

Abstract
Renal cell carcinoma (RCC) is the most common form of kidney cancer. It is categorized into various subtypes, with clear cell RCC (ccRCC) representing about 85% of all RCC tumors. The lack of sensitivity to chemotherapy and radiation therapy prompted research efforts into novel treatment options. The development of targeted therapeutics, including multi-targeted tyrosine kinase inhibitors (TKIs) and mTOR inhibitors, has been a major breakthrough in ccRCC therapy. More recently, other therapeutic strategies, including immune checkpoint inhibitors, have emerged as effective treatment options against advanced ccRCC. Furthermore, recent advances in disease biology, tumor microenvironment, and mechanisms of resistance formed the basis for attempts to combine targeted therapies with newer generation immunotherapies to take advantage of possible synergy. This review focuses on the current status of basic, translational, and clinical studies on mechanisms of resistance to systemic therapies in ccRCC.
Introduction
Renal cell carcinoma (RCC) accounts for more than 330,000 cases of cancer world-wide and more than 140,000 cancer-related deaths each year. The incidence of kidney cancer has risen steadily over several decades and continues to increase. Approximately 65,340 new kidney cancer diagnoses are projected in the United States in 2018 that are expected to result in 14,970 deaths (1). The incidence in men is 1.5–2.0 times greater than in women, and the peak age of incidence is 60–70 years (2). Clear cell renal cell carcinoma (ccRCC) is the most frequent (75–80%) and the best studied subtype of RCC. Papillary RCC and chromophobe RCC represent the most common remaining histologic subtypes with an incidence of 7% - 14% and 6% - 11%, respectively (3). Advanced RCC is a lethal disease portending a 5-year survival of only 11.7% (4). Two distinct groups of patients are at particular risk of death from RCC: Those who present with metastatic disease and those who recur following surgery. Approximately 30% of RCC patients have metastatic disease at initial presentation (5). Recurrence occurs in about 30% of patients after complete resection of the primary tumor (6). This includes 10-25% of patients with localized (pT1-2N0) disease who demonstrate recurrence despite incidental detection and clinically complete surgical resection (7–10). Traditional chemotherapy and radiation therapy are largely ineffective in the treatment of all RCC subtypes (11,12). The lack of sensitivity to chemotherapy and radiation therapy prompted early research efforts into novel treatment options.
Treatment of metastatic RCC: historical and current concepts
Occasional spontaneous tumor regressions and the presence of tumor infiltrating immune cells suggested that adaptive immunity might play an important role in renal malignancy. For more than 20 years, immunotherapy using high-dose IL-2 or Interferon alfa (IFN-α) remained the primary treatment for patients with metastatic RCC (mRCC). Unfortunately, response rates to immunotherapy were disappointing, ranging from 15% to 25% (13).
The field of renal cancer therapy has undergone radical changes in the last decade. The clinical knowledge that ccRCC is a highly vascular cancer and that the Von Hippel Lindau (VHL) protein has an important role in sporadic ccRCC has made anti-angiogenic strategies an attractive approach (14,15). The introduction of therapeutic agents targeting Vascular Endothelial Growth Factor (VEGF) signaling, especially multi-targeted tyrosine kinase inhibitors (TKIs), has been a major breakthrough in ccRCC therapy. Various VEGF receptor TKIs have demonstrated considerable efficacy in RCC. Sunitinib (Sutent) and pazopanib (Votrient) are approved for first-line treatment in mRCC (16), whereas axitinib (Inlyta) and sorafenib (Nexavar) have demonstrated progression-free survival (PFS) benefits as second-line agents (17). As opposed to earlier TKIs, axitinib blocks receptors known to be involved in escape pathways that lead to treatment resistance. After its effectiveness was established, a head-to-head trial of axitinib vs. sorafenib (AXIS) demonstrated the clinical superiority of axitinib over sorafenib (PFS 6.7 mo vs. 4.7mo) in second-line treatment (17). Also, a consistent PFS benefit was demonstrated with bevacizumab + IFN-α (Avastin), a recombinant humanized monoclonal antibody that binds VEGF preventing its interaction with VEGF Receptor (VEGFR), in treatment-naïve mRCC patients (18–20). Most recently, cabozantinib (COMETRIQ), inhibitor of VEGF-R, MET, and AXL, demonstrated PFS and OS advantages over sunitinib, and received FDA-approval for frontline mRCC treatment (21). Cabozantinib appears specifically effective at blocking the combination of angiogenic pathways that emerges following oncogenic VHL inactivation. Lenvatinib (Lenvima) is another TKI targeting VEGF-R1-R3, FGF-R1-4, PDGF-R, RET, and KIT. Phase II analysis demonstrated a PFS superiority of lenvatinib + everolimus vs. either agent alone (22). The successful combination of lenvatinib with everolimus is especially notable because of the historical failure of combining VEGF and mTOR inhibitors due to excessive toxicity.
In addition to the VEGF pathway, the Akt/mTOR mammalian target of rapamycin (mTOR) pathway was identified as a promising therapeutic target for the treatment of mRCC. Single agent activities led to market approval of two mTOR inhibitors, everolimus and temsirolimus, for the treatment of advanced RCC (23).
Improvements in molecular understanding of resistance mechanisms has led to the discovery of many new targetable pathways. Several new agents are under early clinical investigation, and these may play an important future role in combination therapy (20). Trebananib is a Ang/Tie-2 pathway inhibitor. This pathway is responsible for basal angiogenesis and vascular stability following VEGF blockade, so it potentially has significant utility when used in combination with TKIs (24). Dalantercept inhibits ALK-1, which is thought to play a role in vascular bed formation. Other agents are being tested in preclinical models that exhibit dual mTORC1/2 inhibition. Most mTOR inhibitors primarily target mTORC1, and may fail to disrupt the activity of HIF-2α, which is more highly regulated by mTORC2. By more effectively blocking HIF translation, dual mTORC1/2 inhibitors may offer unique treatment possibilities in the future.
Recent advances have led to new treatment approaches in the post-TKI second- or third-line settings. Nivolumab (programmed death (PD-1) checkpoint inhibitor), cabozantinib, and lenvatinib plus everolimus demonstrated promising clinical efficacy and have gained FDA approval in the last two years (25–28).
Mechanisms of primary and acquired resistance to systemic therapeutic agents
Response to anticancer therapy is currently defined by the Response Evaluation Criteria In Solid Tumors (RECIST) criteria as evidence of disease progression despite therapeutic treatment. This progression is defined as an increase of 20% or more in the sum of measurable lesions, the appearance of new lesions, or an unequivocal progression of non-measurable disease such as small lung nodules or bone lesions (29). Resistance to targeted therapeutics can be classified into intrinsic (primary) and acquired (secondary) resistance. Intrinsic resistance can be classified as an immediate inefficacy of therapeutic agents. This type of resistance can be attributed to the presence of resistant tumor clones prior to therapy due to inherited resistance or evolutionary clonal selection. Acquired resistance is characterized by tumor growth after initial tumor regression while the patient is still receiving therapeutic treatment. While the precise mechanisms of resistance to targeted therapeutics are still being elucidated, laboratory and clinical studies have identified several underlying factors contributing to both intrinsic and acquired resistance mechanisms.
Lysosomal sequestration of TKIs
Lysosomes contain about 50 different acid hydrolases, which are optimally active at the acidic pH of 4.6–5.0. To maintain the low pH, lysosomes utilize proton-pumping vacuolar ATPases (30). Lysosomal sequestration is a process in which hydrophobic weak base compounds accumulate within acidic lysosomes (31). Such compounds travel freely across the lysosomal membrane due to their hydrophobic nature. Upon encountering the acidic lysosomal compartment, these compounds become protonated due to their weak base properties and can no longer exit across the lysosomal lipid membrane (31). Most TKIs are membrane-permeable weak bases, and are therefore trapped in their protonated forms in the acidic lysosomal compartment away from their target sites. Lysosomal sequestration as a mechanism of drug resistance is reversible; drug-free cultures of resistant tumor cells result in recovery of drug sensitivity (32).
Several TKIs have been shown to undergo lysosomal sequestration including sunitinib, erlotinib, and pazopanib (32,33). Interestingly, although lysosomal sequestration of sorafenib was not found in renal cancer cells (33), it has been demonstrated in hepatocellular carcinoma (34). Sorafenib does not belong to the same class of hydrophobic, membrane-permeable weak bases as sunitinib; and therefore, a different mechanism could explain its lysosomal sequestration, possibly involving the activity of drug pumps (35). Indeed, the lysosomal sequestration of both sorafenib and sunitinib was reported to be mediated by the ABC transporter P-glycoprotein (P-gp) (34). Treatment with verapamil, a P-gp inhibitor, enhanced the antitumor activity of sorafenib and sunitinib, supporting the role of P-gp in TKIs resistance (34).
