

The neurodegenerative diseases are a major focus of scientific and clinical interest because of their increasing medical and social importance. The neurodegenerative diseases represent some of the greatest challenges for basic science and clinical medicine because of their prevalence, cost, complex biochemistry and pathology, and lack of mechanism-based treatments. These diseases have great impacts not only on individuals, but also on care-givers and society at large.
This heterogeneous group of age-associated, chronic illnesses, including Alzheimer’s disease (AD), Parkinson’s disease (PD), motor neuron disease (MND), the trinucleotide repeat disease (i.e. the autosomal dominant spinocerebellar ataxias, SCA and Huntington’s disease, HD). The prion disorders are usually characterized by: genetic risk factors, onset within certain age ranges, progressive courses, well-defined clinical symptoms, dysfunction and death of specific population of neurons, specific biochemical abnormalities, and presence of intra- and/or extracellular protein. Epidemiology of neurodegenerative disorders is shown in Table 1.
Table 1.
Epidemiology of neurodegenerative diseases in USA*
| Disease | Number of Patients | Prevalence per 100 000 |
|---|---|---|
| AD | 4 000 000 | 1200 |
| PD | 1 000 000 | 300 |
| FTD | 40 000 | 14 |
| ED | 30 000 | 10 |
| ALS(MND) | 20 000 | 7 |
| PSP | 15 000 | 5 |
| SCA | 12 000 | 4 |
| Prion disease | 400 | 01 |
*US population approximately 275 million
FTD=frontotemporal dementia, ALS=amyotrophic lateral sclerosis
PSP=progressive supranuclear palsy (Parkinson “plus” syndrome)
Aetiological frequency (sporadic, genetic and infectious) of neurodegenerative diseases is shown in Table 2.
Table 2.
Sporadic, genetic and infectious etiologies of neurodegenerative disorders
| Etiologic frequency (%) | |||
|---|---|---|---|
| Disease | Sporadic | Genetic | Infectious |
| Priori disease | 85 | >10 | <1 |
| AD | 90 | 10 | |
| PD | 95 | <5 | |
| FTD | 90 | 10 | |
| Pick's disease | 95 | <5 | |
| PSP | 95 | <5 | |
| ALS (MND) | 90 | 10 | |
| HD | 100 | ||
| Spinocerebellar ataxias | 100 |
FTD=frontotemporal dementia, ALS=amyotrophic lateral sclerosis PSP=progressive supranuclear palsy (Parkinson “plus” syndrome)
14.1 Aging and neurodegeneration
Age is the single most important risk factors for degenerative disease of the central nervous system (CNS). As the lifespan of humans continues to increase, an increasing burden of degenerative diseases is emerging. Over four million people suffer from AD in the United States and another million have PD. By age 85, nearly 50% of people exhibit at least one symptom or sign of Parkinsonism.
A close historical relationship has existed regarding attempts to understand the nature of both neurodegenerative diseases and ageing. Some signs of degeneration, such as neuronal loss or even specific pathologic changes, may also occur in ageing in the absence of disease. Healthy ageing is also characterized by changes in neurotransmitters, which could be responsible for some of the changes in cognitive (age-associated memory impairment) or motor abilities in older individuals. Some of these neurochemical changes occur in the same neuronal system affected in neurodegenerative diseases. Moreover, older individuals are at risk for most neurodegenerative disorders.
14.2 Aetiology and pathology
The aetiology and pathogenesis of these devastating diseases remain largely unknown; however, an area of fruitful contribution has been the study of the neurochemistry and synaptic transmission. Knowledge of the different neurotransmitter changes has helped to characterize involvement of specific subpopulations of neurons. The pattern of selective vulnerability of neurons provides important clues to pathogenesis and genetic regulation of neuronal development, and endogenous and exogenous neurotoxins. Our understanding of the degenerative disorders, and development of treatment for them, is one of the fastest expanding areas in the neurosciences. In less than 20 years we have moved from a world where most disorders were minimally investigated and rarely diagnosed, to one where there is now effective symptomatic treatment for some of the illnesses. Well-characterized deficit of certain neurotransmitters have important clinical implications for diagnosis and treatment.
Dopamine replacement brought dramatic progress in the management of patients with Parkinson’s disease. The success of levo-dopa therapy for the treatment of PD is the basis for much of the optimism that other neurodegenerative disorders may be treated with replacement therapy. However, it remains to be determined whether replacement therapy is justified for other disorders. Up to now, the dramatic success of levo-dopa therapy for PD has not been repeated for other neurodegenerative diseases. A similar approach in Alzheimer disease with cholinergic drugs has been less successful but nevertheless helpful.
14.2.1 Glutamate excito-toxicity
iGlutamate (excitatory amino acid) is the major excitatory neurotransmitter in the human nervous system with excito-toxic effects on neurons. It is accepted that glutamate overactivity caused by exogenous or endogenous factors is an aetiological factor in chronic neurodegenerative disease characterized by the slow progressive death of vulnerable neuronal populations or that it contributes to their natural history and progression. Exogenous or endogenous neurotoxic compounds might activate glutamate receptors.
Enhanced glutamate activity may not necessarily be caused by neurotoxins but could also be of genetic or metabolic origin, such as in Huntington’s disease or in aging. Other environmental factors individually or in combination may also facilitate a chronic, progressive, neuronal cell loss related to glutamate overactivity.
Glutamate excito-toxicity could be involved also in toxin-induced dopamine cell death, especially as glutamate receptors do occur in the substantia nigra.
