

Abstract
Rare diseases, in totality, affect a significant proportion of the population and represent an unmet medical need facing the scientific community. However, the treatment of individuals affected by rare diseases is hampered by poorly understood mechanisms preventing the development of viable therapeutics. The discovery and application of cellular reprogramming to create novel induced pluripotent stem cell models of rare diseases has revolutionized the rare disease community. Through developmental and functional analysis of differentiated cell types, these stem cell models carrying patient-specific mutations have become an invaluable tool for rare disease research. In this review article, we discuss the reprogramming of samples from individuals affected with rare diseases to induced pluripotent stem cells, current and future applications for this technology, and how integration of genome editing to rare disease research will help to improve our understanding of disease pathogenesis and lead to patient therapies.
Keywords: Rare disease, induced pluripotent, pluripotency, iPSC, CRISPR, disease modeling
1. Modeling rare diseases
In what can be viewed as a misnomer, rare diseases are not appreciably “rare” when considered collectively. Commonly classified by prevalence, definitions and estimates vary across regions and are often complicated by unclear diagnosis and unique presentations [1]. A rare disease is defined as a condition affecting fewer than 5 in 10,000 people in Europe and fewer than 200,000 people total within the United States, according to the Orphan Drug Regulation 141/2000 and Orphan Drug Act of 1983, respectively. It is estimated that between 8–10% of the population are affected by a rare condition [2]. This translates to >30 million affected individuals in the United States alone and approximately 350 million worldwide [3]. Based on epidemiological and genomic data, estimates from the US National Institutes of Health suggest approximately 7,000 unique rare diseases are present worldwide. However, less than 10% of rare disease patients are treated, reflecting a significant need for development of medical interventions and increased studies to understand disease pathogenesis [2].
Over 80% of rare diseases are considered genetic in origin [2]. A majority of these conditions including neurofibromatosis I, achondrophasia, Friedrich’s ataxia, and many inborn errors of metabolism, are monogenic diseases defined by defects in a single gene. In polygenic disorders, including Fanconi anemia and muscular dystrophies, multiple genes contribute to a single disease. The importance of disease modifiers at additional genetic loci, such as allelic variants of α1-antitrypsin antiprotease (or SERPINA1) associated with portal hypertension in cystic fibrosis or defects arising in regulatory regions of the genome, have also been recognized [4]. Further, approximately half of rare diseases manifest in children and result in developmental malformations accounting for 20% of infant deaths, a leading cause of mortality in this age group [5].
Rare disease research relies heavily upon the modeling of genetic changes and developmental pathways to recapitulate the unique aspects of human disease pathology. Induced pluripotent stem cells (iPSCs) derived from human samples have developed into a viable and complementary biological model to overcome some of the challenges associated with traditional approaches, such as animal models and immortalized cell lines. In this brief review, we discuss the values and challenges in the use of iPSCs for the study of rare diseases, as well as potential uses for iPSCs in translational applications. Due to the many significant publications within this rapidly maturing field, we are forced to limit our discussions to a selected number of publications. We apologize to any authors whose excellent work was not specifically cited here.
2. Induced pluripotent stem cells for modeling rare diseases
The in vitro modeling and analysis of human diseases was revolutionized by the discovery of reprogramming mature cells to pluripotency by Kazutoshi Takahashi and Shinya Yamanaka in 2006. The induction of four transcription factors, KLF4, MYC, POU5F1, and SOX2, was found to allow derivation of embryonic stem cell-like pluripotent cells, now referred to as iPSCs, from mouse and later human somatic cells [6, 7]. The simplicity of these experiments was surprising given the complexity of reprogramming experiments leading up to its discovery. The use of somatic cell nuclear transfer (SCNT) demonstrated in Xenopus laevis by Sir John Gurdon in 1958 and later in mammals with the cloning of “Dolly” the sheep by Wilmut et al. in 1996 suggested complex mechanisms encompassing genetic and epigenetic changes controlled cellular de-differentiation [8, 9]. Therefore, the ability of a quartet of transcription factors to yield pluripotent cells largely indistinguishable from human ES cells was remarkable. This seminal work also opened up new possibilities for the use of iPSCs in disease and gene-specific applications. The Yamanaka studies and subsequent publications from other labs also helped alleviate some of the ethical debates surrounding human pluripotent stem cells by avoiding stem cell isolation from the embryonic inner cell mass.
Since their initial discovery, iPSCs have shown great potential in modeling the pathogenesis of rare diseases. Traditional approaches have often relied upon primary or patient-derived immortalized cell lines to study the etiology and physiology of rare conditions. While primary cell types are readily available from blood or tissue biopsies, disease relevant cell types are not always easily isolated nor may they be propagated indefinitely. Moreover, immortalized cell lines are often not an accurate reflection of their primary culture counterparts, limiting their reliability in functional studies. Similarly, despite being an irreplaceable tool to date for in vivo validation, animal models do not always recapitulate human pathogenesis [10]. There are considerable anatomic, embryonic, and metabolic differences between mice and humans which may reflect difficulties in translating therapeutic discoveries to clinical trials [11].
2.1 Advantages of iPSCs for disease modeling
Patient-derived iPSCs offer an invaluable alternative for modeling rare diseases, directly addressing some of the challenges associated with traditional methods (Figure 1). Along with the capacity to propagate indefinitely, iPSCs have the potential to differentiate into virtually any human cell type given the proper environmental stimuli. By utilizing this pluripotent capacity in iPSCs carrying specific pathogenic mutations, patient-specific iPSCs can model the molecular mechanisms underlying disease pathophysiology. The hope for iPSCs in regenerative medicine and cell therapy applications are further fueled by the potential immune compatibility of iPSC derivatives in autologous settings, suggesting a lessened risk for graft rejection compared to more common allogeneic stem cell-based therapies [12]. Indeed, ongoing clinical studies utilizing iPSCs as a source for transplantable cellular derivatives, such as retinal pigment epithelium for treatment of age-related macular degeneration, have demonstrated tissue engraftment >1 yr. post-transplantation to patients, providing hope for the continued success of regenerative therapies [13].
Figure 1.
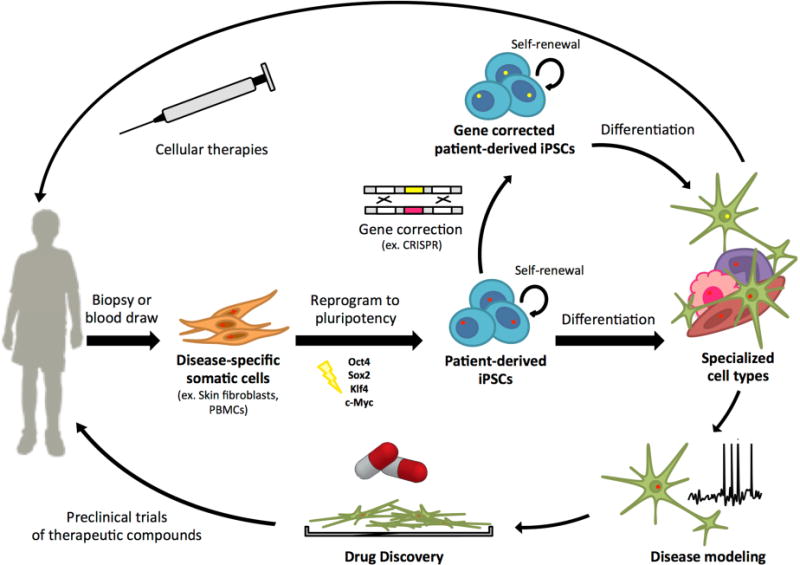
iPSC generation and potential uses of iPSC-derivatives for rare disease studies.