Lysosomal sequestration of hydrophobic weak base TKIs induces lysosomal biogenesis (36). Increased lysosomal biogenesis, in turn, results in augmented lysosomal drug sequestration and multi-drug cross-resistance. Lysosomal biogenesis is regulated by the transcription factor EB (TFEB), which is in turn controlled by mTORC1 (37). These findings provide rationale for combined treatment with TKIs and mTOR inhibitors, though excessive toxicity was reported for TKI/mTOR combination therapy (38,39).
Mutations and modifications of expression level
Mutation rates can vary by as much as three orders of magnitudes between various cancer types and even patients (40). RCC is a complex disease characterized by mutations in many genes. Deactivation of the VHL gene is the most common mutation in RCC. Some studies have reported VHL gene inactivation in >90% of patients with sporadic ccRCC (41). Epigenetic silencing of VHL was present in 7% of ccRCC tumors, which is consistent with the role of epigenetic changes in renal carcinogenesis (42,43). Loss of VHL gene function results in the stabilization and increased expression of hypoxia inducible factors (HIFs) and up-regulation of HIF-responsive genes that mediate angiogenesis and cell growth. Nearly universal loss of VHL function and subsequent HIF activation in the pathogenesis of ccRCC would suggest that TKIs are a highly efficient treatment modality of this type of kidney cancer (44). However, a study by Choueiri et al. demonstrated that VHL mutation status has little effect on patient responses to VEGF-targeted agents in metastatic ccRCC (45). VHL mutations may confer aberrant activation of AKT/mTOR signaling (46) given that VHL inactivation subsequently activates mTOR, which in turn up-regulates HIF and jumpstarts other angiogenic pathways (47). Several clinical trials have evaluated the efficacy and safety of everolimus and temsirolimus in the treatment of mRCC after progression on TKIs. Whereas treatment with mTOR inhibitors offers significantly improved clinical benefits, such treatment rarely yields a complete response and is not curative (48). A potential mechanism that accounts for resistance to everolimus and temsirolimus is mutation in FKBP-12 domain in mTOR, which reduces the binding affinity of mTOR inhibitors (49). The mutational status of the mTOR pathway genes TSC1, TSC2, and REDD1 could also predict response to everolimus or temsirolimus in RCC tumors (50). REDD1 is a HIF-1 target gene that inhibits mTORC1 by activating the TSC1/2 complex under normal conditions. Alterations in TSC1, TSC2, or REDD1 therefore, prevent the inhibition of mTORC1 (48). Mutations in all these genes have been observed in RCC (48).
The aberrant expression of proteins involved in the mTOR signaling pathway may also modulate sensitivity to mTOR inhibitors. Phospholipase D2 (PLD2)-derived phosphatidic acid binds to and activates ribosomal S6 kinase (S6K), which, in turn, activates a feedback loop inactivating PLD2 and decreasing phosphatidic acid levels (51). Phosphatidic acid is required for mTORC1/mTORC2 complex assembly and competes with rapamycin for mTOR binding (52). Critically, PLD2 expression and activity are greatly elevated in RCC tissue specimens as compared with the adjacent normal tissues (53). Taken together, these findings provide a mechanistic link between increased PLD2 activity and resistance to mTOR inhibitors.
A recent study by Adelaiye et al. demonstrated that the overexpression of the histone methyltransferase enhancer of zeste homologue 2 (EZH2) promotes tumor angiogenesis by inactivating anti-angiogenic factors via methylation at their promoter regions, causing resistance to sunitinib (54). Tumors resistant to sunitinib had an increased level of EZH2 expression. However, this increase was reversible upon dose escalation, suggesting that tumor adaptation to sunitinib was dynamic and was likely driven by epigenetic alterations (54).
The p4E-BP1/eIF4E axis represents a critical convergence point for several upstream signaling pathways such as EGF-R/ERK and AKT/mTOR, all of which are targeted in some respect by molecular-targeted therapies used in the treatment of RCC. Levels of phospho-4e-BP1 and total eIF4E are elevated in ccRCC (55). Accordingly, phosphorylated 4E-BP1 has recently been shown to be the single most accurate biomarker for predicting treatment response to mTOR inhibitors (56).
Angiogenic Switch
Inactivation of VHL in RCC cells lead to increased HIF-1 and HIF-2 activities. Interestingly, HIF-2 antagonist PT2399 was more active than sunitinib (p=0.0126) and inhibited tumor growth in several sunitinib-resistant RCC xenograft tumors (57). However, as discussed above, VHL mutation status by itself had little effect on patient responses to VEGF-targeted agents in metastatic ccRCC (45). Furthermore, studies by Bielecka et al. demonstrate that sorafenib and axitinib effectively inhibit the growth of primary and metastatic ccRCC cell lines in normoxia and hypoxia. Only the growth of papillary kidney cancer stem-like cells was inhibited in an oxygen-dependent manner in this study (58). Nevertheless, hypoxia and HIFs may contribute to resistance to targeted therapeutics by up-regulating expression of VEGF, platelet derived growth factor (PDGF), interleukin-6 (IL-6), interleukin-8 (IL-8), transforming growth factor α (TGF-α), erythropoietin (EPO), epidermal growth factor receptor (EGFR), hepatocyte growth factor receptor (HGFR/c-MET), placental growth factor (PlGF), and fibroblast growth factor 2 (FGF2) (59,60). Notably, hypoxia caused by the regression of tumor vasculature during the course of anti-angiogenic treatment may also lead to enhanced expression of various proangiogenic factors (61).
The findings relative to IL-6 and IL-8 are of particular interest given their well-established roles as prognostic and predictive factors associated with resistance to TKIs in ccRCC patients (62–65). In patients receiving pazopanib or sunitinib, high baseline serum IL-6 and IL-8 levels are associated with shorter PFS and/or overall survival (OS) (65–67). IL-8 promotes the expression of VEGF mRNA and protein in endothelial cells by binding to CXCR2, which subsequently leads to the autocrine activation of VEGFR-2, resulting in increased angiogenesis (68). Studies by Huang et al. showed that increased plasma levels of IL-8 were detected in the plasma of mice with sunitinib-resistant tumors compared to mice bearing sunitinib-responsive tumors (62). Treatment with IL-8 neutralizing antibodies reinstated sensitivity to sunitinib (62). Studies by Wu et al. evaluated the predictive value of polymorphisms in IL-8 in sunitinib and pazopanib resistance (69–71). These findings showed that variant alleles of IL-8 are associated with poorer survival outcomes in pazopanib- or sunitinib-treated patients with advanced RCC (69–71). Recent studies by Ishibashi et al. showed that treatment with sorafenib, sunitinib, and pazopanib stimulated the autocrine secretion of IL-6, which consequently lead to the activation of AKT/mTOR and STAT3 signaling, VEGF expression, and TKI resistance in RCC cells (63). Combination therapy with tocilizumab, a humanized antihuman IL-6 receptor (IL-6R) antibody reinstated TKI sensitivity in RCC cells (63). These findings are in agreement with previous studies by Zhu et al. showing that IL-6 signaling inhibition leads to the increased efficacy of sunitinib in cell and animal models of human RCC (64).
Studies by Mizumoto et al. suggested that the mechanism of acquired resistance to sunitinib in RCC cells may be also related to the activation of EGFR (72). Inhibition of EGFR signaling with erlotinib decreased the viability of RCC cells treated with sunitinib (72). The pro-angiogenic function of FGF2 may be also directly relevant for resistance to sunitinib. FGF2 suppresses the anti-angiogenic activity of sunitinib by directly stimulating pro-angiogenic signaling in endothelial cells (73). This is likely to be especially important for RCC, in which FGF2 expression is prominent (73).
