

Abstract
Tranilast is clinically indicated for the treatment of allergic disorders and is also a non-selective blocker of the Transient Receptor Potential Vanilloid 2 (TRPV2) channel. Prior studies have found that it has protective effects in various animal models of cardiac disease. Our laboratory has found that genetic deletion of TRPV2 results in a blunted hypertrophic response to increased afterload, thus this study tested the hypothesis that tranilast through cardiomyocyte TRPV2 blockade can inhibit the hypertrophic response to pressure overload in-vivo via transverse aortic constriction and ex vivo via isolated myocyte studies.
The in vivo studies demonstrated that tranilast blunted the fibrotic response to increased afterload and to a lesser extent the hypertrophic response. After 4 weeks, this blunting was associated with improved cardiac function, though at 8 weeks the cardiac function deteriorated similarly to the control group. Lastly, the in vitro studies demonstrated that tranilast was not inhibiting these responses at the cardiomyocyte level. In conclusion, we demonstrated that tranilast blunting of the fibrotic and hypertrophic response occurs independently of cardiac TRPV2 channels and may be cardioprotective in the short term but not after prolonged administration.
Keywords: TRPV2, tranilast, TAC, hypertrophy, fibrosis
Introduction
Tranilast is a non-selective cation channel blocker that effectively inhibits TRPV2 activation (3) in the gastrointestinal tract (28), pancreas (4), and heart (8). It also inhibits TGF-β1 and platelet derived growth factor (PDGF) mediated collagen synthesis, as well as migration and proliferation of vascular smooth muscle cells (7, 16). It is approved for clinical use in Japan and South Korea for the treatment of allergic disorders such as asthma, allergic rhinitis and atopic dermatitis (3). In the cardiovascular field, it was studied in the prevention of restenosis after percutaneous transluminal coronary where it was well tolerated, but was not found to be successful in decreasing the incidence of restenosis (6, 22, 23).
These basic and translational studies have led investigators to test the use of tranilast on various animal models of heart disease including hypertensive (16, 24), diabetic (10, 15), and ischemic rats (5), a canine model of atrial tachycardia (17), and a hamster model of dilated cardiomyopathy (8). While these studies have shown promise for tranilast-mediated protection, not all demonstrated a positive effect. Furthermore, the precise mechanisms via which it exerts protective effects have not been clarified, but a significant number of these studies demonstrated decreased myocardial fibrosis and/or improved LV remodeling.
Our laboratory has found that TRPV2 plays a crucial role in regulating calcium handling and myocyte contractility (13, 20) and others demonstrated that it also plays an integral role in maintaining cardiac structure (9). Based on these findings we have studied the role that TRPV2 plays in regulating the physiologic and pathologic response to increased afterload and have demonstrated that genetic TRPV2 channel deletion (TRPV2-KO) results in significantly impaired exercise tolerance (18) and a decreased hypertrophic response to transverse aortic constriction (TAC)(12). The blunting of the hypertrophic response upon TRPV2 ablation was also associated with significant improvement of cardiac function and a decrease in maladaptive LV remodeling. Thus, in the current study, we sought to evaluate the therapeutic potential of tranilast as a pharmacological blocker of TRPV2 in the setting of TAC-induced pathological LV hypertrophy and remodeling as these characteristics are common in patients with heart failure with a preserved ejection fraction (HFpEF).
We demonstrate that while tranilast does decrease the development of compensatory mechanisms to increased afterload such as fibrosis and hypertrophy with improved cardiac function in the short term (4 weeks), longer treatment (8 weeks) results in LV dysfunction likely through the blunting of these same mechanisms. Furthermore, we demonstrate that these effects are unlikely to be due to direct inhibition of TRPV2 at the cardiomyocyte level, but rather appear to be independent of the actions of tranilast on the myocytes.
Methods
All animal procedures were performed with the approval of the Institutional Animal Care and Use Committee (IACUC) of the University of Cincinnati and in accordance with the Eighth Edition of the Guide for the Care and Use of Laboratory Animals (2). All mice were wild type (B6129FS2/JF2 background) males 8-20 weeks of age or mice with the pore region of the TRPV2 calcium channel removed in the whole animal (TRPV2-fKO)(11, 12).
TAC Surgery
Transaortic constriction (TAC) was performed on 10-12 week old mice as previously described (12). Briefly, the mice were intubated on isoflurane (2-2.5% to effect), followed by an incision made in the sternal area and the sternum cut at the midline to enter the chest cavity. The thymus was gently retracted to expose the aortic arch. Following isolation of the transverse aorta between the carotid arteries, aortic constriction was performed by tying a 7-0 silk ligature against a 27G needle, the latter being promptly removed to yield a constriction of standard size. For sham procedures, the ligature was not tied against the needle. Hemodynamic variables were measured prior to euthanasia at either 4 or 8 weeks post-surgery.
Tranilast oral treatment
Following TAC surgery mice were treated with oral tranilast. Tranilast treatment, 2mg/day (80mg/kg) (25), was started 10 days after surgery and continued until mice were euthanized 4 or 8 weeks post-surgery. The use of tranilast in this study was investigational to determine its pharmacological potential. Tranilast (Medicalchem Manufacturing Company, Touzeng Town Binhai, Yancheng City, Jiangsu Province, China) is water insoluble, and therefore was mixed in low fat peanut butter (Better’n Peanut Butter™, Wonder Natural Foods Corporation, Watermill, NY), and fed to the mice once a day. TAC control and sham mice were given equivalent amounts of low fat peanut butter containing no tranilast.
Invasive blood pressure (IVP)
IVP measurements were performed on the TAC and exercise mice prior to euthanization as previously described (20) with isoflurane (IH, 2% to effect) as the anesthetic. Briefly, the right femoral artery was cannulated and connected to a low compliance pressure transducer (COBE Cardiovascular, Arvada, CO). A high fidelity, 1.2-French SciSence pressure catheter (SciSence, London, ON, Canada) was inserted into the right carotid artery and advanced into the left ventricle to monitor cardiac performance. Hemodynamic variables were collected and analyzed using a MacLab 4/S system (AD Instruments, Colorado Springs, CO) and Chart Software. Following euthanasia, body weights were taken, whole hearts dissected out and weighed to estimate heart weight/body weight ratio (HW/BW). The hearts were then cut longitudinally, half was fixed with formaldehyde for histopathological analysis and half was flash frozen in N2 for molecular studies.