Stem cell-based models have been successfully used to study disorders of varying genetic origin. Monogenic-based rare disorders are, thus far, the most widely studied using iPSC approaches, particularly when a clear cellular phenotype has been established [14]. Given the genetic basis for most rare disorders, iPSCs are particularly well adapted for this purpose. Additionally, rare childhood diseases of developmental origin can be robustly modeled using directed differentiation assays [15]. However, recapitulating mature cell defects of late onset disorders has proven to be more challenging as some differentiation protocols better reflect immature rather than adult cell types [16, 17]. Several studies have utilized cell stressors, such as hydrogen peroxide or antibiotics, to generate ROS promoting mitochondrial stress to induce cellular aging [11, 18]. A more physiologically relevant approach recently developed involves small molecule inhibition of telomerase activity that demonstrated classical features of aging, including increased DNA damage, ROS, and downregulation of tyrosine hydroxylase [19]. iPSC models of premature aging syndromes, such as Hutchinson-Gilford progeria syndrome, have not only successfully modeled rapid differentiation and stem cell aging, but have also facilitated identification of age-related markers utilized in the understanding of more common, late onset diseases such as Parkinson’s disease [20, 21]. Complex diseases involving multiple or unknown genes, as in Autism spectrum disorder and schizophrenia, have also been successfully developed and modeled using iPSCs [22, 23]. In particular, the study of rare monogenic disorders displaying phenotypic elements of poorly understood polygenetic diseases, holds much promise for mechanistic insight into these complex disorders. For example, a role for brain-specific L1 retrotransposon activity, traditionally associated with Rett syndrome, was recently demonstrated within a schizophrenia derived iPSC model [24].
A principle advantage of iPSC modeling is the ability to construct a model within the context of an individual’s genome, allowing a robust approach to disorders involving unknown loci. Conceivably, patient-derived iPSCs lines from multiple individuals spanning a disease spectrum could be comparatively analyzed within a specific disorder across diverse genetic backgrounds. Furthermore, this approach lends itself to personalized decisions regarding disease course and potential therapeutic options. For example, analyses of type 3 long QT syndrome patient-derived iPSCs were predictive of patient response to specific treatment strategies [25].
More recently efforts have been expanded from modeling cellular phenotypes in traditional two-dimensional differentiation cultures to modeling spatial interactions that mimic human tissues and organ systems. Co-culturing different cellular lineages for interaction effects has led to breakthroughs in modeling diseases such as Amyotrophic lateral sclerosis (ALS), where neuronal toxicity was not evident in the absence of glial cells exhibiting SOD1 mutations [26]. These methods have now been expanded to self-aggregation models of iPSCs inducing three-dimensional organoid structures that can exhibit tissue patterning of all germ layers. Directed stem-cell differentiation for organoid systems has been described for various tissues of all three germ layers: endodermal tissues of the stomach [27], small intestine [28], thyroid [29], liver [30], and lung [31]; ectodermal tissues of the retina [32], inner ear [33], pituitary [34], and cerebrum [35]; and mesodermal cardiac tissue [36, 37]. The importance of cell-cell interactions and the microenvironment in determining cell fate has also been explored [38]. Recently published work has utilized the fusion of organoids induced to differentiate into separate tissues, then fused to model functional interactions. Organoid fusion has been used to study interneuron migration between the ventral and dorsal forebrain [39]. This technique allowed identification of an interneuron migration defect within the neurodevelopmental disorder Timothy syndrome, which holds promise for broader implications within autism spectrum disorder [40]. As additional differentiation protocols are developed and standardized, organoid and fusion assays will become indispensable for understanding spatial organization and tissues morphogenesis in rare diseases. In the future, these assays should allow measurement of network-level activity in spatially organized iPSC-derived tissue, as has been previously shown in two-dimensional models [23].
2.2 Current iPSC models of rare diseases
Cellular reprogramming has now allowed the generation of iPSCs modeling a number of rare diseases, radically accelerating our understanding of disease physiology and pathogenesis, particularly in previously inaccessible tissues such as neurons and cardiomyocytes (Table 1). For example, disorders involving neuronal loss such as ALS, Friedreich’s ataxia, Ataxia-telangiectasia, Niemann-Pick disease Type C1, and Cockayne syndrome have seen new insights in disease mechanisms, largely due to the ability to accurately model disease relevant cell types [26, 41–44]. Likewise, electrophysiology of iPSC-derived cardiomyocytes has successfully aided identification of disease mechanisms within long QT syndrome, as well as Jervell and Lange-Nielsen syndrome [45–47].
Table 1. Human induced pluripotent stem cell models of rare diseases – selected publications of significant interest.
Summary of a subset of published human induced pluripotent stem cell models of rare diseases.
