

Abstract
To investigate the cyanylated cysteine vibrational probe group’s ability to report on binding-induced changes along a protein–protein interface, the probe group was incorporated at several sites in a peptide of the calmodulin (CaM)-binding domain of skeletal muscle myosin light chain kinase. Isothermal titration calorimetry was used to determine the binding thermodynamics between calmodulin and each peptide. For all probe positions, the binding affinity was nearly identical to that of the unlabeled peptide. The CN stretching infrared band was collected for each peptide free in solution and bound to calmodulin. Binding-induced shifts in the IR spectral frequencies were correlated with estimated solvent accessibility based on molecular dynamics simulations. This work generally suggests (1) that site-specific incorporation of this vibrational probe group does not cause major perturbations to its local structural environment and (2) that this small probe group might be used quite broadly to map dynamic protein-binding interfaces. However, site-specific perturbations due to artificial labeling groups can be somewhat unpredictable and should be evaluated on a site-by-site basis through complementary measurements. A fully quantitative, simulation-based interpretation of the rich probe IR spectra is still needed but appears to be possible given recent advances in simulation techniques.
Site-specific vibrational spectroscopy is an attractive approach to study binding interactions between proteins or other biomolecules without the sampling and dynamic constraints of either crystallography or NMR. Vibrational dynamics occur on femtosecond-picosecond (fs-ps) time scales, so vibrational spectral line shapes from ensemble samples inherently report on the dynamic structural distributions of biomolecules. Thus, vibrational spectroscopy should be an ideal technique for viewing binding-induced changes in a protein’s dynamic structure. However, absorption bands from common biomolecular functional groups are crowded into a few mid-infrared frequency ranges, and the assignment of site-specific vibrational bands is normally very difficult: infrared (IR) absorption in the particularly strong amide I region is most often used as a probe of only the total protein secondary structure content.1
One strategy that circumvents the problem of vibrational spectral overlap is using a functional group with a vibrational frequency in an isolated region of the spectrum.2−5 The stretching frequency of the nitrile (C≡N) moiety falls in a clear region of the IR spectrum; using nitriles as site-specific probes in proteins was first implemented by Gai and DeGrado.6 Since that initial example, the nitrile group has been placed at specific sites in many biomolecules;2,6−20 however, there was not a clear indication in many cases of the functional or structural perturbation caused by the presence of the artificial nitrile group.
Carbon-bound nitriles can be effective site-specific probes, but amino acids with carbon-linked nitriles must either be incorporated into proteins synthetically or through novel expression approaches. Another approach is to post-translationally modify a naturally occurring amino acid, cysteine, to add the nitrile group as part of a covalently bound thiocyanate moiety in the artificial amino acid β-thiocyanatoalanine, or cyanylated cysteine (C*).11,14,16,17,21,22 The SC≡N stretching band of C* appears in the IR spectrum between 2153 and 2164 cm–1 in biologically relevant environments.23−25 The C≡N frequency depends on the local electrostatic environment26,27 and also strongly on H-bond donors like water. Frequency shifts of the SC≡N stretching mode indicated changes in water exposure at specific protein sites bound to membranes15 and other proteins,16 and in a dynamically opening enzymatic active site,28 and also in exposure or sequestration of the substrate-carrying arm of acyl carrier proteins.29 Smaller frequency shifts were used to examine electric fields inside folded proteins9 and enzymatic active sites10,12,30 and along the Ras-effector protein–protein interface.31−34 The SC≡N line width depends on the structural heterogeneity of the probe’s environment and on the fs-ps time scale fluctuations of that environment.16,17,23,35 Line widths of the SC≡N band at water-exposed sites display dynamic differences due to nearby order/disorder transitions in the protein,16,17 while line width changes for SC≡N groups that are not uniformly solvent-exposed have been interpreted as evidence for the local distribution of probe environments.14,15
The vibrational lifetime of the C≡N stretching mode in the SCN probe group is quite long, on the order of many 10s to 100s of ps according to recent reports based on pump–probe and other nonlinear IR experiments.35,36 However, the strength of this absorption signal is weak enough that despite some serious effort, clear nonlinear C≡N IR signals from SCN probes in proteins have not been reported except for in one case.35
Previous studies of both C*17,31 and of carbon-bound nitriles18 showed that there is a small perturbation of folded states due to the artificial amino acid. C* caused a minor perturbation to local helical secondary structure17 but did not preclude binding between peptides and membranes.15 C* did also not abrogate binding between two proteins as assessed by the activity of their cooperative enzymatic complex,31,32 except for sites of extreme electrostatic perturbation by the substitution of C* for a native residue. The most relevant function of regulatory proteins is their binding to targets: whether a small probe like C* disrupts this function when placed directly along the binding interface is an open question. In this work, we examine the binding of a ubiquitous and canonical regulatory protein, calmodulin (CaM), to a relatively well-characterized target peptide from skeletal muscle myosin light chain kinase (“M13”; Figure 1),37 and we place the C* probe group directly in the binding interface to assess both the degree of functional perturbation and the ability of the probe to report on the binding event.
Figure 1.
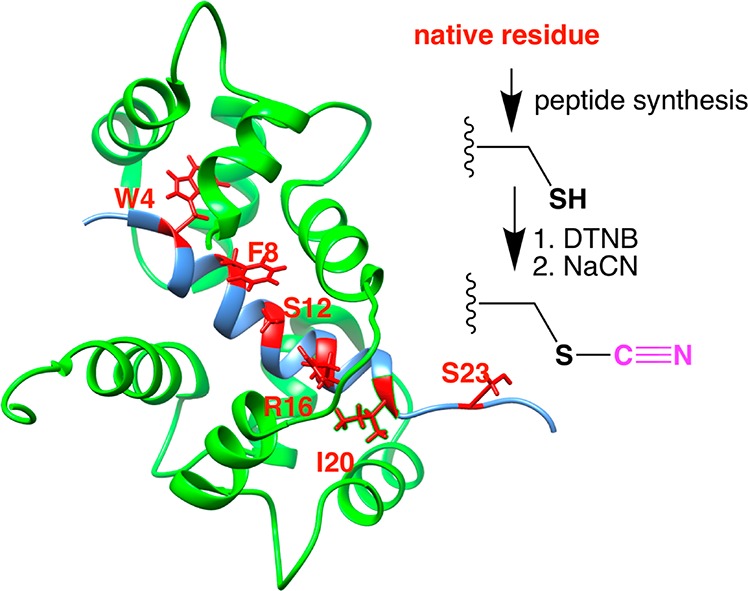
Calcium-saturated calmodulin (green, with Ca2+ ions omitted) bound to M13 (blue) (left). Residues replaced with cyanylated cysteine are highlighted in red. Adapted from coordinates from PDB structure 2BBM.1 Scheme for replacement of each native residue by cyanylated cysteine (C*) (right).
CaM is a particularly interesting regulatory protein due to its promiscuity of interactions and its context-dependent structure.38−40 CaM contains two globular domains connected by a flexible linker; each domain contains two EF-hand helix–loop–helix Ca2+-binding motifs. Ca2+ binding by CaM leads to large conformational changes and increases CaM’s affinity for many target proteins whose functions are regulated by CaM binding.41 The modular structure of CaM and the relatively high proportion of flexible methionine side chains along its binding surfaces allow CaM to assume a variety of target-bound conformations, ranging from partially extended (utilizing only one domain, such as for the Ca2+ pump “C20W” peptide42) to completely collapsed (where both subdomains “wrap around” the target sequence). Successful previous incorporation of C* in helical peptide and protein segments9,15,17,28 suggested that C* could be particularly useful in CaM-target complexes, which usually induce the target peptide to adopt a helical conformation. While CaM serves in this study mainly as a relatively well-characterized model system for protein–protein binding, any new binding-related dynamic information that we uncover would also help to better understand CaM’s unique promiscuity. CaM dynamics have been a subject of substantial interest, with many different experiments performed to examine them: residual conformational entropy in the CaM/M13 complex is suggested by NMR relaxation parameters,43,44 a limited loss of entropy in the bound state has been implicated as a significant thermodynamic driving force for target binding,43,45−49 and some refinement of the overall secondary structure on binding was suggested by FTIR/isotopic labeling experiments focused on the amide backbone of CaM.50
Our specific goals here with the M13 target peptide and calcium-loaded CaM are 3-fold: (1) to measure the perturbation of binding by C* substituted for a range of hydrophobic or otherwise substantially different naturally occurring side chains along the protein–protein-binding interface, (2) to document the response of C* to a range of interfacial environments of varying solvent exposure and structural flexibility, and (3) to provide experimental design guidance and interpretive tools for applying C* and other vibrational labels directly at other binding interfaces. An ancillary goal for future work is to better understand the structural modularity, flexibility, and dynamics of CaM and its targets as they bind to each other. For the CaM-bound M13 peptide, we chose probe sites from hydrophobic “anchor” residues near the peptide’s N-terminus to residues closer to the unstructured and solution-exposed peptide C-terminus. (See Table 1 and Figure 1.) A simulated estimate of solvent-accessible surface area (SASA) for the labeled residues was used to assess possible correlation between SASA and the measured C≡N vibrational frequencies, and both frequencies and line shapes were compared to thermodynamic quantities from isothermal titration calorimetry (ITC) experiments. We found that C* was minimally perturbative at all sites according to ITC. The probe IR mean frequencies were consistent with expectations based on SASA in simulations started from the NMR structure. The probe line widths for some solvent-excluded probes were unexpectedly broad, suggesting that there is additional, new information that could be extracted from the line widths via a more quantitative and simulation-based technique. We conclude that while this probe methodology could be broadly useful for mapping binding interfaces of proteins (since the mean probe frequency shift is driven strongly by solvent exposure), a truly quantitative interpretation of the probe line shapes remains to be achieved.