Histopathology
Hearts were formalin-fixed, routinely processed, paraffin embedded and cut into 6μm sections by the CCHMC Department of Pathology Research Core (Cincinnati, Ohio), and individually stained with Masson’s Trichrome (MT; Masson Trichrome Stain Kit, ThermoScientific, Kalamazoo, MI). Briefly, 10 non-overlapping photomicrographs were taken for each mouse heart (Olympus 1X71; Olympus, Melville, New York) and then randomized before analysis. Quantification of MT-stained sections was performed via color based thresholding assisted by ImageJ (NIH), as previously described (12).
Echocardiography
Echocardiography measurements were taken at baseline and at the conclusion of the 4 week study, the 8 week study had echocardiograms on a bi-weekly basis. All echocardiographic studies were performed as previously described(11). Briefly, mice were anesthetized with isoflurane (2.5%) and placed on the heated stage of the Vevo 2100. Parasternal long axis (PSLAX) and short axis (SAX) images were recorded and then analyzed on a separate work station with VevoStrain software (Vevo 2100, v1.6, Visualsonic, Toronto, Canada). From the M-mode images, left ventricular cavity size for systole and diastole (LV Vol;s and LV Vol;d, respectively) as well as wall thickness of the septal and posterior wall (IVS and PW, respectively) were measured and ejection fraction (%EF) was calculated using the Vevo software.
Cardiomyocyte contractility
Ventricular myocytes from sham and 4 week post-TAC mouse hearts were enzymatically dissociated using Langendorff perfusion as previously described(11, 27). Myocytes were placed in a plexiglass cell chamber filled with Tyrode solution containing 1.8 mM CaCl2 at room temperature (24°C). The solution also contained either vehicle or the indicated treatment drugs, at a concentration of 1μM for probenecid and 100μM for tranilast. Myocytes were excited with field stimulation (Grass S48 stimulator, Grass Instruments, Quincy, MA) with 2 ms 1.5x threshold pulses at a rate of 1 Hz. Steady state myocyte shortening was imaged with a CCD camera and examined using a video-edge detector (Crescent Electronics, Sandy, UT). Data were sampled through an Axon Digidata 1322A board using the PCLAMP 9 software (both by Molecular Devices, Sunnyvale, CA).
Neonatal Rat Ventricular Myocyte Isolation and Cell Culture
Neonatal Rat Ventricular Myocytes (NRVMs) were isolated as previously described (21). Briefly, hearts were removed from Sprague Dawley rats at 1-2 days old, atria were removed, the ventricles were cut into small pieces and digested first in .05% trypsin/EDTA (Corning, Corning NY) overnight, then in collagenase II (Gibco, ThermoFisher, Waltham, MA) for 30 minutes. Cells were then spun at 100 × g followed by a 40 minute pre-plating process on non-treated plates to allow the fibroblasts to adhere. The non-adherent NRVMs were then transferred to cell culture-treated dishes in MEM alpha media (Gibco, ThermoFisher, Waltham, MA) with 10% FBS.
RNA Isolation and qRT-PCR
Total RNA was isolated from whole mouse hearts (Qiazol method, Qiagen, Venlo, Limburg, Netherlands) and cDNA was synthesized (High-Capacity RNA-to-cDNA kit, Applied Biosystems, Carlsbad, CA). Samples were run on Stratagene Mx3005P (Agilent Technologies, Santa Clara, CA) using ABsolute™ Blue qPCR Mix, SYBR Green, ROX (Thermo Scientific, Kalamazoo, MI). Primer sequences are listed in Table 1.
Table 1.
Primer Sequences for qRT-PCR experiments.
| Protein | Abbreviation | Primer Sequence | |
|---|---|---|---|
| Sense | Antisense | ||
| Collagen Type III Alpha 1 chain | mCol3A1 | AACCCTGCTCGGAATTGCAG | TCTGTCCACCAGTGCTTCCG |
| Connective Tissue Growth Factor | mCTGF | CACCTAAAATCGCCAAGCCTG | AGTTCGTGTCCCTTACTTCCTG |
| Fibronectin 1 | mFN1 | ATGTGGACCCCTCCTGATAGT | GCCCAGTGATTTCAGCAAAGG |
| Transforming Growth Factor beta-1 | mTGFβ1 | CAATTCCTGGCGTTACCTTG | CCCTGTATTCCGTCTCCTG |
| Glyceraldehyde 3-phosphate dehydrogenase | mGAPDH | CATGGCCTTCCGTGTTCCTA | CCTGCTTCACCACCTTCTTGAT |
| Atrial Natiuretic Factor | rANF | AGGAGAAGATGCCGGTAG | GCTTTTCAAGAGGGCAGA |
| Glyceraldehyde 3-phosphate dehydrogenase | rGAPDH | ACCACAGTCCATGCCATCAC | TCCACCACCCTGTTGCTGTA |
m, mouse; r, rat
NRVM RNA was isolated using a Macherey-Nagel NucleoSpin RNA kit (Takara Bio USA, Inc, Düran, Germany) and cDNA was synthesized using a BioScript All-in-One cDNA Synthesis SuperMix (Biotool, Stratech Scientific Limited, Suffolk, UK). Samples were run on Stratagene Mx3005P (Agilent Technologies, Santa Clara, CA) using SYBR Green qPCR Master Mix (Biotool, Stratech Scientific Limited, Suffolk, UK) to assess levels of GAPDH and ANF. Results were analyzed using the ΔΔCt method (14). Primers sequences are listed in Table 1. Cells were treated with 10uM phenylephrine (PE±tranilast; 100μM) for 24 hours to induce hypertrophic signaling.
