifton RP, Gunel M, Roeder K, Geschwind DH, Devlin B, State MW. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clapham DE. A STIMulus Package puts orai calcium channels to work. Cell. 2009;136:814–816. doi: 10.1016/j.cell.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 14.Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE, Lewis RS. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136:876–890. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hogan PG. The STIM1-ORAI1 microdomain. Cell Calcium. 2015;58:357–367. doi: 10.1016/j.ceca.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sadaghiani AM, Lee SM, Odegaard JI, Leveson-Gower DB, McPherson OM, Novick P, Kim MR, Koehler AN, Negrin R, Dolmetsch RE, Park CY. Identification of Orai1 channel inhibitors by using minimal functional domains to screen small molecule microarrays. Chem Biol. 2014;21:1278–1292. doi: 10.1016/j.chembiol.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 17.Stoilova-McPhie S, Ali S, Laezza F. Protein-Protein Interactions as New Targets for Ion Channel Drug Discovery. Austin J Pharmacol Ther. 2013;1:5. [PMC free article] [PubMed] [Google Scholar]
- 18.Laezza F, Gerber BR, Lou JY, Kozel MA, Hartman H, Craig AM, Ornitz DM, Nerbonne JM. The FGF14(F145S) mutation disrupts the interaction of FGF14 with voltage-gated Na+ channels and impairs neuronal excitability. J Neurosci. 2007;27:12033–12044. doi: 10.1523/JNEUROSCI.2282-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lou JY, Laezza F, Gerber BR, Xiao M, Yamada KA, Hartmann H, Craig AM, Nerbonne JM, Ornitz DM. Fibroblast growth factor 14 is an intracellular modulator of voltage-gated sodium channels. J Physiol. 2005;569:179–193. doi: 10.1113/jphysiol.2005.097220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laezza F, Lampert A, Kozel MA, Gerber BR, Rush AM, Nerbonne JM, Waxman SG, Dib-Hajj SD, Ornitz DM. FGF14 N-terminal splice variants differentially modulate Nav1.2 and Nav1.6-encoded sodium channels. Mol Cell Neurosci. 2009;42:90–101. doi: 10.1016/j.mcn.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsu WCJ, Scala F, Nenov MN, Wildburger NC, Elferink H, Singh AK, Chesson CB, Buzhdygan T, Sohail M, Shavkunov AS, Panova NI, Nilsson CL, Rudra JS, Lichti CF, Laezza F. CK2 activity is required for the interaction of FGF14 with voltage-gated sodium channels and neuronal excitability. FASEB J. 2016;30:2171–2186. doi: 10.1096/fj.201500161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ali SR, Singh AK, Laezza F. Identification of Amino Acid Residues in Fibroblast Growth Factor 14 (FGF14) Required for Structure-Function Interactions with Voltage-gated Sodium Channel Nav1.6. J Biol Chem. 2016;291:11268–11284. doi: 10.1074/jbc.M115.703868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan H, Pablo JL, Wang C, Pitt GS. FGF14 modulates resurgent sodium current in mouse cerebellar Purkinje neurons. eLife. 2014;3:e04193. doi: 10.7554/eLife.04193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pablo JL, Wang C, Presby MM, Pitt GS. Polarized localization of voltage-gated Na+ channels is regulated by concerted FGF13 and FGF14 action. Proc Natl Acad Sci USA. 2016;113:E2665–2674. doi: 10.1073/pnas.1521194113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shakkottai VG, Xiao M, Xu L, Wong M, Nerbonne JM, Ornitz DM, Yamada KA. FGF14 regulates the intrinsic excitability of cerebellar Purkinje neurons. Neurobiol Dis. 2009;33:81–88. doi: 10.1016/j.nbd.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wozniak DF, Xiao M, Xu L, Yamada KA, Ornitz DM. Impaired spatial learning and defective theta burst induced LTP in mice lacking fibroblast growth factor 14. Neurobiol Dis. 2007;26:14–26. doi: 10.1016/j.nbd.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldfarb M, Schoorlemmer J, Williams A, Diwakar S, Wang Q, Huang X, Giza J, Tchetchik D, Kelley K, Vega A, Matthews G, Rossi P, Ornitz DM, D’Angelo E. Fibroblast growth factor homologous factors control neuronal excitability through modulation of voltage-gated sodium channels. Neuron. 2007;55:449–463. doi: 10.1016/j.neuron.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alshammari MA, Alshammari TK, Laezza F. Improved Methods for Fluorescence Microscopy Detection of Macromolecules at the Axon Initial Segment. Front Cell Neurosci. 