Table 1. Sequences and Abbreviations for Seven Synthetic Variants of the “M13” Peptidea.
| peptide | amino acid sequence |
|---|---|
| M13 | Ac-KRRWKKNFIAVSAANRFKKISSSGAL-NH2 |
| W4C*b | Ac-KRRC*KKNFIAVSAANRFKKISSSGALW-NH2 |
| F8C* | Ac-KRRWKKNC*IAVSAANRFKKISSSGAL-NH2 |
| S12C* | Ac-KRRWKKNFIAVC*AANRFKKISSSGAL-NH2 |
| R16C* | Ac-KRRWKKNFIAVSAANC*FKKISSSGAL-NH2 |
| I20C* | Ac-KRRWKKNFIAVSAANRFKKC*SSSGAL-NH2 |
| S23C* | Ac-KRRWKKNFIAVSAANRFKKISSC*GAL-NH2 |
Which is the CaM-binding domain of skeletal muscle myosin light chain kinase.
An addition of an additional tryptophan residue at the C-terminus was made to the W4C* peptide to facilitate the accurate determination of its concentration in solution.
Materials and Methods
Peptide Synthesis and Purification
All peptides were synthesized on an Applied BioSystems ABI 433A synthesizer using standard fmoc chemistry with either 10× HBTU or HATU activator. Fmoc-labeled amino acids and all peptide synthesis reagents were purchased from Applied BioSystems or Advanced ChemTech and used as received. The PAL resin was used to furnish a C-terminal carboxamide on cleavage; treatment of the resin-bound peptides with acetic anhydride was used to affect N-acetylation. Cleaved peptides were purified via reversed-phased HPLC using a semiprep scale Varian Dynamax C18 column. The eluent was 20–40% acetonitrile in H2O (with 0.1% trifluoroacetic acid as modifier) over 40 min, with each peptide eluting at approximately 30% acetonitrile. Purity and identity of peptides was verified by MALDI-MS (performed either at the Wistar Institute, Philadelphia, PA or in the Biophysics/Molecular Biology Department, University of Pennsylvania, Philadelphia, PA).
Calmodulin Expression and Purification
Mammalian sequence calmodulin was produced recombinantly in Escherichia coli and purified according to extensive previous work.51,52
Cyanylation of Peptides
Lyophilized cysteine-containing peptides purified by HPLC were treated for 20 min with a 0.01 M HCl solution and relyophilized to remove residual trifluoroacetic acid (TFA). The lyophilized peptides were then dissolved in 200 mM HEPES-NaOH buffer, pH 7, and treated with 100× D,L-dithiothreitol (DTT, from Aldrich) to yield the free thiol at each cysteine. DTT was separated from the peptides using a 35 cm column of Sephadex G-10, and the reduced peptide was lyophilized. The peptides were then redissolved in a 250 mM HEPES-NaOH buffer, pH 7, and treated with 3× 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB, from Acros) for 20 min at room temperature to form a mixed disulfide intermediate at cysteine. The samples were then treated with 40× NaCN (Aldrich), and the cyanylated peptides were isolated using the same Sephadex G-10 column equilibrated with 20 mM HEPES-NaOH, pH 7. Peptide-containing fractions in 20 mM HEPES-NaOH were combined and then diluted with a CaCl2 solution to 10 mM HEPES-NaOH and 10 mM CaCl2. Peptides in this buffer were then concentrated about 5× using a stirred ultrafiltration cell (Amicon, Millipore) with a 1000 MWCO membrane (SpectraPor). The presence of the C≡N label was verified using IR spectroscopy (see below). The final sample concentration was 1–2 mM C*-containing peptide in 10 mM HEPES-NaOH and 10 mM CaCl2.
Preparation of Calmodulin-Bound Peptide Samples for Infrared Spectroscopy
Purified calmodulin and peptides were each dissolved in 10 mM HEPES and 10 mM CaCl2 buffer at pH 7.00–7.05 with their concentrations determined via absorption at 280 nm using a Jasco V-570 UV/vis/NIR spectrophotometer. Extinction coefficients were calculated based on the sequences of CaM and the M13 peptide.53 Calmodulin and peptide solutions were combined at a molar ratio of 1.2 protein:1.0 peptide and concentrated to a peptide concentration > 1 mM using a Millipore Amicon 3 mL stirred ultrafiltration cell with a 1000 MWCO ultrafiltration membrane from Spectra-Por. The final total volume was 50–75 μL. These samples were used for IR measurements with no further changes.
Infrared Spectroscopy
About 30 μL of a cyanylated peptide sample from either of the previous two preparations was placed between the windows of a 22 μm CaF2 BioCell (BioTools, Jupiter, FL) inside a BioJack temperature circulating jacket at 22 °C. All spectra were collected using 1024 scans at 2 cm–1 resolution using a Bruker Optics Vertex 70 FTIR spectrometer with a photovoltaic HgCdTe detector. A spectrum of buffer solution was subtracted from each raw protein or peptide spectrum, and a further baseline correction was accomplished by fitting the baseline (outside the region from 2140 to 2185 cm–1) to a polynomial and subtracting the fit.
Line Shape Analysis
C≡N stretching line shapes were analyzed without any assumptions about the underlying symmetry or shape of the bands, since there is no particular reason that the bands should be symmetric about the mean and because line shape asymmetry could result from multiple underlying subdistributions. The mode (most probable, or maximum) frequency of each band was directly drawn from the data. Mean C≡N stretching frequencies were calculated using eq 1 for the first central moment of a distribution:
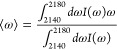 |
1 |
where ω is the frequency in wavenumbers and I (ω) is the absorbance as a function of frequency. The full width at half-maximum was directly extracted from the data, where two points were determined whose intensity was half that of the intensity at the mode frequency.
Isothermal Titration Calorimetry
ITC data were collected on a TA Instruments (New Castle, DE) Nano ITC low-volume instrument equilibrated at 25 °C. The cell was filled with 350 mL of calmodulin at a concentration of 25–50 μM in 10 mM Hepes-NaOH and 10 mM CaCl2, pH 7.04 buffer. The syringe was filled with 50 μL of 200–500 μM peptide in the same batch of buffer (see above). The signal from the first injection was ignored and was followed by 24 × 2 μL injections while stirring at 300 rpm. See the Supporting Information for data and fits from the ITC experiments.
Molecular Dynamics Simulations
The reported NMR structure of the CaM-M13 complex37 was used as the starting structure for MD simulations in GROMACS v. 5.0.354 using the Amber99SB force field55 with a modification that provides Amber parameters for the artificial SCN probe group56 based partly on simulations of methyl thiocyanate in water.25 Simulations were run on either 3 16-processor nodes of TACC’s Stampede supercomputer, which was available through a startup allocation from XSEDE, or the Fock cluster at Haverford College. The SCN probe group was inserted using an automated script that replaces the residue in question with C*, and the SC≡N group’s orientation was allowed to equilibrate with the rest of the protein before any analysis of the production trajectory. After extensive equilibration following the example of Layfield and Hammes–Schiffer56 and further observation of the RMSD and radius of gyration to establish thermal equilibration, simulations with the probe group included were run for 20 ns, and the solvent-accessible surface area of the C* side chain N atom was calculated using the gmx sasa command in GROMACS. Simulated frames were not Boltzmann-weighted by total energy, but rather directly averaged to estimate the mean solvent-accessible surface area for the probe group at each site from the thermally equilibrated simulations.
