

Abstract
Microvesicles (MVs) are emerging as a new mechanism of intercellular communication by transferring cellular lipid and protein components to target cells, yet their function in disease is only now being explored. We found that neutrophil-derived MVs were increased in concentration in synovial fluid from rheumatoid arthritis patients compared to paired plasma. Synovial MVs overexpressed the proresolving, anti-inflammatory protein annexin A1 (AnxA1). Mice deficient in TMEM16F, a lipid scramblase required for microvesiculation, exhibited exacerbated cartilage damage when subjected to inflammatory arthritis. To determine the function of MVs in inflammatory arthritis, toward the possibility of MV-based therapeutics, we examined the role of immune cell–derived MVs in rodent models and in human primary chondrocytes. In vitro, exogenous neutrophil-derived AnxA1+ MVs activated anabolic gene expression in chondrocytes, leading to extracellular matrix accumulation and cartilage protection through the reduction in stress-adaptive homeostatic mediators interleukin-8 and prostaglandin E2. In vivo, intra-articular injection of AnxA1+ MV lessened cartilage degradation caused by inflammatory arthritis. Arthritic mice receiving adoptive transfer of whole neutrophils displayed abundant MVs within cartilage matrix and revealed that MVs, but not neutrophils themselves, can penetrate cartilage. Mechanistic studies support a model whereby MV-associated AnxA1 interacts with its receptor FPR2 (formyl peptide receptor 2)/ALX, increasing transforming growth factor–b production by chondrocytes, ultimately leading to cartilage protection. We envisage that MVs, either directly or loaded with therapeutics, can be harnessed as a unique therapeutic strategy for protection in diseases associated with cartilage degeneration.
INTRODUCTION
In most rheumatoid arthritis (RA) patients, tumor necrosis factor–α (TNF-α) drives synovial inflammation, and in mice, TNF-α overexpression is sufficient to induce a severe form of inflammatory arthritis, culminating in cartilage and bone destruction (1). In a significant proportion of RA patients, TNF blockade arrests joint erosion; however, cartilage damage can progress despite the inflammatory process being well controlled, leading to secondary osteoarthritis and permanent disability. Arresting and, if possible, reversing the cartilage breakdown induced by the inflammatory environment of the arthritic joint is an unmet need.
Microvesicles (MVs; 0.05 to 1 μm), comprising both exosomes and microparticles, are emerging as novel determinants of paracellular communication. Released by most eukaryotic cells, MVs can transfer proteins, lipids, nucleic acids, and cytosolic material (2). MVs originating from different cells differ in composition and biological function; for instance, in human RA, MVs of leukocyte, platelet, and synovial fibroblast origin can be found in synovial fluids, with concentrations positively correlating with disease activity (3, 4). Neutrophils accumulate in large numbers in the synovial space during acute and transient RA flares and contribute to ≥30% of total synovial fluid MVs during active phases of the disease. The “biological impact” of neutrophil MVs is intriguing because these microstructures express >300 proteins that vary in relation to the stimulus and microenvironment leading to their production. It is therefore plausible that neutrophil MV subtypes can elicit distinct downstream effects (5).
The concept that neutrophils are not just short-lived effectors of inflammation but have long-term homeostatic actions is gaining ground. For example, neutrophil apoptotic bodies reprogram M1 macrophages to an M2 phenotype via efferocytosis, and depletion of neutrophils in murine models of ulcerative colitis leads to disease exacerbation (6). Although neutrophil depletion in models of experimental arthritis attenuates inflammatory parameters (7), it also decreases cartilage proteoglycan synthesis (8). Because the interaction between these two cell types in vivo is undescribed, we investigated whether MVs could enable a novel mechanism for the proanabolic effects of neutrophils on cartilage synthesis.
Here, we found that human RA synovial fluids contain abundant neutrophil-derived MVs. Mice lacking TMEM16F (anoctamin 6) produced fewer neutrophil MVs and displayed higher cartilage erosion in experimental arthritis, compared with wild-type animals. When administered into mice with inflammatory arthritis (K/BxN model), MVs protected the cartilage from the loss of sulfated glycosaminoglycans (sGAGs). Building upon these observations that immune cells may interact indirectly with chondrocytes in mature cartilage, we envisage that this MV-chondrocyte interaction can be harnessed as a platform for innovative therapeutic strategies in arthritis and other cartilage degradation diseases.
RESULTS
Rheumatoid synovial fluid neutrophil MVs contain annexin A1
To characterize MVs in human disease, we quantified immune cell–derived MVs in synovial fluid and paired plasma samples of seven RA patients (table S1). Significantly more neutrophil-derived (CD66b+), monocyte-derived (CD14+), and T cell–derived (CD3+) MVs were in the synovial fluid compared with plasma for each patient (Fig. 1A). Synovial MVs were enriched in the proresolving/anti-inflammatory protein annexin A1 (AnxA1) compared to plasma MVs, with more AnxA1+ MVs being of neutrophil origin (CD66b+) (Fig. 1B). Further stratification of a second, larger cohort of RA synovial fluid samples (n = 22) revealed higher numbers of neutrophil MVs (Fig. 1C) compared to MVs of monocyte (CD14+) or T cell origin (CD3+) that also contained AnxA1+ (Fig. 1D). Most MVs in human synovial fluids are therefore of neutrophil origin with the potential to elicit proresolving effects in the context of RA.
Fig. 1. Human RA synovial fluid is rich in AnxA1-positive neutrophil MVs that protect cartilage.
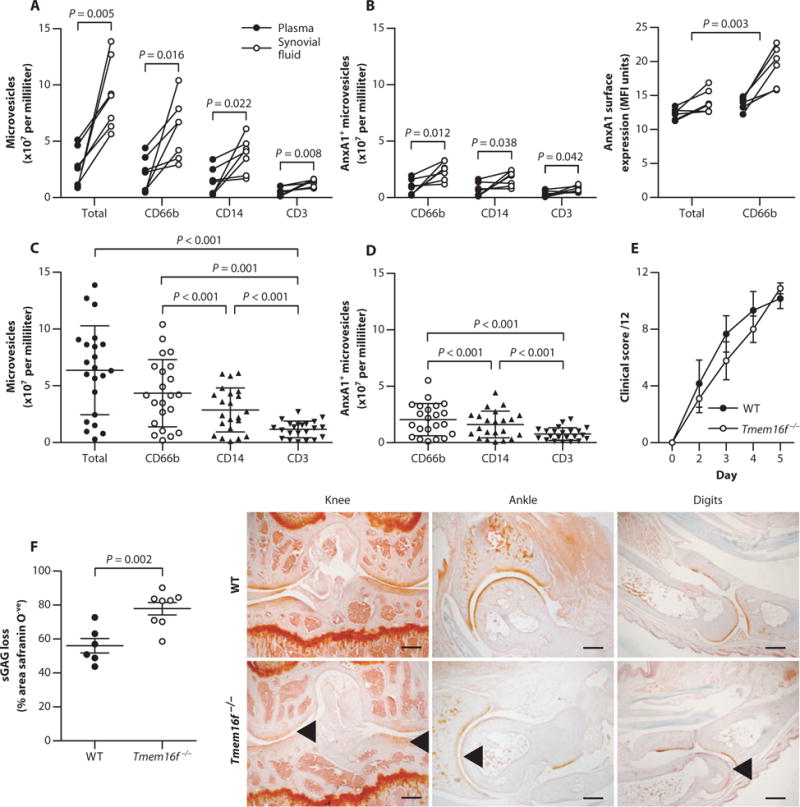
(A and B) CD66b, CD14, and CD3 expression on MVs (A) and the presence and expression levels of AnxA1-positive MVs (B) from paired synovial fluid and plasma of RA patients (n = 7). P values were determined by paired, two-tailed Student’s t test. MFI, mean fluorescence intensity. (C and D) Validation of synovial MV phenotype (C) and AnxA1+ MVs (D) in additional RA patient synovial fluid samples (n = 22). Data are means ± SD. P values were determined by one-way analysis of variance (ANOVA) and Bonferroni multiple comparison test. (E) Mice deficient in the putative phospholipid scramblase TMEM16F (Tmem16f−/−) and wild-type (WT) littermate controls were administered K/BxN serum to induce arthritis. Clinical scores are means of all joints ± SEM (n = 9 Tmem16f−/− and 6 WT mice). (F) Sections from digits, wrists, ankles, and knees at day 5 from mice in (E) were assessed for sGAG loss by safranin O staining. Data are means ± SEM (n = 6 to 9). P value was determined by unpaired two-tailed Student’s t test. Arrowheads indicate sGAG loss. Scale bars, 250 μm.