NFAT-Luciferase Reporter Assays
NRVMs were transfected with 75 ng per well (in a 96-well plate) of NFAT-luciferase reporter plasmid acquired from AddGene (Cambridge, MA) (26). Twenty-four hours post-transfection, cells were treated with 10μM phenylephrine (±tranilast; 100μM) for 24 hours. To quantify luciferase reporter expression, cells were rinsed with sterile PBS and then lysed with 50μl Cell Lysis Buffer (Promega, Madison, WI) for 5 min at room temperature, followed by addition of 100 μl luciferase assay reagent (Promega, Madison, WI); and luminescence was read immediately using a Cytation 5 (BioTek, Winooski, VT).
Data Analysis
Statistical analysis was performed by one-way analysis of variance (ANOVA), two-way ANOVA or two-way repeated measures ANOVA (RM) as appropriate. Where significance was indicated, post-hoc testing was performed using the Holm-Sidak method for comparing individual means and correcting for family-wise error (SigmaPlot v.13.0, Systat Software, Inc., San Jose, CA). Data are presented as means ± SEM, and differences are regarded as significant at P ≤ 0.05.
Results
Four-week treatment with tranilast preserves cardiac function in response to increased afterload
TRPV2-KO mice have a blunted response to TAC (12), thus we sought to establish whether this lack of hypertrophy would be seen in WT mice treated with tranilast. We induced TAC on 30 WT and sham procedure on 14 mice. Immediately following TAC, mice were randomized to control (N=13) or tranilast (N=17). At 4 weeks post-TAC, IVP revealed augmented systolic and diastolic function (as measured by the maximum and minimum rate of contraction) in the TAC tranilast group in comparison to the TAC control group (Figure 1A and B) (P<0.05 for TAC tranilast compared to sham and TAC). Furthermore, function was higher in the treated mice in comparison to sham mice, likely due to the observed hypertrophic response. Echocardiographic measures of cardiac function when compared to baseline demonstrated a similar pattern, as the decrease in EF was worse in the TAC group compared to the TAC tranilast group (Figure 1C, P<0.05 for TAC 4 weeks compared to TAC baseline), though the degree of LV cavity dilation was worse in the treatment group (Figure 1D, P<0.05 for TAC tranilast 4 weeks compared to baseline).
Figure 1. Assessment of cardiac function after 4 weeks of treatment with tranilast.
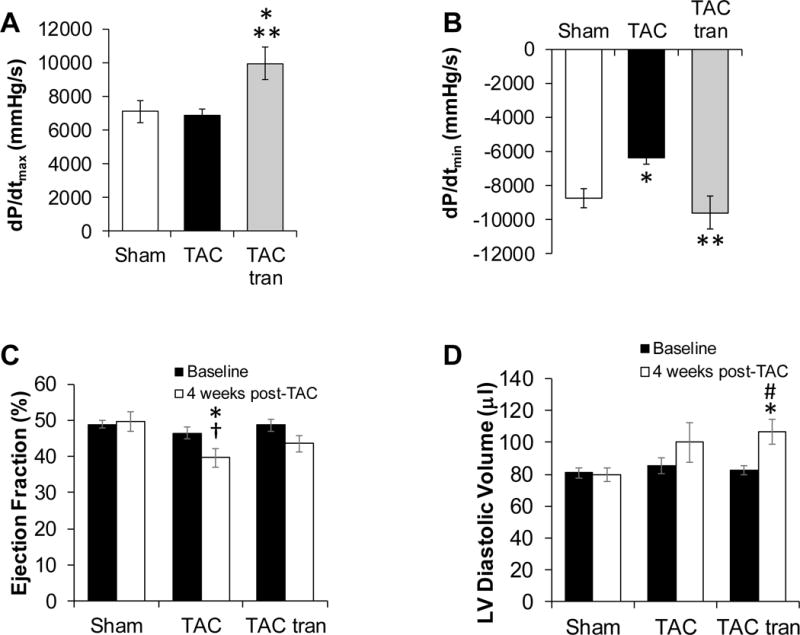
IVP measurements at 4 weeks post-TAC of maximum rate of contraction (dP/dtmax) (A) and maximum rate of relaxation and (dP/dtmin) (B) (*P<0.05 compared to sham and **P<0.05 compared to TAC). Echocardiographic measurements of Ejection Fraction (C) and LV diastolic volume (D) at baseline, and 4 weeks post-surgery (*P<0.05 compared to sham; †P<0.05 TAC baseline vs TAC 4 weeks; #TAC tranilast baseline compared to TAC tranilast 4 weeks). P values are from one-way ANOVA (A and B) and two-way ANOVA (C and D).
Four-week treatment with tranilast blunts the hypertrophic and fibrotic response to increased afterload
The development of hypertrophy in response to TAC was blunted in the tranilast treated group as measured via posterior wall thickness during diastole and LV mass/body weight ratio (Figure 2A and B). Anterior wall thickness demonstrated a similar trend (data not shown). These findings were consistent with the traditional ex vivo measurement of HW/BW ratio (Figure 2C; P<0.05 compared to sham). There was a non-significant decrease in HW:BW ratio between TAC and TAC tranilast. Histological analysis demonstrated that the percent of fibrotic tissue in the tranilast treated group was significantly lower than in the non-treated cohort (Figure 2D and E, P<0.05). These studies suggest that tranilast treatment can blunt the development of hypertrophy and fibrosis and preserve cardiac function in the short term.
Figure 2. Hypertrophic and fibrotic response to 4 weeks treatment with tranilast.