| Disease | Affected gene | Description | References |
|---|---|---|---|
| α1-antitrypsin deficiency | A1AT | Polymerization of A1AT in hepatocyte-like cells, piggyBAC gene correction restores A1AT function | [103] |
| Amyotrophic lateral sclerosis | SOD1 | iPSC generation as model of chronic disease, terminal differentiation into motor neurons, impaired motor neuron maturation | [104, 105] |
| Andersen’s syndrome | KCNJ2 | Decreased osteogenic markers in embryoid bodies | [106] |
| Angelman syndrome | UBE3A | Impaired neuronal maturation, synaptic activity & plasticity, unsilencing paternal allele rescues phenotype | [48] |
| Ataxia oculomotor apraxia type 2 | SETX | Increased DNA DSBs and oxidative damage, genome-wide expression analysis | [54] |
| Ataxia-telangiectasia | ATM | Defective repair of DSBs, impaired neuronal maturation, topoisomerase 1-DNA complex accumulation | [41] |
| Bernard-Soulier syndrome | GPIX | iPSC model derived from peripheral blood mononuclear cells | [59] |
| Chronic granulomatous disease | Multiple genes possibly affected; NADPH oxidase deficiency | Proteoliposome delivery in an iPSC- derived macrophage model restored NADPH activity and oxidative burst; CRISPR gene correction | [56, 107] |
| Cockayne syndrome | ERCC6 | Decreased synchrony and synaptic density, dysregulation of GH/IGF-1 pathway | [43] |
| Coenzyme Q10 deficiency | COQ4 | iPSC-derived skeletal muscle displayed metabolic and respiration dysfunction | [60] |
| Craniometaphyseal dysplasia | ANKH | iPSC model derived from peripheral blood mononuclear cells | [108] |
| Danon disease | LAMP-2 | Impaired mitophagy and respiratory capacity in iPSC-derived cardiomyocytes, confirmed pathophysiology in mouse model precedes heart failure | [109] |
| Fabry disease | GLA | Lysosomal accumulation of globotriaosylceramide in cardiomyocytes prevented by substrate reduction therapy | [57] |
| Familial dysautonomia (Hereditary sensory and autonomic neuropathy III, Riley-Day syndrome) | IKBKAP | Aberrant splicing of neural crest precursors, defective neuronal differentiation and migration, kinetin identified as potential therapeutic | [15] |
| Fanconi anemia | FANCA | Gene correction gives rise to normal hematopoietic progenitors | [110] |
| Fibrodysplasia ossificans progressiva | ALK2 | Increased mineralization of iPSC- derived pericytes, reduced vascular endothelial growth factor receptor 2 and mesenchymal transformation of iPSC-derived endothelial cells | [111] |
| Friedreich’s ataxia | FXN | Trinucleotide expansion and reduced mRNA expression, reversal of phenotype through gene correction | [58] |
| Glanzmann thrombasthenia | ITGA2B | Defective platelet aggregation, CD41 expression and aggregation restored by gene correction | [112] |
| Hutchinson-Gilford progeria syndrome | LMNA | Progerin accumulation and age- associated nuclear envelope and epigenetic changes only upon differentiation | [21] |
| Immunodeficiency, centromeric instability and facial anomalies type I (ICF1) syndrome | DNMT3B | Hypomethylation and altered gene expression in iPSCs and after mesenchymal stem cell differentiation, identified DNMT3B responsible for non-GC methylation | [52] |
| Jervell and LangeNielsen syndrome | KCNQ1 | Severe deficits in delayed rectifier current in iPSC-derived cardiomyocytes | [46, 47] |
| Long-QT syndrome | KCNQ1 | Recapitulation of prolonged action potential of ventricular and atrial myocytes, reduced IKs current and trafficking defect, responsive to arrhythmia drugs | [45] |
| Machado-Joseph disease (Spinocerebellar ataxia type 3) | MJD1 (ATXN3) | Abnormal protein processing, Ca2+- dependent proteolysis of ATXN3 and subsequent aggregation following neuronal excitation | [55] |
| Metachromatic leukodystrophy | ARSA | CNS pathology restored after intracerebral transplantation of ARSA overexpressing iPSC-derived neural stem cells in an animal model | [113] |
| Neuronal ceroid lipofuscinoses (Batten disease) | CLN5 | Accumulation of autoflurorescent storage material and ATP synthase subunit C, abnormal sphingolipid transportation | [64] |
| Neuronopathic Gaucher’s disease | GBA1 | Elevated pro-inflammatory factors secreted by iPSC derived macrophage; iPSC derived neurons exhibit reduced action potential by whole-cell patch-clamping; disease modeling affirms stabilization of the acid-β-glucosidase enzyme with chemical chaperones | [63, 114, 115] |
| Niemann-Pick disease, types A and C1 | SMPD1, NPC1 | Impaired cholesterol trafficking in neural progenitor cells; reduced AMPA receptor calcium influx; reduced sphingomyelin accumulation with hydroxypropyl-β-cyclodextrin treatment | [42, 44, 116] |
| Pelizaeus- Merzbacher disease | PLP1 | Impaired myelination and morphology of iPSC-derived oligodendrocytes, improvement with ER stress response modulators | [117, 118] |
| Phelan-McDermid syndrome | Chromosome 22q13.3 del | Neurodevelopmental disorder, intellectual disability; excitatory synaptic deficits rescued by IGF-1 | [93] |
| Pompe disease | GAA | Lysosomal accumulation of glycogen in iPSC derived cardiomyocytes; identified impaired oxidative stress response and mitochondrial dysfunction | [119, 120] |
| Prader-Willi syndrome | 15q11.2-q13 | Retained DNA methylation following reprogramming, differentiation | [50, 51] |
| Pyruvate kinase deficiency | PKLR | TALEN-mediated gene correction | [121] |
| Retinitis pigmentosa | MERTK | Impaired phagocytosis in retinal pigment epithelium; read through drugs improved phagocytic activity | [122, 123] |
| Rett syndrome | MECP2 | Modeling of X-linked disorders, reduction of dendritic spines and synapses, altered calcium spikes and electrophysiology | [67] |
| Shwachman-Diamond syndrome | SBDS | Protease-mediated autodigestion following pancreatic and hematopoietic differentiation | [61] |
| Smith-Lemli-Opitz syndrome | DHCR7 | Accelerated neural differentiation mediated by inhibition of Wnt/β-catenin signaling | [124] |
| Spinal muscular atrophy | SMN1 | Reduced SMN expression, selective deficit of motor neurons, increased SMN protein levels in response to drug treatment; increased motor neuron cell death mitigated by apoptosis inhibitors | [125, 126] |
| Timothy syndrome | CACNA1C | Defects in calcium influx, cortical neuron differentiation | [68] |
| Williams syndrome | Various; Chr.7 | Increased dendritic spines and longer dendrites in pyramidal neurons, increased apoptosis rescued by Frizzled 9 | [127] |
| Wolman disease (lysosomal acid lipase disease) | LIPA | Lipid accumulation in iPSC-derived NSCs, alleviated by enzyme replacement therapy and responsive to therapeutic compounds | [62] |
While DNA and histone methylation may be reset during reprogramming to pluripotency, disorders of genomic imprinting have been successfully modeled with iPSCs. iPSC studies involving Angelman syndrome [48, 49] and Prader-Willi syndrome [49–51] demonstrated that the methylation status of the paternal and maternal alleles on chromosome 15q11–13 were retained, necessary for recapitulation of these disease phenotypes. Additionally, syndromes associated with hypomethylation, such as ICF syndrome characterized by mutations within a DNA methyltransferase, have also demonstrated relevant epigenetic marks at the iPSC stage and after generation of mesenchymal stem cells [52]. Importantly, retention of allele specific gene expression throughout reprogramming and directed differentiation can be validated through RNA FISH and DNA methylation analysis [53].
Recent studies utilizing iPSC modeling of rare diseases has demonstrated robust cellular phenotypes in a number of cell types, including a large number of studies on various ataxias [41, 54, 55], disorders affecting blood-derived cell types [56–61], and lysosomal storage disorders [42, 44, 62–64]. Based upon strong cellular phenotypes in the iPSC derivatives that reflect patient symptoms, a number of these studies have also evaluated small molecules of interest or gene therapies for potential patient treatment strategies. Recapitulation of identified cellular phenotypes and genetic defects by the iPSC derivatives were critical in allowing effective treatment evaluation within these assays. It is hoped that ongoing and future iPSC-based studies will further help to elucidate disease mechanisms and screen possible treatments for these and additional rare diseases.