Mice with impaired MV production exhibit exacerbated cartilage erosion during inflammatory arthritis
Neutrophil MVs can evoke anti-inflammatory effects (9–11), although their actions in disease are not defined. Thus, to determine whether neutrophil MVs affected joint pathophysiology, a model of neutrophilic arthritis (K/BxN serum transfer) was performed in mice lacking the lipid scramblase TMEM16F (Tmem16f−/−). These mice display abnormal skeletal development, yet normal cartilage (12); their response to arthritic challenge has not been evaluated. The absence of TMEM16F affected neutrophil MV shedding, as evident in acute peritonitis (fig. S1, A to C). When subjected to inflammatory arthritis, Tmem16f−/− mice and wild-type littermates displayed similar clinical scores of inflammation and synovitis (Fig. 1E and fig. S1, D and E). However, an about twofold increase in sGAG loss, an index of cartilage integrity, was measured in Tmem16f−/− tissues (Fig. 1F and fig. S1F), suggesting that the loss of TMEM16F-dependent MV formation may exacerbate cartilage damage.
Neutrophil-derived MVs recapitulate physicochemical characteristics of those found in RA patient synovial fluids
Purified MVs from TNF-α–treated human neutrophils (fig. S2A) were used as a model of RA synovial fluid neutrophil-derived MV, with neutrophil activation confirmed by CD62L and CD11b expression (fig. S2, B and C). TNF-α stimulation led to a greater than twofold increase in MV shedding (referred to as MVTNF) compared to vehicle-treated control cells (MVCtrl) (Fig. 2A). More than 75% of MVs were CD66b+ and bound annexin V, indicating phosphatidylserine exposure, whereas about 60% were positive for phalloidin, confirming filamental actin in the vesicles (Fig. 2B) (8). MVs also expressed myeloid-related protein 8 (MRP8) (~80% positive) and its binding partner MRP14 (Fig. 2C). Flow cytometry (Fig. 2, D and E) and Western blotting (Fig. 2F) detected abundant AnxA1 expression, with larger positive percentages and higher intensities in the MVTNF populations than MVCtrl. However, total protein concentration per MV remained unchanged with TNF-α stimulation (Fig. 2G), suggesting specific enrichment of AnxA1 in MVTNF.
Fig. 2. Generation and characterization of AnxA1-rich MVs from healthy neutrophils.
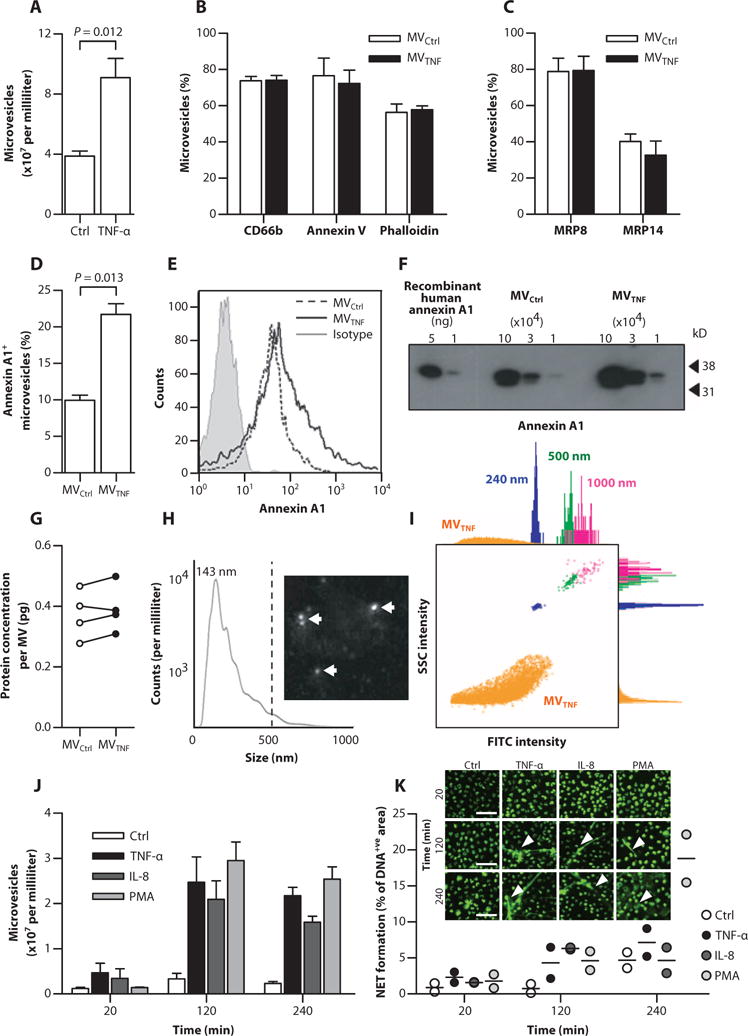
(A) Human circulating neutrophils (106) were stimulated with TNF-α (50 ng/ml) or vehicle (Control); resultant MVTNF or MVCtrl, respective ly, was enumerated. (B) Expression of CD66b, annexin V, and phalloidin. (C) Expression of MRP8 and MRP14. (D and E) The percentage of AnxA1+ MVs (D) and comparative surface expression (E) of AnxA1. Data in (A) to (E) are means ± SEM (n = 4). P values were determined by two-tailed Student’s t test. (F) AnxA1 content in MVTNF by Western blotting. Human recombinant AnxA1 was the positive control. (G) Total protein concentration of paired MVCtrl and MVTNF (n = 4 donors). (H) NTA for absolute MV size (six pooled MVTNF donors). Dotted line indicates 90% of MVs below 412 nm; median was 143 nm. The representative image is a still from the NTA. White arrows indicate MVs. (I) Representative side scatter (SSC)/fluorescence plot from ImageStreamX showing MVs (orange) versus 240- (blue), 500- (green), and 1000-nm (pink) calibration beads, with related histograms. FITC, fluorescein isothiocyanate. (J) MVs quantified at 20 min, 2 hours, and 4 hours after stimulation of parent human neutrophils with TNF-α (50 ng/ml), IL-8 (50 ng/ml), or PMA (100 nM). Data are means ± SEM (n = 3). (K) NET formation (n = 2). Images are representative micrographs used for analysis; white arrowheads indicate NETs. Scale bars, 50 μm.
Nanoparticle tracking analysis (NTA) provided accurate measurements of pooled MVTNF samples, where 90% of the vesicles were <412 nm in diameter (dotted line) with mode frequency at 143 nm (Fig. 2H). Detection of small-diameter vesicles suggested the presence of exosomes, confirmed by the expression of the tetraspanin protein TSG101 (tumor susceptibility gene 101) in MVs from two healthy donors (fig. S3). Using imaging cytometry, we detected MVs with superior precision (Fig. 2I) and enumerated MV formation after stimulation of parent neutrophils with TNF-α, interleukin-8 (IL-8) (an unrelated cell activator), and phorbol myristate acetate (PMA) over 240 min (Fig. 2J). MVs were obtained within 20 min of stimulation, with a peak at 120 min. However, neutrophil extracellular trap (NET) formation was observed microscopically at the two later time points (Fig. 2K). On the basis of these data and previous kinetics data (13), we selected the 20-min time point to generate enriched MV preparations for further experimentation.