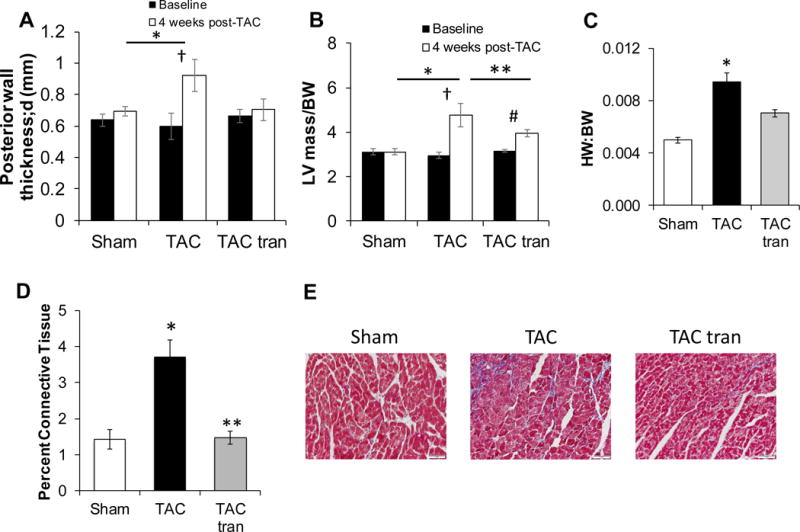
Sham, TAC, and TAC tranilast posterior wall thickness (A) and LV mass/BW (B) was determined using echocardiography (*P<0.05 compared to sham; †P<0.05 TAC baseline vs TAC 4 weeks; **P<0.05 compared to TAC; #P<0.05 TAC tranilast baseline compared to TAC tranilast 4 weeks). HW/BW ratio (C) 4 weeks post-surgery (*P<0.05 compared to sham). Masson’s trichrome stained slides were used to determine the percentage of connective tissue (D; *P<0.05 compared to sham; **P<0.05 compared to TAC) with representative images (E). P values are from two-way ANOVA (A and B) and one-way ANOVA (C and D).
Eight-week treatment with tranilast did not preserve cardiac function in response to increased afterload
In order to establish if the blunting of the hypertrophic and fibrotic response after 4 weeks of tranilast was protective over a longer period of time, we induced TAC on 22 WT and sham procedure on 6 WT mice. 13 TAC mice were randomized to tranilast and 9 to vehicle treatment for 8 weeks with echocardiographic studies performed on all at baseline and every 2 weeks. In contrast to the short-term study, invasive catherization demonstrated no significant difference in systolic and diastolic function between the TAC and TAC tranilast groups as measured by dP/dtmax and dP/dtmin (Figure 3A and B). Furthermore, even though there was a non-significant trend towards improved function in the tranilast treated group at 6 and 8 weeks, both groups showed a similar decrease in EF (Figure 3C; P<0.05 TAC compared to sham) and increase in LV cavity dilation (Figure 3D; P<0.05 TAC tranilast compared to sham at 6 and 8 weeks).
Figure 3. Assessment of cardiac function after 8 weeks of treatment with tranilast.
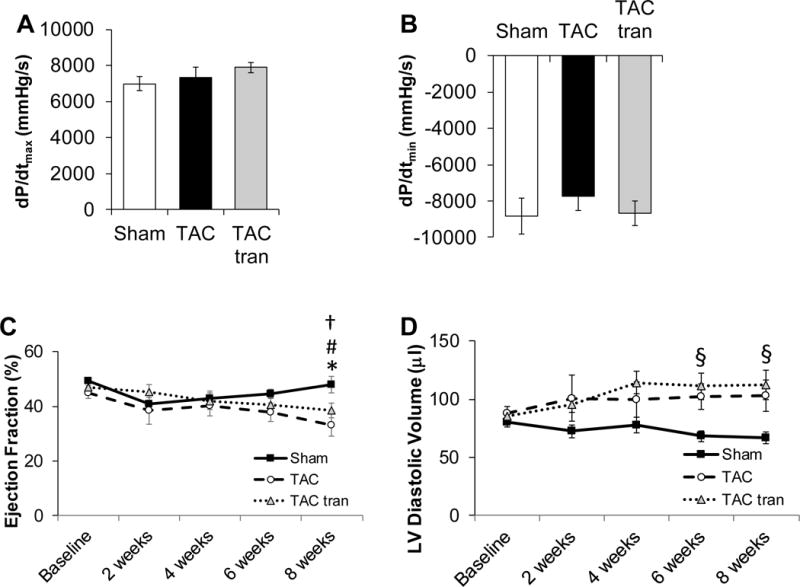
IVP measurements at 8 weeks post-TAC of maximum rate of contraction (dP/dtmax) (A) and maximum rate of relaxation (dP/dtmin) (B). Echocardiographic measurements of Ejection Fraction (C) and LV diastolic volume (D) at baseline, 2, 4, 6, and 8 weeks post-surgery (*P<0.05 TAC compared to sham; †P<0.05 TAC baseline vs TAC 8 weeks; #P<0.05 TAC tranilast baseline compared to TAC tranilast 8 weeks; §p<0.05 TAC tranilast compared to sham). P values are from one-way ANOVA (A and B) and 2-way ANOVA RM (C and D).
Eight-week treatment with tranilast blunts the hypertrophic and fibrotic response to increased afterload
Despite the blunted hypertrophic response that was observed at 2 and 4 weeks in the tranilast group, after 8 weeks there was no significant difference in hypertrophy between the TAC and TAC tranilast groups as measured via posterior wall thickness (Figure 4A; P<0.05 for sham compared to TAC and sham compared to TAC tranilast), LV mass/BW (Figure 4B; P<0.05 for sham compared to TAC and sham compared to TAC tranilast) or HW/BW ratio (Figure 4C) (P<0.05 compared to sham). Via histology, we observed significantly decreased fibrosis in the tranilast treated group (Figure 4D and E; P<0.05 TAC tranilast compared to TAC). Thus, these studies show that while short-term inhibition of the hypertrophic and fibrotic response may preserve cardiac function, longer term blunting of these compensatory mechanisms results in similar degree of cardiac dysfunction and dilation.
Figure 4. Hypertrophic and fibrotic response to 8 weeks treatment with tranilast.