2.3 Challenges in modeling rare diseases with iPSCs
While reprogramming techniques, iPSC generation, and the use of iPSCs have become common practice in some laboratories, significant hurdles still exist in the use of iPSCs for modeling of rare diseases. A number of factors that are challenging for all iPSC research and not specific to rare disease studies, such as poor reprogramming efficiency, the comparatively high economic cost of iPSC research, and the significant technical expertise and training required for high quality iPSC studies, will not be discussed. However, some aspects of an iPSC research program are particularly relevant to rare disease studies.
Even if separate individuals carry identical pathogenic mutations, inter-individual heterogeneity across the rest of the genome will cause iPSC lines to display some degree of heterogeneity [65]. Ideally, multiple patient samples should be reprogrammed and compared in parallel. However, limited access to patients for a particular disease may prevent this. Therefore, analyses of patient iPSCs exhibiting representative clinical phenotypes and mutations, as well as gene correction experiments, are paramount for establishing disease validity. In addition, differentiation to cell types of interest can be limited based upon available protocols. While well- established protocols are available for various neural and cardiac tissues, specific methods may need to be developed for a particular target cell type [15, 66–68].
Concerns have also been raised regarding the potential of iPSCs to continually reflect the epigenetic status of somatic cells used in reprogramming. Tissue-specific methylation status may not always be fully corrected during reprogramming, persists through extended passaging, and impacts differentiation to cell types of interest [69–71]. While more recent studies have better controlled for inter-individual genetic variation and concluded epigenetic effects due to cell type of origin are likely less significant than previously believed, the possibility should still be considered if multiple somatic cell types are available for reprogramming experiments [72].
While the ability to model disease within a given genetic background is often considered advantageous, it can also lead to variability within models in differentiation and functionality. Parallel reprogramming of closely related, heterozygote carriers, as well as genome editing to produce gene-corrected or gene-edited isogeneic cell lines, has been employed to generate appropriate controls to infer mutation-specific effects [73]. Efforts to generate repositories of human iPSCs may also aid in accounting for variability within disease models, as well as generating human leukocyte antigen-matched, immunocompatible and transplantable cell types for replacement therapies [74].
3. Use of CRISPR/genome editing for rare diseases
Advances in genome editing have further expanded the impact of iPSC technology. Targeted double strand breaks (DSB) can be introduced in a site-specific manner, followed by DNA repair via non-homologous end-joining (NHEJ) or homology directed repair (HDR). Error-prone NHEJ is often used to disrupt gene function by introducing insertions and deletions (indels), while HDR utilizes sequence homology flanking the DSB site to knock-in reporters or other sequences of interest. The relative frequency of DNA repair between these two mechanisms has been estimated around 20–50% and 0.5–20% for NHEJ and HDR respectively, and is particularly low in transfection resistant cell lines including iPSCs [75]. To enhance the frequency of HDR mechanisms, regulation of the cell cycle, NHEJ inhibitor SCR7, and HDR enhancer RS-1 have been shown to improve knock-in efficiency up to 5-fold [76–78].
The utilization of the clustered regularly-interspaced short palindromic repeat (CRISPR)/Cas9 system within iPSC systems has helped mainstream the generation of genetic modifications in iPSCs. Previously used nuclease-based targeting methods, such as zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) require labor intensive manipulations and design methodology, while also limiting genomic targeting sites in comparison to CRISPR [79]. First discovered as a prokaryotic defense system against integration of foreign DNA from bacteriophages and plasmids, the power of CRISPR-associated genes to selectively and efficiently alter genomic sequences was quickly realized [80, 81]. Briefly, the Cas9 endonuclease is guided to a region of interest by a 20 base pair single-guide RNA complementary to the genomic region adjacent a targeted protospacer adjacent motif (PAM) site. Variants of the Cas9 which cleave only one DNA strand (nickase) have been utilized in a paired fashion to increase the specificity of DSB and reduce off-targeting changes [82].
As previously mentioned, the use of genome editing is particularly important in generating isogenic controls during disease modeling, particularly for rare diseases with limited sample availability. For diseases in which animal models are not predictive of clinical efficacy, validation of iPSC results through isogenic controls are critical experiments [83]. Two genome editing assays have most commonly been employed to control for genetic variability beyond a gene of interest: gene correction of disease-associated genes in patient-derived iPSCs or introduction of mutations within wild-type iPSCs [84]. Several rare diseases have been modeled in this fashion, including rescue of cholesterol metabolism in Niemann-Pick disease, type C1 [85]. Many publications to date using human iPSC models have generated isogenic, mutation-corrected cell lines utilizing a ‘safe-harbor’ based targeting approach. This strategy has most commonly involved insertion of a normal complementary DNA sequence within the adeno-associated virus site 1 (AAVS1) locus through HDR. For a detailed discussion of characterized safe harbor sites amenable for integration of DNA sequences, please see the following review article [86].
Recent work has also begun to address the potential use of the CRISPR/Cas9 system in germline editing. Ideally, this would allow genetic correction of deleterious human mutations within affected embryos prior to disease initiation, as has been demonstrated in Duchenne muscular dystrophy mice [87]. However, unlike in the mouse, published studies editing human embryos have exhibited mixed results. While studies using triponuclear zygotes demonstrated CRISPR/Cas9 protein could induce cleavage and HDR at separate genomic loci, off-target nuclease activity, mosaicism and poor HDR efficiency were encountered [88, 89]. Recently published work utilized co-injection of CRISPR/Cas9 and sperm to M-phase oocytes, demonstrating significantly attenuated mosaicism and improved gene correction in zygotes through maternal DNA transfer [90]. However, the unclear mechanisms allowing HDR to occur, potential off-target DNA cleavages by the Cas9 protein, loci-specific effects, non-targeting of all embryos and ethical questions regarding whether germline editing is even necessary given the ability to screen for mutant embryos during in vitro fertilization procedures remain open questions regarding the applicability of CRISPR/Cas9 germline editing in rare diseases.
4. Future directions for application of iPSCs in rare disease research
In addition to modeling of pathogenesis and functional deficits in rare diseases, advances in iPSC technology have potentially broad applications in drug discovery, toxicity testing, and clinical applications in the treatment of human disease.
4.1 Use of iPSC derivatives in drug discovery and therapeutic screening
Drug discovery has been plagued in recent years by poor translation of in vitro results and animal study findings into the clinic. Within the rare diseases, animal models may not accurately reflect patient phenotypes. For example, a transgenic mouse model overexpressing the transcription factor TDP-43 commonly used in ALS to model neurodegeneration was found to exhibit reduced gastrointestinal motility and decrease longevity independent of the central nervous system [91]. iPSCs have been utilized as a novel screening platform to develop more accurate predictions during drug development. These advancements have been particularly useful in defining cellular responses resulting from neurological and cardiac dysfunction in rare diseases due to having clearly defined genomic or functional deficits [15, 25, 66, 92, 93]. While not currently amenable to high-throughput assay development due to time and cost, utilization of differentiated organoids for validation of lead compounds in spatially organized tissues of interest will serve as an important screening tool to complement, or serve as an alternative, to animal models [94].