VHL inactivation in ccRCC induces overexpression and activation of the receptor tyrosine kinases MET and AXL (74). Both, MET and AXL signaling have been implicated in clinical resistance to VEGF-targeted therapeutics. Therefore, targeting the MET and AXL represents a logical option after progression on initial VEGF therapy. Cabozantinib, VEGFR, c-MET and AXL inhibitor, was approved by the FDA in April 2016 for the treatment of advanced RCC, pretreated with at least one prior antiangiogenic therapy. Cabozantinib demonstrated clinical efficacy in advanced pretreated RCC, with statistically significant improvements in RR, PFS, and OS (27,75) (Fig. 1). Combination with another c-met inhibitor, crizotinib, not only increased axitinib-mediated inhibition of tumor microvessel density, but also suppressed growth of patient derived xenograft (PDX) RP-R-01 tumors. In this highly proliferative model, concurrent c-met inhibition and VEGFR blockade was required to inhibit tumor growth and improve survival (76). Lastly, the VEGF homologue PlGF binds to VEGFR-1 and displaces VEGF from VEGFR-1, resulting in an increased bioavailability of VEGF. The increase in VEGF bioavailability stimulates VEGFR-2 and thereby promotes angiogenesis (48,77). PlGF also stimulates angiogenesis through other mechanisms, such as up-regulation of VEGF-A, FGF2, and PDGFβ expression, and by recruiting bone marrow-derived angiocompetent myeloid cells, which stimulate angiogenesis through the secretion of pro-angiogenic cytokines (48).
Figure 1. Recent clinical trials in RCC.
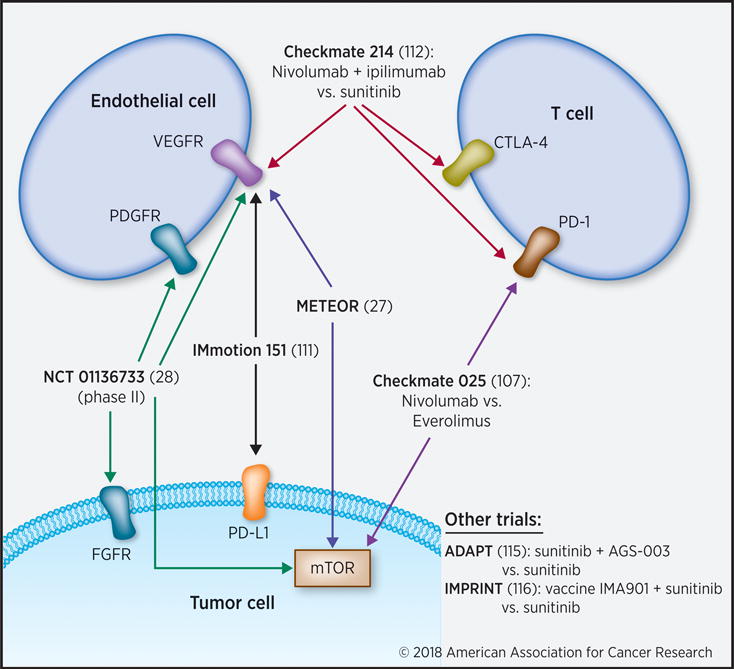
The current treatment landscape for advanced RCC includes combination strategies targeting tumor, endothelial and immune cells.
Bypass or alternative pathways activation
The tumor suppressor PTEN acts as a negative regulator of PI3K/Akt/mTOR pathway. The loss of PTEN function results in the constitutive activation of AKT/mTOR signaling downstream of TKIs cellular targets. PTEN mutations are rare in RCC (78). However, PTEN gene expression has been shown to be down-modulated in a large percentage of RCC (42,79). Our own studies demonstrate that lack of PTEN expression coincides with sunitinib resistance in renal cells, whereas restoration of PTEN expression or pharmacologic inhibition of AKT/mTOR signaling reinstates sensitivity of PTEN-deficient ccRCC cells to sunitinib-mediated apoptosis. (80). It has been reported that reduced expression of PTEN also correlates with a lower activity of bevacizumab (81).
ccRCC is a highly lipogenic tumor. Our recent studies demonstrate that the addition of LDL cholesterol increases activation of PI3K/AKT signaling, which coincides with reduced antitumor activity of clinically relevant TKIs such as sorafenib, pazopanib, lapatinib and sunitinib against ccRCC and endothelial cells. Furthermore, ccRCC xenograft tumors resisted treatment with sunitinib in mice fed a high fat/high cholesterol diet (82). Interestingly, results from the 3-arm phase III Global Advanced Renal Cell Carcinoma Trial (ARCC) demonstrated that increases in cholesterol potentially predicted temsirolimus efficacy. Longer survival in patients treated with temsirolimus was observed in those with larger increases in cholesterol (83). The exact mechanism underlying this phenomenon is not clear. The authors acknowledge that whether the increase in serum cholesterol merely reflects successful targeting of the mTOR pathway, or if it is mechanistically required for the antitumor response, cannot be determined from the results of this study (83).
ATP-binding cassette (ABC) efflux transporters
ABC transporters represent a large superfamily of active transporter proteins that mediate the efflux or uptake of specific substrates across different membranes including the plasma membrane, endoplasmic reticulum, Golgi compartment, peroxisomes, and mitochondria (84). Multiple TKIs have been reported to interact with ABC transporters (e.g. ABCB1, ABCC1, ABCG2, and ABCC10) (84). Investigation by Sato et al. demonstrated that sunitinib induced upregulation of ABC sub-family G member 2 (ABCG2) in 786-O RCC cells; and treatment with elacridar (GF120918) (85), a third generation P-glycoprotein inhibitor, enhanced the cytotoxic effect of sunitinib (86). Another study by Shibayama et al. revealed that sorafenib may serve as a substrate for multidrug resistance-associated protein 2 (MRP2) (87). However, TKIs may also serve as inhibitors of ABC transporters depending on drug concentration, the expression of specific pumps, and affinity for transporters. In general, at low concentrations TKIs display substrate-like properties; and at high concentration they can inhibit the function of pumps (84). A number of TKIs including sunitinib, sorafenib, and pazopanib have been established as inhibitors of various classes of ABC transporters (3,14).
Tumor heterogeneity
RCC tumor specimens from different patients with similar pathological grade and stage can be extremely heterogeneous, displaying different profiles at genomic and transcriptomic levels. This phenomenon drastically limits the utility of prognostic and predictive biomarkers. Furthermore, intratumoral heterogeneity (ITH) presents a considerable therapeutic challenge significantly limiting the efficacy of targeted therapies (88). Varying gene, microRNA, and protein expression signatures can be detected even within the same tumor specimen, and variations in the dysregulation of microRNA has been shown to impact ccRCC pathogenicity (89). Significant molecular heterogeneity was also detected between primary and metastatic lesions, with only a small subset of alterations present in both sites. In one study, post-sunitinib metastatic lesions carried mutations in FLT4, KMT2D, and BMP5, which were not detected in the primary tumor (90). Studies by Hatiboglu et al. demonstrated that although neoadjuvant treatment with sorafenib was clinically active in downsizing tumors in patients with locally confined, non-metastatic ccRCC, such treatment led to an enhanced functional ITH in the residual tumor tissue (91). These results are in agreement with findings by Stewart et al., demonstrating that primary ccRCC tissues from patients treated with sunitinib express greater morphologic heterogeneity compared with tumors from untreated patients (92). Unsupervised analysis did not reveal any significant effect of sunitinib therapy on ITH at a chromosomal or mRNA level. However, results from supervised analyses do show a consistent increase in ITH for both DNA and mRNA of several oncogenes. These findings were supported by protein expression results demonstrating an increase in ITH for selected tumor-specific proteins (92). According to Stewart et al., the results of this study do not support the hypothesis that a single resistant clone predominates from a number of clones after treatment commences; treatment was actually associated with more, rather than less, ITH. As such, the authors speculate that VEGF-targeted therapy may generate a polyclonal outgrowth of tumor cell subclones that can lead to acquired drug resistance (92).
Tumor Microenvironment
The tumor microenvironment is critical for the initiation and maintenance of tumorigenesis (93). Growing evidence also indicates the direct involvement of the tumor microenvironment in the development of resistance to targeted therapeutics. The tumor microenvironment consists of tumor cells, extracellular matrix (ECM), signaling molecules, and stromal cells (e.g. fibroblasts, vascular endothelial cells, pericytes, and immune cells). Myeloid-derived suppressor cells (MDSC) are one of the major components of the tumor microenvironment. Accumulating evidence suggests that MDSC are recruited by tumors to mediate resistance to anti-angiogenic drugs by expressing various pro-angiogenic factors, which stimulate VEGF-independent angiogenesis (94). Studies by Ko et al. demonstrate that unlike the pronounced decline in peripheral blood MDSC observed in RCC patients treated with sunitinib, tumor tissues obtained from sunitinib-treated patients have not demonstrated declines in MDSC (95). MDSC resistance to sunitinib corresponded to compartmental availability of GM-CSF in renal tumors. Treatment with recombinant GM-CSF also conferred sunitinib resistance in vitro and in vivo. GM-CSF-induced sunitinib-resistance in MDSC was STAT5 mediated, as it was negated in STAT5ab(null/null) MDSC (95). Another population of stromal cells, which directly contribute to aberrant tumor angiogenesis and drug resistance are pericytes. Increased pericyte coverage and increased production of VEGF by pericytes enhances survival of endothelial cells, rendering them less responsive to inhibition of VEGF signaling (59). Targeting pericytes and endothelial cells by inhibiting both VEGF and PDGF signaling has proven to be more effective in reducing angiogenesis and tumor growth than targeting either of these pathways alone (59).
