

Abstract
The development of the first enantioselective transition metal-catalyzed allylic alkylation providing access to acyclic products bearing vicinal all-carbon quaternary centers is disclosed. The iridium-catalyzed allylic alkylation reaction proceeds with excellent yields and selectivities for a range of malononitrile-derived nucleophiles and trisubstituted allylic electrophiles. The utility of these sterically congested products is explored through a series of diverse chemo- and diastereoselective product transformations to afford a number of highly valuable, densely functionalized building blocks, including those containing vicinal all-carbon quaternary stereocenters.
Keywords: allylic alkylation, quaternary stereocenter, vicinal all-carbon quaternary centers, enantioselective synthesis, iridium
Won’t you be my neighbor
The first enantioselective transition metal-catalyzed allylic alkylation providing access to acyclic products bearing vicinal all-carbon quaternary centers has been developed. The iridium-catalyzed reaction proceeds with excellent yields and selectivities for a range of malononitrile-derived nucleophiles and trisubstituted allylic electrophiles. The utility of these sterically congested products is explored through a series of diverse chemoselective and diastereoselective product transformations to afford a number of highly valuable, densely functionalized building blocks, including those containing vicinal all-carbon quaternary stereocenters.

The enantioselective preparation of singular all-carbon quaternary stereocenters has been a persistent challenge in the synthetic community and a topic of great interest to our research group.1 However, due to significant progress in this area over recent decades,2 the more formidable challenge of constructing vicinal all-carbon quaternary centers has become the forefront of investigation. A limited number of organic3 and transition metal-catalyzed 4,5 methods for the enantioselective preparation of vicinal all-carbon quaternary stereocenters have been reported,6 with enantioselective transition metal-catalyzed allylic alkylation strategies remaining underexplored.
In 2011, Trost reported an enantioselective palladium-catalyzed allylic alkylation of oxindoles to provide reverse prenylated products containing a homoallylic quaternary stereocenter vicinal to an all-carbon quaternary center (Figure 1A).5a,b In 2014, Ooi5c and Zhang5d each disclosed examples of enantio- and diastereoselective palladium-catalyzed allylic alkylation reactions to form cyclic products bearing vicinal all-carbon quaternary stereocenters (Figure 1B). To date, these three methods represent the only enantioselective transition metal-catalyzed allylic alkylation protocols for the synthesis of vicinal all-carbon quaternary centers7,8 – none of these reports provide access to acyclic products.
Figure 1.

State-of-the-art in the enantioselective synthesis of vicinal all-carbon quaternary centers via transition metal-catalyzed allylic alkylation
Recently, our group reported the first iridium-catalyzed allylic alkylation method to allow for the synthesis of highly enantioenriched allylic quaternary stereocenters. 9 Given that iridium-catalyzed allylic alkylation is well known to facilitate the synthesis of vicinal stereocenters (3°/3° and 3°/4°), 10 we hypothesized that we could utilize our newly developed technology to prepare vicinal all-carbon quaternary centers, with the use of appropriately designed nucleophiles.11 Herein, we report the first enantioselective transition metal-catalyzed allylic alkylation to form acyclic products bearing vicinal all-carbon quaternary centers (Figure 1C).
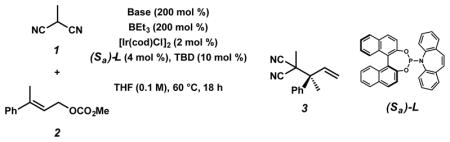
Preliminary studies focused on identifying a suitable catalyst system to promote the reaction of nucleophile 1 and trisubstituted allylic electrophile 2 (Table 1). In designing our optimal nucleophile for the allylic alkylation reaction, we imagined that methylmalononitrile (1) would mimic both the acidity and steric bulk of the previously utilized masked acyl cyanide (MAC) nucleophile9 (Figure 1C, R1 = OMOM) as well as provide versatile functional handles for derivatizations of product 3. We were pleased to find that our hypothesis was valid, and when utilizing our optimized conditions for the iridium-catalyzed allylic alkylation of MAC reagents9 with nucleophile 1, product 3 was obtained in nearly quantitative yield, though in only a moderate 73% ee (Table 1, entry 1). In an effort to improve the enantioselectivity of the transformation we investigated a range of basic additives, as bases have been reported to have a pronounced effect on selectivity in allylic alkylation reactions.12 While addition of LiOt-Bu provided only a slight enhancement in enantioselectivity to 81% ee (entry 2), we were delighted to find that the amine base DABCO afforded product 3 in 92% yield and an excellent 95% ee (entry 3). At this time, the specific role of DABCO remains unknown; however, due to the additive’s drastic effect on enantioselectivity, we hypothesize that DABCO allows for increased equilibration between diastereomers of an iridium π-allyl complex by slowing the rate of nucleophilic attack.13,14 Moreover, while we observed the highest yield for the allylic alkylation reaction using a 1:2 nucleophile to electrophile ratio, the nucleophile and electrophile stoichiometry can be varied (1:1 or 2:1) without affecting reaction selectivity, though yields are diminished (entries 4 and 5).15
Table 1.