Predicting drug toxicity within the context of diverse genetic backgrounds is another advantage of iPSC models. iPSCs derived hepatocyte-like cells generated from patients with various polymorphisms in cytochrome p450 enzymes reliably predicted differences in pharmacokinetics compared to primary hepatic cells [95]. The importance of accurately interpreting drug responsiveness and metabolism to minimize adverse reactions is especially true of rare diseases, where the sporadic population base limits comprehensive drug trials. Patients may be left to rely upon case reports of related syndromes for therapeutic decisions regarding their care. Moreover, the incentive for drug development by pharmaceutical companies is generally unattractive considering a lack of overall market share. However, the Orphan Drug Act of 1983 has raised the FDA approval share for rare conditions to around 35% [2]. The use of iPSCs to augment drug discovery specifically within rare and multifactorial diseases holds much promise, lending itself to precision medicine allowing individualized safety and efficacy analysis. Additionally, iPSC-based screening methods for rare conditions can repurpose previously approved FDA drugs to fast-track treatment for unique applications. Advances in the use of iPSCs in drug discovery and high-throughput screening techniques have been expertly reviewed previously [84, 96].
4.2 Use of iPSC derivatives for clinical applications
Considerable regulatory guidelines and safety profiling is needed before clinical applications of iPSC based therapies can be realized. Cell therapy and transplantation studies must take into consideration potential complications of the specific cell type to be targeted. Regenerative studies have largely been limited to mesenchymal stem cell derivatives, many of which have failed to demonstrate clear mechanisms of actions and poor engraftment [97]. The use of patient-derived iPSCs for rare conditions, particularly in combination with advancements in genome editing capabilities, holds potential for transplantation therapies with more definable outcomes.
Before use in clinical applications, the risk of integration of foreign DNA into iPSC derivatives during the reprogramming progress is an issue of serious consideration. The use of retroviral or lentiviral reprogramming, which have been historically used to induce pluripotency, require integration into the genome and may lead to undesired mutations and aberrant effects on gene expression. Integration-free methods, including episomal plasmids, non-integrating Sendai viruses, modified RNAs, and recombinant proteins have been used for integration-free reprogramming [98–101]. More recently, small molecules have also been used exclusively to chemically induce mouse pluripotency [99]. While no reprogramming method has yet gained consensus within the iPSC field or regulatory agencies, these paradigm shifts toward chemical-based approaches and avoidance of integrating factors in reprogramming techniques will help alleviate some of the safety concerns in the downstream clinical application of iPSC derivatives in rare diseases. In addition, robust pre-clinical data regarding iPSC differentiation protocols and cellular outcomes, administration route and dose, and established natural history studies to predict expected disease progression in the absence of cell therapy should also be well defined. Finally, limited patient availability for clinical trials and the typical absence of control subjects requires the use of imaginative internal controls when available to assist in outcome measures. For example, an ongoing clinical trial for ALS is utilizing human neural progenitors transplanted unilaterally within the lumbar spinal region to assess secondary outcome measures [102].
5. Conclusions
Induced pluripotent stem cell technology offers unique advantages in the modeling of rare diseases. Multiple studies have demonstrated iPSCs carrying patient-specific mutations can recapitulate functional and cellular phenotypes observed in patients. Modeling of rare diseases with iPSCs has also led to novel breakthroughs in understanding the pathogenesis of some rare disorders, leading to clinical trials with pharmaceuticals and cell replacement therapies. Further technological advancements to allow greater discernment of disease processes and the development of new iPSC models will only expand the basic research and clinical applications of iPSCs in the context of rare diseases.
Highlights.
Rare diseases constitute an unmet medical need with few therapeutic options
Induced pluripotent stem cells (iPSCs) represent a new model system to study rare diseases
iPSCs allow mechanistic analysis and evaluation of possible therapeutics
Combining CRISPR/Cas9 gene editing and improved differentiation will improve iPSC disease modeling
Acknowledgments
The authors would like to thank Sanford Research and the Department of Pediatrics in the Sanford School of Medicine at the University of South Dakota for their support.
FUNDING
This work was supported by the National Institutes of Health [grant numbers P20GM103620 and P20GM103548].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de la Paz MP, et al. Rare diseases epidemiology research. Adv Exp Med Biol. 2010;686:17–39. doi: 10.1007/978-90-481-9485-8_2. [DOI] [PubMed] [Google Scholar]
- 2.Melnikova I. Rare diseases and orphan drugs. Nat Rev Drug Discov. 2012;11(4):267–8. doi: 10.1038/nrd3654. [DOI] [PubMed] [Google Scholar]
- 3.EURODIS - Rare Diseases Europe [Internet]. Paris (France); [updated 2014 Sep; cited 2017 Dec 22]. Available from: https://www.eurordis.org/sites/default/files/publications/Fact_Sheet_RD.pdf.
- 4.Bartlett JR, et al. Genetic modifiers of liver disease in cystic fibrosis. Jama. 2009;302(10):1076–83. doi: 10.1001/jama.2009.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kochanek Kenneth D, MA, Murphy Sherry L, BS, Xu Jiaquan, MD, Tejada-Vera B. Deaths: Final Data for 2014. National Vital Statistics Reports. 2016;65(4) [PubMed] [Google Scholar]
- 6.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 8.Campbell KH, et al. Sheep cloned by nuclear transfer from a cultured cell line. Nature. 1996;380(6569):64–6. doi: 10.1038/380064a0. [DOI] [PubMed] [Google Scholar]
- 9.Gurdon JB. The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. J Embryol Exp Morphol. 1962;10:622–40. [PubMed] [Google Scholar]
- 10.Chester N, et al. Stage-specific apoptosis, developmental delay, and embryonic lethality in mice homozygous for a targeted disruption in the murine Bloom’s syndrome gene. Genes Dev. 1998;12(21):3382–93. doi: 10.1101/gad.12.21.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamlin RL, Altschuld RA. Extrapolation from mouse to man. Circ Cardiovasc Imaging. 2011;4(1):2–4. doi: 10.1161/CIRCIMAGING.110.961979. [DOI] [PubMed] [Google Scholar]
- 12.Morizane A, et al. Direct comparison of autologous and allogeneic transplantation of iPSC-derived neural cells in the brain of a non-human primate. Stem Cell Reports. 2013;1(4):283–92. doi: 10.1016/j.stemcr.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mandai M, et al. Autologous Induced Stem-Cell-Derived Retinal Cells for Macular Degeneration. N Engl J Med. 2017;376(11):1038–1046. doi: 10.1056/NEJMoa1608368. [DOI] [PubMed] [Google Scholar]
- 14.Davis RP, et al. Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation. 2012;125(25):3079–91. doi: 10.1161/CIRCULATIONAHA.111.066092. [DOI] [PubMed] [Google Scholar]