Neutrophil MVs enhance chondroprotection by inducing transforming growth factor–β generation
Because Tmem16f−/− mice exhibited exacerbated sGAG loss during arthritis, we modeled adult cartilage matrix turnover using high-density three-dimensional (3D) micromass cultures of the human chondrocyte cell line C28/I2 or primary adult human articular chondrocytes (AHACs) in vitro (14) and tested the effect of neutrophil MVs on extracellular matrix (ECM) metabolism. Treatment of chondrocytes with MVCtrl or MVTNF (1 × 105) yielded no changes in ECM accumulation (Fig. 3A). However, when chondrocytes were first stimulated with IL-1β—a cytokine that induces destruction of articular cartilage (15)—or TNF-α, both MVCtrl and MVTNF afforded protection from sGAG loss, showing an increase in ECM deposition versus vehicle controls (Fig. 3A and fig. S4A). Notably, the effects of neutrophil MVs were dependent on the degree of IL-1β stimulation: larger concentrations of MVs (>50,000) were required to induce an effect at 3 ng/ml IL-1β than at 30 ng/ml IL-1β (fig. S4B). Furthermore, cytokine sequestration was not responsible for increased ECM because MVTNF did not express IL-1 receptor (IL-1R), although about 12 to 15% of isolated neutrophil MVs did express the TNF receptor (TNF-R1, fig. S4C).
Fig. 3. Neutrophil MVs induce ECM accumulation and are chondroprotective in vitro in chondrocyte micromasses.
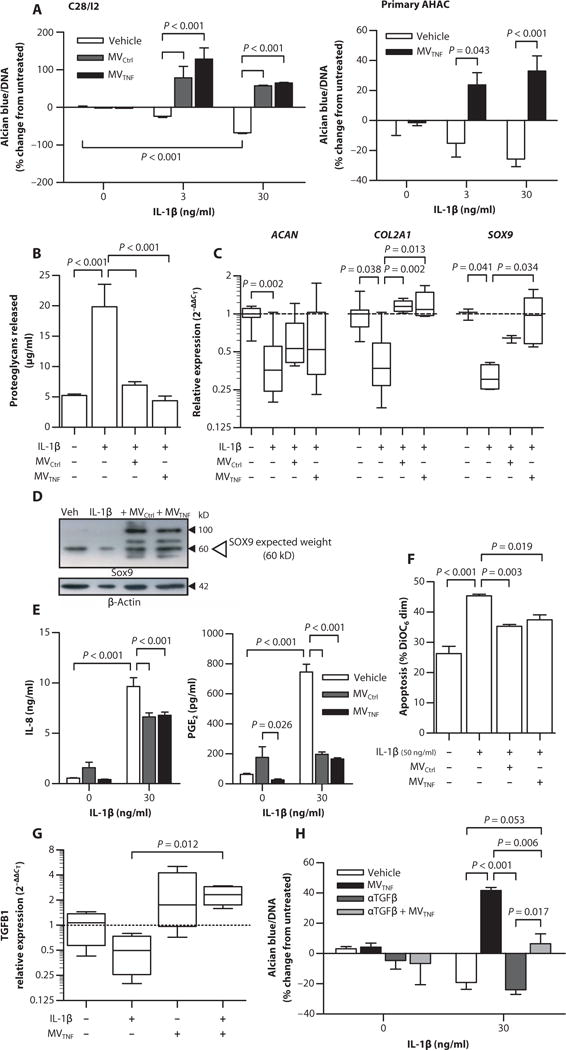
Human C28/I2 chondrocyte and primary AHAC micromass cultures were stimulated with IL-1β (3 or 30 ng/ml) alone or with MVCtrl and MVTNF (105). (A) ECM content normalized to DNA concentration. Data are means ± SEM (n = 6 separate MV and cell culture preparations). (B) ECM degradation in C28/I2 micromasses, quantified as amount of proteoglycans released. Data are means ± SEM (n = 10 separate MVs and 6 cell culture preparations). (C) Relative expression of COL2A1, ACAN, and SOX9 mRNA in C28/I2 micromasses. Data are , median ± interquartile range [n = 3 or 4 separate MV and micromass preparations and relative to housekeeping gene (rpl32) and untreated]. (D) Representative Western blot for SOX9. (E) IL-8 and PGE2 concentrations in C28/I2 micromass superna-tants. Data are means ± SEM (n = 6). (F) C28/I2 chondrocyte apoptosis in micromasses treated as in (A) with IL-1β (50 ng/ml). Data are means ± SEM (n = 4). (G) TGFB1 mRNA expression in C28/I2 mi-cromasses stimulated as in (C). Data are , median ± interquartile range (n = 4) relative to housekeeping gene (RPL32) and untreated. (H) ECM production normalized to DNA content in C28/I2 micromass cultures stimulated as in (A) with or without TGF-β neutralizing antibodies (10 μg/ml; αTGFβ) for 24 hours. Data are means ± SEM (n = 3 separate MV donors). P values were determined by one- or two-way ANOVA and Bonferroni posttest (A, B, E, F, and H) or Kruskal-Wallis test and Dunn’s multiple comparison posttest (C and G).
Without evidence to support direct cytokine sequestration, we investigated whether neutrophil MVs might amplify ECM accumulation by inhibiting its degradation. To test this, we measured the amount of released (enzymatically cleaved) sGAG in the supernatant. As expected, IL-1β treatment induced more proteoglycan release compared to control, an effect abrogated by cotreatment with MVs (1 × 105; Fig. 3B), although no significant down-regulation of MMP13 or ADAMTS5 could be demonstrated at the mRNA level (fig. S5). However, IL-1β treatment of chondrocytes led to reduced expression of ACAN and COL2A1 (the two most abundant cartilage matrix proteins) and SOX9 (the transcription factor driving their expression)—effects partly reversed by cotreatments with either MVCtrl or MVTNF (Fig. 3, C and D). Different molecular weight bands also appeared after MV coculture, yet their identities remain to be determined (Fig. 3D).
These chondrocyte responses to IL-1β were complemented by increased release of IL-8 and prostaglandin E2 (PGE2) (Fig. 3E), which in high levels are associated with cartilage degradation in RA (16). Both MVCtrl and MVTNF significantly attenuated IL-8 (~30% reduction) and PGE2 (>80% inhibition) release (Fig. 3E). Chondrocyte apoptosis is also associated with cartilage loss in animal models and in humans (17, 18). Addition of high levels of IL-1β (50 ng/ml) increased C28/I2 chondrocyte apoptosis (Fig. 3F and fig. S6), but MVCtrl or MVTNF prevented cell death (Fig. 3F).
Coculture of neutrophil MV with monocyte-derived macrophages (MDMs) or dendritic cells induces anti-inflammatory activity, partly via induction of transforming growth factor–β (TGF-β) (10, 11). To determine whether chondroprotection resulted from increased TGF-β synthesis, we assessed the transcription of TGFB1 in C28/I2 human chondrocyte micromasses. TGFB1 mRNA was down-regulated about twofold by IL-1β treatment but significantly up-regulated after coculture with MVTNF, with or without IL-1β (Fig. 3G). Blocking TGF-β resulted in significantly reduced (~50%) anabolic effects exerted by neutrophil MVTNF in the coculture settings (Fig. 3H). These data suggest that chondroprotection results from MV-dependent TGF-β induction.
Collectively, these in vitro studies indicate that neutrophil-derived MVs protect against various sequelae involved in cartilage degradation (chondrocyte apoptosis, PGE2 and IL-8 release, and ECM degradation) by significantly up-regulating the transcripts of genes key to cartilage anabolism after the induction of TGF-β.