Results
Peptide Synthesis and Cyanylation
Six single-cysteine-variant peptides (see Table 1 for sequences, and Figure 1 for the location of the replaced residues in the previously reported NMR structure and the scheme used to make the change to C*) were made via solid-phase peptide synthesis, purified, and cyanylated at the single-cysteine substitution site. All six cysteine-variant peptides were successfully cyanylated with near-quantitative yield. None of the variant peptides exhibited a substantially different solubility compared to the native-sequence peptide. Changing buffers for such short peptides and concentrating the samples for IR spectroscopy proved challenging and was only enabled with some difficulty by the very low MW cutoff of the membranes used for buffer exchange and concentrating (see experimental methods); we have found that working with larger cysteine-variant proteins than the M13 peptide is more practical for this particular probe methodology.
Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) was used to determine for each site how the C* substitution affected the equilibrium between free and bound species. Table 2 presents the calculated binding stoichiometries (n), dissociation constants (Kd), and enthalpies of binding (ΔH) for all seven peptides with CaM under Ca2+-saturated conditions, and Table S1 displays complete binding thermodynamics including free energies, entropies, and changes in binding free energy, binding enthalpy, and binding entropy due to probe substitution. The ITC data and fits to the data appear in the Supporting Information as well. Fitted parameters are generally accurate to 1–2 significant figures only, and the range of error for enthalpies and entropies is relatively large compared to the magnitude of those quantities.
Table 2. Binding Parameters Calculated From ITC Measurements for each M13 Variant Peptide with CaMa.
| peptide | Kd (μM) | ΔH (kJ/mol) | n |
|---|---|---|---|
| M13 | 0.5 | –59 | 0.9 |
| W4C* | 0.3 | –43 | 0.9 |
| F8C* | 1.1 | –27 | 0.9 |
| 0.02 | –26 | 0.3 | |
| S12C* | 2 | –64 | 1.0 |
| R16C* | 2 | –32 | 1.1 |
| I20C* | 1.4 | –72 | 1.0 |
| S23C* | 3 | –65 | 1.0 |
See the Supporting Information for the data and details of these fits.
All of the fitted binding parameters for the interactions between CaM and each variant peptide, especially Kd and n, are in close agreement with the CaM-binding values of the unmodified M13 peptide. The F8C* peptide-CaM interaction behaves somewhat differently, as discussed below, but in all cases the Kd’s determined for the CaM-labeled peptide complexes were within 1 order of magnitude of the Kd for the complex between CaM and the unmodified peptide. The measured dissociation constants indicate that, at the sample concentrations of the IR experiments, all peptides were fully bound to CaM given the slight stoichiometric excess of CaM in the spectroscopic samples. With the possible exception of the F8C* peptide, all CaM-labeled peptide-binding interactions followed a single sigmoidal titration curve with close to the expected n = 1 binding stoichiometry.
The ITC signal-to-noise ratio for the F8C* peptide-CaM interaction was large enough to indicate what appears to be a two-transition titration curve for the F8C* peptide with CaM (Supporting Information). Two transitions were observed whether the peptide or CaM was titrated vs the other species. Neither of the Kd’s determined for the two F8C*-CaM-binding processes is weaker than for the unmodified peptide-CaM interaction, and the two binding processes together add up to an approximate total n = 1 binding stoichiometry.
Infrared Spectroscopy
The IR spectra in the C≡N stretching region of both the unbound and CaM-bound M13 peptide variants are shown in Figure 2. In all cases, there are significant differences between the C≡N stretching bands for the unbound and CaM-bound peptides. Line shape changes for each C≡N stretching band are reported in Table 3, and full line shape parameters for each spectrum can be found in the Supporting Information.
Figure 2.

Infrared absorption spectra for each peptide in the CN stretching region: free (black line) and bound to calmodulin (red line). A, W4C*; B, F8C*; C, S12C*; D, R16C*; E, I20C*; F, S23C*. The left panels (A, B, C) are the most buried locations according to the NMR structure, and the right panels (D, E, F) are predicted to be increasingly solvent-exposed.
Table 3. Line Shape Changes for the CN Stretching Bands (from Figure 2) of Probe-Labeled Peptides Bound to CaMa.
| peptide | Δmode (cm–1) | Δmean (cm–1) | Δfwhm (cm–1) | mean SASA (Å2) |
|---|---|---|---|---|
| W4C* | –8.6 | –6.0 | +3.1 | 0.25 |
| F8C* | –5.8 | –7.4 | +1.1 | 0.01 |
| S12C* | –6.7 | –5.1 | –0.8 | 6.33 |
| R16C* | –1.9 | –1.3 | +4.4 | 6.46 |
| I20C* | –2.9 | –0.8 | –1.5 | 7.92 |
| S23C* | –1.0 | –1.0 | +0.7 | 17.6 |
Full line shape analysis parameters are reported in the Supporting Information. SASA values are calculated for the SCN group’s N atom.
The spectra in Figure 2A–C are for sites that the solution NMR structure37 predicts to have significant hydrophobic character. The W4 “hydrophobic anchor” residue was replaced with C* and upon binding to CaM, the mode frequency of the W4C* probe shifted by −8.6 cm–1 (Figure 2A). The SC≡N stretching frequency also shifted by −5.8 and −6.7 cm–1 for probes at the hydrophobic peptide residues F8C* and S12C*, respectively. In two of these cases, there was also a significant broadening of the C≡N stretching band: for W4C*, the full width at half-maximum (fwhm) increased by about 3 cm–1, and in F8C*, the fwhm broadened by about 1 cm–1.
Figure 2D–F exhibits sites with the probe group in a progressively more water-exposed structural environment. The IR signal from R16C*, which in the M13-CaM NMR structure is pointed roughly toward the linker region between the two domains of CaM, exhibits a small binding-induced frequency shift of −1.9 cm–1 and a large broadening of +4.4 cm–1. The I20C* probe is located near the boundary between solution exposure and the binding interface according to the NMR structure: while the I20C* band does not broaden upon binding, the C≡N frequency still shifts by −1.5 cm–1. The S23C* probe was intended to be a control, since the NMR structure indicates that S23 is completely water-exposed and the peptide is unstructured in that region of the sequence regardless of CaM; however, a small shift of −1.0 cm–1 and broadening of +0.7 cm–1 were still detected for the CaM-bound S23C* site.
Solvent-Exposure Estimates
We used all-atom MD simulations to estimate the solvent exposure of the six labeled sites in the CaM-bound peptide from the NMR structure. The SCN label was placed explicitly at each site, and the solvent-accessible surface area of the C* side chain’s nitrogen atom was evaluated every 1 ps during the simulation. While we started the C* in several different local orientations, it generally collapsed within a few ps to a stable orientation vs its local environment for the remainder of the simulated trajectory. (See Figure S2 for those orientations.) From the MD trajectories, the mean solvent-accessible surface area was calculated. Mean values for SASA appear in the last column of Table 3, and distributions of SASA appear in Figure S3.
Correlation Analysis
Correlation plots comparing the binding-induced frequency shifts and line width changes with four quantities of interest are shown in Figure 3. Those quantities include changes in the mean SASA at each probe site (first row) and thermodynamics measured by ITC including probe-induced changes in the free energy (ΔΔG), enthalpy (ΔΔH), and entropy (ΔΔS) of binding. Since the free energies of binding change very little with each probe placement, there is a strong inverse relationship between ΔΔH and ΔΔS (which is common for biomolecular-binding events and not unique to this case). Notable correlations are observed between the binding-induced CN frequency shift and SASA and between the line width change and ΔΔH and ΔΔS; otherwise there are no other strong correlations evident.
Figure 3.