- 15.Lee G, et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature. 2009;461(7262):402–6. doi: 10.1038/nature08320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bedada FB, Wheelwright M, Metzger JM. Maturation status of sarcomere structure and function in human iPSC-derived cardiac myocytes. Biochim Biophys Acta. 2016;1863(7 Pt B):1829–38. doi: 10.1016/j.bbamcr.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hrvatin S, et al. Differentiated human stem cells resemble fetal, not adult, beta cells. Proc Natl Acad Sci U S A. 2014;111(8):3038–43. doi: 10.1073/pnas.1400709111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Byers B, et al. SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate alpha-synuclein and are susceptible to oxidative stress. PLoS One. 2011;6(11):e26159. doi: 10.1371/journal.pone.0026159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vera E, Bosco N, Studer L. Generating Late-Onset Human iPSC-Based Disease Models by Inducing Neuronal Age-Related Phenotypes through Telomerase Manipulation. Cell Rep. 2016;17(4):1184–1192. doi: 10.1016/j.celrep.2016.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller JD, et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell. 2013;13(6):691–705. doi: 10.1016/j.stem.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu GH, et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature. 2011;472(7342):221–5. doi: 10.1038/nature09879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeRosa BA, et al. Derivation of autism spectrum disorder-specific induced pluripotent stem cells from peripheral blood mononuclear cells. Neurosci Lett. 2012;516(1):9–14. doi: 10.1016/j.neulet.2012.02.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marchetto MC, et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol Psychiatry. 2017;22(6):820–835. doi: 10.1038/mp.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bundo M, et al. Increased l1 retrotransposition in the neuronal genome in schizophrenia. Neuron. 2014;81(2):306–13. doi: 10.1016/j.neuron.2013.10.053. [DOI] [PubMed] [Google Scholar]
- 25.Malan D, et al. Human iPS cell model of type 3 long QT syndrome recapitulates drug-based phenotype correction. Basic Res Cardiol. 2016;111(2):14. doi: 10.1007/s00395-016-0530-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Giorgio FP, et al. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10(5):608–14. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCracken KW, et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature. 2014;516(7531):400–4. doi: 10.1038/nature13863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spence JR, et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature. 2011;470(7332):105–9. doi: 10.1038/nature09691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antonica F, et al. Generation of functional thyroid from embryonic stem cells. Nature. 2012;491(7422):66–71. doi: 10.1038/nature11525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takebe T, et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature. 2013;499(7459):481–4. doi: 10.1038/nature12271. [DOI] [PubMed] [Google Scholar]
- 31.Dye BR, et al. In vitro generation of human pluripotent stem cell derived lung organoids. Elife. 2015;4 doi: 10.7554/eLife.05098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eiraku M, et al. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature. 2011;472(7341):51–6. doi: 10.1038/nature09941. [DOI] [PubMed] [Google Scholar]
- 33.Koehler KR, et al. Generation of inner ear sensory epithelia from pluripotent stem cells in 3D culture. Nature. 2013;500(7461):217–21. doi: 10.1038/nature12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suga H, et al. Self-formation of functional adenohypophysis in three-dimensional culture. Nature. 2011;480(7375):57–62. doi: 10.1038/nature10637. [DOI] [PubMed] [Google Scholar]
- 35.Lancaster MA, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501(7467):373–9. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stevens KR, et al. Scaffold-free human cardiac tissue patch created from embryonic stem cells. Tissue Eng Part A. 2009;15(6):1211–22. doi: 10.1089/ten.tea.2008.0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stevens KR, et al. Physiological function and transplantation of scaffold-free and vascularized human cardiac muscle tissue. Proc Natl Acad Sci U S A. 2009;106(39):16568–73. doi: 10.1073/pnas.0908381106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith Q, et al. Stochasticity and Spatial Interaction Govern Stem Cell Differentiation Dynamics. Sci Rep. 2015;5:12617. doi: 10.1038/srep12617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bagley JA, et al. Fused cerebral organoids model interactions between brain regions. Nat Methods. 2017;14(7):743–751. doi: 10.1038/nmeth.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Birey F, et al. Assembly of functionally integrated human forebrain spheroids. Nature. 2017;545(7652):54–59. doi: 10.1038/nature22330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carlessi L, et al. Functional and molecular defects of hiPSC-derived neurons from patients with ATM deficiency. Cell Death Dis. 2014;5:e1342. doi: 10.1038/cddis.2014.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trilck M, et al. Niemann-Pick type C1 patient-specific induced pluripotent stem cells display disease specific hallmarks. Orphanet J Rare Dis. 2013;8:144. doi: 10.1186/1750-1172-8-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vessoni AT, et al. Cockayne syndrome-derived neurons display reduced synapse density and altered neural network synchrony. Hum Mol Genet. 2016;25(7):1271–80. doi: 10.1093/hmg/ddw008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Long Y, et al. Induced Pluripotent Stem Cells for Disease Modeling and Evaluation of Therapeutics for Niemann-Pick Disease Type A. Stem Cells Transl Med. 2016;5(12):1644–1655. doi: 10.5966/sctm.2015-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moretti A, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363(15):1397–409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- 46.Bellin M, Greber B. Human iPS cell models of Jervell and Lange-Nielsen syndrome. Rare Dis. 2015;3(1):e1012978. doi: 10.1080/21675511.2015.1012978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang M, et al. Recessive cardiac phenotypes in induced pluripotent stem cell models of Jervell and Lange-Nielsen syndrome: disease mechanisms and pharmacological rescue. Proc Natl Acad Sci U S A. 2014;111(50):E5383–92. doi: 10.1073/pnas.1419553111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fink JJ, et al. Disrupted neuronal maturation in Angelman syndrome-derived induced pluripotent stem cells. Nat Commun. 2017;8:15038. doi: 10.1038/ncomms15038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chamberlain SJ, et al. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc Natl Acad Sci U S A. 2010;107(41):17668–73. doi: 10.1073/pnas.1004487107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burnett LC, et al. Induced pluripotent stem cells (iPSC) created from skin fibroblasts of patients with Prader-Willi syndrome (PWS) retain the molecular signature of PWS. Stem Cell Res. 2016;17(3):526–530. doi: 10.1016/j.scr.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang J, et al. Induced pluripotent stem cells can be used to model the genomic imprinting disorder Prader-Willi syndrome. J Biol Chem. 2010;285(51):40303–11. doi: 10.1074/jbc.M110.183392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang K, et al. Selective demethylation and altered gene expression are associated with ICF syndrome in human-induced pluripotent stem cells and mesenchymal stem cells. Hum Mol Genet. 2014;23(24):6448–57. doi: 10.1093/hmg/ddu365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chamberlain SJ, et al. Modeling Genomic Imprinting Disorders Using Induced Pluripotent Stem Cells. Methods Mol Biol. 2016;1353:45–64. doi: 10.1007/7651_2014_169. [DOI] [PubMed] [Google Scholar]
