

Abstract
Objective
Specification of endocrine cell lineages in the developing pancreas relies on extrinsic signals from non-pancreatic tissues, which initiate a cell-autonomous sequence of transcription factor activation and repression switches. The steps in this pathway share reliance on activity-dependent Ca2+ signals. However, the mechanisms by which phasic Ca2+ surges become converted into a dynamic, cell-state-specific and physiologically meaningful code made up by transcription factors constellations remain essentially unknown.
Methods
We used high-resolution histochemistry to explore the coincident expression of secretagogin and transcription factors driving β cell differentiation. Secretagogin promoter activity was tested in response to genetically manipulating Pax6 and Pax4 expression. Secretagogin null mice were produced with their pancreatic islets morphologically and functionally characterized during fetal development. A proteomic approach was utilized to identify the Ca2+-dependent interaction of secretagogin with subunits of the 26S proteasome and verified in vitro by focusing on Pdx1 retention.
Results
Here, we show that secretagogin, a Ca2+ sensor protein that controls α and β cell turnover in adult, is in fact expressed in endocrine pancreas from the inception of lineage segregation in a Pax4-and Pax6-dependent fashion. By genetically and pharmacologically manipulating secretagogin expression and interactome engagement in vitro, we find secretagogin to gate excitation-driven Ca2+ signals for β cell differentiation and insulin production. Accordingly, secretagogin−/− fetuses retain a non-committed pool of endocrine progenitors that co-express both insulin and glucagon. We identify the Ca2+-dependent interaction of secretagogin with subunits of the 26S proteasome complex to prevent Pdx1 degradation through proteasome inactivation. This coincides with retained Nkx6.1, Pax4 and insulin transcription in prospective β cells.
Conclusions
In sum, secretagogin scales the temporal availability of a Ca2+-dependent transcription factor network to define β cell identity.
Keywords: β cell, Ca2+ signaling, Diabetes, Differentiation
Highlights
-
•
Pax4/6 control secretagogin expression during pancreas development.
-
•
Ca2+-dependent interaction of secretagogin with 26S proteasome prevents Pdx1 degradation.
-
•
Secretagogin regulates a transcriptional program required for β cell specification.
1. Introduction
Cellular integrity in Langerhans islets and the functional properties of endocrine cells are programmed during embryonic and early postnatal development and rely on the sequential activation of transcription factors. First, pancreatic and duodenal homeobox 1 (Pdx1) marks pancreatic progenitors, giving rise to both endocrine and exocrine lineages [1]. Subsequently, the Pdx1-driven transient surge of neurogenin 3 (Ngn3) promotes cell fate restriction for endocrine progenitors [2]. The timing and extent of Ngn3 expression are critical for the cellular programming of all endocrine lineages, because its loss results in the complete absence of α, β, δ, and PP cells [2]. Ngn3-dependent expression of paired neuronal differentiation 1 (NeuroD1) leads to the differentiation of pancreatic endocrine cells [3]. The Ngn3-induced endocrine program relies, in large part, on the sustained presence of Pdx1, since expression of NK6 homeobox 1 (Nkx6.1), paired box 6 (Pax6), and paired box 4 (Pax4) is controlled by both Ngn3 and Pdx1 [4], [5], [6], [7], [8]. Coincident and coordinated Pdx1, NeuroD1, Nkx6.1, Pax4, and Pax6 transcriptional activity trigger differentiation of progenitor cells, first towards pancreatic endocrine cells that often co-express both glucagon and insulin [9] and then to differentiated β cells as long as ‘master regulating’ Pdx1 is retained [6]. Pax4-mediated repression of aristaless-related homeobox (Arx) ensures mutually exclusive insulin and glucagon expression in β and α cells, respectively [10]. Lack of either transcription factor will result in Langerhans islets devoid of the corresponding mature endocrine lineage [2], [10]. As such, mutations in the Pdx1 gene are associated with ‘maturity onset diabetes of young’, type 4 (MODY4) and type 2 diabetes [11], while mutations in NeuroD1 and Pax4 are respectively linked to MODY6 [12] and MODY9 in humans [13].
Extracellular cues, mostly derived from mesenchyme activins, fibroblast growth factors (FGFs), and Notch and Hedgehog ligands, shape the spatio-temporal expression of transcription factors to drive specification of endocrine progenitors towards committed α, β, δ, and PP cell fates and their subsequent clonal expansion [14], [15], [16]. Considering the detailed knowledge of both differentiation cues and transcriptional programs, there is, surprisingly, a gap in understanding the cellular events linking signal initiation to transcriptional effectors. Remarkably, activity of these pathways shares reliance on Ca2+ signals [15], [17]. In many cellular niches, chronospecific transients in intracellular Ca2+ control the transcriptional regulation of cell differentiation, proliferation, and apoptosis [17], [18], [19]. However, the mechanisms by which Ca2+ signals are converted into a dynamic and physiologically meaningful code to orchestrate transcription factors constellations in pancreatic endocrine progenitors is elusive. Intracellular Ca2+ oscillations are matched with molecular effectors by Ca2+-sensor proteins, which, upon Ca2+ binding, undergo conformational changes for the phasic activation (or inactivation) of their specific binding partners via protein–protein interactions [20], [21]. Secretagogin (Scgn), a member of the EF-hand Ca2+-sensor superfamily [22], [23], is purportedly the most abundant Ca2+-sensor in pancreatic islets. Recently, secretagogin was identified downstream from excitatory receptors and ion channels [23], [24] and implicated in the regulation of α and β cell proliferation and survival by modulating protein turnover [23], [25]. Even though secretagogin is expressed in fetal pancreas from the inception of lineage segregation [20], [26], its functional significance for endocrine differentiation remains unknown.
Here, we first profiled co-expression patterns of secretagogin and transcription factors crucial for progenitor differentiation in fetal pancreas during secondary transition (that is from embryonic day (E)12.5 to birth, when a massive differentiation wave and lineage allocation occurs) [27]. Next, we combined in silico promoter analysis, luciferase-based promoter activity profiling and biochemistry to show that Scgn expression is antagonistically regulated by Pax4 and Pax6. Once expressed in endocrine pancreatic progenitors, Scgn inhibits proteasome activity upon Ca2+ influx through differentiation-promoting excitatory ion channels, such as TRPV1 [22]. Thereby, Scgn coordinates the retention of Pdx1, thus also tuning its transcriptional targets: Nkx6.1, Pax4, and insulin (Ins). Collectively, Scgn is identified as the first Ca2+-sensor, indispensable to specify β cells and establish a physiologically favored α-to-β cell ratio in Langerhans islets.
2. Materials & methods
2.1. Cell lines
INS-1E cells [28] were cultured at 37 °C in RPMI-1640 medium supplemented with glutamine (2 mM), glucose (11 mM), HEPES (10 mM), heat-inactivated fetal bovine serum (FBS; 5%), sodium pyruvate (1 mM), β-mercaptoethanol (50 μM), penicillin (50 μg/ml), and streptomycin (100 μg/ml). Cells were routinely sub-cultured in 24-well plates up to passage 120 and allowed to reach ∼80% confluence. HEK293T cells (ATCC) were cultured at 37 °C in DMEM supplemented with FBS (5%), penicillin (50 μg/ml), and streptomycin (100 μg/ml). Cells were routinely sub-cultured in 24-well plates and also allowed to reach ∼80% confluence.
2.2. Molecular pharmacology
The effect of (E)-N-[(4-Hydroxy-3-methoxyphenyl)methyl]-8-methyl-6-nonenamide (capsaicin; TRPV1 agonist, 300 nM), N-[2-(4-Chlorophenyl)ethyl]-1,3,4,5-tetrahydro-7,8-dihydroxy-2H-2-benzazepine-2-carbothioamide capsazepine; TRPV1 antagonist, 10 μM), (2R,3S,4R)-3-Hydroxy-2-[(1S)-1-hydroxy-2-methylpropyl]-4-methyl-5-oxo-2-pyrrolidinecarboxy-N-acetyl-l-cysteine thioester (lactacystin; proteasome inhibitor, 5 μM) on mRNA expression and Pdx1 and Ngn3 protein levels was determined.
2.3. Plasmids and transfection
Knock-down of Scgn and Pax4 expression was achieved by the co-application of GIPZ-shRNA constructs carrying shScgn-Ires-EGFP or shPax4-Ires-EGFP, respectively (both 1 μg/ml; Dharmacon) with jetPRIME transfection reagent (Polyplus-transfection SA) for 48 h. GIPZ Lentiviral Empty Vector shRNA carrying EGFP (Dharmacon) was used as a control. Pax6 was over-expressed by transfection of cells with a pmPAX6 construct (gift from M. Busslinger, Research Institute of Molecular Pathology, Vienna, Austria) [29]. A CMV-Ngn3-Ires2-eGFP construct was obtained by cloning mouse Ngn3 (with XhoI and BamhI restriction sites and a Kozak-like sequence near the translation initiation codon attached during PCR) into a pIRES-EGFP vector (Addgene) between its XhoI and BamHI restriction sites. CMV-Scgn constructs were generated by site-directed excision of Ngn3 from a custom-designed CMV-Ngn3-Ires2-Scgn vector. Briefly, Scgn cDNA was amplified from mouse brain total RNA by RT-PCR and cloned into a pCloneJet vector. For cloning, a BsrGI recognition site was added to its 3′ end (the 5′-end of the fragment had a NcoI site: CCATGG-contains the translation initiation sequence). Then, eGFP was replaced by Scgn in the initially constructed CMV-Ngn3-Ires2-eGFP plasmid. Subsequently, we modified the CMV-Ngn3-Ires2-Scgn plasmid by replacing its XhoI-BclI fragment (which contained Ngn3 cDNA, Ires2 and the first 124 bp of Scgn cDNA) with a PCR fragment carrying the complementary (missing) part of Scgn cDNA. To create a Ngn3-luciferase construct, the Ngn3 promoter (∼5,9 kb region up to the endogenous SmaI site) upstream of the ATG was cloned from the C57Bl/6 mouse genome into a pGL3-Basic vector (containing firefly luciferase). All constructs were verified by sequencing. For transfection both in INS-1E and HEK293T cells, all constructs were used at 1 μg/ml concentration together with jetPRIME transfection reagent (Polyplus-transfection SA) and cells were analyzed 48 h after transfection.