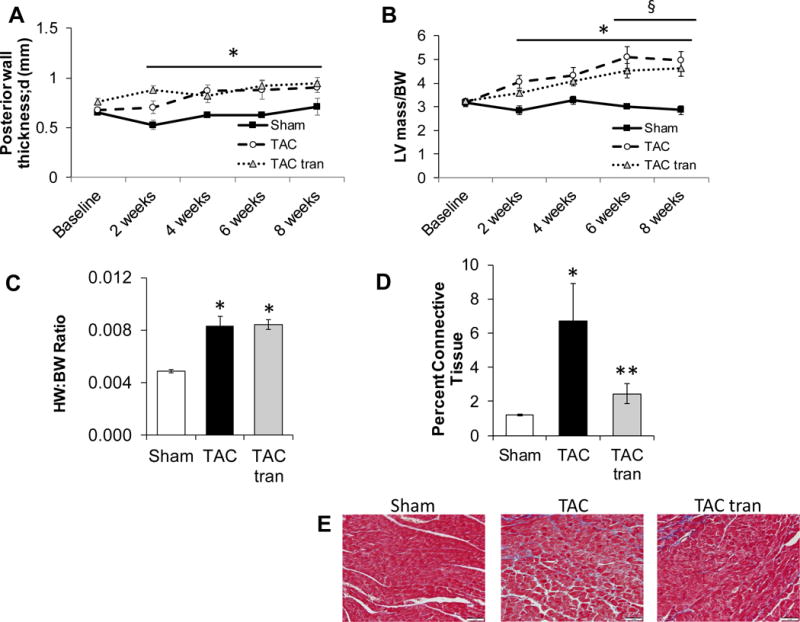
Sham, TAC, and TAC tranilast posterior wall thickness (A) and LV mass/BW (B) was determined using echocardiography (*P<0.05 TAC compared to sham; §P<0.05 TAC tranilast compared to sham). HW/BW ratio (C) 8 weeks post-surgery (*P<0.05 compared to sham). Masson’s trichrome stained slides were used to determine the percentage of connective tissue (D, representative images E; *P<0.05 TAC compared to sham; **P<0.05 TAC versus TAC tranilast). P values are from two-way ANOVA RM (A and B) and one-way ANOVA (C and D).
Tranilast treatment in TRPV2-fKO mice has no effect on hypertrophy
In addition to the WT surgeries, four WT mice underwent sham surgery and eight TRPV2-fKO mice underwent TAC surgery. These control mice were fed tranilast for 4 (sham) and 8 weeks (TAC). The 4 week WT sham tranilast mice had an average LV mass/BW of 3.01±0.24 at baseline and 2.84±0.14 at 4 weeks post-surgery. This was not statistically different from the 4 week WT sham untreated mice, 3.11±0.16 (P=0.71) and 3.03±0.11 (P=0.32), respectively (Figure 2B). The LV mass/BW values of the TRPV2-fKO TAC tranilast treated mice were also not statistically different at baseline (3.23±0.18) or at 8 weeks post-TAC (2.93±0.33) from the TRPV2-fKO TAC mice previously published at baseline (3.23±0.25; P=0.99) and at 8 weeks post-TAC (3.67±0.18, P=0.16) (12).
Tranilast blunted TGF-β1 expression at four but not eight weeks of treatment
We measured various markers of fibrosis and fibrotic signaling at 4 and 8 weeks after TAC. We found that at 4 weeks, the mRNA expression level of TGF-β1 was the only one blunted by tranilast treatment. CTGF trended to increase after TAC and was significantly increased with tranilast treatment. Col3A1 and FN1 were both significantly increased in the TAC and the TAC tranilast groups (Figure 5A). Regarding the 8 week data, TGF-β1 trended higher in the TAC group and was significantly increased in TAC tranilast group; while CTGF, Col3A1, and FN1 were statistically higher in both TAC and TAC tranilast groups in comparison to sham (Figure 5B).
Figure 5.
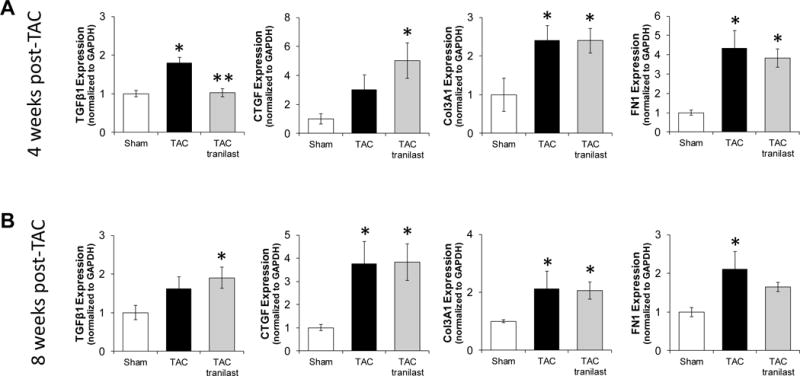
Expression of fibrotic markers in sham, TAC, and TAC tranilast whole mouse hearts. mRNA expression levels were determined for TGF-β1, CTGF, Col3A1, and FN1 at 4 (A) and 8 (B) weeks post-TAC (*P<0.05 compared to sham; **P<0.05 TAC vs TAC tranilast). P values are from one-way ANOVA.
Tranilast does not affect TRPV2-induced cardiomyocyte contractile function under baseline or hypertrophic conditions
We examined whether the observed in vivo effects of tranilast were mediated by its direct effects on TRPV2 in cardiomyocytes. After stimulation with probenecid, a known TRPV2 agonist, isolated myocytes from both sham and TAC mice (4 weeks) exhibited an increase in fractional shortening (FS; P<0.05 for probenecid vs control for sham and TAC), though the TAC myocytes demonstrated significantly higher contractility consistent with greater TRPV2 activity (Figure 6A, middle columns; P<0.05 TAC probenecid compared to sham probenecid). Interestingly, pretreatment with tranilast did not cause significant changes in probenecid-induced contractility in either the TAC or sham mice (Figure 6A middle and right columns). Thus, this data suggests that tranilast does not appear to be significantly altering the myocyte response to a known, potent TRPV2 agonist. Similarly, treatment with probenecid significantly increased myocyte shortening rate (dL/dtmax, 6B) in both sham and TAC myocytes and increased the relengthening rate (dL/dtmin, 6C) in TAC myocytes compared to controls (P<0.05 compared to control). Treatment with both probenecid and tranilast had the same shortening and relengthening rates in sham and TAC myocytes as probenecid alone.
Figure 6. Effect of tranilast on isolated myocytes.