- 54.Becherel OJ, et al. A new model to study neurodegeneration in ataxia oculomotor apraxia type 2. Hum Mol Genet. 2015;24(20):5759–74. doi: 10.1093/hmg/ddv296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koch P, et al. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature. 2011;480(7378):543–6. doi: 10.1038/nature10671. [DOI] [PubMed] [Google Scholar]
- 56.Brault J, et al. Therapeutic effects of proteoliposomes on X-linked chronic granulomatous disease: proof of concept using macrophages differentiated from patient-specific induced pluripotent stem cells. Int J Nanomedicine. 2017;12:2161–2177. doi: 10.2147/IJN.S128611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Itier JM, et al. Effective clearance of GL-3 in a human iPSC-derived cardiomyocyte model of Fabry disease. J Inherit Metab Dis. 2014;37(6):1013–22. doi: 10.1007/s10545-014-9724-5. [DOI] [PubMed] [Google Scholar]
- 58.Liu J, et al. Generation of induced pluripotent stem cell lines from Friedreich ataxia patients. Stem Cell Rev. 2011;7(3):703–13. doi: 10.1007/s12015-010-9210-x. [DOI] [PubMed] [Google Scholar]
- 59.Lopez-Onieva L, et al. Induced pluripotent stem cells derived from Bernard-Soulier Syndrome patient’s peripheral blood cells with a p.Phe55Ser mutation in the GPIX gene. Stem Cell Res. 2017;20:10–13. doi: 10.1016/j.scr.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 60.Romero-Moya D, et al. Genetic Rescue of Mitochondrial and Skeletal Muscle Impairment in an Induced Pluripotent Stem Cells Model of Coenzyme Q10 Deficiency. Stem Cells. 2017;35(7):1687–1703. doi: 10.1002/stem.2634. [DOI] [PubMed] [Google Scholar]
- 61.Tulpule A, et al. Pluripotent stem cell models of Shwachman-Diamond syndrome reveal a common mechanism for pancreatic and hematopoietic dysfunction. Cell Stem Cell. 2013;12(6):727–36. doi: 10.1016/j.stem.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aguisanda F, et al. Neural stem cells for disease modeling of Wolman disease and evaluation of therapeutics. Orphanet J Rare Dis. 2017;12(1):120. doi: 10.1186/s13023-017-0670-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun Y, et al. Properties of neurons derived from induced pluripotent stem cells of Gaucher disease type 2 patient fibroblasts: potential role in neuropathology. PLoS One. 2015;10(3):e0118771. doi: 10.1371/journal.pone.0118771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Uusi-Rauva K, et al. Induced Pluripotent Stem Cells Derived from a CLN5 Patient Manifest Phenotypic Characteristics of Neuronal Ceroid Lipofuscinoses. Int J Mol Sci. 2017;18(5) doi: 10.3390/ijms18050955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hu BY, et al. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc Natl Acad Sci U S A. 2010;107(9):4335–40. doi: 10.1073/pnas.0910012107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lahti AL, et al. Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture. Dis Model Mech. 2012;5(2):220–30. doi: 10.1242/dmm.008409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marchetto MC, et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143(4):527–39. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pasca SP, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. 2011;17(12):1657–62. doi: 10.1038/nm.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim K, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467(7313):285–90. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ohi Y, et al. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nat Cell Biol. 2011;13(5):541–9. doi: 10.1038/ncb2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim K, et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat Biotechnol. 2011;29(12):1117–9. doi: 10.1038/nbt.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Burrows CK, et al. Genetic Variation, Not Cell Type of Origin, Underlies the Majority of Identifiable Regulatory Differences in iPSCs. PLoS Genet. 2016;12(1):e1005793. doi: 10.1371/journal.pgen.1005793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Merkle FT, Eggan K. Modeling human disease with pluripotent stem cells: from genome association to function. Cell Stem Cell. 2013;12(6):656–68. doi: 10.1016/j.stem.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 74.Nakajima F, Tokunaga K, Nakatsuji N. Human leukocyte antigen matching estimations in a hypothetical bank of human embryonic stem cell lines in the Japanese population for use in cell transplantation therapy. Stem Cells. 2007;25(4):983–5. doi: 10.1634/stemcells.2006-0566. [DOI] [PubMed] [Google Scholar]
- 75.Li S, et al. Efficient generation of hiPSC neural lineage specific knockin reporters using the CRISPR/Cas9 and Cas9 double nickase system. J Vis Exp. 2015;99:e52539. doi: 10.3791/52539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Song J, et al. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat Commun. 2016;7:10548. doi: 10.1038/ncomms10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maruyama T, et al. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol. 2015;33(5):538–42. doi: 10.1038/nbt.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin S, et al. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. 2014;3:e04766. doi: 10.7554/eLife.04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vasileva EA, et al. Genome-editing tools for stem cell biology. Cell Death Dis. 2015;6:e1831. doi: 10.1038/cddis.2015.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32(4):347–55. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482(7385):331–8. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- 82.Ran FA, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154(6):1380–9. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McNeish J, et al. From Dish to Bedside: Lessons Learned While Translating Findings from a Stem Cell Model of Disease to a Clinical Trial. Cell Stem Cell. 2015;17(1):8–10. doi: 10.1016/j.stem.2015.06.013. [DOI] [PubMed] [Google Scholar]
- 84.Suh W. A new era of disease modeling and drug discovery using induced pluripotent stem cells. Arch Pharm Res. 2017;40(1):1–12. doi: 10.1007/s12272-016-0871-0. [DOI] [PubMed] [Google Scholar]
- 85.Maetzel D, et al. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells. Stem Cell Reports. 2014;2(6):866–80. doi: 10.1016/j.stemcr.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sadelain M, Papapetrou EP, Bushman FD. Safe harbours for the integration of new DNA in the human genome. Nat Rev Cancer. 2011;12(1):51–8. doi: 10.1038/nrc3179. [DOI] [PubMed] [Google Scholar]
- 87.Long C, et al. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science. 2014;345(6201):1184–1188. doi: 10.1126/science.1254445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kang X, et al. Introducing precise genetic modifications into human 3PN embryos by CRISPR/Cas-mediated genome editing. J Assist Reprod Genet. 2016;33(5):581–588. doi: 10.1007/s10815-016-0710-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liang P, et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell. 2015;6(5):363–372. doi: 10.1007/s13238-015-0153-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ma H, et al. Correction of a pathogenic gene mutation in human embryos. Nature. 2017;548(7668):413–419. doi: 10.1038/nature23305. [DOI] [PubMed] [Google Scholar]
- 91.Hatzipetros T, et al. C57BL/6J congenic Prp-TDP43A315T mice develop progressive neurodegeneration in the myenteric plexus of the colon without exhibiting key features of ALS. Brain Res. 2014;1584:59–72. doi: 10.1016/j.brainres.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 92.Sinnecker D, et al. Modeling long-QT syndromes with iPS cells. J Cardiovasc Transl Res. 2013;6(1):31–6. doi: 10.1007/s12265-012-9416-1. [DOI] [PubMed] [Google Scholar]