Optimization of reaction parametersa
| ||||
|---|---|---|---|---|
| Entry | Nuc:Elec | Base | Yield (%)b | ee (%)c |
| 1 | 1:2 | – | >99 | 73 |
| 2 | 1:2 | LiOt-Bu | >99 | 81 |
| 3 | 1:2 | DABCO | 92 | 95 |
| 4 | 1:1 | DABCO | 67 | 95 |
| 5 | 2:1 | DABCO | 41 | 94 |
Reactions performed on 0.1 mmol scale.
1H NMR yield based on internal standard.
Determined by chiral SFC analysis.
TBD = 1,3,4-triazabicyclo[4.4.0]dec-5-ene, DABCO = 1,4-diazabicyclo[2.2.2]octane.
With the optimal conditions identified (Table 1, entry 3), we explored the substrate scope for this new transformation. With respect to nucleophile 4, the process is tolerant of a wide variety of substituted malononitrile derivatives (Table 2).16 Specifically, we were pleased to find that increasing the steric bulk on the nucleophile results in formation of the corresponding ethyl-substituted 5a and benzyl-substituted 5b products in only slightly decreased yields (76% and 69%, respectively) and no significant loss in enantioselectivity. 17 Interestingly, phenyl-substituted nucleophile 4c gives full conversion to the linear product (SI-1)18 in the allylic alkylation reaction rather than branched product 5c. Additionally, olefinic substituents on the nucleophile are tolerated under the reaction conditions provided the olefin is at least di-substituted;19 methallyl-substituted product 5d can be prepared in 33% yield and 92% ee while prenyl-substituted product 5e can be constructed in an excellent 92% yield with 96% ee. We reason that increased olefin substitution decreases the affinity of the olefin to bind to the catalyst,20 thus leading to increased yields. Furthermore, we were delighted to discover that carbonyl-containing product 5f can be obtained in a moderate 52% yield with an excellent 96% ee. However, we noted that other Lewis basic functionalities, specifically heteroaromatic substituents, are not tolerated on nucleophile 4. Finally, fluorinated product 5g can be accessed in a moderate 50% yield with 91% ee.
Table 2.
Nucleophile substrate scopea

|
Reactions performed on 0.2 mmol scale.
Isolated yield.
Determined by chiral SFC. [d] 99% conversion by 1H NMR to linear product SI-1.
Pleased to find the reaction amenable to a range of nucleophilic substrates, we sought to further examine the scope of the transformation by exploring the diversity of substitution permitted on trisubstituted allylic electrophile 6 (Table 3). Gratifyingly, we observed that a series of para-substituted allylic electrophiles bearing both electron-donating (–p-Me, –p-OMe) and withdrawing groups (–p-Ph, –p-F) on the aromatic ring furnish the corresponding products 7a–d in consistently excellent enantioselectivities (>94% ee) when subjected to the reaction conditions utilizing methylmalononitrile (1) as the nucleophile. In evaluating the effect of meta-substitution, we found that products 7e–h (–m-Me, –m-Cl, –m-NO2, 2-Np) can be obtained with similarly high enantiocontrol (>93% ee). Generally, we noted that electron-rich electrophiles (i.e., 6a, 6b, 6e) provide the corresponding allylic alkylation products in slightly diminished yields (67–80% versus 84–99%) as compared to electron-poor electrophiles (i.e., 6c, 6d, 6f, 6g). At this time, ortho-substitution (i.e., –o-Me) is not tolerated under the reaction conditions and no conversion to product 7i is observed. However, we were pleased to discover that the reaction is amenable to bis-alkyl-substitution on the allylic electrophile allowing for access to product 7j in 65% yield and 84% ee, though as an inseparable mixture (1:1.5) of branched and linear isomers. Additionally, reverse prenylation can be effected to produce achiral product 7k in 61% yield. Finally, extension of the alkyl chain on the allylic electrophile to an ethyl group leads to a decreased yield with 7l isolated in 50% yield but with no loss in selectivity (96% ee).
Table 3.
Electrophile substrate scopea

|
Reactions performed on 0.2 mmol scale.
Isolated yield.