MVs penetrate cartilage ex vivo and in vivo
To determine the mechanism behind MV-induced chondroprotection in a physiological setting where chondrocytes are surrounded by the dense, avascular, and cell-impenetrable cartilage matrix, fluorescent neutrophil MVs were cocultured for 18 hours with rat cartilage explants. The location of MVs was confirmed by immunostaining for anti-human MRP8 (Fig. 4A), which was highly expressed in MVCtrl and MVTNF (Fig. 2C). Migration of human MVs into cartilage was observed in both vehicle- and IL-1β–treated explants (Fig. 4A); however, the distance of penetration was significantly higher in IL-1β–stimulated explants (Fig. 4B). Synthetic fluorescent microcapsules of comparable size and AnxA1 content did not migrate into the cartilage, irrespective of cartilage treatment (Fig. 4A), suggesting that MV entry is not by passive diffusion. Coculture of sonicated (ruptured) MVTNF with IL-1β–stimulated rat femoral heads failed to reduce sGAGloss, in contrast to whole MVTNF (Fig. 4C), indicating that the intact MV structures are required for chondroprotection.
Fig. 4. MVs enter cartilage ex vivo and in vivo to deliver AnxA1.
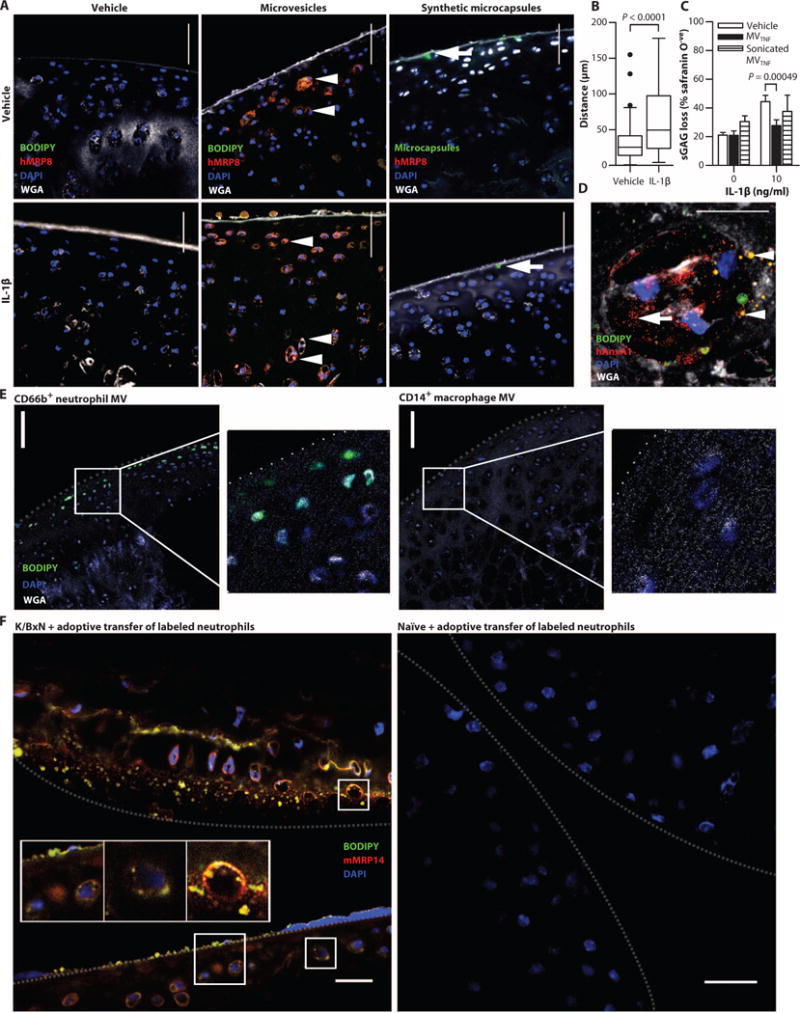
Rat femoral head cartilage explants were stimulated with IL-1β (10 ng/ml, 3 days incubation before the addition of MVs) or vehicle (Veh) [phosphate-buffered saline (PBS)] before coculture with fluorescently labeled MVTNF (5 × 105) or synthetic fluorescent microcapsules (containing AnxA1). (A) Sections were stained for MRP8, 4′,6-diamidino-2-phenylindole (DAPI), and wheat germ agglutinin (WGA). Arrowheads indicate MVTNF; arrows indicate microcapsules. (B) The distance of MV migration was measured. Data are Tukey’s box and whiskers showing outliers (n = 140 or 166 MVs measured over at >10 individual micrographs). P values were determined by Mann-Whitney t test. Scale bars, 50 μm. (C) Murine cartilage explants were stimulated with IL-1β as in (A) or vehicle (saline) before coculture with MVTNF or sonicated MVTNF. sGAG loss was quantified by staining with safranin O. Data are means ± SEM (n = 3 or 6). P values were determined by two-way ANOVA with Bonferroni posttest. (D) Rat cartilage explants treated as in (A) stained for human AnxA1. Arrowheads indicate intact MVs; arrow indicates AnxA1 staining that is no longer MV-associated. Scale bar, 10 μm. Movie S1 shows related z-stack images. (E) Murine cartilage explants stimulated with IL-1β as in (A) and treated with fluorescently labeled neutrophil- or macrophage-derived MVs (5 × 105). Sections were stained with DAPI and WGA. Scale bars, 100 μm; insets, higher magnification. Dotted line indicates ar ticular surface. Images are representative of n = 3 separate MV preparations. (F) Representative wrists from K/BxN or naïve mice (n = 3 per group) receiving fluorescently labeled whole neutrophils (green) intravenously. Sections were stained for MRP14 and DAPI. Scale bars, 20 μm. Dotted lines indicate opposing articular surfaces.
Sections from rat cartilage explants treated with IL-1β and cocultured with MVTNF were stained for AnxA1 (Fig. 4D). Neutrophil-derived MVs colocalized with human AnxA1+ staining and accumulated preferentially within the chondrocyte lacunae. Congruently, diffuse staining of human AnxA1 was observed, suggesting delivery to the chondrocyte cytoplasm. Movie S1 shows serial z-stack images of cartilage-resident chondrocytes that have engulfed human neutrophil-derived AnxA1+ MVs.
We next tested the cartilage penetration of other myeloid-derived vesicles, abundant in RA synovial fluid. Incubation of cartilage explants with MDM-derived (CD14+) MVs indicated lack of cartilage penetration or even adhesion to the articular surface, whereas neutrophil-derived (CD66b+) MVs were abundant within the matrix (Fig. 4E). Thus, it appears that the selective interaction between MVs and cartilage matrix may involve a directed chemotactic response. We measured the expres sion levels of the IL-8 receptor CXCR2 on MVs because chondrocytes up-regulate IL-8 upon insult (19). Neutrophil-derived MVs expressed CXCR2, CXCR1, and CXCR4 (fig. S7A). However, a neutralizing CXCR2 antibody did not affect the ability of MVTNF to migrate into cartilage explants (fig. S7B). Congruently, MVs generated from Cxcr2−/− mice entered cartilage to the same extent as wild-type MVs (fig. S7B). Thus, chemoattractant receptor(s) other than CXCR2 (expressed by neutrophils and their offspring MV, but not by MDM or MDM MV) may be required for cartilage-directed migration. Candidate adhesion molecules ICAM-1 (intercellular adhesion molecule–1) and PECAM (platelet/endothelial cell adhesion molecule) were not expressed on CD66b+ MVs (fig. S7A). The specific mechanism of neutrophil-derived MV uptake into cartilage is not yet clear, but we can rule out CXCR2, ICAM-1, and PECAM.