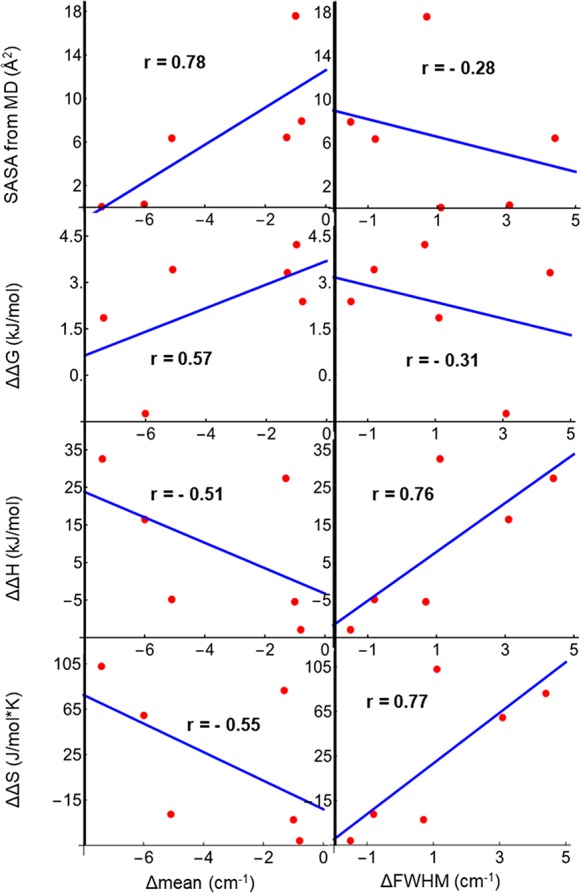
Linear correlation plots between the change in mean CN stretching frequency (first column) or change in fwhm (second column) with simulated SASA and probe-induced changes in binding free energy, binding enthalpy, and binding entropy. Correlation coefficients are displayed on each plot.
Discussion
Nonperturbation of Binding by Probe Groups
The experimental ITC data (Table 2 and Table S1) indicate that in all cases the probe is only minimally perturbative to the functional binding of CaM to this target peptide. This directly functional measurement demonstrates that C* is an innocent observer of this protein–protein interface rather than a strong participant in its structural environment. This is a very different result than might be expected for other, larger probe groups for other spectroscopies (i.e., spin labels or fluorescent dyes) and strongly supports the future placement of C* and possibly other vibrational probes directly inside binding interfaces.
There are small changes in the binding enthalpy (ΔΔH) and binding entropy (ΔΔS) associated with each probe placement (values in Table S1), and these changes do not appear to be predictable or systematic based on the NMR structure or the presumed environment of the native side chains and the C* residues that replace them. The overall binding free energies change very little, so ΔΔH and ΔΔS (here determined mainly from the ITC injection integrations) largely balance each other out in a manner similar to what has been widely documented in other systems.57−59 Each of the probes has a different solvent exposure in the bound complex (SASA estimates in Table 3), and solvation does not appear to be a major factor in the ΔΔH and ΔΔS values. Despite CaM’s highly conserved sequence, CaM binding has been generally shown (see below) to be somewhat mutation-tolerant, but our ITC data for both labels on the M13 peptide in this work and labels on CaM62 show that C* is functionally nonperturbative to its environment in this particular complex.
For W4C*, the large change in both hydrophobicity and residue volume at the W4 label site does not lead to a large difference in the CaM-peptide-binding affinity. This is surprising given the “anchor” identity of W4 in complex formation and the changes in both size and hydrophobicity between the native tryptophan and artificial C* side chains at that site. (However, it was previously shown that the W4 residue is not necessary for binding in this complex.60)
For the F8C*-labeled peptide, ITC indicates a label-induced change in binding behavior between the peptide and CaM. Without further structural data it is difficult to determine the origin of this change, which apparently leads to two distinguishable binding processes, although the major transition has a similar (a factor of 2 weaker) CaM affinity as the unlabeled peptide. Incomplete cyanylation is not the explanation for this behavior given the yield of the cyanylation reaction for that peptide, which was near unity for all samples, and repetition with separately cyanylated batches of F8C* yielding similar results even with the titrant and titrate solutions reversed in the ITC experiment.
The S12C* substitution is slightly more conservative than replacing the hydrophobic W4 and F8 sites, and the binding is weakened by a factor of 4 only, which corresponds to favoring the dissociated complex by less than an additional 2 kJ/mol. A similarly small effect on binding affinity was observed for the R16C* substitution. The replacement of Arg by C* decreases the positive charge on the peptide, and the change we observe in binding agrees with previously reported small binding effects in CaM from Arg to Gln substitutions.61
A small effect (Kd increases by a factor of 3) was observed for I20C*, and the largest perturbation observed was only a factor of 6 for S23C*. The S23C* probe was expected to be least perturbative due to its lack of proximity to the binding interface in the reported NMR structure; this suggests that the observed difference in binding parameters might be influenced by effects on the free peptide rather than changes in the bound complex. The enthalpy change upon binding is the same at S23C* as for the probe-free peptide, implying that the observed effect on affinity has an entropic origin, which could come from either the free or bound species.
While all of the observed probe-induced thermodynamic perturbations in binding are small, the largest perturbations do NOT appear at sites structurally implicated as “most important” in the bound state. When considered along with similar recent measurements for labels placed on CaM,62 the data in Table 2 strongly indicate that the C* probe residue can be placed directly along binding interfaces without strong perturbation of the binding.
IR Probe Frequencies
The binding-induced C≡N frequency changes (visually in Figure 2 and quantitatively in Figure 3 and Table 3) are all consistent with semiquantitative expectations based on the NMR structure of the bound complex (Figure 1). Probes in hydrophobic pockets in the complex (W4C*, F8C*, and S12C*) exhibit large binding-induced red-shifts, and more solvent-exposed positions (R16C*, I20C* and S23C*) exhibit smaller red-shifts. Despite the complicated solvatochromism of the SC≡N vibration15,26,63 and associated recent theoretical discussion,24,25,64−66 the correlation of the C≡N frequencies observed here with solvent exposure (indicated in the first frame of Figure 3) indicates that the frequencies of C* probes qualitatively map out binding interfaces in protein–protein interactions. The C≡N frequency (quantified by either the mean or the mode frequency) in these experiments reports first on solvent exposure. However, neither the IR mean nor mode frequencies correlates perfectly with SASA, so there is more information in the IR label spectra than just a simple readout of solvent exposure. (See the discussion of line shapes below.)
The large IR frequency changes observed in this complex are especially dramatic compared to previous results with the same probe group in other systems. The binding-induced red-shifts of the C≡N band for the most buried sites in the CaM-peptide complex are at least as large as those observed for probe groups on the amphiphilic face of a helical antimicrobial peptide when it binds to phospholipid membranes15 and are all more dramatic than any of the changes observed for the same probe group along the Ras/effector protein–protein interface.31,32 The large changes in C≡N frequency at W4C*, F8C*, and S12C* suggest nearly complete solvent exclusion at buried sites where the probe residues in a hydrophobic environment, without the immediate need for further temperature-dependent measurements like those proposed by Adhikary et al.67 to tease out the local presence of water when nitrile frequencies are more ambiguous. While the S12C* probe has a larger mean SASA according to our MD calculations, that nonzero value mainly comes from a few simulated frames where the probe becomes solvent-exposed (Figure S3) and it spends the majority of its time away from the solvent. The R16C* and I20C* probes sample a large range of possible SASA values in our simulations (Figure S3), from completely solvent-excluded to strongly solvent-exposed, and the S23C* site is mainly solvent-exposed. The smaller C≡N shifts observed by the Webb group in the Ras/effector systems likely indicated a substantial residual solvent “lubrication” of the interface even when the two partners are bound together, whereas in the CaM/M13 complex it appears that water is nearly completely excluded from the three most hydrophobic label sites (and our SASA distributions agree with that assessment).
While Figure 3 indicates correlation between the binding-induced IR frequency shifts and solvent-accessible surface areas of the probes, there are generally not clear correlations between the mean C≡N frequency and thermodynamic binding changes introduced by the probe: this further indicates that the C* probe groups are not functionally perturbing in any systematic way. It should be possible to introduce C* probes along binding interfaces between many species and use changes in mean or mode frequency to map which residues are bound and which remain solvent-exposed.