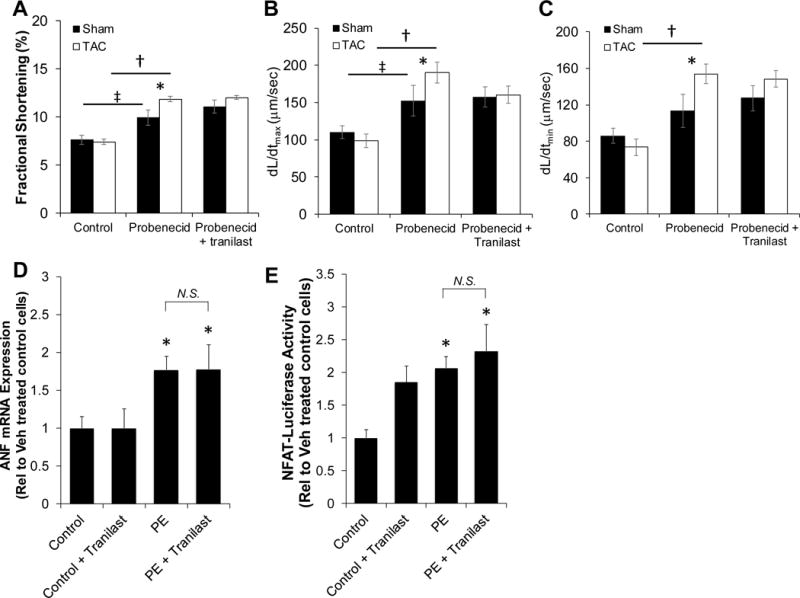
Fractional shortening (A) was measured in sham and TAC myocytes treated with probenecid +/− tranilast (‡P<0.05 sham control compared to sham probenecid; *P<0.05 sham probenecid compared to TAC probenecid; †P<0.05 TAC control compared to TAC probenecid). Rate of contraction (B) (dL/dtmax) and rate of relengthening (C) (dL/dtmin) were measured in Sham and TAC myocytes treated with probenecid +/− tranilast (‡P<0.05 sham control compared to sham probenecid; *P<0.05 sham probenecid compared to TAC probenecid; †P<0.05 TAC control compared to TAC probenecid). ANF mRNA expression (D) was determined in control and phenyl (PE) treated NRVMs +/− tranilast (*P<0.05 compared to control). An NFAT-Luciferase assay was used to measure the activation of NFAT in NRVMs (E; *P<0.05 compared to control). P values are from two-way ANOVA (A, B, and C) and one-way ANOVA (D and E).
Tranilast does not directly affect myocyte hypertrophic signaling
To further determine whether the observed hypertrophy-reducing effects of tranilast were due to direct actions on myocytes, we utilized an in vitro model of myocyte hypertrophy using phenylephrine (PE) treatment of primary neonatal rat neonatal ventricular myocytes (NRVMs) as previously described(21). Results showed that tranilast did not inhibit PE-induced (10 μM for 24 hours) expression of ANF, a known gene marker of cardiac hypertrophy (Figure 6D; P<0.05 compared to control). We also observed similar results with the inability of tranilast to reduce PE-induced transcriptional activation of NFAT (Figure 6E; P<0.05 compared to control).
Discussion
In this study, we found that tranilast modulated the fibrotic, and to a lesser extent the hypertrophic, response to pathological stress. Hemodynamically, the blunting of these responses preserved cardiac function over a 4 week, but not 8 week period following TAC-induced LV pressure overload. This effect did not appear to be mediated by a direct effect on the cardiac myocytes and was at least partially mediated by decreased development of fibrosis.
Tranilast has been tested in a number of cardiac disease animal models with varying results. For example, Betge and colleagues evaluated late onset oral treatment with tranilast following large myocardial infarction (MI) in rats where they treated the animals with tranilast 28 days following an induced MI in the rat. There was no significant difference in survival rate and collagen content in the tranilast treated rats compared to the control group (1).
The studies that have found a positive effect of tranilast on the development of cardiomyopathy used animal models of diabetic (10, 15) and dilated cardiomyopathy (8). The former focused on the effects of tranilast on TGF-β signaling while the latter concluded that its effect was secondary to removal of sarcolemmal TRPV2 with a subsequent reduction in the phosphorylation of calmodulin-dependent protein kinase II (CaMKII). These studies demonstrated reduced development of fibrosis as the likely direct factor influencing the improved cardiac outcomes. Similarly, our laboratory previously demonstrated that TAC induced TRPV2 upregulation and translocation, while the functional deletion of TRPV2 reduced the development of myocyte hypertrophy and fibrosis (12). This was consistent with a study by Ward and colleagues that found that tranilast treatment affected the expression levels of TGF-β isoforms in rats after arterial injury in a dose dependent manner (25). Our study into the expression of various markers of fibrosis indicated that tranilast was also able to decrease TGF-β1 expression 4 weeks, but not 8 weeks, after TAC.
This study adds a translational component to the body of literature regarding TRPV2 and the role of tranilast in the development of hypertrophy and fibrosis in animal models, and it is the first one to test the role of tranilast in a TAC model and to evaluate its effects over different time periods. While TAC is an appropriate model to induce hypertrophy, there are other models of hypertrophy that have also tested the use of tranilast. Umemura and colleagues administered tranilast for 4 weeks to spontaneously hypertensive rats and assessed the impact on the cardiac collagen matrix associated with increased myocardial stiffness (24). Consistent with our findings, they demonstrated that tranilast treatment significantly suppressed increases in LV collagen concentration, LV weight/body weight ratios in the spontaneously hypertensive rats while not affecting systolic, diastolic BP, heart rate, or cardiac output.
This study confirms the positive effects of tranilast on cardiac function and remodeling over a 4-week period, but brings into question the long-term use of tranilast in pressure overload conditions. The blunting of the compensatory mechanisms that occur in response to pressure overload appear in this study to have positive short term outcomes as it pertains to cardiac function, but not over longer term. This is likely secondary to the need of the myocardium to develop hypertrophy and/or dilation in order to overcome the higher pressures as per Laplace’s law, where increased wall thickness and/or dimensions are required to maintain physiologic pressure in response to increased tension (i.e afterload) (19). This finding has important implications, as the targeting of hypertrophic signaling has been considered for years as a potential avenue for the treatment and prevention of left ventricular hypertrophy, diastolic dysfunction and HFpEF, but has not been able to be taken to the bedside (6, 22, 23).