Tumor endothelial cells express the Notch ligand Delta-like 4 (Dll4). The Dll4-Notch pathway functions as a key negative regulator of physiological and pathological angiogenesis downstream of VEGF (96). Dll4 is predominately found in the developing endothelium, with an almost 9-fold increased expression reported within the vasculature of ccRCC, as compared to normal kidneys (97). Miles et al. demonstrated the potent anti-tumor efficacy of combined treatment with anti-Dll4 and anti-VEGF therapy in a sunitinib-resistant metastatic ccRCC model (96). Notch signaling also plays a crucial role in the maintenance of “stemness” by cancer stem-like cells in RCC. Pharmacological blockade of Notch signaling reinstates the sensitivity of human RCC cells to sorafenib (98).
Tumor-associated fibroblasts can also contribute to resistance to targeted therapies via increased expression of PDGF-C, which could overcome the inhibition of VEGF-mediated angiogenesis (99).
Strategies to reverse or overcome TKI resistance
One of the most common approaches in cancer treatment is the use of combinations of agents with different mechanisms of action and molecular targets. Pharmacological inhibition of PI3K/Akt/mTOR signaling reinstates sensitivity to sunitinib and sorafenib in ccRCC cells with aberrant AKT activity (80,82). Sensitivity to sunitinib in PTEN-negative 786-O RCC cells was also successfully reinstated using temsirolimus (80). A meta-analysis of 22 randomized clinical trials supports earlier observations that combining targeted therapies is a promising strategy against advanced RCC. Yet, while efficacy may be potentiated, so can toxicity, as is seen in many combinations of TKIs and mTOR inhibitors. (38,39,100,101). Bevacizumab, for example, is an effective agent in combination with IFN; but combinations with sunitinib, sorafenib, and temsirolimus were all abandoned due to compounded toxicities (25).
Designing treatment sequences with different TKIs has also demonstrated positive outcomes in patients with advanced RCC. This approach relies on the fact that TKIs have varying target profiles and different affinities for common targets. For example, a randomized phase II study demonstrated clinical activity of axitinib in patients with sorafenib-refractory metastatic RCC and in patients who have been treated with additional prior therapies, including sunitinib, cytokines, temsirolimus, or bevacizumab plus IFN-α (102). The sequential use of TKIs followed by mTOR inhibitors, specifically everolimus, has also been established for the systemic treatment of metastatic RCC (103).
New options for immunotherapy
Immune checkpoint inhibitors have recently emerged as an effective treatment against advanced RCC. As opposed to therapies selectively targeting angiogenic pathways, immune checkpoint (PD1-PD-L1/CTLA4) inhibitors work to directly reverse the adaptive camouflage tumor cells deploy to avoid host immunity. PD-1 is a molecule recognized by its ligand PD-L1 on antigen presenting cells (APCs) and tumor cells. This interaction acts as an immune checkpoint and promotes T-cell tolerance (20). Interestingly, preclinical studies have shown that targeted mRCC therapies have immunomodulatory effects, such as promoting T-cell infiltration into tumors and increasing tumor cell antigenicity (104). These findings formed the basis for attempts to combine targeted antiangiogenic therapies with newer generation immunotherapies to take advantage of possible synergy (25).
It was previously demonstrated that ccRCC tumors have varying degrees of PD-L1 expression. An early study of ccRCC tumors after nephrectomy demonstrated that patients with tumors expressing PD-L1 had a significantly lower 5-year CSS (41.9%) vs. those not expressing PD-L1 (82.9%) (105). The degree of PD-L1 expression on mRCC cells directly correlates with aggressive pathologic features (25).
CTLA-4 is a receptor checkpoint inhibitor expressed on T cells, and ligand binding promotes immune tolerance. A Phase II study of ipilumumab (anti-CTLA-4 mAB) in mRCC demonstrated partial responses; and intriguingly, those patients who had autoimmune related side effects (Grade 3/4) tended to see the most tumor regression, indicating that characteristics of host immunity can play a significant role in tumor control (106). Despite the significant toxicities from this agent, there may still be a role for combination therapies with ipilimumab, including for treatment of non-ccRCC tumors.
The clinical success of checkpoint inhibition in other solid tumors led to several trials demonstrating the efficacy of agents such as nivolumab (anti PD-1 mAB).
Single agent IO
Checkmate 025 was a randomized phase III clinical trial comparing nivolumab to everolimus in patients who failed initial antiangiogenic therapy. The median OS for nivolumab vs. everolimus was 25mo vs. 19.6mo, with a ORR of 25% vs. 5%, respectively, favoring nivolumab over everolimus. Importantly, Grade 3/4 treatment side effects appeared to be lower in the nivolumab group (19% for nivolumab vs 37% for everolimus), which makes this an attractive treatment option (107) (Fig. 1).
Other agents are in the early phases of clinical study. Atezolizumab, an IgG mAB against PD-L1 (as opposed to nivolumab which targets PD-1), showed efficacy in phase I studies of patients with advanced RCC, with OS of 28.9 months and 15% ORR (108). Pembrolizumab, another anti-PD1 agent and anti-PDL1 therapies durvalumab and avelumab are being studied as monotherapy in trials NCT02212730, NCT02669914, NCT02493751, respectively.
IO+ VEGFR TKIs
A phase 1 trial was conducted to investigate the combination of escalating doses of nivolumab with sunitinib or pazopanib. While the pazopanib arm was closed due to hepatotoxicity, the sunitinib combination arm was dose escalated. Although toxicity was increased, high response rates were observed in this study (52% in sunitinib arm and 45% in pazopanib arm) (109). Similarly, the combination of nivolumab with cabozantinib is currently being studied in NCT02496208. Pembrolizumab (anti-PD-1) is currently under phase I/II study as a combination drug with several VEGF-targeted therapies in both the first-line and post-TKI spaces (110). These include a phase III first-line trial of pembrolizumab with axitinib (NCT02853331), and other trials of pembrolizumab in combination with lenvatinib (NCT02501096), ziv-aflibercept (NCT02298959), and bevacizumab (NCT02348008). A 3-armed phase III study (IMmotion 151) is currently underway evaluating the efficacy of atezolizumab + bevacizumab, atezolizumab-alone, and sunitinib in first-line therapy. Early findings appear to indicate that patients with positive PD-L1 expression experience a higher PFS, with response rates of 25-46% (111) (Fig. 1). AVELIN Renal 101, a randomized multicenter, phase 3 study (NCT02684006) comparing the combination with sunitinib in treatment-naïve patients with mRCC, began enrollment in March 2016.
IO+ IO combinations
The results of the phase III, randomized, open-label CheckMate-214 study evaluating the combination of nivolumab and ipilimumab compared to sunitinib in patients with previously untreated advanced or metastatic RCC were recently reported at ESMO 2017. The study met its primary endpoint of OS compared to sunitinib in intermediate and poor risk patients in the front line setting. Pembrolizumab is also being evaluated in combination with ipilimumab as one of the three arms of a phase I/II study in patients with metastatic RCC or melanoma (NCT02089685). Anti PD-L1 therapy durvalumab is also evaluated in combination with anti CTLA4 antibody tremelimumab (NCT01975831). Checkmate 214 is a phase III randomized trial of nivolumab + ipilimumab vs. sunitinib in previously untreated mRCC (112) (Fig. 1). The recently-announced results are very promising: After 17.5 months follow-up, the ORR for nivolumab + ipilimumab vs. sunitinib was 41.6% vs. 26.5%, respectively, with 9.4% of combined immune-blockade patients achieving complete response (113).
As with targeted therapies, there is evidence that primary resistance to PD-L1 inhibition may form in some tumor microenvironments, with adaptive resistance developing in others. The mechanisms behind these resistance patterns are still unclear; but as with TKIs and mTOR inhibitors, combination therapy will likely be helpful in preventing evasion of host immunity (114).