2016;10:5. doi: 10.3389/fncel.2016.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tempia F, Hoxha E, Negro G, Alshammari MA, Alshammari TK, Panova-Elektronova N, Laezza F. Parallel fiber to Purkinje cell synaptic impairment in a mouse model of spinocerebellar ataxia type 27. Front Cell Neurosci. 2015;9:205. doi: 10.3389/fncel.2015.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Re J, Wadsworth PA, Laezza F. Intracellular Fibroblast Growth Factor 14: Emerging Risk Factor for Brain Disorders. Front Cell Neurosci. 2017;11:103. doi: 10.3389/fncel.2017.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsu WJ, Wildburger NC, Haidacher SJ, Nenov MN, Folorunso O, Singh AK, Chesson BC, Franklin WF, Cortez I, Sadygov RG, Dineley KT, Rudra JS, Taglialatela G, Lichti CF, Denner L, Laezza F. PPARgamma agonists rescue increased phosphorylation of FGF14 at S226 in the Tg2576 mouse model of Alzheimer’s disease. Exp Neurol. 2017;295:1–17. doi: 10.1016/j.expneurol.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brusse E, de Koning I, Maat-Kievit A, Oostra BA, Heutink P, van Swieten JC. Spinocerebellar ataxia associated with a mutation in the fibroblast growth factor 14 gene (SCA27): A new phenotype. Mov Disord. 2006;21:396–401. doi: 10.1002/mds.20708. [DOI] [PubMed] [Google Scholar]
- 33.van Swieten JC, Brusse E, de Graaf BM, Krieger E, van de Graaf R, de Koning I, Maat-Kievit A, Leegwater P, Dooijes D, Oostra BA, Heutink P. A mutation in the fibroblast growth factor 14 gene is associated with autosomal dominant cerebellar ataxia [corrected] Am J Hum Genet. 2003;72:191–199. doi: 10.1086/345488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verbeek EC, Bakker IM, Bevova MR, Bochdanovits Z, Rizzu P, Sondervan D, Willemsen G, de Geus EJ, Smit JH, Penninx BW, Boomsma DI, Hoogendijk WJ, Heutink P. A fine-mapping study of 7 top scoring genes from a GWAS for major depressive disorder. PLoS One. 2012;7:e37384. doi: 10.1371/journal.pone.0037384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vadasz C, Smiley JF, Figarsky K, Saito M, Toth R, Gyetvai BM, Oros M, Kovacs KK, Mohan P, Wang R. Mesencephalic dopamine neuron number and tyrosine hydroxylase content: Genetic control and candidate genes. Neuroscience. 2007;149:561–572. doi: 10.1016/j.neuroscience.2007.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hsu WC, Nilsson CL, Laezza F. Role of the axonal initial segment in psychiatric disorders: function, dysfunction, and intervention. Front Psychiatry. 2014;5:109. doi: 10.3389/fpsyt.2014.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ali S, Shavkunov A, Panova N, Stoilova-McPhie S, Laezza F. Modulation of the FGF14:FGF14 homodimer interaction through short peptide fragments. CNS Neurol Disord: Drug Targets. 2014;13:1559–1570. doi: 10.2174/1871527313666141126103309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu WC, Nenov MN, Shavkunov A, Panova N, Zhan M, Laezza F. Identifying a kinase network regulating FGF14:Nav1.6 complex assembly using split-luciferase complementation. PLoS One. 2015;10:e0117246. doi: 10.1371/journal.pone.0117246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shavkunov A, Panova N, Prasai A, Veselenak R, Bourne N, Stoilova-McPhie S, Laezza F. Bioluminescence methodology for the detection of protein-protein interactions within the voltage-gated sodium channel macromolecular complex. Assay Drug Dev Technol. 2012;10:148–160. doi: 10.1089/adt.2011.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shavkunov AS, Wildburger NC, Nenov MN, James TF, Buzhdygan TP, Panova-Elektronova NI, Green TA, Veselenak RL, Bourne N, Laezza F. The fibroblast growth factor 14. voltage-gated sodium channel complex is a new target of glycogen synthase kinase 3 (GSK3) J Biol Chem. 2013;288:19370–19385. doi: 10.1074/jbc.M112.445924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anger T, Madge DJ, Mulla M, Riddall D. Medicinal chemistry of neuronal voltage-gated sodium channel blockers. J Med Chem. 2001;44:115–137. doi: 10.1021/jm000155h. [DOI] [PubMed] [Google Scholar]
- 42.Jukic M, Kikelj D, Anderluh M. Isoform selective voltage-gated sodium channel modulators and the therapy of pain. Curr Med Chem. 2014;21:164–186. doi: 10.2174/09298673113206660257. [DOI] [PubMed] [Google Scholar]