IR Probe Line Shapes
A clear feature of the C* spectra for W4C*, F8C*, and S12C* is a broadening of the C≡N band upon binding to CaM. The line width increase on binding is somewhat different than prior observations of this probe group in both protein systems and small-molecule solvatochromic experiments, where red-shifts due to solvent exclusion (i.e., moving from water to tetrahydrofuran) have been accompanied by substantial narrowing of the C≡N band.9,14,15,23 The large line widths of the C≡N bands from water-excluded probes in Figure 2A–C suggests that the inside of this peptide–protein-binding interface is not as structurally or electrostatically homogeneous as either the interior of a lipid bilayer15 or a nonpolar solvent like THF.16
There is a correlation between the change in fwhm and the perturbation of the binding enthalpy and entropy (last two frames of Figure 3, second column) by the probe. ΔΔH and ΔΔS are strongly negatively correlated with each other. Probe placements that lead to broad C≡N line shapes generally increase the binding entropy while weakening the exothermic nature of the binding process. The F8C* and W4C* peptides present two examples where reduction in side chain volume leads to slightly different thermodynamic outcomes: for W4C*, the binding strengthens mainly due to a greater binding entropy, while for F8C* both the binding enthalpy and entropy increase and the result is a slightly weaker binding. In each case, the binding entropy increases due to the smaller and slightly more flexible probe side chain, but the overall binding affinity stays approximately the same due to slightly diminished enthalpically favorable hydrophobic interactions. For the more conservative S12C* substitution, the binding enthalpy is essentially unchanged and the binding entropy slightly decreases.
A reasonable assumption informed by the long vibrational lifetime of the SCN probe group35,36 is that the large line widths at the three hydrophobic label sites are due to a distribution of nonaqueous structural environments around the C* side chain. The long lifetime means that most of the C≡N line width for C* always comes from the inhomogeneous frequency distribution, which for this probe group is determined by hydrogen bonding from water, the instantaneous response of the C≡N frequency to the local electric field, and dispersion and exchange-repulsion effects associated with the close proximity of local solvent and functional groups.23,26,63 While local dynamics and conformational entropy around the probe, which could include either the orientation of the probe in its environment or fluctuations of that environment, could be the source of the large line widths in Figure 2A–C, the exact physical root of the inhomogeneous frequency distribution is not clear.
More detailed MD simulations and frequency calculations following recent precedents30,56,63,68−70 might be able to identify the exact source of this broadening. More extensive MD sampling could provide representative ensembles for the structures around each of the probe sites, and a much more direct physical connection to the C≡N frequency than estimated SASA is also needed to simulate the IR line shapes quantitatively. Further details of the line shapes in Figure 2 might also be revealed through multidimensional IR experiments, which remain extremely challenging for this probe group. The physical factors underlying the line shapes in Figure 2 are not completely clear, and we present the data in Figure 2 as significant and physically meaningful observations that pose a challenge for fully quantitative interpretation. Such a physical and quantitative interpretation would be a large step forward in understanding and implementing vibrational probe groups more broadly, and we have recently taken major steps toward such an interpretive methodology.71
The groups of Boxer,9,27,72,73 Webb,33,34 and Bagchi74 used MD calculations to simulate electric field effects on nitrile vibrational signals, with mixed results. While those reports assumed the electric field to be the sole determinant of the C≡N frequency, Layfield and Hammes-Schiffer,30,56 attempted with some success to model results from C≡N groups in enzymatic active sites using MD simulations with the C* probe group and a QM/MM approach to calculating the fluctuating frequency that uses force-field parametrization, QM approximation, and line shape calculation techniques laid out by Corcelli.69,70 The effective fragment potential (EFP) approach of Blasiak et al.64−66 suggested that local quantum mechanical effects like dispersion and exchange repulsion contribute more heavily to nitrile solvatochromism.63 We have not attempted to use purely electrostatic measures to interpret our results for two main reasons: (a) CaM’s binding to its targets is not generally thought to be electrostatically driven, and (b) the well-documented, complicated nature of nitrile solvatochromism63,68 strongly suggests that a number of other nonelectrostatic factors (beyond empirical ideas like “H-bonding or non H-bonding environments”) should drive the frequency in this (and most other) cases. Recently published results that compared the QM/MM and EFP approaches in the context of some of the results from Figure 2 indicate that the EFP approach quantitatively reproduces the probe line shapes and provides a clear structural and dynamic explanation for the relatively broad CaM-bound line shape at W4C*, and solvation provides an nuanced piece of the physical picture.71
Current Guidelines for Application of This Probe
A few other studies have evaluated label-induced perturbations of nitrile probe groups. Zimmerman et al. noted that p-cyanophenylalanine led to a decrease in folding stability on a site-dependent basis in a two-state folding SH3 domain.75 Crystallographic studies have also been used to show the structural nonperturbativity of unnatural amino acids with vibrational probes.9,76 Webb and co-workers used C* in an enzymatically active context, so an activity assay was their perturbation measure.31,32 The helical propensity of model single-helical peptides without tertiary contacts, as measured by far-UV circular dichroism, was reduced by the placement of C* throughout the sequence.17 In this system with two binding partners, ITC measurements are the most direct way to address binding perturbation, and our data clearly indicate that C* causes very little thermodynamic perturbation even when substituted for large hydrophobic residues such as Phe, Trp, and Ile.
A unique quality of the C* probe as compared to labels for other spectroscopies is its smaller size than the majority of naturally occurring amino acid side chains, and thus it can be placed in their stead without causing added steric hindrance; rather in many cases, there could be fewer direct residual contacts or van der Waals interactions without large disruption of the binding free energy. The perturbations introduced by introduction of C* in proteins should still be carefully evaluated on a site-by-site basis without strongly preconceived notions of the anticipated thermodynamic or structural outcomes of probe placement. This need for continuing evaluation is less related to the C* probe group itself and more that prediction of site-specific consequences of single-residue mutations on binding affinity requires knowledge of the effects on both free and bound states, which is not always at hand.
However, some conclusions do emerge here for “appropriate use” of this probe group along binding interfaces. The ITC data and IR data together suggest that there is no major perturbation of the average bound structure by the probe placement at any of the selected sites (and a similar lack of perturbation was recently observed from the other side of the same interface62), so assessing site-specific binding qualitatively through shifts in the mean or mode frequency should be possible in many systems, including those that are less structurally understood than the CaM-M13 complex. Despite its polarity, C* can apparently be placed in relatively hydrophobic environments without functional consequences. Based on our results with CaM and on other previous work by our group and others, we suggest the following guidelines for appropriate use of the C* probe side chain:
-
1.
C* may be substituted for all neutral amino acids larger than C* (with careful evaluation of the thermodynamic and/or structural consequences of the label introduction).
-
2.
C* may be substituted for alanine in helical sequences, with anticipated minor destabilization of the helix.17
-
3.
C* should not be substituted for charged amino acids in general.
Guideline 3 above is drawn partly from the Webb group’s results, where large electrostatic changes due to the probe group at a few sites disrupted the functional cooperation between Ras and its effectors.31−33 The R16C* mutation in the CaM/M13 system here was not fatal in line with other previous work,61 but such electrostatic perturbations can also have large effects on the conformational ensembles of disordered domains in particular77 (a class of domains that includes many CaM-binding sequences) and should generally be avoided.
Calmodulin/Target Dynamics
CaM’s reported bound complexes tend to be tightly bound and well-structured, but they also maintain some degree of site-specific conformational entropy.45 Whether that means that some of these complexes might be characterized as “fuzzy” with a very large conformational distribution in the bound complex remains to be seen (and is not likely in the case of the well-characterized and strongly collapsed CaM/M13 complex), but site-specific probe groups like C* will certainly have a central role to play in understanding the dynamics and structural distributions of regulatory proteins like CaM bound to both small and large targets. The data set presented in Figure 2, along with complementary data from probes on CaM,62 show that vibrational probes can reveal new details of the protein–protein interactions for CaM and other protein-binding species.
While the C* label does not perturb the binding free energy to any appreciable extent, the broad line shapes at bound sites in Figure 2, and the (small) changes in binding entropy at these sites suggested by ITC, hint at the flexibility of CaM’s binding interface and its tolerance for sequence variations in target sequences. The lack of perturbation from the W4C* substitution implies that the Trp side chain is not critical for the interaction between M13 and CaM, despite its “anchor” identity in the Ikura scheme for assessing and predicting CaM/target-binding motifs.39 The lack of perturbation at the natively charged R16C* site is in line with previously reported insensitivity to charge deletions,61 an effect originating in charge regulation upon complex formation between biomacromolecules of high and opposite charge.78
There is clearly some new and interesting information about local structural dynamics about this otherwise very well-characterized bound complex in the probe frequencies and line shapes in Figure 2. However, without an explicitly quantitative connection to MD simulations, we are not at this point able to clearly identify the exact dynamics that appear in the IR spectra. We are currently working on that connection, and C* labels on CaM62 also report interesting and similarly ill-characterized structural dynamics that should be functionally relevant to better understanding CaM’s unique regulatory promiscuity.