2.4. Secretagogin−/− mice
Secretagogin−/– (Scgn−/–) mice [30] were custom-generated at MMRRC (University of California, Davis, Mouse Biology Program) using the “two-in-one” targeting strategy [31], which generates full knock-outs by expressing a termination signal after exon 3 of the secretagogin gene. The ensuing truncated protein terminates before the first EF-hand domain, excluding Ca2+-binding activity. All animals (including Trpv1−/− mice, Jackson #3770) were kept under standard housing conditions with a 12h/12h light/dark cycle and food and water available ad libitum. Experiments on live animals conformed to the European Communities Council Directive (2010/63/EU) and were ethically approved (66.009/0145-WF/II/3b/2014) and regulated by applicable local laws (Tierversuchsgesetz 2012, BGBI, Nr. 114/2012, Austria). Particular effort was directed towards minimizing the number of animals used and their suffering during experiments.
2.5. In silico transcription factor binding site prediction and firefly luciferase assay
A 1,400-bp fragment (−1,400 to +1) of the 5′ flanking region of the murine (NC_000079.6) Scgn gene was analyzed for potential transcription factor binding sites using the open-label PROMO software (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_6.4) [23]. For activity-determination of the murine Scgn promoter, its 1,400-bp 5′ flanking region was custom-synthesized (Eurofins Genomics) and cloned into a pGL3-Basic vector containing a firefly luciferase cassette (Promega) as previously described [23]. Subsequently, INS-1E cells cultured in 96-well plates (50,000 cells/well) were co-transfected with this firefly luciferase construct, pCMV-green Renilla luciferase (ThermoFisher; as control) and either a GIPZ shRNA Pax4 construct or a pmPax6 construct using jetPRIME transfection reagent (Polyplus-transfection SA). GIPZ Lentiviral Empty Vector shRNA served as negative control. The effect of ectopic secretagogin expression (CMV-Scgn) on the activity of the murine Ngn3 promoter was assessed in HEK293 cells cultured in 96-well plates (50,000 cells/well). Cells were co-transfected with custom designed Ngn3-firefly luciferase construct (Ngn3-Luc), pCMV-green Renilla luciferase vector (ThermoFisher; as control) and CMV-Scgn. Forty eight h later, firefly and Renilla luciferase luminescence was measured using the Dual-Luciferase® Reporter Assay System (Promega) on a GloMax® luminometer (Promega). Data were expressed as the ratio of firefly-to-Renilla luciferase luminescence intensity and normalized to values from unstimulated control cells.
2.6. RNA isolation and gene expression analysis
Total RNA from INS-1E cells and embryonic pancreata (E13.5) was isolated using the Aurum Total RNA Mini Kit (Bio-Rad) followed by DNase digestion and verification of RNA integrity on 2% agarose gels (500 ng RNA). cDNA was prepared by reverse transcription with random primers using a High-capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative PCR analysis (CFX Connect, BioRad) was performed using 50 ng of cDNA template, iTaq Universal SYBR Green Supermix (BioRad) and specific primer pairs (Table S3).
2.7. Western blotting
Proteins were extracted from INS-1E and HEK293 cells using a radioimmunoprecipitation assay buffer containing (in mM): 50 Tris (pH 7.4), 150 NaCl, 10 NaF, 5 EDTA (pH 8.0), 1 Na3VO4, 1 PMSF, 1% TX-100, pepstatin A (5 μg/μl), leupeptin (10 μg/μl), and aprotinine (2 μg/μl). Protein lysates were centrifuged at 12,000 g at 4 °C for 10 min. For the analysis of total protein levels, lysates were pre-incubated with carbocyanine (Cy) 5 labelling buffer (GE Healthcare) for 10 min. Samples were denatured in loading buffer (GE Healthcare) and boiled at 95 °C for 5 min. Proteins were probed by loading 20 μg aliquots under denaturing conditions on SDS-PAGE (13.5% or 8–18% gradient gels), followed by wet transfer onto PVDF membranes. Primary antibodies were listed in Table S4. After exposure to Cy3-or Cy5-conjugated secondary antibodies (GE Healthcare, 1:2,500; 1h), target proteins were visualized using an automated Amersham WB system (GE Healthcare).
2.8. Proteasome activity assay
Proteasome activity in INS-1E cell homogenates (50 μg) was measured for 30 min with Proteasome Activity Assay Kit (Abnova) according to the manufacturer's instructions 48 h after either overexpression or knock-down of Scgn.
2.9. Tissue preparation, immunocytochemistry and immunohistochemistry
For immunocytochemistry, cells were plated on 12-mm coverslips coated with poly-d-lysine (0.001%), washed in 0.1 M phosphate buffer (PB, pH 7.4) and immersion fixed in 4% paraformaldehyde (PFA) for 20 min. Mice were transcardially perfused with ice-cold 0.1 M PB, followed by 4% PFA in PB (100 ml at 3 ml/min flow speed). Pancreata were rapidly dissected and post-fixed in 4% PFA overnight. Embryonic (E13.5) pancreata were dissected and immersion fixed in 4% PFA for 3 h. After equilibrating in 30% sucrose for 48–72 h, tissues were cryosectioned at a thickness of 14 μm (P0 torsos) or 8 μm (embryonic pancreata; Leica CM1850) and thaw-mounted onto SuperFrost+ glass slides. Antigen retrieval was performed prior to immunostaining by incubating sections in citrate buffer (10 mM, pH 6.0) supplemented with Tween-20 (0.5%) at 95 °C for 5 min. After rinsing in 0.1M PB, specimens were exposed to a blocking solution composed of 0.1M PB, 10% normal donkey serum, 5% bovine serum albumin (BSA) and 0.3% TX-100 for 3 h followed by overnight incubation with select combinations of primary antibodies (Table S4). Cy2,3 or 5-conjugated secondary antibodies (1:300, Jackson ImmunoResearch) were applied in 0.1M PB supplemented with 2% BSA (20–22 °C, 2 h). Nuclei were routinely counterstained with Hoechst 33,342 (1:20,000; Sigma). Cells and pancreas sections were imaged on a Zeiss LSM880 laser-scanning microscope equipped with a Plan-Apochromat 20x/air objective at 1.0–2.5x optical zoom. Images were acquired in the ZEN2010 software package. Quantitative analysis was performed using ImageJ1.45 with appropriate plug-ins. Multi-panel images were assembled in CorelDraw X7 (Corel Corp.).
2.10. Pull-down assay
Sixty μg of His6-tagged secretagogin [23] and embryonic (E13.5) pancreas (n = 36 from 5 litters) homogenate (300 μg of protein) were mixed in binding buffer (pH 7.4) containing (in mM): 25 Tris–HCl, 300 NaCl, 0.1 CaCl2, 10 imidazole to obtain 100 μl total sample volume, and incubated together with 40 μl of pre-equilibrated Ni-NTA beads in Spin-X Centrifuge Tube Filter 0.22 μm cellulose acetate columns (Costar) at 20–22 °C for 30 min. In control experiments, His6-tagged secretagogin was replaced with 60 μg of BSA. Flow-through fractions were removed after spinning at 1,500 g for 30 s. Beads were washed (3x) with 100 μl buffer (in mM): 25 Tris–HCl, 300 NaCl, 0.1 CaCl2, 50 imidazole. Proteins were eluted with 70 μl elution buffer composed of (in mM): 25 Tris–HCl, 300 NaCl, 0.1 CaCl2, and 300 imidazole. Samples were stored at −80 °C until processing for mass spectrometry.
2.11. LC-MS/MS analysis of protein complexes, bioinformatics
Filter-aided sample preparation (FASP), LC-MS/MS in-line analysis on an Ultimate 3000 nanoRSLC system (ThermoFisher) coupled to a Thermo Q-Exactive Plus mass spectrometer (ThermoFisher), database search via the open-source software MaxQuant 1.5.3.30 [32] and statistical processing using the Perseus statistical package (version 1.5.4.1) of His6-secretagogin (n = 3) and control pull-downs (n = 3) were carried out as described previously [23] with minor modifications. Proteins were identified against the UniProt mouse (Mus musculus) reference proteome database (as of October 2017; 60,177 entries). Secretagogin pull-downs were compared to control samples via two-tailed Student's t-test applying permutation-based FDR equal to 0.01. Significant hits identified by at least two peptides and matching the criteria of the minimum fold-change (s0 = 2) in the target pull-down were considered as specific for secretagogin.
2.12. Statistics
All experiments were performed in triplicate unless stated otherwise. Data from overexpression and silencing experiments in INS-1E cells were analysed using one/two-way ANOVA followed by pair-wise comparisons where appropriate. Student's t-test (independent group design) was used to statistically evaluate data on pancreas morphology, qPCR-based and immunohistochemical analysis of transcription factor expression. Data were expressed as means ± s.d. Fold changes represent the percentage change from the mean control value in individual experiments. A p value of <0.05 was considered statistically significant.
2.13. Accession numbers
The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD009516.
3. Results
3.1. Pax4/6 controls secretagogin expression during pancreas development
To determine a possible role for Scgn in the regulation of cell fate progression in endocrine pancreas, we first analyzed its co-expression pattern with that of a set of transcription factors relevant for the differentiation of pancreatic progenitors. We investigated mouse pancreata at the beginning of the secondary transition, when the differentiation of endocrine progenitors and their allocation to distinct cell lineages take place [27]. At E13.5, Scgn is present in a circumscribed subset of Ngn3+ endocrine progenitors (4 ± 1%; Figure 1A,F). Significantly, many more Nkx6.1+ and NeuroD1+ progenitors, which label both committed endocrine precursors and immature β cells during this developmental stage [3], [33], were Scgn+ (31 ± 4% and 39 ± 10%, respectively; Figure 1B–C,F). At the same time, we detected Scgn in the majority of Arx+ cells (78 ± 14%; Figure 1D,F), as well as in nearly all Pax6+ endocrine cells (85 ± 6%, Figure 1E–F). Even though Pax4 is expected to be present in endocrine progenitors at E13.5 [34] and, like Pax6, could co-exist with Scgn, we were unable to confirm this given the lack of reliable Pax4 antibodies for multiple labelling histochemistry. Nevertheless, our present data suggest that Scgn expression coincides with progenitor commitment to specific endocrine lineages, and is progressively increased upon β cell maturation.
Figure 1.