- 93.Shcheglovitov A, et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503(7475):267–71. doi: 10.1038/nature12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bredenoord AL, Clevers H, Knoblich JA. Human tissues in a dish: The research and ethical implications of organoid technology. Science. 2017;355(6322) doi: 10.1126/science.aaf9414. [DOI] [PubMed] [Google Scholar]
- 95.Takayama K, et al. Prediction of interindividual differences in hepatic functions and drug sensitivity by using human iPS-derived hepatocytes. Proc Natl Acad Sci U S A. 2014;111(47):16772–7. doi: 10.1073/pnas.1413481111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ko HC, Gelb BD. Concise review: drug discovery in the age of the induced pluripotent stem cell. Stem Cells Transl Med. 2014;3(4):500–9. doi: 10.5966/sctm.2013-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Trounson A, DeWitt ND. Pluripotent stem cells progressing to the clinic. Nat Rev Mol Cell Biol. 2016;17(3):194–200. doi: 10.1038/nrm.2016.10. [DOI] [PubMed] [Google Scholar]
- 98.Yu J, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324(5928):797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hou P, et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science. 2013;341(6146):651–4. doi: 10.1126/science.1239278. [DOI] [PubMed] [Google Scholar]
- 100.Woltjen K, et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458(7239):766–70. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fusaki N, et al. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85(8):348–62. doi: 10.2183/pjab.85.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Deng N, et al. Dishevelled interacts with p65 and acts as a repressor of NF-kappaB-mediated transcription. Cell Res. 2010;20(10):1117–27. doi: 10.1038/cr.2010.108. [DOI] [PubMed] [Google Scholar]
- 103.Yusa K, et al. Targeted gene correction of alpha1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478(7369):391–4. doi: 10.1038/nature10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dimos JT, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321(5893):1218–21. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- 105.Ho R, et al. ALS disrupts spinal motor neuron maturation and aging pathways within gene co-expression networks. Nat Neurosci. 2016;19(9):1256–67. doi: 10.1038/nn.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pini J, et al. Modeling Andersen’s Syndrome in Human Induced Pluripotent Stem Cells. Stem Cells Dev. 2016;25(2):151–9. doi: 10.1089/scd.2015.0258. [DOI] [PubMed] [Google Scholar]
- 107.Flynn R, et al. CRISPR-mediated genotypic and phenotypic correction of a chronic granulomatous disease mutation in human iPS cells. Exp Hematol. 2015;43(10):838–848 e3. doi: 10.1016/j.exphem.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen IP, et al. Induced pluripotent stem cell reprogramming by integration-free Sendai virus vectors from peripheral blood of patients with craniometaphyseal dysplasia. Cell Reprogram. 2013;15(6):503–13. doi: 10.1089/cell.2013.0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hashem SI, et al. Impaired mitophagy facilitates mitochondrial damage in Danon disease. J Mol Cell Cardiol. 2017;108:86–94. doi: 10.1016/j.yjmcc.2017.05.007. [DOI] [PubMed] [Google Scholar]
- 110.Raya A, et al. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature. 2009;460(7251):53–9. doi: 10.1038/nature08129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cai J, et al. Induced Pluripotent Stem Cells to Model Human Fibrodysplasia Ossificans Progressiva. Stem Cell Reports. 2015;5(6):963–70. doi: 10.1016/j.stemcr.2015.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hu L, et al. Modeling Glanzmann thrombasthenia using patient specific iPSCs and restoring platelet aggregation function by CD41 overexpression. Stem Cell Res. 2017;20:14–20. doi: 10.1016/j.scr.2017.02.003. [DOI] [PubMed] [Google Scholar]
- 113.Meneghini V, et al. Generation of Human Induced Pluripotent Stem Cell-Derived Bona Fide Neural Stem Cells for Ex Vivo Gene Therapy of Metachromatic Leukodystrophy. Stem Cells Transl Med. 2017;6(2):352–368. doi: 10.5966/sctm.2015-0414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tiscornia G, et al. Neuronopathic Gaucher’s disease: induced pluripotent stem cells for disease modelling and testing chaperone activity of small compounds. Hum Mol Genet. 2013;22(4):633–45. doi: 10.1093/hmg/dds471. [DOI] [PubMed] [Google Scholar]
- 115.Panicker LM, et al. Gaucher iPSC-derived macrophages produce elevated levels of inflammatory mediators and serve as a new platform for therapeutic development. Stem Cells. 2014;32(9):2338–49. doi: 10.1002/stem.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rabenstein M, et al. Decreased calcium flux in Niemann-Pick type C1 patient-specific iPSC-derived neurons due to higher amount of calcium-impermeable AMPA receptors. Mol Cell Neurosci. 2017 doi: 10.1016/j.mcn.2017.06.007. [DOI] [PubMed] [Google Scholar]
- 117.Nevin ZS, et al. Modeling the Mutational and Phenotypic Landscapes of Pelizaeus-Merzbacher Disease with Human iPSC-Derived Oligodendrocytes. Am J Hum Genet. 2017;100(4):617–634. doi: 10.1016/j.ajhg.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shimojima K, et al. Reduced PLP1 expression in induced pluripotent stem cells derived from a Pelizaeus-Merzbacher disease patient with a partial PLP1 duplication. J Hum Genet. 2012;57(9):580–6. doi: 10.1038/jhg.2012.71. [DOI] [PubMed] [Google Scholar]
- 119.Sato Y, et al. Disease modeling and lentiviral gene transfer in patient-specific induced pluripotent stem cells from late-onset Pompe disease patient. Mol Ther Methods Clin Dev. 2015;2:15023. doi: 10.1038/mtm.2015.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sato Y, et al. Metabolomic Profiling of Pompe Disease-Induced Pluripotent Stem Cell-Derived Cardiomyocytes Reveals That Oxidative Stress Is Associated with Cardiac and Skeletal Muscle Pathology. Stem Cells Transl Med. 2017;6(1):31–39. doi: 10.5966/sctm.2015-0409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Garate Z, et al. Generation of a High Number of Healthy Erythroid Cells from Gene-Edited Pyruvate Kinase Deficiency Patient-Specific Induced Pluripotent Stem Cells. Stem Cell Reports. 2015;5(6):1053–66. doi: 10.1016/j.stemcr.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lukovic D, et al. Human iPSC derived disease model of MERTK-associated retinitis pigmentosa. Sci Rep. 2015;5:12910. doi: 10.1038/srep12910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ramsden CM, et al. Rescue of the MERTK phagocytic defect in a human iPSC disease model using translational read-through inducing drugs. Sci Rep. 2017;7(1):51. doi: 10.1038/s41598-017-00142-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Francis KR, et al. Modeling Smith-Lemli-Opitz syndrome with induced pluripotent stem cells reveals a causal role for Wnt/beta-catenin defects in neuronal cholesterol synthesis phenotypes. Nat Med. 2016;22(4):388–96. doi: 10.1038/nm.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ebert AD, et al. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 2009;457(7227):277–80. doi: 10.1038/nature07677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sareen D, et al. Inhibition of apoptosis blocks human motor neuron cell death in a stem cell model of spinal muscular atrophy. PLoS One. 2012;7(6):e39113. doi: 10.1371/journal.pone.0039113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chailangkarn T, Muotri AR. Modeling Williams syndrome with induced pluripotent stem cells. Neurogenesis (Austin) 2017;4(1):e1283187. doi: 10.1080/23262133.2017.1283187. [DOI] [PMC free article] [PubMed] [Google Scholar]