- 43.Musa H, Kline CF, Sturm AC, Murphy N, Adelman S, Wang C, Yan H, Johnson BL, Csepe TA, Kilic A, Higgins RS, Janssen PM, Fedorov VV, Weiss R, Salazar C, Hund TJ, Pitt GS, Mohler PJ. SCN5A variant that blocks fibroblast growth factor homologous factor regulation causes human arrhythmia. Proc Natl Acad Sci USA. 2015;112:12528–12533. doi: 10.1073/pnas.1516430112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shoichet BK. Screening in a spirit haunted world. Drug Discovery Today. 2006;11:607–615. doi: 10.1016/j.drudis.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pohjala L, Tammela P. Aggregating behavior of phenolic compounds–a source of false bioassay results? Molecules. 2012;17:10774–10790. doi: 10.3390/molecules170910774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kennedy CR, Lin S, Jacobsen EN. The Cation-pi Interaction in Small-Molecule Catalysis. Angew Chem, Int Ed. 2016;55:12596–12624. doi: 10.1002/anie.201600547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.D’Ascenzo M, Podda MV, Fellin T, Azzena GB, Haydon P, Grassi C. Activation of mGluR5 induces spike after-depolarization and enhanced excitability in medium spiny neurons of the nucleus accumbens by modulating persistent Na+ currents. J Physiol. 2009;587:3233–3250. doi: 10.1113/jphysiol.2009.172593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goldfarb M. Voltage-gated sodium channel-associated proteins and alternative mechanisms of inactivation and block. Cell Mol Life Sci. 2012;69:1067–1076. doi: 10.1007/s00018-011-0832-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dover K, Solinas S, D’Angelo E, Goldfarb M. Long-term inactivation particle for voltage-gated sodium channels. J Physiol. 2010;588:3695–3711. doi: 10.1113/jphysiol.2010.192559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grieco TM, Malhotra JD, Chen C, Isom LL, Raman IM. Open-channel block by the cytoplasmic tail of sodium channel beta4 as a mechanism for resurgent sodium current. Neuron. 2005;45:233–244. doi: 10.1016/j.neuron.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 51.Shavkunov AS, Ali SR, Panova-Elektronova NI, Laezza F. Split-luciferase complementation assay to detect channel-protein interactions in live cells. Methods Mol Biol. 2015;1278:497–514. doi: 10.1007/978-1-4939-2425-7_33. [DOI] [PubMed] [Google Scholar]
- 52.Goetz R, Dover K, Laezza F, Shtraizent N, Huang X, Tchetchik D, Eliseenkova AV, Xu CF, Neubert TA, Ornitz DM, Goldfarb M, Mohammadi M. Crystal structure of a fibroblast growth factor homologous factor (FHF) defines a conserved surface on FHFs for binding and modulation of voltage-gated sodium channels. J Biol Chem. 2009;284:17883–17896. doi: 10.1074/jbc.M109.001842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frenkel YV, Clark AD, Jr, Das K, Wang YH, Lewi PJ, Janssen PA, Arnold E. Concentration and pH dependent aggregation of hydrophobic drug molecules and relevance to oral bioavailability. J Med Chem. 2005;48:1974–1983. doi: 10.1021/jm049439i. [DOI] [PubMed] [Google Scholar]
- 54.(2005) Dynamic Light Scattering for Pharmaceutical Drug Development Screening for Promiscuous Inhibitors by Malvern Instruments, Malvern Instruments.
- 55.Small-Molecule Drug Discovery Suite 2016-4. Schrödinger, LLC; New York: 2016. [Google Scholar]
- 56.Tubert-Brohman I, Sherman W, Repasky M, Beuming T. Improved docking of polypeptides with Glide. J Chem Inf Model. 2013;53:1689–1699. doi: 10.1021/ci400128m. [DOI] [PubMed] [Google Scholar]
- 57.Crofton EJ, Nenov MN, Zhang Y, Scala F, Page SA, McCue DL, Li D, Hommel JD, Laezza F, Green TA. Glycogen synthase kinase 3 beta alters anxiety-, depression-, and addiction-related behaviors and neuronal activity in the nucleus accumbens shell. Neuropharmacology. 2017;117:49–60. doi: 10.1016/j.neuropharm.2017.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nenov MN, Tempia F, Denner L, Dineley KT, Laezza F. Impaired firing properties of dentate granule neurons in an Alzheimer’s disease animal model are rescued by PPARgamma agonism. J Neurophysiol. 2015;113:1712–1726. doi: 10.1152/jn.00419.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.