Pax4 and Pax6 control secretagogin levels during embryonic pancreas development. (A–E) Co-immunolabeling of secretagogin (Scgn) and transcription factors crucial for the development of the endocrine pancreas in E13.5 mouse fetuses. Secretagogin was detected in a subset of Ngn3+ cells even if the majority of Ngn3+ cells did not co-express Scgn (A). Notably, most secretagogin+ progenitors belonged to the pool of either Nkx6.1+ (B) or NeuroD1+ (C) cells with more than half of Nkx6.1+/NeuroD1+ progenitors being Scgn− (B, C). Arx, marking α cell fate, was largely co-expressed with Scgn (D). Likewise, Scgn immunoreactivity was found in almost all Pax6+ cells (E). Hoechst 33,342 (pseudo-coloured in grey) was used as nuclear counterstain. Scale bars = 10 μm or 4 μm (for numbered inserts). Representative images are shown. (F) Quantitative analysis of Scgn co-expression with Ngn3, Nkx6.1, NeuroD1, Arx and Pax6 in embryonic pancreas. The percentage ratio of Scgn+ and transcription factor (TF)+ dual-labelled cells was expressed by using the total number of TF+ cells as denominator. n > 100 cells from n = 3 pancreata were counted. (G) Schematic timeline of TF levels crucial for β cell specification. Cell differentiation, maturation, and maintenance of β cell identity are underpinned by the transcriptional activity of Nkx6.1, NeuroD1, Pax4, and Pdx1 [6]. Pancreas development commences at ∼ E9 with expression of Pdx1 in pancreatic progenitors [1]. Subsequently, Pdx1-driven transient presence of Ngn3 promotes development of endocrine progenitors [2]. While Nkx6.1 expression represses pre-acinar cell fate [33], Ngn3-dependent NeuroD1 activity facilitates of the differentiation of pancreatic endocrine cells [3]. This is then followed by Ngn3-and Pdx1-mediated expression of Nkx6.1, Pax6, and Pax4[4], [5], [6], [7], [8]. Coincident Pdx1, NeuroD1, Nkx6.1, Pax4, and Pax6 transcriptional activity establishes the cohort of pancreatic endocrine cells, which co-express glucagon and insulin [9]. High Pax4 activity represses glucagon expression, thus promoting β cell fate [10]. Secretagogin (Scgn) appears in the endocrine pancreas at the beginning of the secondary transition (∼E13), when α vs. β cell fate decisions are dictated by TFs. (H-H1) In silico prediction of Pax4 and Pax6 TF binding sites within the murine secretagogin promoter (up to −1,400 bps). (H1) Overexpression of Pax6 (pmPax6), as well as Pax4 knock-down (shPax4) significantly increase Scgn promoter activity in INS-1E cells. Promoter activity was defined as a ratio of firefly-to-Renilla luciferase chemiluminescence. (I) Pax6 overexpression and shRNA-mediated silencing of Pax4 (shPax4) increase Scgn mRNA levels in vitro. TATA-binding protein (Tbp), a house keeping gene, was used to normalize gene expression. (J–J1) Pax6 overexpression and shPax4 increase secretagogin protein content in INS-1E cells. Quantitative data reflect fold changes that had been normalized to total protein content. (K) Schema of the upstream regulation of secretagogin (Scgn) expression. Pdx1 initially expressed in pancreatic progenitors drives Ngn3 expression to promote differentiation of pancreatic endocrine progenitors. Subsequent expression and opposing actions of Pax4 and Pax6 in these cells control Scgn level. Representative images and immunoblots are shown. Data were expressed as means ± s.d. from triplicate experiments. ***p < 0.001, **p < 0.01, *p < 0.05 calculated with pair-wise comparisons/one-way ANOVA.
Yet, Pax4 and Pax6 expression, reported as early as E9.5, precedes that of Scgn (Figure 1G) [26], [34], [35]. Therefore, we hypothesized that Pax4 and/or Pax6 could control Scgn transcription, as they do for numerous endocrine-specific genes, including glucagon (Gcgn), insulin (Ins), somatostatin (Sst), and pancreatic polypeptide (Ppy) [36], [37]. We have previously analyzed the mouse Scgn promoter (−1,400 to +1) in silico for Sp1 binding sites [23]. Here, we show that it also contains multiple consensus sequences for Pax4 and Pax6 binding (Figure 1H, Table S1). Pax6 is known to act as a transcriptional activator [38], whereas its homolog, Pax4, represses gene transcription [39] (Figure 1K). To confirm their involvement in the regulation of Scgn transcription, we have generated a reporter construct in which the mouse Scgn promoter (Scgn-full, its 1,400 bps 5′ flanking region [23]) drives firefly luciferase. Overexpression of Pax6 (Figure 1A–B1) or knock-down of Pax4 expression (Fig. S1C) in INS-1E cells significantly increased Scgn promoter activity measured as the ratio of firefly-to-Renilla luciferase luminescence (both p < 0.001 vs. control; Figure 1H1). Increased Scgn mRNA (p < 0.05 [shPax4] and p < 0.01 [Pax6] vs. control; Figure 1I) and protein levels (p < 0.05 [shPax4] and p < 0.05 [Pax6] vs. control; Figure 1J,J1) were interpreted as Pax4-mediated repression and Pax6-mediated enhancement of Scgn transcription and translation in vitro (Figure 1K).
3.2. Secretagogin is required for the expression of β cell-specific transcription factors
Considering i) Scgn's role in β cell fate determination [23], ii) α cell hyperplasia in islets of adult Scgn−/– mice [23], iii) the relevance of Ca2+ signals for pancreas organogenesis [40] and iv) early expression of Scgn in fetal pancreas (Figure 1A–G) [26], we hypothesised that Scgn could contribute to defining the size and transition of endocrine pancreatic progenitor pools. First, we took advantage of Scgn−/– mouse embryos [30] to define the effect of Scgn loss-of-function on endocrine cell differentiation. To distinguish different endocrine progenies and their developmental trajectories, we performed co-immunolabeling of transcription factor pairs relevant for endocrine commitment and fate progression at E13.5 (Figure 2A–E1). While we did not observe any change in the total number of Pdx1+ cells (Figure 2A,A1,F), the number of Ngn3+ progenitors substantially decreased in Scgn−/– pancreata (p < 0.05; 52 ± 20 [Scgn−/–] vs. 71 ± 6 cells per section [wild-type]; Figure 2A,A1,F). Moreover, Scgn−/– pancreata were characterized by reduced Ngn3 immunoreactivity in resident cells (p < 0.05; Figure 2H), even though Ngn3 mRNA expression remained unaffected (Figure 2I). Concomitantly, the number of Ngn3+/Pdx1+ progenitors decreased significantly in Scgn−/– mice (p < 0.05; 34 ± 15 [Scgn−/–] vs. 60 ± 7 cells per section [wild-type]; Figure 2F).
Figure 2.

Impaired differentiation of pancreatic endocrine progenitors in Scgn−/−fetuses. (A–E1) Immunohistochemical analysis of embryonic (E13.5) pancreata of Scgn−/– and wild-type littermates reveals significant differences in the expression patterns of transcription factors (TFs) regulating endocrine pancreas development. Reduced immunoreactivity for Ngn3 in significantly less Ngn3 (positive)+ progenitors is seen in Scgn−/– pancreata (A–A1). Additionally, Scgn−/– fetuses contain less NeuroD1+ (B–B1) and Nkx6.1+ (C–C1) endocrine progenitors. This is compatible with the reduced ratio of insulin+-to-NeuroD1+ cells (D–D1). The number of Arx+ cells is comparable between Scgn−/– and wild-type pancreata. Nevertheless, the ratio of cells expressing glucagon and also immunolabelled for Arx is increased in Scgn−/– mice (E–E1). (F) Even though the number of Pdx1+ progenitors in E13.5 pancreata of Scgn−/– mice remains unchanged, significantly less Ngn3+ cells are produced. This also decreases the number of Ngn3+/Pdx1+ progenitors in Scgn−/– mice relative to their wild-type littermates. (G–G1) Quantitative analysis of pancreatic cells at consecutive stages of differentiation towards committed α and β cell lineages in Scgn−/– and wild-type pancreata at E13.5. The likelihood of finding NeuroD1+ or Nkx6.1+ cells that had co-expressed Pdx1 in Scgn−/– mice is significantly lower than in wild-type littermates, suggesting impaired β cell differentiation. Notwithstanding, the ratio of Arx+ α cell precursors to Pdx1+ cells remains unchanged (G). (G1) Impaired allocation of endocrine progenitors to the β cell lineage is reinforced by the reduced ratio of insulin+/NeuroD1+ cells in Scgn−/– mice. As such, the increased proportion of glucagon (Gcgn)+/Arx+ cells implies favoured α cell fate choices in Scgn−/– mice. (H) Reduced immunoreactivity of nuclear Pdx1, Ngn3, and Nkx6.1 (but not NeuroD1 and Arx) in E13.5 pancreata of Scgn−/– mice as compared to wild-type littermates. (I) Ngn3, NeuroD1, and Pax6 mRNA levels were unchanged in E13.5 pancreata of Scgn−/– mice relative to wild-type littermates. In Scgn−/– pancreata, mRNA levels of most β cell-specific genes (Pax4, Nkx6.1, Pdx1 and Ins were significantly down-regulated, while Arx and Gcgn, α cell-specific genes, increased. Tbp mRNA was used to normalize gene expression. Representative images are shown. Hoechst 33,342 was used as nuclear counterstain (pseudo-coloured in grey). Scale bars = 25 μm and 10 μm (inserts). Quantitative data from triplicate experiments were expressed as means ± s.d.; n ≥ 3 pancreata/genotype; n ≥ 100 cells/genotype (G); ***p < 0.001, **p < 0.01, *p < 0.05 (Student's t-test).
We hypothesized that co-expression of Ngn3 and Scgn demarcates a short-lived developmental interface when progenitor cells transit towards specific endocrine lineages. Conceivably, Scgn regulation of Ngn3 would be poised to facilitate terminal differentiation of Ngn3+ endocrine progenitors. To define the molecular basis of any such regulatory interplay, we used HEK293T cells and measured the effect of ectopic Scgn over-expression (CMV-Scgn) on Ngn3 promoter activity, as well as ensuing Ngn3 protein levels when co-expressing CMV-Scgn and CMV-Ngn3-IRES2-eGFP constructs (CMV-Ngn3). Scgn did not affect Ngn3 promoter activity (Fig. S2A). Yet, we observed significantly increased Ngn3 protein levels, as if disrupting protein degradation by the proteasome inhibitor, lactacystin (p < 0.05; Figure 2B–B2). We have previously linked Scgn to the regulation of protein turnover in adult β cells [23] Therefore, we infer these data as Scgn stabilizing Ngn3 either via affecting its post-translational modification or by interacting with the protein degradation machinery. Concurrently, this observation implies either the premature differentiation of Ngn3+ cells resulting from Ngn3 loss [41] or impaired lineage commitment of Ngn3+ progenitors in the absence of Scgn [42].