Determined by chiral HPLC or SFC.
Absolute stereochemistry determined by single-crystal X-ray analysis; the absolute stereochemistry of all other compounds was assigned by analogy.
Combined isolated yield of inseparable linear and branched products (1:1.5 ratio).
With the general reactivity trends for the allylic alkylation reaction explored, we sought to demonstrate the utility of these sterically congested products (Figure 2). Though hydrogenation of the olefin in allylic alkylation product 3 using palladium catalysis proved problematic due to competing reduction of the nitrile groups, we found that treatment of 3 with Wilkinson’s catalyst under a hydrogen atmosphere chemoselectively reduces the olefin to furnish 8 in 92% yield. Additionally, ozonolysis of olefin 3 proceeds smoothly to give enantioenriched aldehyde 9 in 93% yield,21 wherein the aldehyde moiety can serve as a valuable functional handle for further manipulation (e.g., reductive aminations, allylations, and olefinations). Allylic alkylation product 5f was subjected to a two-step ozonolysis/aldol condensation process to deliver a densely functionalized, enantioenriched cyclopentene in 43% yield, which can then undergo diastereoselective hydration of the bis-nitrile functionality to provide amide 10 in 1:11 dr. Chiral cyclopentenes have been demonstrated to be key building blocks in a number of complex molecule total syntheses. 22 Finally, enantioenriched lactone 11 bearing vicinal all-carbon quaternary stereocenters can be accessed in 65% yield and 1:2.5 dr from allylic alkylation product 3 via ozonolysis followed by reductive quenching. The transformations forming products 10 and 11 showcase that the diastereotopic nitrile functionalities of the allylic alkylation products are amenable to diastereoselective differentiation to afford vicinal all-carbon quaternary stereocenters, which are otherwise difficult to prepare.
Figure 2.

(a) RhCl(PPh3)3, H2 (balloon), benzene, 23 °C, 18 h, 92% yield; (b) O3, pyridine, CH2Cl2, −78 °C, 4 min, 93% yield; (c) i. O3, pyridine, CH2Cl2, −78°C, 4 min, ii. p-TsOH, benzene, reflux, 18 h, 47% yield; (d) NaOH, EtOH/H2O (1:1), 60 °C, 18 h, 38% yield. 1:11 dr; (e) i. O3, MeOH, −78 °C, 0.5 h, ii. NaBH4, 0 °C, 3 h, 65% yield, 1:2.5 dr.
In conclusion, we have developed the first enantioselective transition metal-catalyzed allylic alkylation reaction for the preparation of acyclic products bearing vicinal all-carbon quaternary centers. Key to the success of this new reaction is the use of DABCO in combination with triethylborane and our unique catalyst prepared from [Ir(cod)Cl]2, (Sa)-L, and TBD. The developed method proceeds with moderate to excellent yields and high levels of enantioselectivity for a wide range of substitution on both the malononitrile-derived nucleophile and the trisubstituted allylic electrophile. Furthermore, the allylic alkylation products can be transformed by chemo- and diastereoselective methods to a number of highly valuable, densely functionalized building blocks, including those containing vicinal all-carbon quaternary stereocenters. Further exploration of this catalyst system is underway and will be reported in due course.
Supplementary Material
Acknowledgments
The NIH-NIGMS (R01GM080269) and Caltech are thanked for support of our research program. J.C.H. thanks the Camille and Henry Dreyfus postdoctoral program, and S.E.S. thanks the NIH-NIGMS for a predoctoral fellowship (F31GM120804). Dr. Michael Takase and Dr. Lawrence Henling are acknowledged for assistance with X-ray analysis. Dr. Mona Shahgholi and Naseem Torian are thanked for mass spectrometry assistance. Dr. David VanderVelde is thanked for assistance with NMR analysis.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.For select reviews, see: Douglas CJ, Overman LE. Proc Natl Acad Sci USA. 2004;101:5363–5367. doi: 10.1073/pnas.0307113101.Das JP, Marek I. Chem Commun. 2011;47:4593–4623. doi: 10.1039/c0cc05222a.Quasdorf KW, Overman LE. Nature. 2014;516:181–191. doi: 10.1038/nature14007.Corey EJ, Guzman-Perez A. Angew Chem Int Ed. 1998;37:388–401. doi: 10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V.Angew Chem. 1998;110:402–415.Christoffers J, Mann A. Angew Chem Int Ed. 2001;40:4591–4597. doi: 10.1002/1521-3773(20011217)40:24<4591::aid-anie4591>3.0.co;2-v.Angew Chem. 2001;113:4725–4732.Trost BM, Jiang C. Synthesis. 2006:369–396.Denissova I, Barriault L. Tetrahedron. 2003;59:10105–10146.Ling T, Rivas F. Tetrahedron. 2016;72:6729–6777.Liu Y, Han SJ, Liu WB, Stoltz BM. Acc Chem Res. 2015;48:740–751. doi: 10.1021/ar5004658.