Conclusions
Isothermal titration calorimetry experiments on complex formation between CaM and site-specifically C*-labeled variants of the M13 peptide indicate that there is only a very small site-dependent perturbation of the CaM/peptide-binding equilibrium by the artificial C* side chain. Sites where C* is substituted for hydrophobic residues along the binding interface are the least thermodynamically perturbed as compared to the unlabeled complex, and this suggests that C* may be used to replace hydrophobic residues along protein–protein interfaces without major perturbation of the binding interaction. In all cases investigated here, the IR spectra of labeled peptides report on the bound complex only.
The frequencies of the IR C≡N stretching bands of the C* probe groups are correlated with solvent exposure predicted by MD simulations based on the reported NMR structure. Broadening of the C≡N infrared line shapes with formation of the complex is likely due to structural fluctuations and dynamics around the probe groups, and especially broad line shapes are observed at sites where a large hydrophobic residue was replaced by the smaller C*. These probe placements also lead to increased entropy of binding and decreased in binding enthalpy as measured by ITC. The exact dynamics leading to the broad line shapes in the bound complex are not clear and could only be identified through a more quantitative connection between MD simulations and experimental line shapes. Such a connection would provide a highly significant step forward for the physical interpretability of all vibrational probe groups.
An important methodological conclusion is that C* may be used to replace almost any neutral amino acid on binding interfaces, but careful site-specific evaluation of the thermodynamic consequences is still necessary since the perturbative effects of the labeling group are not necessarily predictable even using previously determined structural information. The C≡N stretching frequency of the probe group first reports the site-specific solvent exposure along this protein–protein interface, but the line shape appears to report on local structural dynamics. The C* probe group could provide new and interesting information about the dynamic structures of protein–protein interfaces, and of CaM-target complexes in particular, but interpreting the spectral information quantitatively will be best accomplished with the aid of high-level MD and/or frequency simulations.
Acknowledgments
C.H.L. especially acknowledges continuing advice and support from Professor Karin Akerfeldt, whose intellectual contributions were central to the formulation of this work, and Bashkim Kokona who helped with MALDI characterization of some of the peptides.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.8b00283.
Data and analysis of isothermal titration calorimetry traces for all seven CaM-peptide complexes, full line shape analysis of all infrared spectra, and some quantities extracted from MD simulations of SCN-labeled complexes (PDF)
This work was supported by grants R15-GM088749-01 and R15-GM088749-02 from the National Institute of General Medical Sciences and NSF CAREER grant CHE-1150727. S.L. acknowledges Swedish Research Council, VR 2014-05664.
The authors declare no competing financial interest.
Supplementary Material
References
- Krimm S.; Bandekar J. (1986) Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. Adv. Protein Chem. 38, 181–364. 10.1016/S0065-3233(08)60528-8. [DOI] [PubMed] [Google Scholar]
- Lindquist B. A.; Furse K. E.; Corcelli S. A. (2009) Nitrile groups as vibrational probes of biomolecular structure and dynamics: an overview. Phys. Chem. Chem. Phys. 11, 8119–8132. 10.1039/b908588b. [DOI] [PubMed] [Google Scholar]
- Waegele M. M.; Culik R. M.; Gai F. (2011) Site-Specific Spectroscopic Reporters of the Local Electric Field, Hydration, Structure, and Dynamics of Biomolecules. J. Phys. Chem. Lett. 2, 2598–2609. 10.1021/jz201161b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.; Cho M. (2013) Infrared Probes for Studying the Structure and Dynamics of Biomolecules. Chem. Rev. 113, 5817–5847. 10.1021/cr3005185. [DOI] [PubMed] [Google Scholar]
- Ma J.; Pazos I. M.; Zhang W.; Culik R. M.; Gai F. (2015) Site-Specific Infrared Probes of Proteins. Annu. Rev. Phys. Chem. 66, 357–377. 10.1146/annurev-physchem-040214-121802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getahun Z.; Huang C. Y.; Wang T.; De Leon B.; DeGrado W. F.; Gai F. (2003) Using nitrile-derivatized amino acids as infrared probes of local environment. J. Am. Chem. Soc. 125, 405–411. 10.1021/ja0285262. [DOI] [PubMed] [Google Scholar]
- Jo H.; Culik R. M.; Korendovych I. V.; DeGrado W. F.; Gai F. (2010) Selective Incorporation of Nitrile-Based Infrared Probes into Proteins via Cysteine Alkylation. Biochemistry 49, 10354–10356. 10.1021/bi101711a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waegele M. M.; Tucker M. J.; Gai F. (2009) 5-Cyanotryptophan as an infrared probe of local hydration status of proteins. Chem. Phys. Lett. 478, 249–253. 10.1016/j.cplett.2009.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fafarman A. T.; Boxer S. G. (2010) Nitrile Bonds as Infrared Probes of Electrostatics in Ribonuclease S. J. Phys. Chem. B 114, 13536–13544. 10.1021/jp106406p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fafarman A. T.; Sigala P. A.; Schwans J. P.; Fenn T. D.; Herschlag D.; Boxer S. G. (2012) Quantitative, directional measurement of electric field heterogeneity in the active site of ketosteroid isomerase. Proc. Natl. Acad. Sci. U. S. A. 109, E299–E308. 10.1073/pnas.1111566109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fafarman A. T.; Webb L. J.; Chuang J. I.; Boxer S. G. (2006) Site-specific conversion of cysteine thiols into thiocyanate creates an IR probe for electric fields in proteins. J. Am. Chem. Soc. 128, 13356–13357. 10.1021/ja0650403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigala P. A.; Fafarman A. T.; Bogard P. E.; Boxer S. G.; Herschlag D. (2007) Do ligand binding and solvent exclusion alter the electrostatic character within the oxyanion hole of an enzymatic active site?. J. Am. Chem. Soc. 129, 12104–12105. 10.1021/ja075605a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb L. J.; Boxer S. G. (2008) Electrostatic fields near the active site of human aldose reductase: 1. New inhibitors and vibrational stark effect measurements. Biochemistry 47, 1588–1598. 10.1021/bi701708u. [DOI] [PubMed] [Google Scholar]
- Alfieri K. N.; Vienneau A. R.; Londergan C. H. (2011) Using Infrared Spectroscopy of Cyanylated Cysteine To Map the Membrane Binding Structure and Orientation of the Hybrid Antimicrobial Peptide CM15. Biochemistry 50, 11097–11108. 10.1021/bi200903p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon H. A.; Alfieri K. N.; Clark C. A. A.; Londergan C. H. (2010) Cyanylated Cysteine: A Covalently Attached Vibrational Probe of Protein-Lipid Contacts. J. Phys. Chem. Lett. 1, 850–855. 10.1021/jz1000177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischak C. G.; Longhi S.; Snead D. M.; Costanzo S.; Terrer E.; Londergan C. H. (2010) Probing Structural Transitions in the Intrinsically Disordered C-Terminal Domain of the Measles Virus Nucleoprotein by Vibrational Spectroscopy of Cyanylated Cysteines. Biophys. J. 99, 1676–1683. 10.1016/j.bpj.2010.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein L.; Stetz M. A.; McMahon H. A.; Londergan C. H. (2010) The Effects of alpha-Helical Structure and Cyanylated Cysteine on Each Other. J. Phys. Chem. B 114, 4931–4936. 10.1021/jp101447r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann J.; Thielges M. C.; Yu W.; Dawson P. E.; Romesberg F. E. (2011) Carbon-Deuterium Bonds as Site-Specific and Nonperturbative Probes for Time-Resolved Studies of Protein Dynamics and Folding. J. Phys. Chem. Lett. 2, 412–416. 10.1021/jz200012h. [DOI] [Google Scholar]