Impaired specification of Scgn−/– pancreatic progenitors towards the β cell lineage is further supported by the decreased ratio of NeuroD1+/Pdx1+ and Nkx6.1+/Pdx1+ cells (p < 0.01 and p < 0.01 respectively; Figure 2B–C1,G1). As for the transcription factors themselves, Nkx6.1, but neither NeuroD1 protein (p < 0.01; Figure 2H) nor mRNA content (p < 0.05; Figure 2I), was significantly reduced in Scgn−/– mice relative to those in wild-type pancreata, pointing to deregulation at the transcriptional level. Likewise, Pdx1 immunoreactivity (p < 0.01; Figure 2H) and Pdx1 and Pax4 mRNA expression (p < 0.001 and p < 0.05 respectively; Figure 2I) were reduced in Scgn−/– pancreata. Reduced levels of these β cell-specific transcription factors [6], [10], [33], [43] coincided with a significant reduction of Ins mRNA expression (p < 0.05; Figure 2I).
Scgn−/– mice show α cell hyperplasia in adulthood. Therefore, we have reason to believe that impaired β cell development during the intrauterine period might give rise to more α cells. Indeed, Arx and Gcgn mRNAs were up-regulated in Scgn−/– pancreata (both p < 0.001; Figure 2I). Even though neither the number of Arx+ cells nor their Arx immunoreactivity changed per se upon Scgn deletion (Figure 2E–E1,G,H), an increased ratio of Gcgn+/Arx+ cells (p < 0.05; Figure 2E–E1,G1) suggests premature transition towards glucagon-containing α cells. In sum, our data indicate that manipulating Scgn levels in endocrine progenitors shifts the balance of α/β cell fate decisions with Scgn loss leading to disrupting β cell differentiation.
3.3. Secretagogin loss-of-function in vivo stalls α-and-β lineage segregation
To seek further insights into Scgn's role in cell fate decisions of endocrine progenitors, we co-localized insulin and glucagon in E13.5 pancreas. Despite substantial changes in Ins and glucagon (Gcgn) mRNA expression (Figure 2I), absolute numbers of α/glucagon+ and β/insulin+ cells were not affected by Scgn loss-of-function (Figure 3A,B,E). Nevertheless, many more insulin+ cells also were glucagon+ in Scgn−/– fetal pancreata than in wild-type controls (p < 0.001; 48 ± 24% [Scgn−/–] vs. 6 ± 15% [wild-type] of total Ins+ cells; Figure 3A–A1,B–B1). This phenotype persisted in newborn (P0) Scgn−/- mice (p < 0.001; 8.4 ± 4% [Scgn−/–] vs. 0.9 ± 1% [wild-type] of total Ins+ cells; Figure 3C–C1,D-D1). By birth, significantly increased numbers of α cells (p < 0.01; Figure 3F) manifested in Scgn−/– mice. At the same time, we found reduced Pdx1/insulin co-expression in both E13.5 (p < 0.01; Figure 3A–B2) and P0 pancreas (p < 0.01; Figure 3C–D2), which is compatible with lesser Pdx1 mRNA expression and nuclear localization in Scgn−/– mice (Figure 2H,I). Cumulatively, these data suggest that Pdx1 loss in Scgn−/– progenitors facilitates α cell fate choices for precursors that had initially expressed both hormones in vivo.
Figure 3.

Secretagogin regulates the lineage commitment of pancreatic progenitors. (A–A2) In wild-type (Scgn+/+) mice, the co-expression of insulin (Ins+) and glucagon (Gcgn+) is unlikely (A1), with ∼50% of Ins+ cells co-expressing Pdx1 on E13.5. (B–B2) In Scgn−/– mice, however, ∼50% of Ins+ cells co-express Gcgn (B1) while losing Pdx1 (B2). (C–C2) By birth in wild-type (Scgn+/+) mice, the existence of Ins+/Gcgn+ cells is negligible (C1). The majority of Ins+ cells is Pdx1+, too. (D–D2) In newborn Scgn−/– mice, a contingent of Ins+/Gcgn+ cells remains (D1) together with ∼75% of Ins+ cells lacking Pdx1 (D2). (E) α and β cell numbers in E13.5 Scgn−/– pancreata are indistinguishable from those of wild-type littermates. (F) In newborn Scgn−/– mice, pancreatic islets contain many more α cells. (G) Proposed mechanism of Scgn's role in the control of α- or β-cell lineage. Scgn, when co-expressed with Pdx1, could regulate Pdx1 levels by either increasing Pdx1 transcription or stabilizing it. Consequently, Pdx1 can promote expression of β cell-specific genes (Pax4, Nkx6.1, insulin and Pdx1) while simultaneously repressing glucagon (Gcgn) expression. Additionally, Pax4 inhibits both Arx and Gcgn transcription to favour β cell identity. Representative images are shown. Hoechst 33,342 was used as nuclear counterstain (pseudo-coloured in grey). Scale bars = 15 μm and 8 μm (inserts) (B) and 10 μm and 3 μm (inserts) (D). Quantitative data from n ≥ 3 pancreata/genotype were expressed as means ± s.d.; cell ratios (A1, A2, B1, B2, C1, C2, D1, D2) were presented as percentages of the total number of Ins+ cells. **p < 0.01 (Student's t-test).
3.4. Pdx1 availability and transcriptional activity in β cells are controlled by intracellular Ca2+
Pdx1 is considered a ‘master regulator’ of early endocrine pancreas development [1] with its late expression being restricted to β cells [6]. In β cells, Pdx1 controls the transcription of many β cell-specific genes, most prominently Nkx6.1, Pax4 and Ins, as well as its own expression, thus being mandatory for β cell fate progression [4], [6], [7], [44], [45]. Here, we hypothesized that impaired cell fate decisions are due to the premature loss of Pdx1 during the second wave of its functional role, when Pdx1 and Scgn are co-expressed (Figure 3G). We used INS-1E cells, a β-cell model abundantly expressing Pdx1 (Figure 4A), to determine the effect of Scgn deficiency on Pdx1 protein levels. Using shScgn-EGFP, we silenced Scgn expression (Fig. S3) and determined nuclear Pdx1 localization in EGFP− vs. EGFP+ cells. Scgn silencing significantly reduced Pdx1 levels in Scgn-deficient EGFP+ INS-1E cells, as compared to EGFP− (mock-transfected) cells (p < 0.01; Figure 4A–A4). Retention of Pdx1 in mock-transfected cells (Figure 4A–A1,A4) rendered experimental bias through EGFP over-expression alone unlikely.
Figure 4.

TRPV1-mediated activation of Ca2+ signaling increases Pdx1 in pancreatic β cells. (A–A4) shRNA-mediated Scgn knock-down significantly reduces Pdx1 levels in INS-1E cells (EGFP+ cells; arrowheads), as compared to cells transfected with a mock construct (EGFP+; arrowheads). (A4) Quantitative analysis of Pdx1 signal intensity in INS-1E cells transfected with either a control construct or shScgn. Cells with successful transfection were selected by their EGFP fluorescence (arrowheads). Open arrowheads point to un-transfected cells. (B–B3) Capsaicin (TRPV1 agonist; 300 nM for 4 h) increases (B1) while capsazepine (TRPV1 antagonist; 10 μM for 4 h) decreases (B2) the level of nuclear Pdx1 immunoreactivity in INS-1E cells. (B3) Quantitative analysis of Pdx1 immunoreactivity in INS-1E cells probed pharmacologically. (C–C2) Capsaicin (300 nM for 4 h) failed to rescue Pdx1 reduced by Scgn knock-down in INS-1E cells (EGFP+ cells; arrowheads) relative to non-transfected cells (EGFP−). Representative images for control and shScgn group are shown in Fig. S4. (C2) Quantitative analysis of Pdx1 immunoreactivity in non-transfected (EGFP−) or shScgn-transfected (EGFP+) INS-1E cells upon capsaicin stimulation. (D–D2) Reduced Pdx1 immunoreactivity in pancreata from adult Trpv1−/− mice compared to wild-type littermates. (E–F) Under Ca2+-free conditions (6 h), Scgn overexpression (48 h) fails to increase either Pax4 (E) or Nkx6.1 (F) mRNA levels. Tbp mRNA was used to normalize gene expression. Representative images are shown. Open boxes reflect the general location of insets. Hoechst 33,342 was used as nuclear counterstain (pseudo-coloured in grey). Scale bars = 5 μm (A3, B2, C1), 10 μm (D1). Quantitative data from triplicate experiments were normalized to those in control and expressed as means ± s.d.; n ≥ 30 cells/group; ***p < 0.001, **p < 0.01, *p < 0.05 (pair-wise comparisons after two-way ANOVA or Student's t-test in D2).
As a Ca2+-sensor protein, Scgn could control Pdx1 levels in a phasic manner in response to transient surges in intracellular Ca2+. We have previously shown that TRP family members fine-tune Ca2+-dependent transcriptional elements in pancreatic β cells to promote Scgn expression and, consequently, increase β cell survival [23]. Moreover, we linked TRPV1 activity in developmental pancreas to the control of cell pool sizes in Langerhans islets [24]. To determine if Pdx1 protein levels are subject to changes in intracellular Ca2+, we applied capsaicin (TRPV1 agonist; 300 nM) or capsazepine (TRPV1 antagonist, 10 μM) to INS-1E cells and analysed Pdx1 expression. To minimize the possibility that TRPV1-driven induction of Scgn expression would bias the outcome of these experiments, we treated INS-1E cells with TRPV1 ligands for 4 h only, an interval known to be too short to affect the translation of Scgn [23]. Under these conditions, the intensity of nuclear Pdx1 immunoreactivity was significantly increased by capsaicin (p < 0.05; Figure 4B,B1,B3), whereas reduced by capsazepine (p < 0.05; Figure 4B,B2,B3). To define whether Scgn is causal to regulating Pdx1 levels upon agonist-induced TRPV1-mediated Ca2+ influx [23], [46], we applied capsaicin to INS-1E cells with shRNA-mediated Scgn silencing. Capsaicin failed to reinstate Pdx1 protein content, measured as the intensity of nuclear Pdx1 immunoreactivity brought about by Scgn silencing in EGFP+ cells (p < 0.05; Figure 4C–C2, Fig. S4). These data together with a substantial decrease in Pdx1 immunoreactivity in β cells of Trpv1−/− mice (vs. wild-type, p < 0.05; Figure 4D–D2) identify TRPV1 as a regulator of Pdx1 availability, and more broadly, Scgn to regulate Pdx1 availability in a Ca2+-dependent manner.