To determine whether MVs enter cartilage in pathophysiological settings in vivo, we injected systemically (intravenously) intact fluorescent murine neutrophils into mice with active inflammatory arthritis (day 3 after K/BxN serum transfer). Wrists were collected after 24 hours and costained for murine MRP14. MRP14+ MVs were detected within the cartilage of arthritic mice, yet no MVs or MRP14 staining was evident in the wrists of nonarthritic mice administered with exogenous labeled neutrophils (Fig. 4F). We posit that neutrophils migrate into inflamed joints to release MVs locally, which can penetrate the sGAG-depleted cartilage matrix.
A functional distinction between whole neutrophils and their offspring MVs was demonstrated using neutrophil–C28/I2 chondrocyte coculture with Trans-well filters (fig. S8A). Direct neutrophil contact with chondrocytes inhibited anabolism and induced cell death (fig. S8B). In contrast, restricting neutrophil migration while still allowing MV contact with chondrocytes, by the use of membrane pore sizes 200 and 400 nm, yielded chondroprotection, as evidenced by ECM deposition in the presence of IL-1β, similar to our other in vitro studies (Fig. 3A and fig. S4B).
Cartilage protection by MVs requires AnxA1 and its receptor FPR2/ALX
Because AnxA1 is elevated in neutrophil-derived MVs in RA synovial fluid, a feature reproduced with MVTNF, we questioned whether this protein played an essential role in the chondroprotective properties defined here. Indeed, MVs lacking AnxA1 lost >80% of their proanabolic effects when added to C28/I2 chondrocyte micro-mass culture (Fig. 5A and fig. S9). We next established whether the AnxA1 receptor formyl peptide receptor 2 (FPR2)/ALX is involved in the protective properties of MVs. Application of the selective FPR2/ALX antagonist WRW4 to C28/I2 chondrocytes in addition to MVTNF abrogated the rescue of IL-1β–induced sGAG loss normally mediated by MVTNF, without any direct biological actions (Fig. 5B). Chondrocytes expressed FPR2/ALX with a moderate increase after stimulation with IL-1β (Fig. 5C). The presence of endogenous FPR2/ALX and AnxA1 in chondrocytes (C28/I2 and primary AHAC), with frequent colocalization, was confirmed by electron microscopy (Fig. 5D). To further investigate the impact of the AnxA1 and FPR2/ALX on MV-induced chondroprotection, we incubated MVs with cartilage explants isolated from mice lacking FPR1 and FPR2/3 (the ortholog of human FPR2/ALX). The degradative effect of IL-1β was similar in cartilage explants from all animals; however, application of MVTNF was effective only in wild-type and Fpr1−/− but not Fpr2/3−/− explants (Fig. 5E).
Fig. 5. Requirement of AnxA1 and its receptor FPR2/ALX for MV-induced chondroprotection.
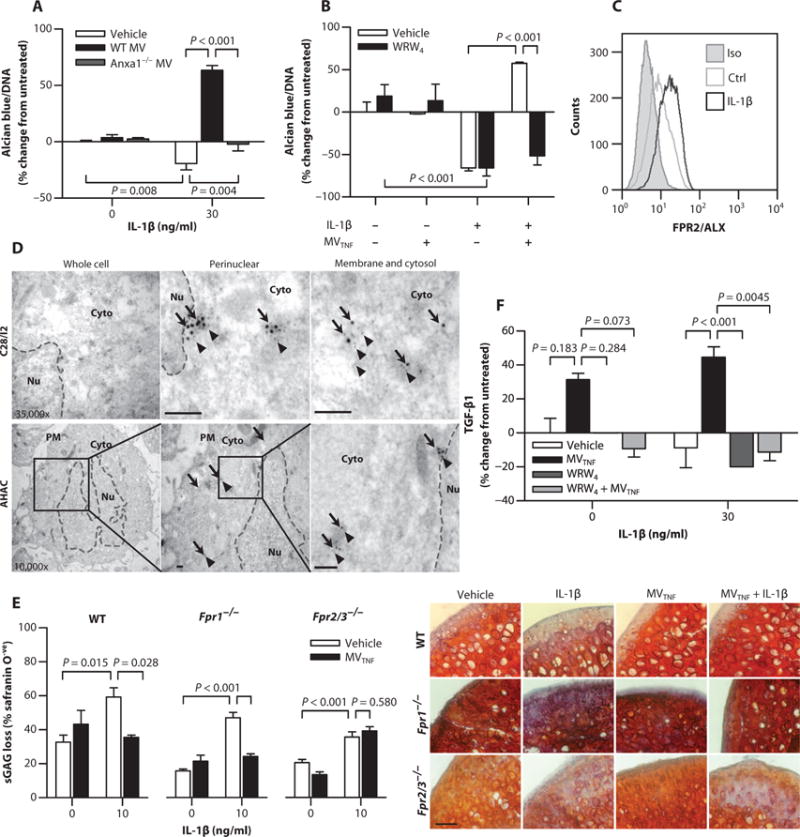
(A) ECM deposition normalized to DNA content in C28/I2 micromass cultures stimulated with IL-1β and cocultured with WT or Anxa1−/−mouse MVTNF (105). (B) ECM deposition normalized to DNA content in C28/I2 micromass cultures stimulated with a combination of IL-1β (30 ng/ml), human neutrophil-derived MVTNF (105), and WRW4 (10 μM, 10 min before MV addition). Data in (A) and (B) are means ± SEM (n = 4 separate MV and cell culture preparations). (C) FPR2/ALX expression in C28/I2 micromasses [stimulated as in (A)]. Data are representative of n = 3. (D) Immunogold labeling of AnxA1 (arrows) and FPR2/ALX (arrowheads) in C28/I2 and AHAC micromasses. Black boxes indicate panel magnified to the right for AHAC. Dashed lines, nuclear envelope. PM, plasma membrane; Cyto, cytoplasm; Nu, nucleus. Scale bars, 100 nm. (E) Cartilage explants from WT, Fpr1−/−, or Fpr2/3−/− mice were stimulated with IL-1β with or without 105 MVTNFs every other day for 7 days. Sections were stained with safranin O. Representative images of micrographs used for analysis are shown. Scale bars, 200 μm. Data are means ± SEM (n = 3 to 6). (F) Total TGF-β1 content in supernatants of C28/I2 micromasses treated as in (B). Data are means ± SEM (n = 8 separate MV donors). P values were determined by two-way ANOVA and Bonferroni posttest (A, B, E, and F).
To determine whether chondroprotection resulted from the induction of TGF-β downstream of FPR2/ALX, we assayed C28/I2 culture supernatant after IL-1β and MV stimulation, with or without the FPR2/ALX antagonist. TGF-β levels were increased in C28/I2 chondrocyte micromasses after costimulation with IL-1β and MVTNF (Fig. 5F). However, the addition of WRW4 reduced TGF-β concentrations to levels comparable to IL-1β stimulation alone, suggesting FPR2/ALX engagement upstream of TGF-β production. Altogether, these data indicate that AnxA1 and FPR2/ALX are responsible for the protective properties of neutrophil MVs.
MVs protect from cartilage degradation in vivo in experimental inflammatory arthritis
To translate these findings, we administered MVs to a mouse model of K/BxN arthritis, in which the knee joint is markedly affected (20), providing a favorable site for intra-articular injection of MVs (21). MVTNFs were injected into the right knee 3 days after arthritis induction, just before peak arthritis. Saline was administered to contralateral knees as a control. Cartilage integrity was determined on day 5. Nonarthritic mice retained sGAG, regardless of treatment, but arthritic mice experienced a marked reduction in sGAG content that was prevented in MVTNF-treated joints (Fig. 6A). Confirming the nonredundant role of FPR2/ALX, MVs lost efficacy when coadministered with WRW4 or injected into Fpr2/3−/− (Fig. 6, A and B). Similarly, in an adaptive immunity–dependent RA model, elicited by glucose-6-phosphate isomerase (GPI) immunization (22), MVTNF injected into the right knee 21 days after arthritis induction (before peak arthritis) prevented sGAG content loss (~25%) compared with controls (saline, ~40%) (Fig. 6C).