- 2.For select reviews, see: Peterson EA, Overman LE. Proc Natl Acad Sci USA. 2004;101:11943–11948. doi: 10.1073/pnas.0402416101.Long R, Huang J, Gong J, Yang Z. Nat Prod Rep. 2015;32:1584–1601. doi: 10.1039/c5np00046g.
- 3.For recent examples, see: Uyeda C, Rötheli AR, Jacobsen EN. Angew Chem Int Ed. 2010;49:9753–9756. doi: 10.1002/anie.201005183.Angew Chem. 2010;122:9947–9950.Gao L, Hwang GS, Ryu DH. J Am Chem Soc. 2011;133:20708–20711. doi: 10.1021/ja209270e.Jolit A, Walleser PM, Yap GPA, Tius MA. Angew Chem Int Ed. 2014;53:6180–6183. doi: 10.1002/anie.201403587.Angew Chem. 2014;126:6294–6297.Ohmatsu K, Ando Y, Ooi T. J Am Chem Soc. 2013;135:18706–18709. doi: 10.1021/ja411647x.
- 4.For reports of transition metal-catalyzed methods, excluding allylic alkylation protocols, see: DeAngelis A, Dmitrenko O, Yap GPA, Fox JM. J Am Chem Soc. 2009;131:7230–7231. doi: 10.1021/ja9026852.Zhang H, Hong L, Kang H, Wang R. J Am Chem Soc. 2013;135:14098–14101. doi: 10.1021/ja408336v.Cao ZY, Wang X, Tan C, Zhao XL, Zhou J, Ding K. J Am Chem Soc. 2013;135:8197–8200. doi: 10.1021/ja4040895.Zheng J, Lin L, Dai L, Tang Q, Liu X, Feng X. Angew Chem Int Ed. 2017;56:13107–13111. doi: 10.1002/anie.201705943.Angew Chem. 2017;129:13287–13291.
- 5.For reports of enantioselective transition metal-catalyzed allylic alkylation methods, see: Trost BM, Malhotra S, Chan WH. J Am Chem Soc. 2011;133:7328–7331. doi: 10.1021/ja2020873.Trost BM, Chan WH, Malhotra S. Chem Eur J. 2017;23:4405–4414. doi: 10.1002/chem.201605810.Ohmatsu K, Imagawa N, Ooi T. Nat Chem. 2014;6:47–51. doi: 10.1038/nchem.1796.Khan A, Yang L, Xu J, Jin LY, Zhang YJ. Angew Chem Int Ed. 2014;53:11257–11260. doi: 10.1002/anie.201407013.Angew Chem. 2014;126:11439–11442.
- 6.For select reports of the preparation of vicinal all-carbon quaternary centers but not stereocenters, see: Ramachary DB, Reddy PS, Shruthi KS, Madhavachary R, Reddy PVG. Eur J Org Chem. 2016:5220–5226.Trost BM, Cramer N, Silverman SM. J Am Chem Soc. 2007;129:12396–12397. doi: 10.1021/ja075335w.
- 7.For reports of enantiospecific transition metal-catalyzed allylic alkylation methods, see: Kawatsura M, Sato M, Tsuji H, Ata F, Itoh T. J Org Chem. 2011;76:5485–5488. doi: 10.1021/jo2007169.Huang X, Wu S, Wu W, Li P, Fu C, Ma S. Nat Commun. 2016 doi: 10.1038/ncomms12382.
- 8.For reports of enantio- and diastereoselective preparation of vicinal all-carbon quaternary stereocenters via two sequential allylic alkylations, see: Ghosh S, Bhunia S, Kakde BN, De S, Bisai A. Chem Commun. 2014;50:2434–2437. doi: 10.1039/c3cc49064e.Trost BM, Osipov M. Angew Chem Int Ed. 2013;52:9176–9181. doi: 10.1002/anie.201302805.Angew Chem. 2013;125:9346–9351.
- 9.Shockley SE, Hethcox JC, Stoltz BM. Angew Chem Int Ed. 2017;56:11545–11548;. doi: 10.1002/anie.201707015. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2017;129:11703–11706. [Google Scholar]
- 10.For a recent review, see: Hethcox JC, Shockley SE, Stoltz BM. ACS Catal. 2016;6:6207–6213. doi: 10.1021/acscatal.6b01886.