- Thielges M. C.; Axup J. Y.; Wong D.; Lee H. S.; Chung J. K.; Schultz P. G.; Fayer M. D. (2011) Two-Dimensional IR Spectroscopy of Protein Dynamics Using Two Vibrational Labels: A Site-Specific Genetically Encoded Unnatural Amino Acid and an Active Site Ligand. J. Phys. Chem. B 115, 11294–11304. 10.1021/jp206986v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gai X. S.; Fenlon E. E.; Brewer S. H. (2010) A Sensitive Multispectroscopic Probe for Nucleic Acids. J. Phys. Chem. B 114, 7958–7966. 10.1021/jp101367s. [DOI] [PubMed] [Google Scholar]
- Degani Y.; Degani C. (1979) Subunit-Selective Chemical Modifications of Creatine Kinase- Evidence for Asymmetrical Association of the Subunits. Biochemistry 18, 5917–5923. 10.1021/bi00593a024. [DOI] [PubMed] [Google Scholar]
- Degani Y.; Neumann H.; Patchornik A. (1970) Selective Cyanylation of Sulfhydryl Groups. J. Am. Chem. Soc. 92, 6969–6971. 10.1021/ja00726a043. [DOI] [PubMed] [Google Scholar]
- Maienschein-Cline M. G.; Londergan C. H. (2007) The CN stretching band of aliphatic thiocyanate is sensitive to solvent dynamics and specific solvation. J. Phys. Chem. A 111, 10020–10025. 10.1021/jp0761158. [DOI] [PubMed] [Google Scholar]
- Oh K.-I.; Choi J.-H.; Lee J.-H.; Han J.-B.; Lee H.; Cho M. (2008) Nitrile and thiocyanate IR probes: Molecular dynamics simulation studies. J. Chem. Phys. 128, 154504. 10.1063/1.2904558. [DOI] [PubMed] [Google Scholar]
- Choi J.-H.; Oh K.-I.; Lee H.; Lee C.; Cho M. (2008) Nitrile and thiocyanate IR probes: Quantum chemistry calculation studies and multivariate least-square fitting analysis. J. Chem. Phys. 128, 134506. 10.1063/1.2844787. [DOI] [PubMed] [Google Scholar]
- Fafarman A. T.; Sigala P. A.; Herschlag D.; Boxer S. G. (2010) Decomposition of Vibrational Shifts of Nitriles into Electrostatic and Hydrogen-Bonding Effects. J. Am. Chem. Soc. 132, 12811–12813. 10.1021/ja104573b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagchi S.; Fried S. D.; Boxer S. G. (2012) A Solvatochromic Model Calibrates Nitriles’ Vibrational Frequencies to Electrostatic Fields. J. Am. Chem. Soc. 134, 10373–10376. 10.1021/ja303895k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londergan C. H.; Baskin R.; Bischak C. G.; Hoffman K. W.; Snead D. M.; Reynoso C. (2015) Dynamic Asymmetry and the Role of the Conserved Active-Site Thiol in Rabbit Muscle Creatine Kinase. Biochemistry 54, 83–95. 10.1021/bi5008063. [DOI] [PubMed] [Google Scholar]
- Johnson M. N. R.; Londergan C. H.; Charkoudian L. K. (2014) Probing the Phosphopantetheine Arm Conformations of Acyl Carrier Proteins Using Vibrational Spectroscopy. J. Am. Chem. Soc. 136, 11240–11243. 10.1021/ja505442h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. T.; Layfield J. P.; Stewart R. J.; French J. B.; Hanoian P.; Asbury J. B.; Hammes-Schiffer S.; Benkovic S. J. (2014) Probing the Electrostatics of Active Site Microenvironments along the Catalytic Cycle for Escherichia coli Dihydrofolate Reductase. J. Am. Chem. Soc. 136, 10349–10360. 10.1021/ja5038947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford A. J.; Ensign D. L.; Webb L. J. (2010) Vibrational Stark Effect Spectroscopy at the Interface of Ras and Rap1A Bound to the Ras Binding Domain of RalGDS Reveals an Electrostatic Mechanism for Protein-Protein Interaction. J. Phys. Chem. B 114, 15331–15344. 10.1021/jp106974e. [DOI] [PubMed] [Google Scholar]
- Stafford A. J.; Walker D. M.; Webb L. J. (2012) Electrostatic Effects of Mutations of Ras Glutamine 61 Measured Using Vibrational Spectroscopy of a Thiocyanate Probe. Biochemistry 51, 2757–2767. 10.1021/bi201225p. [DOI] [PubMed] [Google Scholar]
- Ragain C. M.; Newberry R. W.; Ritchie A. W.; Webb L. J. (2012) Role of Electrostatics in Differential Binding of RaIGDS to Rap Mutations E30D and K31E Investigated by Vibrational Spectroscopy of Thiocyanate Probes. J. Phys. Chem. B 116, 9326–9336. 10.1021/jp303272y. [DOI] [PubMed] [Google Scholar]
- Walker D. M.; Hayes E. C.; Webb L. J. (2013) Vibrational Stark effect spectroscopy reveals complementary electrostatic fields created by protein-protein binding at the interface of Ras and Ral. Phys. Chem. Chem. Phys. 15, 12241–12252. 10.1039/c3cp51284c. [DOI] [PubMed] [Google Scholar]
- van Wilderen L. J. G. W.; Kern-Michler D.; Müller-Werkmeister H.; Bredenbeck J. (2014) Vibrational dynamics and solvatochromism of the label SCN in various solvents and hemoglobin by time dependent IR and 2D-IR spectroscopy. Phys. Chem. Chem. Phys. 16, 19643–19653. 10.1039/C4CP01498G. [DOI] [PubMed] [Google Scholar]
- Levin D. E.; Schmitz A. J.; Hines S. M.; Hines K. J.; Tucker M. J.; Brewer S. H.; Fenlon E. E. (2016) Synthesis and evaluation of the sensitivity and vibrational lifetimes of thiocyanate and selenocyanate infrared reporters. RSC Adv. 6, 36231–36237. 10.1039/C5RA27363C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikura M.; Clore G. M.; Gronenborn A. M.; Zhu G.; Klee C. B.; Bax A. (1992) Solution Structure of a Calmodulin-Target Peptide Complex by Multidimensional NMR. Science 256, 632–638. 10.1126/science.1585175. [DOI] [PubMed] [Google Scholar]
- Valeyev N. V.; Heslop-Harrison P.; Postlethwaite I.; Kotov N. V.; Bates D. G. (2008) Multiple calcium binding sites make calmodulin multifunctional. Mol. BioSyst. 4, 66–73. 10.1039/B713461D. [DOI] [PubMed] [Google Scholar]
- Yap K. L.; Ames J. B.; Swindells M. B.; Ikura M. (1999) Diversity of conformational states and changes within the EF-hand protein superfamily. Proteins: Struct., Funct., Genet. 37, 499–507. . [DOI] [PubMed] [Google Scholar]
- Hoeflich K. P.; Ikura M. (2002) Calmodulin in Action. Cell 108, 739–742. 10.1016/S0092-8674(02)00682-7. [DOI] [PubMed] [Google Scholar]
- Vetter S. W.; Leclerc E. (2003) Novel aspects of calmodulin target recognition and activation. Eur. J. Biochem. 270, 404–414. 10.1046/j.1432-1033.2003.03414.x. [DOI] [PubMed] [Google Scholar]
- Elshorst B.; Hennig M.; Forsterling H.; Diener A.; Maurer M.; Schulte P.; Schwalbe H.; Griesinger C.; Krebs J.; Schmid H.; Vorherr T.; Carafoli E. (1999) NMR solution structure of a complex of calmodulin with a binding peptide of the Ca2+ pump. Biochemistry 38, 12320–12332. 10.1021/bi9908235. [DOI] [PubMed] [Google Scholar]
- Wand A. J. (2013) The dark energy of proteins comes to light: conformational entropy and its role in protein function revealed by NMR relaxation. Curr. Opin. Struct. Biol. 23, 75–81. 10.1016/j.sbi.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gsponer J.; Christodoulou J.; Cavalli A.; Bui J. M.; Richter B.; Dobson C. M.; Vendruscolo M. (2008) A Coupled Equilibrium Shift Mechanism in Calmodulin-Mediated Signal Transduction. Structure 16, 736–746. 10.1016/j.str.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick K. K.; Marlow M. S.; Valentine K. G.; Wand A. J. (2007) Conformational entropy in molecular recognition by proteins. Nature 448, 325–U3. 10.1038/nature05959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasinath V.; Sharp K. A.; Wand A. J. (2013) Microscopic Insights into the NMR Relaxation-Based Protein Conformational Entropy Meter. J. Am. Chem. Soc. 135, 15092–15100. 10.1021/ja405200u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick K.; Sharp K.; Warischalk N.; Wand A. (2008) Re-evaluation of the model-free analysis of fast internal motion in proteins using NMR relaxation. J. Phys. Chem. B 112, 12095–12103. 10.1021/jp8038576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasinath V.; Valentine K. G.; Wand A. J. (2013) A C-13 Labeling Strategy Reveals a Range of Aromatic Side Chain Motion in Calmodulin. J. Am. Chem. Soc. 135, 9560–9563. 10.1021/ja4001129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlow M. S.; Dogan J.; Frederick K. K.; Valentine K. G.; Wand A. J. (2010) The role of conformational entropy in molecular recognition by calmodulin. Nat. Chem. Biol. 6, 352–358. 10.1038/nchembio.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.; Fabian H.; Mantsch H. H.; Vogel H. J. (1994) Isotope-Edited Fourier Transform Infrared Spectroscopy Studies of Calmodulin’s Interaction with Its Target Peptides. Biochemistry 33, 10883–10888. 10.1021/bi00202a006. [DOI] [PubMed] [Google Scholar]