Fig. 6. Neutrophil MVs protect from cartilage degradation in models of inflammatory arthritis.
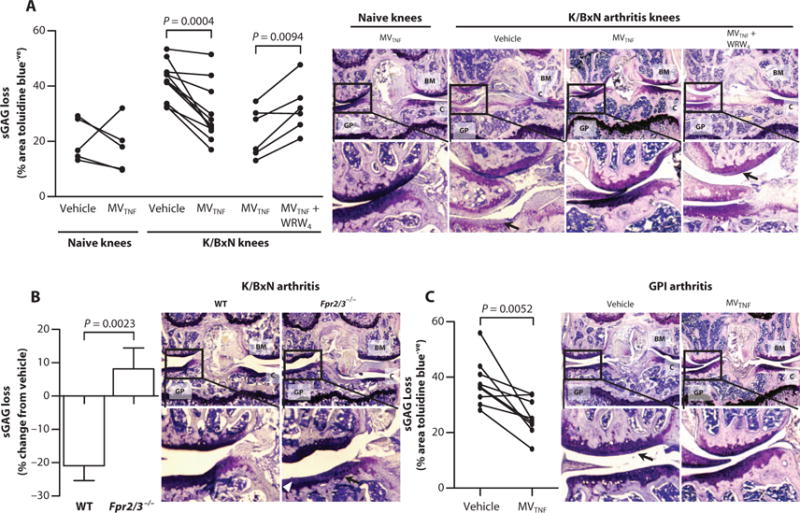
(A and B) Mice received arthritogenic K/BxN serum (100 μl) on days 0 and 2; on day 3, vehicle (left knee; PBS, 5 μl) or MVTNF (right knee; 3 × 104 in 5 μl) with or without WRW4 (10 μM) was injected intra-articularly into WT (A) or Fpr2/3−/− mice (B). On day 5, knee joints were sectioned and stained with toluidine blue for sGAG content, and representative images are shown; black arrows in (A) indicate sGAG loss, and white arrowheads in (B) indicate cartilage surface fibrillations. Data are calculated from three sections per paired knees with three images per section (n = 5 to 9). P values were determined by two-way repeated-measures ANOVA and Bonferroni posttest. (C) DBA-1 female mice (n = 9) were immunized with GPI peptide on day 0 and administered vehicle intra-articularly in the left knee (PBS, 5 μl) or MVTNF in the right knee (3 × 104 in 5 μl) intra-articularly on day 21. Knee joint sections were stained with toluidine blue, and representative images are shown; black arrows indicate sGAG loss. Data are from three sections per knee with three 20× images captured per section. P values were determined by paired, two-tailed Student’s t test. In (A) to (C): BM, bone marrow; GP, growth plate; C, cartilage.
DISCUSSION
We have uncovered chondroprotective properties of neutrophil-derived MVs, which directly interact with chondrocytes, revealing a potential new therapeutic approach for arthritis. Previous reports suggest pathogenic roles for MVs of platelet (23) and immune-cell origin (24) in the RA joint, focusing particularly on the synovial tissue as the target. However, MVs of neutrophil origin exert anti-inflammatory actions on macrophages and microvascular beds (9, 11). We propose a model where neutrophils are recruited to the joint during the active flares of arthritis and release high numbers of MVs. These MVs enter the cartilage and bind the FPR2/ALX receptor, inducing TGF-β1 production and ECM deposition, while protecting chondrocytes from apoptosis. Furthermore, these MVs carry a complex cargo that includes the tissue-protective, proresolving protein AnxA1, opening up the possibility of modifying MVs to deliver other therapeutics.
Dogma dictates that cartilage is an immune cell–impenetrable, dense, avascular matrix through which metabolites from the synovial fluid must diffuse. Indeed, direct coculture of neutrophils with chondrocytes resulted in chondrocyte death. We provide evidence that neutrophil-derived MVs are instead able to cross this barrier to deliver AnxA1, a proresolving protein, and promote chondrocyte anabolism. Hence, cargo delivery via MVs through the impenetrable matrix may provide a mechanism of excluding damaging cell-cell contact while providing protection from cartilage degradation. MVs were active solely in inflammatory settings, likely through FPR2/ALX up-regulation, to promote anabolic gene transcription and accumulation of ECM in vitro and ex vivo in cartilage explants. These results were confirmed in vivo where selective presence of neutrophil MVs was observed in arthritic but not naïve joints after adoptive transfer of whole cells. Injection of MVs intra-articularly induced chondroprotection, whereas mice lacking TMEM16F, having inadequate production of endogenous MVs, experienced greater cartilage erosion, without differences in synovitis.
Human AnxA1 and MRP8 colocalized in part with an apparent vesicular form in rodent midzone cartilage, suggesting that a proportion of MVs may enter the chondrocyte in their entirety and release their content after fusion. Although studies determining the kinetics of endogenous human MV migration into cartilage are not feasible, there are reports that neutrophil-derived proteins are selectively present within human cartilage during arthritis. MRP8 protein, but not mRNA, is detected abundantly in human arthritic cartilage (25), and this protein is highly enriched in neutrophil MVs, as detected by proteomic analysis (8) and confirmed in this study. We speculate that the source of this myeloid-restricted protein within arthritic cartilage is neutrophil MVs; thus, although indirect, there is evidence that MV migration into cartilage may be a physiological process occurring in humans.
A higher degree of MV penetration was observed in inflammatory settings, which may result from the extent of cartilage damage and/or the presence of chemoattractant factors released by inflamed chondrocytes. Neutrophil-derived MVs contain several cytoskeletal proteins, a finding that may explain their ability to migrate in response to chemokine gradients (8). However, we could not identify the chemo-attractant operative in our settings. A plausible candidate was IL-8 (or murine counterpart KC) because it is produced by human and rodent chondrocytes both in steady state and upon catabolism (19). However, we found that the IL-8 receptor CXCR2, albeit present on neutrophil MVs, was dispensable for matrix entry. Presently, the mechanism of MV migration is unclear, yet a degree of selectivity toward neutrophil MVs—rather than MDM-derived MVs—allows them to enter cartilage. Expression of some neutrophil-derived specific receptor, adhesion molecule, or matrix metalloproteinase may allow MV-matrix interactions, though any involvement of ICAM-1 and PECAM was excluded by this study.
Mediators of resolution regulate several phenomena, ultimately repairing injured tissue. An important effector of resolution is AnxA1, which activates tissue repair programs in the context of gut (26) and eye epithelia (27). AnxA1 lacks a signaling peptide (28) yet is found abundantly in inflammatory exudates, including RA synovial fluids (29), with its main mechanism of secretion likely to be via the release of MV. We found that neutrophil MVs in synovial fluid of RA patients were rich in AnxA1 compared to those circulating in plasma. This finding could reflect higher cell concentrations within the synovial fluid, but the context-specific activation of the cells leads to changes in their protein expression: AnxA1 gene promoter activation occurs in neutrophils during transmigration (30), explaining the AnxA1-enriched MV in RA exudates and the higher degree of expression at the single-vesicle level. Calcium (Ca2+) chelation by AnxA1 promotes binding to phosphatidylserine and, subsequently, a conformational rearrangement with extrusion of the N-terminal region (28), enabling interaction with its receptor (31). Thus, Ca2+-dependent MV generation (13, 32), reliant on TMEM16F, provides the molecular requirements for externalization of active AnxA1. It is not clear from our data whether chondroprotection requires surface-expressed AnxA1 to bind FPR2/ALX or the engulfment of the entire particle. Certainly, surface-expressed AnxA1 would be in the correct orientation (with Ca2+ and bound to phospholipids) for FPR2/ALX binding; however, micrographs suggest engulfment and dismantling of the MVs; congruently, sonicated MVs do not evoke chondroprotective actions.