Next, we examined if genetically manipulating Scgn in INS-1E cells also affects the expression of genes downstream from Pdx1 and being relevant to acquiring and/or maintaining β cell identity; reminiscent to data from Scgn−/– fetal pancreata (Figure 2J). Indeed, Scgn knock-down (Fig. S3) significantly and coincidently reduced NeuroD1 (p < 0.01; Fig. S5A), Pax4 (p < 0.05; Fig. S5B), Nkx6.1 (p < 0.01; Fig. S5C), Pdx1 (p < 0.05; Fig. S5D) and Ins (p < 0.01; Fig. S5E) mRNA levels. Conversely, Scgn over-expression (CMV-Scgn; Fig. S3) up-regulated NeuroD1 (p < 0.01; S5A), Pax4 (p < 0.05; S5B), Nkx6.1 (p < 0.01; S5C), Pdx1 (p < 0.05; S5D) and Ins (p < 0.05; S5E). Moreover, Scgn-mediated enhancement of the expression of β-cell specific transcription factors relied on the presence of Ca2+, since neither Pax4 (Figure 4E) nor Nkx6.1 (Figure 4F) modulation was seen when Scgn was over-expressed in Ca2+-free conditions.
3.5. Secretagogin protects Pdx1 from proteasomal degradation
Given that Pdx1 availability is primarily regulated at the protein level via its phosphorylation, ubiquitination and subsequent proteasomal degradation [47], [48], we aimed to determine if and how Scgn could participate in regulating Pdx1 stability. In doing so, we first analysed Scgn's protein interactome in fetal pancreas. By using His6-tagged recombinant Scgn [23] in the presence of Ca2+ (100 μM), we isolated its putative interacting partners from E13.5 pancreata and captured their molecular identity by mass spectrometry. Interestingly, next to previously described interacting partners (participating in vesicle-mediated transport, cytoskeletal organization and members of chaperonin-containing complex [23]), we found 15 subunits of the 26S proteasome (Figure 5A, Table S2), making a case for Scgn-dependent modification of protein degradation. Regardless of whether this putative interaction was direct or indirect, Scgn silencing significantly increased (p < 0.01 vs. control; Figure 5B), while its overexpression decreased (p < 0.01 vs. control; Figure 5B) proteolytic activity of the proteasome when probing INS-1E cells.
Figure 5.

Secretagogin controls Pdx1 by inhibiting the 26S proteasome. (A) Secretagogin's molecular interactome at E13.5. Recombinant His6-tagged secretagogin was used as bait with data adhering to gene ontology (GO) classifiers. Bracketed numbers refer to the number of proteins per GO cluster. The identity, discovery rate and peptide fragmentation data for all interacting proteins from our LC-MS/MS analyses are listed in Table S2. (B) Scgn silencing by shRNA (48 h) increases, whereas Scgn overexpression brought about by CMV-Scgn (48 h) reduces proteasome activity. (C-C4) Proteasome inhibition by lactacystin (5 μM, 6 h) rescues Pdx1 in shScgn-transfected INS-1E cells (EGFP+). Representative images are shown. Hoechst 33,342 was used as nuclear counterstain (pseudo-colored in gray). Scale bars = 5 μm. (C4) Quantitative analysis of Pdx1 signal intensity in lactacystin-treated INS-1E cells transfected with either mock or shScgn constructs. EGFP (arrowheads) indicates successful transfection. Open arrowheads point to non-transfected cells. (D–D2) Proteasome inhibition by lactacystin (‘Lact.’, 5 μM, 6 h) rescues Pax4 (D) and Nkx6.1 (D1) expression levels otherwise down-regulated by shScgn. Tbp mRNA level was used to normalize gene expression. Quantitative data from triplicate experiments were normalized to those in control and expressed as means ± s.d.; n ≥ 30 cells/group; *p < 0.05 (pair-wise comparisons after two-way ANOVA).
To demonstrate that reduced Pdx1 levels upon Scgn loss-of-function are due to Scgn's interaction with subunit(s) of the 26S proteasome, we applied lactacystin (5 μM, 6 h) after silencing Scgn expression in INS-1E cells. Lactacystin, an irreversible proteasome inhibitor, rescued Pdx1 protein in shScgn transfected EGFP+ cells (p < 0.01; Figure 5C–C4).
Although, inhibition of proteasomal degradation per se can significantly affect gene expression [49], we performed another proof-of-principle experiment: after application of lactacystin we analyzed the mRNA levels of those Pdx1-controlled β cell-specific genes, which were down-regulated upon Scgn knock-down (Fig. S5). We determined the lactacystin-mediated rescue of Pax4 (p < 0.05 vs. [control shRNA + lactacystin] and p < 0.05 vs. [shScgn]; Figure 5D) and Nkx6.1 (p < 0.05; Figure 5D1). Overall, our data suggest that, in the presence of Ca2+, Scgn-mediated control of the proteasomal degradation of Pdx1 serves as a regulatory junction to allocate pancreatic endocrine progenitors to the β cell lineage.
4. Discussion
Our study identifies Scgn as a Ca2+ sensor instructing pancreas development through its control of the protein degradation machinery to initiate and propagate transcription factor cascades. In the context of cellular diversification of the endocrine pancreas, our findings are of appeal to integrate extracellular differentiation-promoting signals (‘community effect’), autocrine activity, and the time-locking of transcription factor availability for specific cell lineages to emerge. Moreover, we uncover a self-regulating expressional cycle with Pax4 and Pax6 positioned upstream to control Scgn expression. This can produce an additional level of precision between transcriptional codes for fate progression and tissue complexity defined as receptor-mediated Ca2+ signaling. We did not attempt to characterize either the exact Scgn fold structure or the 26S proteasome subunit mediating direct Scgn binding. Still, our simultaneous loss-of-function and rescue experiments unequivocally show that Scgn inhibits Pdx1 proteolysis in a Ca2+-dependent fashion. Thus, Scgn serves as a transcriptional ‘circuit breaker’ to (dis-)allow the propagation of the Pdx1-Nkx6.1-Ins cascade needed to produce mature β cells. Furthermore, our results are important to conceptualize switches in β-to-α cell identity associated with diabetes, inherently associated with Scgn deficiency [23].
No single cell in the body has a solitary Ca2+ sensor protein. Instead, compartmentalization/activity-dependent partitioning, differential affinity for Ca2+ binding, and protein-specific interactome segregate members Ca2 binding proteins functionally. More broadly, Scgn is a member of the EF-hand superfamily of Ca2+-binding protein with significant homology to calretinin and calbindin D28k [50]. Previous expression profiling of gene regulatory networks relevant for the development of the endocrine pancreas showed enrichment of the Ca2+-binding protein ALG2, a Ca2+-dependent activator (Cadps), as well as Ca2+/calmodulin-dependent protein kinase 2b (Camk2b), reinforcing the relative importance of Ca2+ signaling in endocrine diversification [40]. The reliance of stem and progenitor proliferation on Ca2+-mediated signaling events is exemplified by the temporal synchrony of intracellular Ca2+ operations in closely-situated ‘small world’ progenitor networks driving their differentiation [51], as well as determining the spatiotemporal pattern of Ca2+ oscillations in β cell network in adult [52]. Analogous to Scgn in the control of cell fate [53], [54], calcineurin also impacts β cell proliferation and survival in humans and rodents [53], [55], [56]. This is because upon Ca2+ influx, calcineurin's B subunit binds Ca2+ at its EF-hands, thereby changing conformation and losing its autoinhibitory activity. Subsequently, calmodulin binding to calcineurin's A subunit induces its dephosphorylase activity. Mice with β cell-specific deletion of calcineurin develop age-dependent type 2 diabetes due to loss of β cell mass [53]. Notably, over-expression of calmodulin in β cells is equally detrimental, probably because of the overt disruption of signal transduction cascades (e.g., ectopic activation of calmodulin-dependent kinases and phosphatases), manifesting as β cell loss and diabetes [57]. These findings from diabetic animal models [23], [58] thus support that Ca2+-binding proteins are required for β cell physiology. An element of novelty in our present data is that, unlike calcineurin and calmodulin that tune cell proliferation and apoptosis [56], [57], Scgn instead modulates the proper allocation of progenitor cells to specific endocrine cell lineages.
Even though Ca2+-sensors are fundamental for cellular physiology, their expressional regulation is unexpectedly understudied. A reason for this is that hundred(s) of Ca2+-binding proteins are predicted in the mammalian genome (through EF-hand homology domains in their open-reading frames; J. Mulder, personal communication), many awaiting functional characterization. For Scgn in β cells, promoter activation upon glucose priming has been shown [22]. However, the extent of promoter activation was insufficient to explain on-off switches in Scgn availability. Here, we show that antagonistic action of the homeobox and paired domain-containing transcription factors Pax4 and Pax6 efficiently gates Scgn transcription. Pax6 is broadly expressed in many cell lineages (except PP cells) of both fetal and adult pancreas [35]. Therefore, it can qualify as a favored transcriptional activator for the persistent positive control of Scgn transcription. In contrast, Pax4 expression, and consequently Pax4-mediated repression, is much more dynamic: Pax4 broadly marks endocrine progenitors before becoming restricted to the β/δ lineage [10] and extinguished in mature (healthy) β cells [37]. These spatial–temporal differences in Pax6 and Pax4 action suggest that Pax4 is a transcriptional lever ‘on-off’ regulating Scgn expression to tune the physiologically-favoured balance of α and β cells. In adult islets, Scgn is no longer subjected to Pax4-mediated inhibition (but maintained through Pax6-mediated transcriptional activation), thereby being steadily expressed to prime the SNARE exocytosis machinery for insulin secretion [22], [23].
Pancreatic organogenesis, on the one hand, relies on extrinsic signals of non-pancreatic origin (such as, growth factors, endocannabinoids, endovanilloids) [14], [16], [24] that prime stem cell proliferation and differentiation. On the other hand, the precise coupling of receptor-mediated, extracellular signal-evoked transduction events to cell-intrinsic transcription factor activation is required as a cell-intrinsic mechanism to broadcast and titrate the physiological efficacy extracellular differentiation cues. With a sequence of transcription factor switches operating during β cell development [2], [3], [4], [6], [10], it is of primary importance to limit transcription factor availability to specific temporal windows. Transcription factors that reside in the cytosol at non-stimulated conditions undergo fast nuclear import upon their activation by post-translational modifications, most commonly phosphorylation [59]. To ensure their short-lived action, proteolytic degradation is a fast and efficient strategy to cease transcription factor action [47], [60]. In β cells, Pdx1 and Ngn3 are subject to post-translational modifications that reduce their half-life [41], [47], [48]. Even so, molecular determinants instructing the capture and degradation of transcription factors by the proteasome are of significant appeal, since any such regulatory interplay likely integrates the temporal activation of more than one signaling cascade, thus being poised to drive cell-fate progression by switching-off transcription factors. One classical cytosolic signal integrator is Ca2+. Combinatorial activity of the many Ca2+-permeable channels expressed by β cells [23], [61], [62], [63] can rapidly increase intracellular Ca2+ with Ca2+ extrusion or its organellar store-operated sequestration [64], [65] tuning the brevity of Ca2+-dependent signal transduction. As one of the signaling arcs, Ca2+ regulates proteasome activity [66], [67]. To initiate signal transduction, Ca2+ binds Ca2+-binding proteins, which function either as buffers (with low nM Kd) or sensors (with μM Kd) [50]. Scgn is one such Ca2+ sensor with a calculated 25 μM Kd [68], thereby primed to respond only to substantial Ca2+ surges. Moreover, Scgn transcription itself is sensitive to Ca2+ influx through ligand-gated ion channels [23], which is a means to load β cells with the Ca2+ sensor, if and when needed. Here, we show that receptor-mediated Ca2+ transients (including TRPV1 [46]) are equally sufficient to mobilize pre-existing Scgn pools to engage their protein interactome [20] in developing β cells. Subunits of the 26S proteasome are a fate-determining domain of the Scgn interactome. This interaction, however, only occurs upon significant Ca2+ load. We suggest that activation of ligand- or voltage-gated Ca2+-permeable channels other than TRPV1 can use the regulatory principle shown here, thus qualifying Scgn as a central hub to control transcription factor availability crucial for the development of β cells and beyond (e.g., endocrine progenitors, neurons) [19], [40].