- 11.Diastereoselective reverse prenylation of indoles via iridium-catalyzed allylic alkylation has been reported, see: Ruchti J, Carreira EM. J Am Chem Soc. 2014;136:16756–16759. doi: 10.1021/ja509893s.Müller JM, Stark CBW. Angew Chem Int Ed. 2016;55:4798–4802. doi: 10.1002/anie.201509468.Angew Chem. 2016;128:4877–4881.
- 12.For select examples, see: Kanayama T, Yoshida K, Miyabe H, Takemoto Y. Angew Chem Int Ed. 2003;42:2054–2056. doi: 10.1002/anie.200250654.Angew Chem. 2003;115:2100–2102.Jiang X, Chen W, Hartwig JF. Angew Chem Int Ed. 2016;55:5819–5823. doi: 10.1002/anie.201600235.Angew Chem. 2016;128:5913–5917.Huo X, He R, Zhang X, Zhang W. J Am Chem Soc. 2016;138:11093–11096. doi: 10.1021/jacs.6b06156.Chen W, Hartwig JF. J Am Chem Soc. 2013;135:2068–2071. doi: 10.1021/ja311363a.Chen W, Hartwig JF. J Am Chem Soc. 2014;136:377–382. doi: 10.1021/ja410650e.Krautwald S, Schafroth MA, Sarlah D, Carreira EM. J Am Chem Soc. 2014;136:3020–3023. doi: 10.1021/ja5003247.Sandmeier T, Krautwald S, Zipfel HF, Carreira EM. Angew Chem Int Ed. 2015;54:14363–14367. doi: 10.1002/anie.201506933.Angew Chem. 2015;127:14571–14575.Liu WB, Reeves CM, Virgil SC, Stoltz BM. J Am Chem Soc. 2013;135:10626–10629. doi: 10.1021/ja4052075.Hethcox JC, Shockley SE, Stoltz BM. Angew Chem Int Ed. 2016;55:16092–16095. doi: 10.1002/anie.201609960.Angew Chem. 2016;128:16326–16329.
- 13.DABCO has been previously utilized in iridium-catalyzed allylic alkylation leading to higher yields but slower rates of reaction, see: Bondzic BP, Farwick A, Liebich J, Eilbracht P. Org Biomol Chem. 2008;6:3723–3731. doi: 10.1039/b809143a.
- 14.Previous reports have hypothesized that facile equilibration between diastereomers of the iridium π-allyl complex leads to higher selectivities, see: Bartels B, Helmchen G. Chem Commun. 1999:741–742.Alexakis A, Polet D. Org Lett. 2004;6:3529–3532. doi: 10.1021/ol048607y.Polet D, Alexakis A, Tissot-Croset K, Corminboeuf C, Ditrich K. Chem Eur J. 2006;12:3596–3609. doi: 10.1002/chem.200501180.d) ref. 12h.
- 15.Excess electrophile is unable to be quantitatively recovered as a competing elimination reaction takes place to form a diene byproduct.
- 16.To date, only functionalized malononitrile nucleophiles have been successfully utilized in the reported allylic alkylation reaction, despite studies into other bis-electron-withdrawing group-functionalized nucleophiles (e.g., malonates and 1,3-diketones).
- 17.Branched substitution is not tolerated in the allylic alkylation reaction as isopropyl-substituted 4 returns only starting material.
-
18.Linear product (SI-1):

- 19.Allyl-substituted 4 returns only starting material.
- 20.Schurig V. Inorg Chem. 1986;25:945–949. [Google Scholar]
- 21.Willand-Charnley R, Fisher TJ, Johnson BM, Dussault PH. Org Lett. 2012;14:2242–2245. doi: 10.1021/ol300617r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.For select examples, see: Hu X, Xu S, Maimone TJ. Angew Chem Int Ed. 2017;56:1624–1628. doi: 10.1002/anie.201611078.Angew Chem. 2017;129:1646–1650.Brown B, Hegedus LS. J Org Chem. 2000;65:1865–1872. doi: 10.1021/jo9919759.Suzuki M, Yanagisawa A, Noyori R. J Am Chem Soc. 1988;110:4718–4726.Schnermann MJ, Overman LE. Angew Chem Int Ed. 2012;51:9576–9580. doi: 10.1002/anie.201204977.Angew Chem. 2012;124:9714–9718.Hong AY, Stoltz BM. Angew Chem Int Ed. 2012;51:9674–9678. doi: 10.1002/anie.201205276.Angew Chem. 2012;124:9812–9816.Alishetty S, Shih HP, Han C-C. Org Lett. 2018 doi: 10.1021/acs.orglett.8b00510.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.