Vaccines have been used effectively in other solid tumor malignancies, and new options have recently emerged for RCC. AGS-003 is a dendritic cell vaccine prepared with amplified tumor RNA that can prime the immune response. A randomized phase III trial (ADAPT) of sunitinib + AGS-003 vs. sunitinib-alone for previously untreated mRCC was launched after promising phase II study results, which found that the magnitude of increased effector/memory T cell production correlated with improved OS (115) (Fig. 1). IMA901 is a multipeptide vaccine utilizing a combination of HLA class I & II-binding tumor-associated peptides that underwent a phase III randomized study for first line therapy in combination with sunitinib. Unfortunately, it appeared that the magnitude of immune response was not sufficient to demonstrate an OS difference when IMA901 was used in conjunction with sunitinib, though this doesn’t completely close the door on this strategy in the future (116) (Fig. 1).
Conclusions and perspectives
Targeted therapies are nowadays the standard treatment options for renal cancer that have changed the treatment landscape of advanced ccRCC compared with the cytokines era. However, nearly all patients treated with currently approved targeted drugs will eventually experience disease progression. Furthermore, a significant number of RCC patients are primarily refractory to targeted therapeutics, showing neither disease stabilization nor clinical benefits. Newer agents that augment tumor/immune-system interactions and that concurrently target multiple oncogenic pathways hold great promise. Further characterization of the RCC oncogenic pathways and the ongoing clinical trials should help to optimize the management of patients with advanced RCC.
Acknowledgments
This work was supported in part by the National Institutes of Health Grants RO3CA216173 to P. Makhov, and RO3CA212949 to V.M. Kolenko, and the Department of Defense Prostate Cancer Research Program (PCRP) Impact Award PC160049 to V.M. Kolenko.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Bhatt JR, Finelli A. Landmarks in the diagnosis and treatment of renal cell carcinoma. Nature reviews Urology. 2014;11:517–25. doi: 10.1038/nrurol.2014.194. [DOI] [PubMed] [Google Scholar]
- 3.Shuch B, Amin A, Armstrong AJ, Eble JN, Ficarra V, Lopez-Beltran A, et al. Understanding pathologic variants of renal cell carcinoma: distilling therapeutic opportunities from biologic complexity. Eur Urol. 2015;67:85–97. doi: 10.1016/j.eururo.2014.04.029. [DOI] [PubMed] [Google Scholar]
- 4.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 5.Motzer RJ, Bukowski RM, Figlin RA, Hutson TE, Michaelson MD, Kim ST, et al. Prognostic nomogram for sunitinib in patients with metastatic renal cell carcinoma. Cancer. 2008;113:1552–8. doi: 10.1002/cncr.23776. [DOI] [PubMed] [Google Scholar]
- 6.Nerich V, Hugues M, Paillard MJ, Borowski L, Nai T, Stein U, et al. Clinical impact of targeted therapies in patients with metastatic clear-cell renal cell carcinoma. OncoTargets and therapy. 2014;7:365–74. doi: 10.2147/OTT.S56370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flanigan RC, Campbell SC, Clark JI, Picken MM. Metastatic renal cell carcinoma. Current treatment options in oncology. 2003;4:385–90. doi: 10.1007/s11864-003-0039-2. [DOI] [PubMed] [Google Scholar]
- 8.Rini BI. Metastatic renal cell carcinoma: many treatment options, one patient. J Clin Oncol. 2009;27:3225–34. doi: 10.1200/JCO.2008.19.9836. [DOI] [PubMed] [Google Scholar]
- 9.Rini BI. New strategies in kidney cancer: therapeutic advances through understanding the molecular basis of response and resistance. Clin Cancer Res. 2010;16:1348–54. doi: 10.1158/1078-0432.CCR-09-2273. [DOI] [PubMed] [Google Scholar]
- 10.Sorbellini M, Kattan MW, Snyder ME, Reuter V, Motzer R, Goetzl M, et al. A postoperative prognostic nomogram predicting recurrence for patients with conventional clear cell renal cell carcinoma. J Urol. 2005;173:48–51. doi: 10.1097/01.ju.0000148261.19532.2c. [DOI] [PubMed] [Google Scholar]
- 11.Goyal R, Gersbach E, Yang XJ, Rohan SM. Differential diagnosis of renal tumors with clear cytoplasm: clinical relevance of renal tumor subclassification in the era of targeted therapies and personalized medicine. Archives of pathology & laboratory medicine. 2013;137:467–80. doi: 10.5858/arpa.2012-0085-RA. [DOI] [PubMed] [Google Scholar]
- 12.Rini BI, Campbell SC, Escudier B. Renal cell carcinoma. Lancet. 2009;373:1119–32. doi: 10.1016/S0140-6736(09)60229-4. [DOI] [PubMed] [Google Scholar]
- 13.Rini BI, McDermott DF, Hammers H, Bro W, Bukowski RM, Faba B, et al. Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of renal cell carcinoma. Journal for immunotherapy of cancer. 2016;4:81. doi: 10.1186/s40425-016-0180-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaelin WG., Jr The von Hippel-Lindau gene, kidney cancer, and oxygen sensing. Journal of the American Society of Nephrology : JASN. 2003;14:2703–11. doi: 10.1097/01.asn.0000092803.69761.41. [DOI] [PubMed] [Google Scholar]
- 15.Sosman JA, Puzanov I, Atkins MB. Opportunities and obstacles to combination targeted therapy in renal cell cancer. Clin Cancer Res. 2007;13:764s–9s. doi: 10.1158/1078-0432.CCR-06-1975. [DOI] [PubMed] [Google Scholar]
- 16.Motzer RJ, McCann L, Deen K. Pazopanib versus sunitinib in renal cancer. N Engl J Med. 2013;369:1970. doi: 10.1056/NEJMc1311795. [DOI] [PubMed] [Google Scholar]
- 17.Motzer RJ, Escudier B, Tomczak P, Hutson TE, Michaelson MD, Negrier S, et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: overall survival analysis and updated results from a randomised phase 3 trial. Lancet Oncol. 2013;14:552–62. doi: 10.1016/S1470-2045(13)70093-7. [DOI] [PubMed] [Google Scholar]
- 18.Rini BI, Halabi S, Rosenberg JE, Stadler WM, Vaena DA, Ou SS, et al. Bevacizumab plus interferon alfa compared with interferon alfa monotherapy in patients with metastatic renal cell carcinoma: CALGB 90206. J Clin Oncol. 2008;26:5422–8. doi: 10.1200/JCO.2008.16.9847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Escudier B, Pluzanska A, Koralewski P, Ravaud A, Bracarda S, Szczylik C, et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet. 2007;370:2103–11. doi: 10.1016/S0140-6736(07)61904-7. [DOI] [PubMed] [Google Scholar]
- 20.Tsao CK, Liaw B, He C, Galsky MD, Sfakianos J, Oh WK. Moving beyond vascular endothelial growth factor-targeted therapy in renal cell cancer: latest evidence and therapeutic implications. Ther Adv Med Oncol. 2017;9:287–98. doi: 10.1177/1758834016687261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choueiri TK, Halabi S, Sanford BL, Hahn O, Michaelson MD, Walsh MK, et al. Cabozantinib Versus Sunitinib As Initial Targeted Therapy for Patients With Metastatic Renal Cell Carcinoma of Poor or Intermediate Risk: The Alliance A031203 CABOSUN Trial. J Clin Oncol. 2017;35:591–7. doi: 10.1200/JCO.2016.70.7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Motzer RJ, Hutson TE, Ren M, Dutcus C, Larkin J. Independent assessment of lenvatinib plus everolimus in patients with metastatic renal cell carcinoma. The Lancet Oncology. 2016;17:e4–5. doi: 10.1016/S1470-2045(15)00543-4. [DOI] [PubMed] [Google Scholar]
- 23.Guo H, German P, Bai S, Barnes S, Guo W, Qi X, et al. The PI3K/AKT Pathway and Renal Cell Carcinoma. Journal of genetics and genomics = Yi chuan xue bao. 2015;42:343–53. doi: 10.1016/j.jgg.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atkins MB, Gravis G, Drosik K, Demkow T, Tomczak P, Wong SS, et al. Trebananib (AMG 386) in Combination With Sunitinib in Patients With Metastatic Renal Cell Cancer: An Open-Label, Multicenter, Phase II Study. J Clin Oncol. 2015;33:3431–8. doi: 10.1200/JCO.2014.60.6012. [DOI] [PubMed] [Google Scholar]