There is emerging evidence linking neutrophil MVs to the induction of TGF-β (10, 11)—a key mediator of chondrocyte homeostasis that stimulates type II collagen and sGAG deposition (33) and down-regulates cartilage-degrading enzymes (15). We found that MVTNF reversed TGF-β1 down-regulation by IL-1β, with levels higher than untreated chondrocytes. In support of a mechanism for production of TGF-β downstream of AnxA1-FPR2/ALX signaling, AnxA1 activates TGF-β production via FPRs in macrophages fed with apoptotic neutrophils (34). The augmentation of chondrocyte TGF-β production during MV coculture represents a protective mechanism against IL-1β–induced damage.
Using MVs as novel therapeutic tools in inflammatory diseases is a promising clinical approach because they can be enriched with proteins (35) or bioactive lipid mediators and their precursors (36). Relevantly, recent studies have used nanoparticle-based formulations to deliver peptide fragments of AnxA1 to resolve inflammation in murine models of atherosclerosis (37). Mesenchymal stem cells may be viable delivery vehicles for MVs because they naturally release exosomes that suppress hypoxia in mice (38). MVs are also amenable to targeting by incorporation of organ-specific homing peptides (39); moreover, because the sequence directing mRNAs to the pathway of MV secretion has been elucidated, MVs could be enriched for specific RNA species, enhancing their ability to deliver desired or therapeutic mRNA to target cells (40). Nevertheless, the future development of MV-based therapies requires the transition from mouse to human clinical studies. In support of advocating MV treatment in clinical trials, recent meta-analyses of platelet-rich plasma (PRP) administration into the joints of patients with mild degenerative chondral lesions demonstrated promising efficacy compared to hyaluronic acid treatment (41). We speculate that the MV component of PRP may be responsible for these effects, considering on average the presence of over 2 × 106 MVs per milliliter of plasma (13). Thus, a reasonable next step in ongoing PRP clinical studies would be to treat more advanced chondrodegenerative conditions, from trauma-induced damage to rheumatoid or osteoarthritis, to establish the translational potential of MVs.
In summary, the results presented indicate that neutrophil MVs, enriched in AnxA1, enter cartilage inducing FPR2/ALX signaling, which causes TGF-β production, matrix deposition, and chondrocyte homeostasis. These findings advocate the delivery of autologous MVs, either native or engineered to contain bioactive molecules, to chondrocytes in situ. Our study sheds additional light on the functional and cellular associations between immune system and cartilage biology, delineating new strategies for innovative drug discovery with the goal of treating damaged cartilage of arthritic joints.
MATERIALS AND METHODS
Study design
The goal of this study was to investigate the role of neutrophil-derived MV during inflammatory arthritis and models of chondrocyte ECM turnover. We enumerated and performed phenotypic analyses on paired RA patient samples (n = 7) and a larger cohort of synovial fluid samples (n = 22). Because no previous data were available to estimate group variances, the entire biobank was analyzed to power the data as highly as possible. These human studies were approved by the Multi-centre Research Ethics Committee (reference 07/Q0605/29) (table S1). Several in vitro and ex vivo approaches were used to determine the pro-anabolic function of human MV in chondrocyte coculture {prepared according to approved protocols: Research Ethics Committee [P/00/029 ELCHA (East London and The City Health Authority)]; volunteers gave written informed consent to blood collection, and procedures were approved by East London and The City Local Research Ethics Committee (REC Ref. 05/Q0603/34 ELCHA, London, UK)}.
The chondroprotective effects of MV were examined in vivo in the K/BxN mouse model of arthritis by either knocking out Tmem16f or administering exogenous MVs. For one-way experiments, on the basis of the variance of pilot data (for 80% power and an α of 0.05), five mice per group were required to detect a mean difference of 25% sGAG loss between groups (considered a priori to be the minimal biologically significant effect; Cohen’s d of ~1.5). For multifactorial experiments, at least six mice per group were required (for 80% power, α of 0.05 and smallest partial η2 of ~0.3). All experiments were approved and performed under Home Office regulations (Scientific Procedures Act 1986). Mice were randomized to different groups, but the experimenter was not blinded to the group identities until after the administration of MVs. All samples were processed and analyzed in a blinded manner.
MV characterization and in vitro generation
Human RA synovial fluid MVs
Human RA synovial fluid (tables S1 to S3) was centrifuged at 3000g for 25 min at 4°C and treated with hyaluronidase (2 mg/ml; type I-S, 620 U/mg) for 30 min at room temperature. Samples were clarified by centrifugation at 10,000g for 10 min at room temperature. MVs were stained with antibodies directly in synovial fluid diluted 1:1 in double-sterile filtered (0.22-μm filter) PBS. Antibody targets (CD14, CD3, CD66b, and AnxA1) and preparations are described in Supplementary Materials and Methods.
Human neutrophil-derived MVs
Neutrophils were isolated from healthy human volunteers by density gradient centrifugation and then stimulated with recombinant TNF-α, IL-8, or phorbol myristate. MVs were then pelleted and stained for CD66b, AnxA1, TSG101, MRP8, MRP14, ICAM-1, PECAM, CXCR1, CXCR2, CXCR4, IL-1R1, TNF-R1, and annexin V and further characterized as described in Supplementary Materials and Methods.
Mouse MV generation
MV isolation from F Tmem16f−/−, C57BL/6 Anxa1−/−, and Cxcr2−/− mice is described in Supplementary Materials and Methods.
Chondrocyte micromass culture and assays
Chondrocyte micromasses were formed from differentiated C28/I2 human chondrocytes and primary AHAC in culture. They were characterized by Alcian blue staining, proteoglycan, PGE2, IL-8, and TGF-β release, gene expression, apoptosis, flow cytometry, electron microscopy, and Western blotting, as described in Supplementary Materials and Methods.
Ex vivo cartilage explants
Cartilage explants were collected from male Wistar rats or C57BL/6 mice (wild-type, Fpr1−/−, or Fpr2/3−/−) within 30 min of sacrifice, treated for 3 days with recombinant mouse IL-1β (10 ng/ml, 3 days), and co-cultured with BODIPY-Maleimide–labeled human neutrophil MV or macrophage MV. Cryosections (6 μm) were stained with anti-MRP8, anti-AnxA1, DAPI, and WGA and visualized by confocal microscopy, or sGAG loss was quantified by safranin O staining.
In vivo arthritis
The induction of K/BxN arthritis and GPI-induced arthritis models is described in Supplementary Materials and Methods. Tmem16f −/− and wild-type littermate control mice (males) undergoing a 5-day K/BxN arthritis were scored daily, and ankle thickness was measured. Wild-type or Fpr2/3−/− mice (C57BL/6) undergoing a 5-day K/BxN arthritis were administered MVTNF (3 × 104; 5-μl final volume) or PBS (5-μl final volume) intra-articularly on day 3, and joints were harvested for histological analysis on day 5. Wild-type female DBA-1 mice undergoing 25-day GPI arthritis were administered MVTNF or PBS as above on day 21 before joint collection on day 25. Joints were decalcified in formic acid (10% v/v) and paraffin-embedded. Sections (10 μm; at least three per joint per mouse) were stained with toluidine blue stain or safranin O, and toluidine blue– or safranin O– positive percentage area was calculated from micrographs in a blinded manner using ImageJ.
A further cohort of mice undergoing K/BxN arthritis (as above) received adoptive transfer of BODIPY-Maleimide–labeled neutrophils intravenously on day 3. Joints were harvested for immunofluorescence staining on day 4 without decalcification.
Statistical analyses
Statistical comparisons were performed with mean ± SEM for continuous variables of experiments conducted at least three times, where n is the biological replicate (human donors or mice). Data were analyzed (two-tailed) using either paired or unpaired Student’s t test, one-way ANOVA with Bonferroni posttest, Kruskal-Wallis followed by Dunn’s multiple comparison, or two-way ANOVA with Bonferroni posttest where appropriate. In all cases, P < 0.05 was taken as significant. Analyses were performed using GraphPad Prism 5 or SPSS 22 software.