The pathophysiology underlying HFpEF is related to abnormalities in diastolic function due to relaxation and ventricular stiffness associated with development of left ventricular hypertrophy and development of fibrosis. Tranilast has been studied with regards to its effect on fibrosis by Hocher and colleagues who used tranilast in rats with renovascular hypertension. In that study, tranilast did not alter blood pressure, but using a computer aided image analysis system it was demonstrated tranilast had an effect on preventing cardiac fibrosis and attenuating left ventricular fibrosis, most likely due to the decrease in the proliferation of cardiac non-myocyte cells (8). Furthermore, Martin and colleagues evaluated the role of tranilast in cardiac matrix deposition in cultured cardiac fibroblasts and then assessed effects on diabetic rats. They demonstrated a 58% reduction in TGF-β1 induced 3[H]-hydroxyproline incorporation with high dose tranilast (400 mg/kg). Additionally, after 16 weeks, the control rats showed increased cardiac fibrosis and evidence of TGF-β1 activation compared to those given tranilast, which, despite persistent hyperglycemia and hypertension, had attenuated cardiac fibrosis in association with a reduction of TGF-β1 activation (15).
Lastly, this study suggests that tranilast has no significant direct effect on myocyte function or hypertrophic signaling in vitro. Our prior work demonstrated that a functional TRPV2 genetic deletion also inhibits the development of hypertrophy in response to TAC, but the deletion model used was not myocyte specific (12). Thus, in light of these current findings, it appears that tranilast’s blunting of cardiac hypertrophy and fibrosis are at least partially mediated by non-myocyte specific effects. Elucidation of the full mechanisms by which tranilast mediates cardiac fibrosis is beyond the scope of this body of work, but, future studies will identify the role of TRPV2 in cardiac fibroblasts, myocyte-fibroblast interactions, and extracellular matrix production.
Conclusion
In summary, this study demonstrates that short term blunting of cardiac fibrosis in response to increased afterload with tranilast has beneficial effects that are not sustained after longer-term therapy. Furthermore, these effects do not appear to be mediated by the drug’s effects on TRPV2 in the cardiomyocyte. Regardless of the mechanism of action, we show that tranilast may be an appropriate therapeutic option for the prevention of cardiac dysfunction associated with HFpEF.
Footnotes
The authors report no relevant conflicts of interest.
References
- 1.Betge S, Kunz C, Figulla H, Jung C. Late onset oral treatment with tranilast following large myocardial infarction has no beneficial effects on cardiac remodeling and mortality in rats. Exp Ther Med. 2014;8:1789–1796. doi: 10.3892/etm.2014.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Committee. Guide for the Care and Use of Laboratory Animals: Eighth Edition. Guide for the Care and Use of Laboratory Animals. 2011:118. [Google Scholar]
- 3.Darakhshan S, Pour AB. Tranilast: A review of its therapeutic applications. Pharmacol Res. 2015;91:15–28. doi: 10.1016/j.phrs.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 4.Hisanaga E, Nagasawa M, Ueki K, Kulkarni RN, Mori M, Kojima I. Regulation of calcium-permeable TRPV2 channel by insulin in pancreatic beta-cells. Diabetes. 2009;58:174–84. doi: 10.2337/db08-0862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hocher B, Godes M, Olivier J, Weil J, Eschenhagen T, Slowinski T, Neumayer H-H, Bauer C, Paul M, Pinto YM. Inhibition of left ventricular fibrosis by tranilast in rats with renovascular hypertension. [Online] J Hypertens. 2002;20:745–51. doi: 10.1097/00004872-200204000-00034. http://www.ncbi.nlm.nih.gov/pubmed/11910312 [17 Aug. 2017] [DOI] [PubMed] [Google Scholar]
- 6.Holmes DR, Savage M, LaBlanche J-M, Grip L, Serruys PW, Fitzgerald P, Fischman D, Goldberg S, Brinker JA, Zeiher AM, Shapiro LM, Willerson J, Davis BR, Ferguson JJ, Popma J, King SB, Lincoff AM, Tcheng JE, Chan R, Granett JR, Poland M. Results of Prevention of REStenosis with Tranilast and its Outcomes (PRESTO) trial. [Online] Circulation. 2002;106:1243–50. doi: 10.1161/01.cir.0000028335.31300.da. http://www.ncbi.nlm.nih.gov/pubmed/12208800 [17 Aug. 2017] [DOI] [PubMed] [Google Scholar]
- 7.Ishibashi S, Ikeda U, Ihara T, Shimada K. Tranilast inhibits contraction and Ca2+ movement of porcine coronary arteries. [Online] Atherosclerosis. 1997;130:113–9. doi: 10.1016/s0021-9150(96)06053-4. http://www.ncbi.nlm.nih.gov/pubmed/9126655 [18 Aug. 2017] [DOI] [PubMed] [Google Scholar]
- 8.Iwata Y, Ohtake H, Suzuki O, Matsuda J, Komamura K, Wakabayashi S. Blockade of sarcolemmal TRPV2 accumulation inhibits progression of dilated cardiomyopathy. Cardiovasc Res. 2013;99:760–768. doi: 10.1093/cvr/cvt163. [DOI] [PubMed] [Google Scholar]
- 9.Katanosaka Y, Iwasaki K, Ujihara Y, Takatsu S, Nishitsuji K, Kanagawa M, Sudo A, Toda T, Katanosaka K, Mohri S, Naruse K. TRPV2 is critical for the maintenance of cardiac structure and function in mice. Nat Commun. 2014;5:3932. doi: 10.1038/ncomms4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelly DJ, Zhang Y, Connelly K, Cox AJ, Martin J, Krum H, Gilbert RE. Tranilast attenuates diastolic dysfunction and structural injury in experimental diabetic cardiomyopathy. AJP Hear Circ Physiol. 2007;293:H2860–H2869. doi: 10.1152/ajpheart.01167.2006. [DOI] [PubMed] [Google Scholar]