- 25.Zarrabi K, Fang C, Wu S. New treatment options for metastatic renal cell carcinoma with prior anti-angiogenesis therapy. J Hematol Oncol. 2017;10:38. doi: 10.1186/s13045-016-0374-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Motzer RJ, Rini BI, McDermott DF, Redman BG, Kuzel TM, Harrison MR, et al. Nivolumab for Metastatic Renal Cell Carcinoma: Results of a Randomized Phase II Trial. J Clin Oncol. 2015;33:1430–7. doi: 10.1200/JCO.2014.59.0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choueiri TK, Escudier B, Powles T, Tannir NM, Mainwaring PN, Rini BI, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2016;17:917–27. doi: 10.1016/S1470-2045(16)30107-3. [DOI] [PubMed] [Google Scholar]
- 28.Motzer RJ, Hutson TE, Glen H, Michaelson MD, Molina A, Eisen T, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015;16:1473–82. doi: 10.1016/S1470-2045(15)00290-9. [DOI] [PubMed] [Google Scholar]
- 29.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 30.Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol. 2007;8:622–32. doi: 10.1038/nrm2217. [DOI] [PubMed] [Google Scholar]
- 31.MacIntyre AC, Cutler DJ. The potential role of lysosomes in tissue distribution of weak bases. Biopharmaceutics & drug disposition. 1988;9:513–26. doi: 10.1002/bod.2510090602. [DOI] [PubMed] [Google Scholar]
- 32.Gotink KJ, Broxterman HJ, Labots M, de Haas RR, Dekker H, Honeywell RJ, et al. Lysosomal sequestration of sunitinib: a novel mechanism of drug resistance. Clin Cancer Res. 2011;17:7337–46. doi: 10.1158/1078-0432.CCR-11-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gotink KJ, Rovithi M, de Haas RR, Honeywell RJ, Dekker H, Poel D, et al. Cross-resistance to clinically used tyrosine kinase inhibitors sunitinib, sorafenib and pazopanib. Cellular oncology. 2015;38:119–29. doi: 10.1007/s13402-015-0218-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Colombo F, Trombetta E, Cetrangolo P, Maggioni M, Razini P, De Santis F, et al. Giant Lysosomes as a Chemotherapy Resistance Mechanism in Hepatocellular Carcinoma Cells. PloS one. 2014;9:e114787. doi: 10.1371/journal.pone.0114787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Azijli K, Gotink KJ, Verheul HMW. The Potential Role of Lysosomal Sequestration in Sunitinib Resistance of Renal Cell Cancer. Journal of Kidney Cancer and VHL. 2015;2:195–203. doi: 10.15586/jkcvhl.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhitomirsky B, Assaraf YG. Lysosomal sequestration of hydrophobic weak base chemotherapeutics triggers lysosomal biogenesis and lysosome-dependent cancer multidrug resistance. Oncotarget. 2015;6:1143–56. doi: 10.18632/oncotarget.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campbell MT, Millikan RE, Altinmakas E, Xiao L, Wen SJ, Siefker-Radtke AO, et al. Phase I trial of sunitinib and temsirolimus in metastatic renal cell carcinoma. Clin Genitourin Cancer. 2015;13:218–24. doi: 10.1016/j.clgc.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patel PH, Senico PL, Curiel RE, Motzer RJ. Phase I study combining treatment with temsirolimus and sunitinib malate in patients with advanced renal cell carcinoma. Clin Genitourin Cancer. 2009;7:24–7. doi: 10.3816/CGC.2009.n.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–8. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu SL, Chang A, Perazella MA, Okusa MD, Jaimes EA, Weiss RH, et al. The Nephrologist’s Tumor: Basic Biology and Management of Renal Cell Carcinoma. Journal of the American Society of Nephrology : JASN. 2016;27:2227–37. doi: 10.1681/ASN.2015121335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cancer Genome Atlas Research N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–9. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shenoy N, Vallumsetla N, Zou Y, Galeas JN, Shrivastava M, Hu C, et al. Role of DNA methylation in renal cell carcinoma. Journal of hematology & oncology. 2015;8:88. doi: 10.1186/s13045-015-0180-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hsieh JJ, Manley BJ, Khan N, Gao J, Carlo MI, Cheng EH. Overcome tumor heterogeneity-imposed therapeutic barriers through convergent genomic biomarker discovery: A braided cancer river model of kidney cancer. Seminars in cell & developmental biology. 2017;64:98–106. doi: 10.1016/j.semcdb.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choueiri TK, Vaziri SA, Jaeger E, Elson P, Wood L, Bhalla IP, et al. von Hippel-Lindau gene status and response to vascular endothelial growth factor targeted therapy for metastatic clear cell renal cell carcinoma. J Urol. 2008;180:860–5. doi: 10.1016/j.juro.2008.05.015. discussion 5-6. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Y, Kwok-Shing Ng P, Kucherlapati M, Chen F, Liu Y, Tsang YH, et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell. 2017;31:820–32.e3. doi: 10.1016/j.ccell.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morais C. Sunitinib resistance in renal cell carcinoma. J Kidney Cancer VHL. 2014;1:1–11. doi: 10.15586/jkcvhl.2014.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duran I, Lambea J, Maroto P, Gonzalez-Larriba JL, Flores L, Granados-Principal S, et al. Resistance to Targeted Therapies in Renal Cancer: The Importance of Changing the Mechanism of Action. Targeted oncology. 2017;12:19–35. doi: 10.1007/s11523-016-0463-4. [DOI] [PubMed] [Google Scholar]
- 49.Carew JS, Kelly KR, Nawrocki ST. Mechanisms of mTOR inhibitor resistance in cancer therapy. Targeted oncology. 2011;6:17–27. doi: 10.1007/s11523-011-0167-8. [DOI] [PubMed] [Google Scholar]
- 50.Kucejova B, Pena-Llopis S, Yamasaki T, Sivanand S, Tran TA, Alexander S, et al. Interplay between pVHL and mTORC1 pathways in clear-cell renal cell carcinoma. Mol Cancer Res. 2011;9:1255–65. doi: 10.1158/1541-7786.MCR-11-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kornakiewicz A, Solarek W, Bielecka ZF, Lian F, Szczylik C, Czarnecka AM. Mammalian Target of Rapamycin Inhibitors Resistance Mechanisms in Clear Cell Renal Cell Carcinoma. Curr Signal Transd T. 2013;8:210–8. doi: 10.2174/1574362409666140206222746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Foster KG, Fingar DC. Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J Biol Chem. 2010;285:14071–7. doi: 10.1074/jbc.R109.094003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao Y, Ehara H, Akao Y, Shamoto M, Nakagawa Y, Banno Y, et al. Increased activity and intranuclear expression of phospholipase D2 in human renal cancer. Biochem Biophys Res Commun. 2000;278:140–3. doi: 10.1006/bbrc.2000.3719. [DOI] [PubMed] [Google Scholar]
- 54.Adelaiye R, Ciamporcero E, Miles KM, Sotomayor P, Bard J, Tsompana M, et al. Sunitinib dose escalation overcomes transient resistance in clear cell renal cell carcinoma and is associated with epigenetic modifications. Molecular cancer therapeutics. 2015;14:513–22. doi: 10.1158/1535-7163.MCT-14-0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Campbell L, Jasani B, Griffiths DF, Gumbleton M. Phospho-4e-BP1 and eIF4E overexpression synergistically drives disease progression in clinically confined clear cell renal cell carcinoma. American journal of cancer research. 2015;5:2838–48. [PMC free article] [PubMed] [Google Scholar]
- 56.Nishikawa M, Miyake H, Harada K, Fujisawa M. Expression level of phosphorylated-4E-binding protein 1 in radical nephrectomy specimens as a prognostic predictor in patients with metastatic renal cell carcinoma treated with mammalian target of rapamycin inhibitors. Med Oncol. 2014;31:792. doi: 10.1007/s12032-013-0792-4. [DOI] [PubMed] [Google Scholar]