Motor neuron disease (MND, ALS): motor neurons are activated by stimulation of cell surface glutamate receptors and the excitatory signal is terminated by active removal of glutamate from the synaptic cleft by transporter proteins which are largely located on perisynaptic glial cell. It is known that excessive stimulation of neuronal glutamate receptors (excito-toxicity) can injure neurons by mechanisms, which include derangement of intracellular calcium homeostasis and excessive free radical production. A body of circumstantial evidence has implicated glutamate-mediated toxicity as a contributory factor to motor neuron disease (ALS). Antiglutamate therapy is the only therapy that has some effect in prolonging survival in patients with MND and in a transgenic mouse model of familial MND.
14.3 Protein deposition and neurodegenerative disorders
Twenty years ago there was little understanding of the causes of neurodegeneration and the term degenerative disease was used as a wastebasket for illnesses of unknown aetiology. But today, it is clear that the misprocessing of proteins causes neurodegenerative diseases. In each disease, one or more specific proteins have been identified that are misprocessed. This results in the accumulation of one or more particular proteins. The proteins that accumulate in the CNS of patients with neurodegenerative disease were initially identified by purifying these polypeptides from the brains of affected humans or animals. Protein deposition and neurodegenerative disorders are shown in Table 3.
Table 3.
Protein deposition and neurodegenerative diseases
| Disease | Protein | Aggregate |
|---|---|---|
| Prion disease | PrPSc | PrP amyloid |
| Alzheimer ásease | Aß | Aß amyloid |
| tau | PHF in MFT | |
| FTD | tau | straight filaments PHF |
| Pick's disease | tau | Pick bodies |
| Parkinson's disease | ą synuclein | Levy bodies |
| PSP | tau | straight filaments PHF |
| ALS(MND) | neurofilament | neuronal aggregates |
| HD | Huntingdon | nuclear inclusions |
| Spinocerebellar ataxias 1 | ataxin 1 | nuclear inclusions |
| Spinocerebellar ataxias 2 | ataxin 2 | cytoplasmic inclusions |
| Macado-Joseph disease | ataxin 3 | nuclear inclusions |
NFT=neurofibrillary tangles; PHF=paired helical filaments
Subsequently, molecular genetics was used to identify the genes responsible for the familial forma of AD and PD as well as ALS and FTD. Similarly, molecular genetic investigations of HD and spinocerebellar ataxias have led to the identification of the genes responsible for the pathogenesis of these illnesses.
Prions are infectious proteins. In mammals, prions reproduce by recruiting the normal, cellular isoform of the prion protein (PrPSc). A major feature that distinguishes prions from viruses is the finding that both PrP isoforms are encoded by a chromosomal gene. In humans, the PrP gene is designated PRNP and is located on the short arm of chromosome 20.
Of all the studies on neurodegenerative diseases, the discovery of prions has been most unexpected. The finding that prion can act as an infectious pathogen and cause degeneration of the CNS was unprecedented. The prion concept not only explained how a disease could be both infectious and genetic, but it has also created new disease paradigms and revolutionized thinking in biology. The prion concept readily explains how a disease can be manifest as a inherited or sporadic disease as well as infectious illness. Moreover, the hallmark common to all of the prion diseases, whether sporadic, dominantly inherited, or acquired by infection, is that they involve the aberrant metabolism of the prion protein.
14.4 The future therapeutic strategies
Neurochemical alterations are probably secondary to neuronal death and dysfunction, but their characterizations may help to better understand the pathogenesis of neurodegeneration and design new approaches in therapeutic strategies. But the widespread neuropathology in most of neurodegenerative disorders makes replacement therapy an approach that is unlikely to be as successful as in PD. But the time has come to develop drugs that interrupt the disease processes. In some of the neurodegenerative disorders, it may be most efficacious to develop drugs that specifically block the misprocessing of a particular protein while in others, drugs that enhance the clearance of an aberrant protein of fragment may prove to be more useful.
Regardless of the therapeutic approach, the need for early detection of neurodegeneration will be important so that drugs can be given before significant damage to the CNS has occurred.
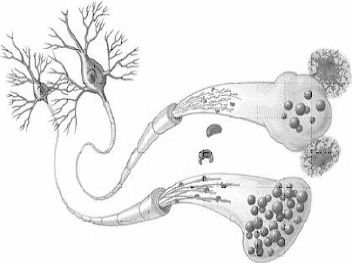
Neurons in Alzheimer's Disease. This comparison of a normal cell (lower) and AD-affected cell shows the telltale microscopic signs of the disease - neuritic plaques, neurofibrillary tangles and deformed boutons which block normal synaptic function. http://www.mc.vanderbilt.edu/health/health_lib/alzheimer.html
References
- 1.Balter M. Tracking the human fallout from “mad cow disease”. Science 2000; 289:1452. [DOI] [PubMed] [Google Scholar]
- 2.Bennett DA, Beckett LA, Murray AM, et al. Prevalence of Parkinsonian signs and associated mortality in a community population in older people, N Eng J Med 1996;334:71. [DOI] [PubMed] [Google Scholar]
- 3.De Jong JMBV. The World Federation of Neurology classification of spinal muscular atrophy and other disorders of motor neuron. In Handbook of Clinical Neurology; Vol. 15 Amsterdam: Elsevier Science; 1991. [Google Scholar]
- 4.Margolis RI, McInnis MG, Rosenblatt A, Ross CA. Trinucleotid repeat expansion and neuropsychiatric disease. Arc Gen Psychiatry 1999; 56:1019. [DOI] [PubMed] [Google Scholar]
- 5.Scolding N. Contemporary treatment in neurology. Buterworth, Heineman (Oxford) 2001. [Google Scholar]