- 11.Koch SE, Gao X, Haar L, Jiang M, Lasko VM, Robbins N, Cai W, Brokamp C, Varma P, Tranter M, Liu Y, Ren X, Lorenz JN, Wang H-S, Jones WK, Rubinstein J. Probenecid: novel use as a non-injurious positive inotrope acting via cardiac TRPV2 stimulation. J Mol Cell Cardiol. 2012;53:134–44. doi: 10.1016/j.yjmcc.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koch SE, Mann A, Jones S, Robbins N, Alkhattabi A, Worley MC, Gao X, Lasko-Roiniotis VM, Karani R, Fulford L, Jiang M, Nieman M, Lorenz JN, Rubinstein J. Transient receptor potential vanilloid 2 function regulates cardiac hypertrophy via stretch-induced activation. J Hypertens. 2017;35:602–611. doi: 10.1097/HJH.0000000000001213. [DOI] [PubMed] [Google Scholar]
- 13.Koch SE, Tranter M, Robbins N, Luther K, Singh U, Jiang M, Ren X, Tee T, Smith L, Varma P, Jones WK, Rubinstein J. Probenecid as a noninjurious positive inotrope in an ischemic heart disease murine model. J Cardiovasc Pharmacol Ther. 2013;18:280–9. doi: 10.1177/1074248412469299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 15.Martin J, Kelly DJ, Mifsud SA, Zhang Y, Cox AJ, See F, Krum H, Wilkinson-Berka J, Gilbert RE. Tranilast attenuates cardiac matrix deposition in experimental diabetes: role of transforming growth factor-beta. Cardiovasc Res. 2005;65:694–701. doi: 10.1016/j.cardiores.2004.10.041. [DOI] [PubMed] [Google Scholar]
- 16.Miyazawa K, Kikuchi S, Fukuyama J, Hamano S, Ujiie A. Inhibition of PDGF- and TGF-β1-induced collagen synthesis, migration and proliferation by tranilast in vascular smooth muscle cells from spontaneously hypertensive rats. Atherosclerosis. 1995;118:213–221. doi: 10.1016/0021-9150(95)05607-6. [DOI] [PubMed] [Google Scholar]
- 17.Nakatani Y, Nishida K, Sakabe M, Kataoka N, Sakamoto T, Yamaguchi Y, Iwamoto J, Mizumaki K, Fujiki A, Inoue H. Tranilast prevents atrial remodeling and development of atrial fibrillation in a canine model of atrial tachycardia and left ventricular dysfunction. J Am Coll Cardiol. 2013;61:582–8. doi: 10.1016/j.jacc.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 18.Naticchioni M, Karani R, Smith MA, Onusko E, Robbins N, Jiang M, Radzyukevich T, Fulford L, Gao X, Apel R, Heiny J, Rubinstein J, Koch SE. Transient Receptor Potential Vanilloid 2 Regulates Myocardial Response to Exercise. PLoS One. 2015;10:e0136901. doi: 10.1371/journal.pone.0136901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Regen DM. Calculation of left ventricular wall stress. [Online] Circ Res. 1990;67:245–52. doi: 10.1161/01.res.67.2.245. http://www.ncbi.nlm.nih.gov/pubmed/2198115 [1 Sep. 2017] [DOI] [PubMed] [Google Scholar]
- 20.Rubinstein J, Lasko VM, Koch SE, Singh VP, Carreira V, Robbins N, Patel AR, Jiang M, Bidwell P, Kranias EG, Jones WK, Lorenz JN. Novel role of transient receptor potential vanilloid 2 in the regulation of cardiac performance. Am J Physiol Heart Circ Physiol. 2014;306:H574–84. doi: 10.1152/ajpheart.00854.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Slone S, Anthony SR, Wu X, Benoit JB, Aube J, Xu L, Tranter M. Activation of HuR downstream of p38 MAPK promotes cardiomyocyte hypertrophy. Cell Signal. 2016;28:1735–41. doi: 10.1016/j.cellsig.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tamai H, Katoh K, Yamaguchi T, Hayakawa H, Kanmatsuse K, Haze K, Aizawa T, Nakanishi S, Suzuki S, Suzuki T, Takase S, Nishikawa H, Katoh O. The impact of tranilast on restenosis after coronary angioplasty: the Second Tranilast Restenosis Following Angioplasty Trial (TREAT-2). [Online] Am Heart J. 2002;143:506–13. doi: 10.1067/mhj.2002.120770. http://www.ncbi.nlm.nih.gov/pubmed/11868058 [17 Aug. 2017] [DOI] [PubMed] [Google Scholar]
- 23.Tamai H, Katoh O, Suzuki S, Fujii K, Aizawa T, Takase S, Kurogane H, Nishikawa H, Sone T, Sakai K, Suzuki T. Impact of tranilast on restenosis after coronary angioplasty: Tranilast Restenosis Following Angioplasty Trial (TREAT) Am Heart J. 1999;138:968–975. doi: 10.1016/s0002-8703(99)70025-6. [DOI] [PubMed] [Google Scholar]
- 24.Umemura K, Kikuchi S, Suzuki Y, Nakashima M. Inhibitory Effect of Tranilast on Hypertrophic Collagen Production in the Spontaneously Hypertensive Rat Heart. Jpn J Pharmacol. 1998;78:161–167. doi: 10.1254/jjp.78.161. [DOI] [PubMed] [Google Scholar]
- 25.Ward MR, Sasahara T, Agrotis A, Dilley RJ, Jennings GL, Bobik A. Inhibitory effects of tranilast on expression of transforming growth factor-β isoforms and receptors in injured arteries. Atherosclerosis. 1998;137:267–275. doi: 10.1016/s0021-9150(97)00275-x. [DOI] [PubMed] [Google Scholar]
- 26.Wilkins BJ, Dai Y-S, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–8. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 27.Yan S, Chen Y, Dong M, Song W, Belcher SM, Wang H-S. Bisphenol A and 17β-Estradiol Promote Arrhythmia in the Female Heart via Alteration of Calcium Handling. PLoS One. 2011;6:e25455. doi: 10.1371/journal.pone.0025455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yashiro M, Chung YS, Sowa M. Tranilast (N-(3,4-dimethoxycinnamoyl) anthranilic acid) down-regulates the growth of scirrhous gastric cancer. [Online] Anticancer Res. 17:895–900. [date unknown]. http://www.ncbi.nlm.nih.gov/pubmed/9137424 [18 Aug. 2017] [PubMed] [Google Scholar]