More specifically, Ca2+-bound Scgn interacts with components of 26S proteasome and inhibits its activity. The proteasome subunits we have identified as Scgn-interacting ones (Psmc1-5 and Pcmd1-6, Pcmd11, 12) mostly belong to the 19S regulatory complex. Accordingly, they are implicated in binding polyubiquitinated proteins and cleaving the ubiquitin chains off, thus mediating substrate recognition and processing prior to transport into the catalytic chamber of the 20S core [69], [70]. Previously, we have uncovered [23] that Scgn can also inhibit the enzymatic activity of USP7 and USP9X ubiquitin carboxyl-terminal hydrolases and, consequently, increase the stability of their substrates. By combining these data, we infer that Scgn participates in both the presentation and processing of polyubiquitinated proteins destined to degradation. Here, we support this hypothesis by focusing on the Scgn-mediated protection of Pdx1. However, other transcription factors involved in endocrine differentiation might well be processed through this pathway, Ngn3 being a prime example. Ngn3 expression in endocrine progenitors is transient (Figure 1G) to safe-keep the balance of proliferation and differentiation [41], which is due to the short half-life of this protein [60]. Here, we show that Ngn3 levels at E13.5 are post-translationally regulated. One might therefore speculate that Scgn also contributes to protecting (or facilitating) Ngn3 degradation. If so, by stabilizing Ngn3, Scgn could modulate the pool size of endocrine progenitors [41].
Overall, our study defines how a Ca2+-sensor protein can integrate upstream community- or activity-driven extracellular signals to facilitate commitment to, and progression of, the differentiation program required for β cell fate in fetal pancreas. These results suggest that modulating Ca2+-dependent signal transduction at signaling nodes more specific than calmodulin or calcineurin could effectively be used to prime β cell differentiation as part of preventive or restorative cell-replacement strategies in type 1 and 2 diabetes.
Funding statement
This work was supported by the Swedish Research Council (T.Ha., T. Hö.), Hjärnfonden (T.Ha.), the Novo Nordisk Foundation (K.M., T. Ha., T. Hö.), the European Research Council (‘Secret-Cells’, 2015-AdG-695,136; T. Ha.) and intramural funds of the Medical University of Vienna (T.Ha.).
Author contributions
K.M. and T. Ha. conceived the study design; G.L., T. Hö., and T. Ha. procured funding for experimental work; K.M., E.S., and R.S. performed experiments. R.S. also provided unique reagents. K.M. and T. Ha. wrote the manuscript, which was proof-read and approved by all authors.
Acknowledgements
The authors thank M. Busslinger (Research Institute of Molecular Pathology, Vienna, Austria) for providing the pmPAX6 construct, L. Wagner (University Clinic for Internal Medicine III, General Hospital Vienna, Austria) for providing anti-secretagogin antibody, Z. Máté for plasmid design and construction (Institute of Experimental Medicine, Hungarian Academy of Sciences, Budapest, Hungary), A. Gavalas and E. Rodriguez (Paul Langerhans Institute Dresden at the University Clinic Carl Gustav Carus of TU Dresden, Germany) for training in isolating and culturing embryonic pancreata, G. Tortoriello for help in generating secretagogin−/- mice and S. Kubicek (CeMM Research Centre for Molecular Medicine, Austrian Academy of Sciences, Vienna, Austria) for constructive discussions.
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.molmet.2018.05.019.
Conflict of interest
T.Ha. declares support from GW Pharmaceuticals on projects unrelated to the focus of this report. All other authors declare no conflict of interest.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
In silico transcription factor binding site prediction in the mouse Scgn promoter. Predicted Pax4 and Pax6 transcription factors binding sites within a 1,400-bp fragment (−1,400 to +1) of the 5′ flanking region of the murine Scgn gene (NC_000079.6). Analysis was performed by the open-source PROMO software.
Protein interactome for secretagogin when used as bait. Proteins precipitated with His6-tagged secretagogin as bait from homogenates of E13.5 wild-type mouse pancreata were identified by liquid chromatography-tandem mass spectrometry and functionally clustered by gene ontology (GO) profiling.
Primer sequences used in this study. qPCR reactions were performed with primer pairs amplifying short fragments for each gene. Primer pairs were custom-designed to efficiently anneal to homologous nucleotide sequences from mouse or rat.
List of antibodies used in this study. Commercially available primary antibodies were used for immunohistochemistry and Western blotting. Abbreviations: Arx, Aristaless-related homeobox; NeuroD1, Neuronal differentiation 1; Ngn3, Neurogenin 3; Nkx6.1, NK6 homeobox 1; Pax6, Paired box 6; Pdx1, Pancreatic and duodenal homeobox 1.
figs1.

Validation of Pax6 overexpression and Pax4 silencing in INS-1E cells. (A) Transfection of INS-1E cells with a construct carrying pmPax6 allowed for the successful overexpression of Pax6. Silencing of Pax4 (shPax4) did not affect Pax6 mRNA levels. (B–B1) Western blotting confirmed the overexpression of Pax6 with pmPax6. Silencing of Pax4 did not affect Pax6 protein levels. Cy5-labeling of the total protein load was used to normalize Pax6 data. Representative full-length immunoblot is shown. (C) A significant decrease in Pax4 mRNA expression 48 h after transfecting INS-1E cells with a construct delivering shPax4. Pax6 overexpression did not affect Pax4 levels. Tbp mRNA expression was used to normalize gene expression levels. Data were expressed as means ± s.d. from triplicate experiments, **p < 0.01, *p < 0.05 (one-way ANOVA).
figs2.

Secretagogin increases Ngn3 protein level without affecting Ngn3 promoter activity. (A) Ectopic expression of Scgn (CMV-Scgn; 48 h) in HEK293T cells does not affect the activity of the Ngn3 promoter. Promoter activity was defined as the ratio of firefly-to-Renilla luciferase chemiluminescence. (B–B1) HEK293T cells where transfected with Ngn3-expression constructs (CMV-Ngn3). Scgn co-expression (CMV-Scgn; 48 h) significantly increased Ngn3 protein levels as shown by Western blotting. Rescuing Ngn3 degradation by proteasome inactivation through lactacystin (5 μM, 6 h) was used as positive control. Representative immunoblot is shown. Total protein load (Cy5) was used to normalize Ngn3 protein levels. Data were expressed as means ± s.d. from triplicate experiments, *p < 0.05 (Student's t-test in A or one-way ANOVA in B–B1).2
figs3.

Validation of Pax6 overexpression and Pax4 silencing in INS-1E cells. (A) Significantly decreased Scgn mRNA expression was observed 48 h after transfection of INS-1E cells with a vector carrying shScgn. In contrast, transfection of INS-1E cells with CMV-Scgn (48 h) significantly increased Scgn mRNA content. (B–B1) Western blotting confirmed successful Scgn silencing and overexpression by shScgn and CMV-Scgn, respectively. Total protein load (Cy5) was used to normalize protein expression. Representative immunoblot is shown. Tbp mRNAs were used to normalize gene expression. Data were expressed as means ± s.d. from triplicate experiments, ***p < 0.001, *p < 0.05 (one-way ANOVA).3
figs4.
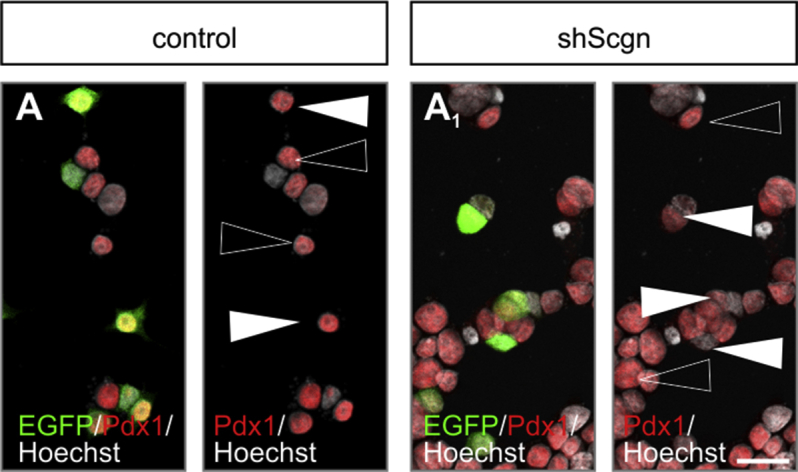
Secretagogin silencing reduces nuclear Pdx1 levels. (A–A1) shRNA-mediated Scgn silencing reduces nuclear Pdx1 immunoreactivity in INS-1E cells (EGFP+ cells; arrowheads in A1) relative to cells transfected with a control construct (EGFP+; arrowheads in A). Open arrowheads point to non-transfected cells with unchanged Pdx1 signal. Hoechst 33,342 was used as a nuclear counterstain (pseudo-coloured in grey). Scale bar = 5 μm. Quantitative analysis is shown in Figure 4C2.4
figs5.