- Fallon J. L.; Quiocho F. A. (2003) A closed compact structure of native Ca2+-calmodulin. Structure 11, 1303–U7. 10.1016/j.str.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Finn B. E.; Evenas J.; Drakenberg T.; Waltho J. P.; Thulin E.; Forsen S. (1995) Calcium-Induced Structural Changes and Domain Autonomy in Calmodulin. Nat. Struct. Mol. Biol. 2, 777–783. 10.1038/nsb0995-777. [DOI] [PubMed] [Google Scholar]
- Pace C. N.; Vajdos F.; Fee L.; Grimsley G.; Gray T. (1995) How to Measure and Predict the Molar Absorption Coefficient of a Protein. Protein Sci. 4, 2411–2423. 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk S.; Pall S.; Schulz R.; Larsson P.; Bjelkmar P.; Apostolov R.; Shirts M. R.; Smith J. C.; Kasson P. M.; van der Spoel D.; Hess B.; Lindahl E. (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854. 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. (2006) Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins: Struct., Funct., Genet. 65, 712–725. 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layfield J. P.; Hammes-Schiffer S. (2013) Calculation of Vibrational Shifts of Nitrile Probes in the Active Site of Ketosteroid Isomerase upon Ligand Binding. J. Am. Chem. Soc. 135, 717–725. 10.1021/ja3084384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug R. R.; Hunter W. G.; Grieger R. A. (1976) Statistical interpretation of enthalpy–entropy compensation. Nature 261, 566–567. 10.1038/261566a0. [DOI] [Google Scholar]
- Gallicchio E.; Kubo M. M.; Levy R. M. (1998) Entropy–Enthalpy Compensation in Solvation and Ligand Binding Revisited. J. Am. Chem. Soc. 120, 4526–4527. 10.1021/ja974061h. [DOI] [Google Scholar]
- Reynolds C. H.; Holloway M. K. (2011) Thermodynamics of Ligand Binding and Efficiency. ACS Med. Chem. Lett. 2, 433–437. 10.1021/ml200010k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay W. A.; Gradwell M. J.; Bayley P. M. (1995) Role of the N-terminal region of the skeletal muscle myosin light chain kinase target sequence in its interaction with calmodulin. Protein Sci. 4, 2375–2382. 10.1002/pro.5560041116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- André I.; Kesvatera T.; Jönsson B.; Åkerfeldt K. S.; Linse S. (2004) The Role of Electrostatic Interactions in Calmodulin-Peptide Complex Formation. Biophys. J. 87, 1929–1938. 10.1529/biophysj.104.040998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly K. L.; Dalton S. R.; Wai R. B.; Ramchandani K.; Xu R. J.; Linse S.; Londergan C. H. (2018) Conformational Ensembles of Calmodulin Revealed by Nonperturbing Site-Specific Vibrational Probe Groups. J. Phys. Chem. A 122, 2947–2955. 10.1021/acs.jpca.8b00475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasiak B.; Ritchie A. W.; Webb L. J.; Cho M. (2016) Vibrational solvatochromism of nitrile infrared probes: beyond the vibrational Stark dipole approach. Phys. Chem. Chem. Phys. 18, 18094–18111. 10.1039/C6CP01578F. [DOI] [PubMed] [Google Scholar]
- Blasiak B.; Lee H.; Cho M. (2013) Vibrational solvatochromism: Towards systematic approach to modeling solvation phenomena. J. Chem. Phys. 139, 044111. 10.1063/1.4816041. [DOI] [PubMed] [Google Scholar]
- Blasiak B.; Cho M. (2014) Vibrational solvatochromism. II. A first-principle theory of solvation-induced vibrational frequency shift based on effective fragment potential method. J. Chem. Phys. 140, 164107. 10.1063/1.4872040. [DOI] [PubMed] [Google Scholar]
- Blasiak B.; Cho M. (2015) Vibrational solvatochromism. III. Rigorous treatment of the dispersion interaction contribution. J. Chem. Phys. 143, 164111. 10.1063/1.4934667. [DOI] [PubMed] [Google Scholar]
- Adhikary R.; Zimmermann J.; Dawson P. E.; Romesberg F. E. (2015) Temperature Dependence of CN and SCN IR Absorptions Facilitates Their Interpretation and Use as Probes of Proteins. Anal. Chem. 87, 11561–11567. 10.1021/acs.analchem.5b03437. [DOI] [PubMed] [Google Scholar]
- Błasiak B.; Londergan C. H.; Webb L. J.; Cho M. (2017) Vibrational Probes: From Small Molecule Solvatochromism Theory and Experiments to Applications in Complex Systems. Acc. Chem. Res. 50, 968–976. 10.1021/acs.accounts.7b00002. [DOI] [PubMed] [Google Scholar]
- Lindquist B. A.; Corcelli S. A. (2008) Nitrile groups as vibrational probes: Calculations of the C N infrared absorption line shape of acetonitrile in water and tetrahydrofuran. J. Phys. Chem. B 112, 6301–6303. 10.1021/jp802039e. [DOI] [PubMed] [Google Scholar]
- Lindquist B. A.; Haws R. T.; Corcelli S. A. (2008) Optimized Quantum Mechanics/Molecular Mechanics Strategies for Nitrile Vibrational Probes: Acetonitrile and para-Tolunitrile in Water and Tetrahydrofuran. J. Phys. Chem. B 112, 13991–14001. 10.1021/jp804900u. [DOI] [PubMed] [Google Scholar]
- Xu R. J.; Blasiak B.; Layfield J. P.; Cho M.; Londergan C. H. (2018) A Direct, Quantitative Connection Between Molecular Dynamics Simulations and Vibrational Probe Line shapeshapes. J. Phys. Chem. Lett. 9, 2560–2567. 10.1021/acs.jpclett.8b00969. [DOI] [PubMed] [Google Scholar]
- Fried S. D.; Wang L.-P.; Boxer S. G.; Ren P.; Pande V. S. (2013) Calculations of the Electric Fields in Liquid Solutions. J. Phys. Chem. B 117, 16236–16248. 10.1021/jp410720y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried S. D.; Boxer S. G. (2015) Measuring Electric Fields and Noncovalent Interactions Using the Vibrational Stark Effect. Acc. Chem. Res. 48, 998–1006. 10.1021/ar500464j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb P.; Haldar T.; Kashid S. M.; Banerjee S.; Chakrabarty S.; Bagchi S. (2016) Correlating Nitrile IR Frequencies to Local Electrostatics Quantifies Noncovalent Interactions of Peptides and Proteins. J. Phys. Chem. B 120, 4034–4046. 10.1021/acs.jpcb.6b02732. [DOI] [PubMed] [Google Scholar]
- Zimmermann J.; Thielges M. C.; Seo Y. J.; Dawson P. E.; Romesberg F. E. (2011) Cyano Groups as Probes of Protein Microenvironments and Dynamics. Angew. Chem., Int. Ed. 50, 8333–8337. 10.1002/anie.201101016. [DOI] [PubMed] [Google Scholar]
- Dippel A. B.; Olenginski G. M.; Maurici N.; Liskov M. T.; Brewer S. H.; Phillips-Piro C. M. (2016) Probing the effectiveness of spectroscopic reporter unnatural amino acids: a structural study. Acta Crystallogr. Sect. Struct. Biol. 72, 121–130. 10.1107/S2059798315022858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao A. H.; Crick S. L.; Vitalis A.; Chicoine C. L.; Pappu R. V. (2010) Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. U. S. A. 107, 8183–8188. 10.1073/pnas.0911107107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund M.; Jönsson B. (2005) On the Charge Regulation of Proteins. Biochemistry 44, 5722–5727. 10.1021/bi047630o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.