- 57.Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature. 2016;539:112–7. doi: 10.1038/nature19796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bielecka ZF, Malinowska A, Brodaczewska KK, Klemba A, Kieda C, Krasowski P, et al. Hypoxic 3D in vitro culture models reveal distinct resistance processes to TKIs in renal cancer cells. Cell Biosci. 2017;7:71. doi: 10.1186/s13578-017-0197-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Joosten SC, Hamming L, Soetekouw PM, Aarts MJ, Veeck J, van Engeland M, et al. Resistance to sunitinib in renal cell carcinoma: From molecular mechanisms to predictive markers and future perspectives. Biochim Biophys Acta. 2015;1855:1–16. doi: 10.1016/j.bbcan.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 60.Rajabi M, Mousa SA. The Role of Angiogenesis in Cancer Treatment. Biomedicines. 2017;5:34. doi: 10.3390/biomedicines5020034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Buczek M, Escudier B, Bartnik E, Szczylik C, Czarnecka A. Resistance to tyrosine kinase inhibitors in clear cell renal cell carcinoma: from the patient’s bed to molecular mechanisms. Biochim Biophys Acta. 2014;1845:31–41. doi: 10.1016/j.bbcan.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 62.Huang D, Ding Y, Zhou M, Rini BI, Petillo D, Qian CN, et al. Interleukin-8 mediates resistance to antiangiogenic agent sunitinib in renal cell carcinoma. Cancer Res. 2010;70:1063–71. doi: 10.1158/0008-5472.CAN-09-3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ishibashi K, Haber T, Breuksch I, Gebhard S, Sugino T, Kubo H, et al. Overriding TKI resistance of renal cell carcinoma by combination therapy with IL-6 receptor blockade. Oncotarget. 2017;8:55230–45. doi: 10.18632/oncotarget.19420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhu Y, Liu H, Xu L, An H, Liu W, Liu Y, et al. p21-activated kinase 1 determines stem-like phenotype and sunitinib resistance via NF-kappaB/IL-6 activation in renal cell carcinoma. Cell death & disease. 2015;6:e1637. doi: 10.1038/cddis.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Harmon CS, DePrimo SE, Figlin RA, Hudes GR, Hutson TE, Michaelson MD, et al. Circulating proteins as potential biomarkers of sunitinib and interferon-alpha efficacy in treatment-naive patients with metastatic renal cell carcinoma. Cancer chemotherapy and pharmacology. 2014;73:151–61. doi: 10.1007/s00280-013-2333-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu Y, Tran HT, Lin Y, Martin AM, Zurita A, Sternberg CN, et al. Baseline (BL) IL-6, IL-8, and VEGF as Predictive and Prognostic Markers for Overall Survival (OS) in Metastatic Renal Cell Carcinoma (mRCC) Patients (pts) Treated in a Phase III Trial of Pazopanib (PAZO) Versus Placebo (PL) European journal of cancer. 2011;47:S170–S. [Google Scholar]
- 67.Tran HT, Liu Y, Zurita AJ, Lin Y, Baker-Neblett KL, Martin AM, et al. Prognostic or predictive plasma cytokines and angiogenic factors for patients treated with pazopanib for metastatic renal-cell cancer: a retrospective analysis of phase 2 and phase 3 trials. The Lancet Oncology. 2012;13:827–37. doi: 10.1016/S1470-2045(12)70241-3. [DOI] [PubMed] [Google Scholar]
- 68.Martin D, Galisteo R, Gutkind JS. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J Biol Chem. 2009;284:6038–42. doi: 10.1074/jbc.C800207200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu CF, Bing NX, Ball HA, Rajagopalan D, Sternberg CN, Hutson TE, et al. Pazopanib efficacy in renal cell carcinoma: evidence for predictive genetic markers in angiogenesis-related and exposure-related genes. J Clin Oncol. 2011;29:2557–64. doi: 10.1200/JCO.2010.32.9110. [DOI] [PubMed] [Google Scholar]
- 70.Xu CF, Johnson T, Choueiri TK, Deen KC, Xue ZY, Spraggs CF, et al. Association of IL8 polymorphisms with overall survival in patients with renal cell carcinoma in COMPARZ (pazopanib versus sunitinib phase III study) Journal of Clinical Oncology. 2013;31 [Google Scholar]
- 71.Xu CF, Johnson T, Garcia-Donas J, Choueiri TK, Sternberg CN, Davis ID, et al. IL8 polymorphisms and overall survival in pazopanib- or sunitinib-treated patients with renal cell carcinoma. Br J Cancer. 2015;112:1190–8. doi: 10.1038/bjc.2015.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mizumoto A, Yamamoto K, Nakayama Y, Takara K, Nakagawa T, Hirano T, et al. Induction of epithelial-mesenchymal transition via activation of epidermal growth factor receptor contributes to sunitinib resistance in human renal cell carcinoma cell lines. J Pharmacol Exp Ther. 2015;355:152–8. doi: 10.1124/jpet.115.226639. [DOI] [PubMed] [Google Scholar]
- 73.Welti JC, Gourlaouen M, Powles T, Kudahetti SC, Wilson P, Berney DM, et al. Fibroblast growth factor 2 regulates endothelial cell sensitivity to sunitinib. Oncogene. 2011;30:1183–93. doi: 10.1038/onc.2010.503. [DOI] [PubMed] [Google Scholar]
- 74.Rankin EB, Fuh KC, Castellini L, Viswanathan K, Finger EC, Diep AN, et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc Natl Acad Sci U S A. 2014;111:13373–8. doi: 10.1073/pnas.1404848111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Singh H, Brave M, Beaver JA, Cheng J, Tang S, Zahalka E, et al. U.S. Food and Drug Administration Approval: Cabozantinib for the Treatment of Advanced Renal Cell Carcinoma. Clin Cancer Res. 2017;23:330–5. doi: 10.1158/1078-0432.CCR-16-1073. [DOI] [PubMed] [Google Scholar]
- 76.Ciamporcero E, Miles KM, Adelaiye R, Ramakrishnan S, Shen L, Ku S, et al. Combination strategy targeting VEGF and HGF/c-met in human renal cell carcinoma models. Mol Cancer Ther. 2015;14:101–10. doi: 10.1158/1535-7163.MCT-14-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Loges S, Schmidt T, Carmeliet P. “Antimyeloangiogenic” Therapy for Cancer by Inhibiting PIGF. Clinical Cancer Research. 2009;15:3648–53. doi: 10.1158/1078-0432.CCR-08-2276. [DOI] [PubMed] [Google Scholar]
- 78.van der Mijn JC, Mier JW, Broxterman HJ, Verheul HM. Predictive biomarkers in renal cell cancer: insights in drug resistance mechanisms. Drug Resist Updat. 2014;17:77–88. doi: 10.1016/j.drup.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 79.Brenner W, Farber G, Herget T, Lehr HA, Hengstler JG, Thuroff JW. Loss of tumor suppressor protein PTEN during renal carcinogenesis. International journal of cancer. 2002;99:53–7. doi: 10.1002/ijc.10303. [DOI] [PubMed] [Google Scholar]
- 80.Makhov PB, Golovine K, Kutikov A, Teper E, Canter DJ, Simhan J, et al. Modulation of Akt/mTOR Signaling Overcomes Sunitinib Resistance in Renal and Prostate Cancer Cells. Mol Cancer Ther. 2012;11:1510–7. doi: 10.1158/1535-7163.MCT-11-0907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tsavachidou-Fenner D, Tannir N, Tamboli P, Liu W, Petillo D, Teh B, et al. Gene and protein expression markers of response to combined antiangiogenic and epidermal growth factor targeted therapy in renal cell carcinoma. Ann Oncol. 2010;21:1599–606. doi: 10.1093/annonc/mdp600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Naito S, Makhov P, Astsaturov I, Golovine K, Tulin A, Kutikov A, et al. LDL cholesterol counteracts the antitumour effect of tyrosine kinase inhibitors against renal cell carcinoma. Br J Cancer. 2017;116:1203–7. doi: 10.1038/bjc.2017.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee CK, Marschner IC, Simes RJ, Voysey M, Egleston B, Hudes G, et al. Increase in cholesterol predicts survival advantage in renal cell carcinoma patients treated with temsirolimus. Clin Cancer Res. 2012;18:3188–96. doi: 10.1158/1078-0432.CCR-11-3137. [DOI] [PubMed] [Google Scholar]
- 84.Beretta GL, Cassinelli G, Pennati M, Zuco V, Gatti L. Overcoming ABC transporter-mediated multidrug resistance: The dual role of tyrosine kinase inhibitors as multitargeting agents. European journal of medicinal chemistry. 2017;142:271–89. doi: 10.1016/j.ejmech.2017.07.062. [DOI] [PubMed] [Google Scholar]
- 85.Hyafil F, Vergely C, Du Vignaud P, Grand-Perret T. In vitro and in vivo reversal of multidrug resistance by GF120918, an acridonecarboxamide derivative. Cancer Res. 1993;53:4595–602. [PubMed] [Google Scholar]
- 86.Sato H, Siddig S, Uzu M, Suzuki S, Nomura Y, Kashiba T, et al. Elacridar enhances the cytotoxic effects of sunitinib and prevents multidrug resistance in renal carcinoma cells. European journal of pharmacology. 2015;746:258–66. doi: 10.1016/j.ejphar.2014.11.021. [DOI] [PubMed] [Google Scholar]