Ca2+-dependent secretagogin-mediated control of gene expression with relevance to acquiring and maintaining β cell-like identity in INS-1E cells. (A–D) Secretagogin knock-down by shRNA (48 h) reduces, while secretagogin overexpression (CMV-Scgn; 48 h) up-regulates RNA expression of NeuroD1 (A), Pax4 (B), Nkx6.1 (C) and Pdx1 (D) in INS-1E cells. (E) Decreased Ins mRNA levels are seen upon transfection of INS-1E cells with shRNA targeting Scgn. In turn, transfection with CMV-Scgn significantly increases Ins RNA expression. Tbp mRNA was used as internal control. Data were expressed as means ± s.d. from triplicate experiments, ***p < 0.001, **p < 0.01, *p < 0.05 (one-way ANOVA).5
References
- 1.Oliver-Krasinski J.M., Kasner M.T., Yang J., Crutchlow M.F., Rustgi A.K., Kaestner K.H. The diabetes gene Pdx1 regulates the transcriptional network of pancreatic endocrine progenitor cells in mice. Journal of Clinical Investigation. 2009;119:1888–1898. doi: 10.1172/JCI37028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gradwohl G., Dierich A., LeMeur M., Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proceedings of the National Academy of Sciences of the U S A. 2000;97:1607–1611. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gasa R., Mrejen C., Leachman N., Otten M., Barnes M., Wang J. Proendocrine genes coordinate the pancreatic islet differentiation program in vitro. Proceedings of the National Academy of Sciences of the U S A. 2004;101:13245–13250. doi: 10.1073/pnas.0405301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watada H., Mirmira R.G., Leung J., German M.S. Transcriptional and translational regulation of beta-cell differentiation factor Nkx6.1. Journal of Biological Chemistry. 2000;275:34224–34230. doi: 10.1074/jbc.M004981200. [DOI] [PubMed] [Google Scholar]
- 5.Smith S.B., Gasa R., Watada H., Wang J., Griffen S.C., German M.S. neurogenin3 and hepatic nuclear factor 1 cooperate in activating pancreatic expression of Pax4. Journal of Biological Chemistry. 2003;278:38254–38259. doi: 10.1074/jbc.M302229200. [DOI] [PubMed] [Google Scholar]
- 6.Gao T., McKenna B., Li C., Reichert M., Nguyen J., Singh T. Pdx1 maintains beta cell identity and function by repressing an alpha cell program. Cell Metabolism. 2014;19:259–271. doi: 10.1016/j.cmet.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brink C., Gruss P. DNA sequence motifs conserved in endocrine promoters are essential for Pax4 expression. Developmental Dynamics. 2003;228:617–622. doi: 10.1002/dvdy.10405. [DOI] [PubMed] [Google Scholar]
- 8.Qin Y., Xiao L., Zhan X.B., Zhou H.X. Pdxl and its role in activating Ngn3 and Pax6 to induce differentiation of iPSCs into islet beta cells. Genetics and Molecular Research. 2015;14:8892–8900. doi: 10.4238/2015.August.3.12. [DOI] [PubMed] [Google Scholar]
- 9.Jeon J., Correa-Medina M., Ricordi C., Edlund H., Diez J.A. Endocrine cell clustering during human pancreas development. Journal of Histochemistry and Cytochemistry. 2009;57:811–824. doi: 10.1369/jhc.2009.953307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collombat P., Mansouri A., Hecksher-Sorensen J., Serup P., Krull J., Gradwohl G. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes and Development. 2003;17:2591–2603. doi: 10.1101/gad.269003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stoffers D.A., Ferrer J., Clarke W.L., Habener J.F. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nature Genetics. 1997;17:138–139. doi: 10.1038/ng1097-138. [DOI] [PubMed] [Google Scholar]
- 12.Horikawa Y., Enya M., Mabe H., Fukushima K., Takubo N., Ohashi M. NEUROD1-deficient diabetes (MODY6): identification of the first cases in Japanese and the clinical features. Pediatric Diabetes. 2017;19:236–242. doi: 10.1111/pedi.12553. [DOI] [PubMed] [Google Scholar]
- 13.Sujjitjoon J., Kooptiwut S., Chongjaroen N., Semprasert N., Hanchang W., Chanprasert K. PAX4 R192H and P321H polymorphisms in type 2 diabetes and their functional defects. Journal of Human Genetics. 2016;61:943–949. doi: 10.1038/jhg.2016.80. [DOI] [PubMed] [Google Scholar]
- 14.Hebrok M., Kim S.K., St J.B., McMahon A.P., Melton D.A. Regulation of pancreas development by hedgehog signaling. Development. 2000;127:4905–4913. doi: 10.1242/dev.127.22.4905. [DOI] [PubMed] [Google Scholar]
- 15.Raya A., Kawakami Y., Rodriguez-Esteban C., Ibanes M., Rasskin-Gutman D., Rodríguez-León J. Notch activity acts as a sensor for extracellular calcium during vertebrate left-right determination. Nature. 2004;427:121–128. doi: 10.1038/nature02190. [DOI] [PubMed] [Google Scholar]
- 16.Seymour P.A., Shih H.P., Patel N.A., Freude K.K., Xie R., Lim C.J. A Sox9/Fgf feed-forward loop maintains pancreatic organ identity. Development. 2012;139:3363–3372. doi: 10.1242/dev.078733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shibata H., Yasuda H., Sekine N., Mine T., Totsuka Y., Kojima I. Activin A increases intracellular free calcium concentrations in rat pancreatic islets. FEBS Letters. 1993;329:194–198. doi: 10.1016/0014-5793(93)80220-o. [DOI] [PubMed] [Google Scholar]
- 18.Kawano S., Shoji S., Ichinose S., Yamagata K., Tagami M., Hiraoka M. Characterization of Ca(2+) signaling pathways in human mesenchymal stem cells. Cell Calcium. 2002;32:165–174. doi: 10.1016/s0143416002001240. [DOI] [PubMed] [Google Scholar]
- 19.Gu X., Spitzer N.C. Distinct aspects of neuronal differentiation encoded by frequency of spontaneous Ca2+ transients. Nature. 1995;375:784–787. doi: 10.1038/375784a0. [DOI] [PubMed] [Google Scholar]
- 20.Nelson M.R., Chazin W.J. An interaction-based analysis of calcium-induced conformational changes in Ca2+ sensor proteins. Protein Science. 1998;7:270–282. doi: 10.1002/pro.5560070206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen X., Li H., Ou Y., Tao W., Dong A., Kong J. The secondary structure of calcineurin regulatory region and conformational change induced by calcium/calmodulin binding. Journal of Biological Chemistry. 2008;283:11407–11413. doi: 10.1074/jbc.M708513200. [DOI] [PubMed] [Google Scholar]
- 22.Wagner L., Oliyarnyk O., Gartner W., Nowotny P., Groeger M., Kaserer K. Cloning and expression of secretagogin, a novel neuroendocrine- and pancreatic islet of Langerhans-specific Ca2+-binding protein. Journal of Biological Chemistry. 2000;275:24740–24751. doi: 10.1074/jbc.M001974200. [DOI] [PubMed] [Google Scholar]
- 23.Malenczyk K., Girach F., Szodorai E., Storm P., Segerstolpe A., Tortoriello G. A TRPV1-to-secretagogin regulatory axis controls pancreatic beta-cell survival by modulating protein turnover. The EMBO Journal. 2017;36:2107–2125. doi: 10.15252/embj.201695347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malenczyk K., Keimpema E., Piscitelli F., Calvigioni D., Bjorklund P., Mackie K. Fetal endocannabinoids orchestrate the organization of pancreatic islet microarchitecture. Proceedings of the National Academy of Sciences of the U S A. 2015;112:E6185–E6194. doi: 10.1073/pnas.1519040112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferdaoussi M., Fu J., Dai X., Manning Fox J.E., Suzuki K., Smith N. SUMOylation and calcium control syntaxin-1A and secretagogin sequestration by tomosyn to regulate insulin exocytosis in human β cells. Scientific Reports. 2017;7:248. doi: 10.1038/s41598-017-00344-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Willmann S.J., Mueller N.S., Engert S., Sterr M., Burtscher I., Raducanu A. The global gene expression profile of the secondary transition during pancreatic development. Mechanisms of Development. 2016;139:51–64. doi: 10.1016/j.mod.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 27.Benitez C.M., Goodyer W.R., Kim S.K. Deconstructing pancreas developmental biology. Cold Spring Harbor Perspectives in Biology. 2012;4 doi: 10.1101/cshperspect.a012401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merglen A., Theander S., Rubi B., Chaffard G., Wollheim C.B., Maechler P. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology. 2004;145:667–678. doi: 10.1210/en.2003-1099. [DOI] [PubMed] [Google Scholar]
- 29.Czerny T., Schaffner G., Busslinger M. DNA sequence recognition by Pax proteins: bipartite structure of the paired domain and its binding site. Genes and Development. 1993;7:2048–2061. doi: 10.1101/gad.7.10.2048. [DOI] [PubMed] [Google Scholar]
- 30.Hanics J., Szodorai E., Tortoriello G., Malenczyk K., Keimpema E., Lubec G. Secretagogin-dependent matrix metalloprotease-2 release from neurons regulates neuroblast migration. Proceedings of the National Academy of Sciences of the U S A. 2017;114:E2006–E2015. doi: 10.1073/pnas.1700662114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skarnes W.C., Rosen B., West A.P., Koutsourakis M., Bushell W., Iyer V. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474:337–342. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 33.Schaffer A.E., Taylor B.L., Benthuysen J.R., Liu J., Thorel F., Yuan W. Nkx6.1 controls a gene regulatory network required for establishing and maintaining pancreatic Beta cell identity. PLoS Genetics. 2013;9:e1003274. doi: 10.1371/journal.pgen.1003274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sosa-Pineda B., Chowdhury K., Torres M., Oliver G., Gruss P. The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature. 1997;386:399–402. doi: 10.1038/386399a0. [DOI] [PubMed] [Google Scholar]
- 35.Sander M., Neubuser A., Kalamaras J., Ee H.C., Martin G.R., German M.S. Genetic analysis reveals that PAX6 is required for normal transcription of pancreatic hormone genes and islet development. Genes and Development. 1997;11:1662–1673. doi: 10.1101/gad.11.13.1662. [DOI] [PubMed] [Google Scholar]
- 36.St-Onge L., Sosa-Pineda B., Chowdhury K., Mansouri A., Gruss P. Pax6 is required for differentiation of glucagon-producing alpha-cells in mouse pancreas. Nature. 1997;387:406–409. doi: 10.1038/387406a0. [DOI] [PubMed] [Google Scholar]