- 87.Shibayama Y, Nakano K, Maeda H, Taguchi M, Ikeda R, Sugawara M, et al. Multidrug resistance protein 2 implicates anticancer drug-resistance to sorafenib. Biological & pharmaceutical bulletin. 2011;34:433–5. doi: 10.1248/bpb.34.433. [DOI] [PubMed] [Google Scholar]
- 88.Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England journal of medicine. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gowrishankar B, Ibragimova I, Zhou Y, Slifker MJ, Devarajan K, Al-Saleem T, et al. MicroRNA expression signatures of stage, grade, and progression in clear cell RCC. Cancer Biol Ther. 2014;15:329–41. doi: 10.4161/cbt.27314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dietz S, Sultmann H, Du Y, Reisinger E, Riediger AL, Volckmar AL, et al. Patient-specific molecular alterations are associated with metastatic clear cell renal cell cancer progressing under tyrosine kinase inhibitor therapy. Oncotarget. 2017;8:74049–57. doi: 10.18632/oncotarget.18200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hatiboglu G, Hohenfellner M, Arslan A, Hadaschik B, Teber D, Radtke JP, et al. Effective downsizing but enhanced intratumoral heterogeneity following neoadjuvant sorafenib in patients with non-metastatic renal cell carcinoma. Langenbeck Arch Surg. 2017;402:637–44. doi: 10.1007/s00423-016-1543-8. [DOI] [PubMed] [Google Scholar]
- 92.Stewart GD, O’Mahony FC, Laird A, Eory L, Lubbock AL, Mackay A, et al. Sunitinib Treatment Exacerbates Intratumoral Heterogeneity in Metastatic Renal Cancer. Clin Cancer Res. 2015;21:4212–23. doi: 10.1158/1078-0432.CCR-15-0207. [DOI] [PubMed] [Google Scholar]
- 93.Gupta V, Bassi DE, Simons JD, Devarajan K, Al-Saleem T, Uzzo RG, et al. Elevated expression of stromal palladin predicts poor clinical outcome in renal cell carcinoma. PLoS One. 2011;6:e21494. doi: 10.1371/journal.pone.0021494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A. 2009;106:6742–7. doi: 10.1073/pnas.0902280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ko JS, Rayman P, Ireland J, Swaidani S, Li G, Bunting KD, et al. Direct and differential suppression of myeloid-derived suppressor cell subsets by sunitinib is compartmentally constrained. Cancer Res. 2010;70:3526–36. doi: 10.1158/0008-5472.CAN-09-3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Miles KM, Seshadri M, Ciamporcero E, Adelaiye R, Gillard B, Sotomayor P, et al. Dll4 blockade potentiates the anti-tumor effects of VEGF inhibition in renal cell carcinoma patient-derived xenografts. PLoS One. 2014;9:e112371. doi: 10.1371/journal.pone.0112371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Patel NS, Li JL, Generali D, Poulsom R, Cranston DW, Harris AL. Up-regulation of delta-like 4 ligand in human tumor vasculature and the role of basal expression in endothelial cell function. Cancer Res. 2005;65:8690–7. doi: 10.1158/0008-5472.CAN-05-1208. [DOI] [PubMed] [Google Scholar]
- 98.Xiao W, Gao Z, Duan Y, Yuan W, Ke Y. Notch signaling plays a crucial role in cancer stem-like cells maintaining stemness and mediating chemotaxis in renal cell carcinoma. J Exp Clin Cancer Res. 2017;36:41. doi: 10.1186/s13046-017-0507-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Crawford Y, Kasman I, Yu L, Zhong C, Wu X, Modrusan Z, et al. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell. 2009;15:21–34. doi: 10.1016/j.ccr.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 100.Azad NS, Posadas EM, Kwitkowski VE, Steinberg SM, Jain L, Annunziata CM, et al. Combination targeted therapy with sorafenib and bevacizumab results in enhanced toxicity and antitumor activity. J Clin Oncol. 2008;26:3709–14. doi: 10.1200/JCO.2007.10.8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Feldman DR, Baum MS, Ginsberg MS, Hassoun H, Flombaum CD, Velasco S, et al. Phase I trial of bevacizumab plus escalated doses of sunitinib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:1432–9. doi: 10.1200/JCO.2008.19.0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rini BI, Wilding G, Hudes G, Stadler WM, Kim S, Tarazi J, et al. Phase II study of axitinib in sorafenib-refractory metastatic renal cell carcinoma. J Clin Oncol. 2009;27:4462–8. doi: 10.1200/JCO.2008.21.7034. [DOI] [PubMed] [Google Scholar]
- 103.Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–56. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 104.Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H, et al. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J Immunol. 2015;194:950–9. doi: 10.4049/jimmunol.1401686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Thompson RH, Kuntz SM, Leibovich BC, Dong H, Lohse CM, Webster WS, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow-up. Cancer Res. 2006;66:3381–5. doi: 10.1158/0008-5472.CAN-05-4303. [DOI] [PubMed] [Google Scholar]
- 106.Yang JC, Hughes M, Kammula U, Royal R, Sherry RM, Topalian SL, et al. Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J Immunother. 2007;30:825–30. doi: 10.1097/CJI.0b013e318156e47e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. The New England journal of medicine. 2015;373:1803–13. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.McDermott DF, Sosman JA, Sznol M, Massard C, Gordon MS, Hamid O, et al. Atezolizumab, an Anti-Programmed Death-Ligand 1 Antibody, in Metastatic Renal Cell Carcinoma: Long-Term Safety, Clinical Activity, and Immune Correlates From a Phase Ia Study. J Clin Oncol. 2016;34:833–42. doi: 10.1200/JCO.2015.63.7421. [DOI] [PubMed] [Google Scholar]
- 109.Amin A, Plimack ER, Infante JR, Ernstoff MS, Rini BI, McDermott DF, et al. Nivolumab (anti-PD-1; BMS-936558, ONO-4538) in combination with sunitinib or pazopanib in patients (pts) with metastatic renal cell carcinoma (mRCC) Journal of Clinical Oncology. 2014;32:5010. [Google Scholar]
- 110.Ross K, Jones RJ. Immune checkpoint inhibitors in renal cell carcinoma. Clin Sci (Lond) 2017;131:2627–42. doi: 10.1042/CS20160894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McDermott DF, Atkins MB, Motzer RJ, Rini BI, Escudier BJ, Fong L, et al. A phase II study of atezolizumab (atezo) with or without bevacizumab (bev) versus sunitinib (sun) in untreated metastatic renal cell carcinoma (mRCC) patients (pts) Journal of Clinical Oncology. 2017;35:431. [Google Scholar]
- 112.Hammers HJ, Plimack ER, Sternberg C, McDermott DF, Larkin JMG, Ravaud A, et al. CheckMate 214: A phase III, randomized, open-label study of nivolumab combined with ipilimumab versus sunitinib monotherapy in patients with previously untreated metastatic renal cell carcinoma. Journal of Clinical Oncology. 2015;33:TPS4578–TPS. [Google Scholar]
- 113.Escudier B, Tannir NM, McDermott DF, Frontera OA, Melichar B, Plimack ER, et al. LBA5CheckMate 214: Efficacy and safety of nivolumab + ipilimumab (N+I) v sunitinib (S) for treatment-naïve advanced or metastatic renal cell carcinoma (mRCC), including IMDC risk and PD-L1 expression subgroups. Annals of Oncology. 2017;28:mdx440.029–mdx440.029. [Google Scholar]
- 114.O’Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev. 2017;52:71–81. doi: 10.1016/j.ctrv.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 115.Figlin RA, Wood CG, Group AS ADAPT: An ongoing international phase III randomized trial of autologous dendritic cell immunotherapy (AGS-003) plus standard treatment in advanced renal cell carcinoma (RCC) Journal of Clinical Oncology. 2014;32:449. [Google Scholar]
- 116.Rini BI, Stenzl A, Zdrojowy R, Kogan M, Shkolnik M, Oudard S, et al. IMA901, a multipeptide cancer vaccine, plus sunitinib versus sunitinib alone, as first-line therapy for advanced or metastatic renal cell carcinoma (IMPRINT): a multicentre, open-label, randomised, controlled, phase 3 trial. The Lancet Oncology. 2016;17:1599–611. doi: 10.1016/S1470-2045(16)30408-9. [DOI] [PubMed] [Google Scholar]