- 37.Smith S.B., Ee H.C., Conners J.R., German M.S. Paired-homeodomain transcription factor PAX4 acts as a transcriptional repressor in early pancreatic development. Molecular and Cellular Biology. 1999;19:8272–8280. doi: 10.1128/mcb.19.12.8272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Czerny T., Busslinger M. DNA-binding and transactivation properties of Pax-6: three amino acids in the paired domain are responsible for the different sequence recognition of Pax-6 and BSAP (Pax-5) Molecular and Cellular Biology. 1995;15:2858–2871. doi: 10.1128/mcb.15.5.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fujitani Y., Kajimoto Y., Yasuda T., Matsuoka T.A., Kaneto H., Umayahara Y. Identification of a portable repression domain and an E1A-responsive activation domain in Pax4: a possible role of Pax4 as a transcriptional repressor in the pancreas. Molecular and Cellular Biology. 1999;19:8281–8291. doi: 10.1128/mcb.19.12.8281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu G., Wells J.M., Dombkowski D., Preffer F., Aronow B., Melton D.A. Global expression analysis of gene regulatory pathways during endocrine pancreatic development. Development. 2004;131:165–179. doi: 10.1242/dev.00921. [DOI] [PubMed] [Google Scholar]
- 41.Azzarelli R., Hurley C., Sznurkowska M.K., Rulands S., Hardwick L., Gamper I. Multi-site Neurogenin3 phosphorylation controls pancreatic endocrine differentiation. Developmental Cell. 2017;41:274–286. doi: 10.1016/j.devcel.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gannon M., Ables E.T., Crawford L., Lowe D., Offield M.F., Magnuson M.A. pdx-1 function is specifically required in embryonic beta cells to generate appropriate numbers of endocrine cell types and maintain glucose homeostasis. Developmental Biology. 2008;314:406–417. doi: 10.1016/j.ydbio.2007.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor B.L., Liu F.F., Sander M. Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Reports. 2013;4:1262–1275. doi: 10.1016/j.celrep.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohneda K., Mirmira R.G., Wang J., Johnson J.D., German M.S. The homeodomain of PDX-1 mediates multiple protein-protein interactions in the formation of a transcriptional activation complex on the insulin promoter. Molecular and Cellular Biology. 2000;20:900–911. doi: 10.1128/mcb.20.3.900-911.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marshak S., Benshushan E., Shoshkes M., Havin L., Cerasi E., Melloul D. Functional conservation of regulatory elements in the pdx-1 gene: PDX-1 and hepatocyte nuclear factor 3beta transcription factors mediate beta-cell-specific expression. Molecular and Cellular Biology. 2000;20:7583–7590. doi: 10.1128/mcb.20.20.7583-7590.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fagelskiold A.J., Kannisto K., Bostrom A., Hadrovic B., Farre C. Insulin-secreting INS-1E cells express functional TRPV1 channels. Islets. 2012;4:56–63. doi: 10.4161/isl.18915. [DOI] [PubMed] [Google Scholar]
- 47.Fagelskiold A.J., Kannisto K., Bostrom A., Hadrovic B., Farre C., Eweida M. Phosphorylation marks IPF1/PDX1 protein for degradation by glycogen synthase kinase 3-dependent mechanisms. Journal of Biological Chemistry. 2006;281:6395–6403. doi: 10.1074/jbc.M511597200. [DOI] [PubMed] [Google Scholar]
- 48.Ardestani A., Paroni F., Azizi Z., Kaur S., Khobragade V., Yuan T. MST1 is a key regulator of beta cell apoptosis and dysfunction in diabetes. Nature Medicine. 2014;20:385–397. doi: 10.1038/nm.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fleming J.A., Lightcap E.S., Sadis S., Thoroddsen V., Bulawa C.E., Blackman R.K. Complementary whole-genome technologies reveal the cellular response to proteasome inhibition by PS-341. Proceedings of the National Academy of Sciences of the U S A. 2002;99:1461–1466. doi: 10.1073/pnas.032516399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alpar A., Attems J., Mulder J., Hokfelt T., Harkany T. The renaissance of Ca2+-binding proteins in the nervous system: secretagogin takes center stage. Cellular Signalling. 2012;24:378–387. doi: 10.1016/j.cellsig.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Malmersjo S., Rebellato P., Smedler E., Planert H., Kanatani S., Liste I. Neural progenitors organize in small-world networks to promote cell proliferation. Proceedings of the National Academy of Sciences of the U S A. 2013;110:E1524–E1532. doi: 10.1073/pnas.1220179110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Markovic R., Stozer A., Gosak M., Dolensek J., Marhl M., Rupnik M.S. Progressive glucose stimulation of islet beta cells reveals a transition from segregated to integrated modular functional connectivity patterns. Scientific Reports. 2015;5:7845. doi: 10.1038/srep07845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heit J.J., Apelqvist A.A., Gu X., Winslow M.M., Neilson J.R., Grabtree T.R. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature. 2006;443:345–349. doi: 10.1038/nature05097. [DOI] [PubMed] [Google Scholar]
- 54.Bollo M., Paredes R.M., Holstein D., Zheleznova N., Camacho P., Lechleiter J.D. Calcineurin interacts with PERK and dephosphorylates calnexin to relieve ER stress in mammals and frogs. PLoS One. 2010;5:e11925. doi: 10.1371/journal.pone.0011925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Soleimanpour S.A., Crutchlow M.F., Ferrari A.M., Raum J.C., Groff D.N., Rankin M.M. Calcineurin signaling regulates human islet {beta}-cell survival. Journal of Biological Chemistry. 2010;285:40050–40059. doi: 10.1074/jbc.M110.154955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goodyer W.R., Gu X., Liu Y., Bottino R., Crabtree G.R., Kim S.K. Neonatal beta cell development in mice and humans is regulated by calcineurin/NFAT. Developmental Cell. 2012;23:21–34. doi: 10.1016/j.devcel.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu W., Niwa T., Miura Y., Horio F., Teradaira S., Ribar T.J. Calmodulin overexpression causes Ca(2+)-dependent apoptosis of pancreatic beta cells, which can be prevented by inhibition of nitric oxide synthase. Laboratory Investigation. 2002;82:1229–1239. doi: 10.1097/01.lab.0000027921.01548.c5. [DOI] [PubMed] [Google Scholar]
- 58.Bazwinsky-Wutschke I., Wolgast S., Muhlbauer E., Peschke E. Distribution patterns of calcium-binding proteins in pancreatic tissue of non-diabetic as well as type 2 diabetic rats and in rat insulinoma beta-cells (INS-1) Histochemistry and Cell Biology. 2010;134:115–127. doi: 10.1007/s00418-010-0721-y. [DOI] [PubMed] [Google Scholar]
- 59.Nardozzi J.D., Lott K., Cingolani G. Phosphorylation meets nuclear import: a review. Cell Communication and Signaling. 2010;8:32. doi: 10.1186/1478-811X-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roark R., Itzhaki L., Philpott A. Complex regulation controls Neurogenin3 proteolysis. Biol. Open. 2012;1:1264–1272. doi: 10.1242/bio.20121750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qian F., Huang P., Ma L., Kuznetsov A., Tamarina N., Philipson L.H. TRP genes: candidates for nonselective cation channels and store-operated channels in insulin-secreting cells. Diabetes. 2002;51(Suppl. 1):S183–S189. doi: 10.2337/diabetes.51.2007.s183. [DOI] [PubMed] [Google Scholar]
- 62.Cheng H., Beck A., Launay P., Gross S.A., Stokes A.J., Kinet J.P. TRPM4 controls insulin secretion in pancreatic beta-cells. Cell Calcium. 2007;41:51–61. doi: 10.1016/j.ceca.2006.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Braun M., Ramracheya R., Bengtsson M., Zhang Q., Karanauskaite J. Voltage-gated ion channels in human pancreatic beta-cells: electrophysiological characterization and role in insulin secretion. Diabetes. 2008;57:1618–1628. doi: 10.2337/db07-0991. [DOI] [PubMed] [Google Scholar]
- 64.Dufer M., Haspel D., Krippeit-Drews P., Kelm M., Ranta F., Nitschke R. The KATP channel is critical for calcium sequestration into non-ER compartments in mouse pancreatic beta cells. Cellular Physiology and Biochemistry. 2007;20:65–74. doi: 10.1159/000104154. [DOI] [PubMed] [Google Scholar]
- 65.Graves T.K., Hinkle P.M. Ca(2+)-induced Ca(2+) release in the pancreatic beta-cell: direct evidence of endoplasmic reticulum Ca(2+) release. Endocrinology. 2003;144:3565–3574. doi: 10.1210/en.2002-0104. [DOI] [PubMed] [Google Scholar]
- 66.Djakovic S.N., Schwarz L.A., Barylko B., DeMartino G.N., Patrick G.N. Regulation of the proteasome by neuronal activity and calcium/calmodulin-dependent protein kinase II. Journal of Biological Chemistry. 2009;284:26655–26665. doi: 10.1074/jbc.M109.021956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kawahara H., Yokosawa H. Intracellular calcium mobilization regulates the activity of 26 S proteasome during the metaphase-anaphase transition in the ascidian meiotic cell cycle. Developmental Biology. 1994;166:623–633. doi: 10.1006/dbio.1994.1342. [DOI] [PubMed] [Google Scholar]
- 68.Rogstam A., Linse S., Lindqvist A., James P., Wagner L., Berggard T. Binding of calcium ions and SNAP-25 to the hexa EF-hand protein secretagogin. Biochemical Journal. 2007;401:353–363. doi: 10.1042/BJ20060918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gomes A.V. Genetics of proteasome diseases. Scientifica (Cairo.) 2013;2013:637629. doi: 10.1155/2013/637629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sha Z., Peth A., Goldberg A.L. Keeping proteasomes under control–a role for phosphorylation in the nucleus. Proceedings of the National Academy of Sciences of the U S A. 2011;108:18573–18574. doi: 10.1073/pnas.1115315108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In silico transcription factor binding site prediction in the mouse Scgn promoter. Predicted Pax4 and Pax6 transcription factors binding sites within a 1,400-bp fragment (−1,400 to +1) of the 5′ flanking region of the murine Scgn gene (NC_000079.6). Analysis was performed by the open-source PROMO software.
Protein interactome for secretagogin when used as bait. Proteins precipitated with His6-tagged secretagogin as bait from homogenates of E13.5 wild-type mouse pancreata were identified by liquid chromatography-tandem mass spectrometry and functionally clustered by gene ontology (GO) profiling.
Primer sequences used in this study. qPCR reactions were performed with primer pairs amplifying short fragments for each gene. Primer pairs were custom-designed to efficiently anneal to homologous nucleotide sequences from mouse or rat.
List of antibodies used in this study. Commercially available primary antibodies were used for immunohistochemistry and Western blotting. Abbreviations: Arx, Aristaless-related homeobox; NeuroD1, Neuronal differentiation 1; Ngn3, Neurogenin 3; Nkx6.1, NK6 homeobox 1; Pax6, Paired box 6; Pdx1, Pancreatic and duodenal homeobox 1.