Supplementary Material
Fig. S1. Tmem16f knockout mice have reduced MV shedding and exacerbated sGAG loss in experimental arthritis.
Fig. S2. Characterization of parent neutrophils used to generate offspring MVs.
Fig. S3. Presence of tetraspanin TSG101 in neutrophil MVs.
Fig. S4. Concentration-dependent chondroprotection of MV against cytokine stimulation of chondrocytes.
Fig. S5. Selective effect of MV treatment on chondrocyte matrix degrading enzyme mRNA expression.
Fig. S6. MV treatment protects from chondrocyte apoptosis.
Fig. S7. MV chemokine receptor and adhesion molecule expression.
Fig. S8. Neutrophils induce ECM accumulation in chondrocytes only when prohibited from direct cell-to-cell contact.
Fig. S9. Generation of neutrophil MVs from AnxA1−/− mice.
Table S1. RA patient demographics.
Table S2. RA patient clinical profiles.
Table S3. RA patient treatment history.
Movie S1. MVs penetrate cartilage ex vivo to deliver AnxA1.
Acknowledgments
We thank H. B. Patel and R. Jones (William Harvey Research Institute) for help with arthritis models, J. Dalli for conceptual insight, and G. Sukhorukov (Queen Mary University of London) for providing microcapsules.
Funding:
This work was supported by The Wellcome Trust (program 086867/Z/08 and 101604/Z/13/Z), FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo; 2012/21603-2), and CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico; 308144/2014-7) (to S.M.O.), and The Nuffield Foundation (Oliver Bird Studentship to S.E.H.). L.V.N. is supported by an Arthritis Research UK Career Development Fellowship (19909), and S.E.H. is supported by an Arthritis Research UK Foundation Fellowship (20842). L.Y.J. is an investigator of the Howard Hughes Medical Institute. The Medical Research Council and Arthritis Research UK provided support for the development of the biobank that provided the samples for these studies.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/7/315/315ra190/DC1) Materials and Methods
Author contributions:
S.E.H. designed and performed in vitro, ex vivo, and in vivo experiments and wrote the manuscript. H.R.J. performed MV expression analyses, in vitro experiments, and statistical analyses, and contributed to the manuscript. L.V.N. designed and performed in vivo experiments and contributed to the manuscript. C.D.G. and S.M.O. performed electron microscopy analyses. A.N. and C.P. provided clinical samples and data. E.C. and P.R.S. performed NET experiments. F.D. and C.P. provided conceptual expertise and edited the manuscript. A.K. and L.Y.J. provided mice and data and edited the manuscript. M.P. provided mice, coordinated the project, and wrote the manuscript.
Competing interests:
The authors declare that they have no competing interests.
Data and materials availability:
The data for this study have been deposited in the database dbGAP (database of Genotypes and Phenotypes).
REFERENCES AND NOTES
- 1.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G. Transgenic mice expressing human tumour necrosis factor: A predictive genetic model of arthritis. EMBO J. 1991;10:4025–4031. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Headland SE, Norling LV. The resolution of inflammation: Principles and challenges. Semin Immunol. 2015;27:149–160. doi: 10.1016/j.smim.2015.03.014. [DOI] [PubMed] [Google Scholar]
- 3.Berckmans RJ, Nieuwland R, Kraan MC, Schaap MC, Pots D, Smeets TJ, Sturk A, Tak PP. Synovial microparticles from arthritic patients modulate chemokine and cytokine release by synoviocytes. Arthritis Res Ther. 2005;7:R536–R544. doi: 10.1186/ar1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cloutier N, Tan S, Boudreau LH, Cramb C, Subbaiah R, Lahey L, Albert A, Shnayder R, Gobezie R, Nigrovic PA, Farndale RW, Robinson WH, Brisson A, Lee DM, Boilard E. The exposure of autoantigens by microparticles underlies the formation of potent inflammatory components: The microparticle-associated immune complexes. EMBO Mol Med. 2013;5:235–249. doi: 10.1002/emmm.201201846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalli J, Montero-Melendez T, Norling LV, Yin X, Hinds C, Haskard D, Mayr M, Perretti M. Heterogeneity in neutrophil microparticles reveals distinct proteome and functional properties. Mol Cell Proteomics. 2013;12:2205–2219. doi: 10.1074/mcp.M113.028589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, Bayless AJ, Scully M, Saeedi BJ, Golden-Mason L, Ehrentraut SF, Curtis VF, Burgess A, Garvey JF, Sorensen A, Nemenoff R, Jedlicka P, Taylor CT, Kominsky DJ, Colgan SP. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity. 2014;40:66–77. doi: 10.1016/j.immuni.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eyles JL, Hickey MJ, Norman MU, Croker BA, Roberts AW, Drake SF, James WG, Metcalf D, Campbell IK, Wicks IP. A key role for G-CSF–induced neutrophil production and trafficking during inflammatory arthritis. Blood. 2008;112:5193–5201. doi: 10.1182/blood-2008-02-139535. [DOI] [PubMed] [Google Scholar]
- 8.Gresnigt MS, Joosten LAB, Verschueren I, van der Meer JWM, Netea MG, Dinarello CA, van de Veerdonk FL. Neutrophil-mediated inhibition of proinflammatory cytokine responses. J Immunol. 2012;189:4806–4815. doi: 10.4049/jimmunol.1103551. [DOI] [PubMed] [Google Scholar]
- 9.Dalli J, Norling LV, Renshaw D, Cooper D, Leung KY, Perretti M. Annexin 1 mediates the rapid anti-inflammatory effects of neutrophil-derived microparticles. Blood. 2008;112:2512–2519. doi: 10.1182/blood-2008-02-140533. [DOI] [PubMed] [Google Scholar]
- 10.Eken C, Gasser O, Zenhaeusern G, Oehri I, Hess C, Schifferli JA. Polymorphonuclear neutrophil-derived ectosomes interfere with the maturation of monocyte-derived dendritic cells. J Immunol. 2008;180:817–824. doi: 10.4049/jimmunol.180.2.817. [DOI] [PubMed] [Google Scholar]
- 11.Gasser O, Schifferli JA. Activated polymorphonuclear neutrophils disseminate anti-inflammatory microparticles by ectocytosis. Blood. 2004;104:2543–2548. doi: 10.1182/blood-2004-01-0361. [DOI] [PubMed] [Google Scholar]
- 12.Ehlen HWA, Chinenkova M, Moser M, Munter HM, Krause Y, Gross S, Brachvogel B, Wuelling M, Kornak U, Vortkamp A. Inactivation of anoctamin-6/Tmem16f, a regulator of phosphatidylserine scrambling in osteoblasts, leads to decreased mineral deposition in skeletal tissues. J Bone Miner Res. 2013;28:246–259. doi: 10.1002/jbmr.1751. [DOI] [PubMed] [Google Scholar]
- 13.Headland SE, Jones HR, D’Sa ASV, Perretti M, Norling LV. Cutting-edge analysis of extracellular microparticles using ImageStreamX imaging flow cytometry. Sci Rep. 2014;4:5237. doi: 10.1038/srep05237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greco KV, Iqbal AJ, Rattazzi L, Nalesso G, Moradi-Bidhendi N, Moore AR, Goldring MB, Dell’Accio F, Perretti M. High density micromass cultures of a human chondrocyte cell line: A reliable assay system to reveal the modulatory functions of pharmacological agents. Biochem Pharmacol. 2011;82:1919–1929. doi: 10.1016/j.bcp.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Beuningen HM, van der Kraan PM, Arntz OJ, van den Berg WB. Protection from interleukin 1 induced destruction of articular cartilage by transforming growth factor β: Studies in anatomically intact cartilage in vitro and in vivo. Ann Rheum Dis. 1993;52:185–191. doi: 10.1136/ard.52.3.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nah SS, Choi IY, Lee CK, Oh JS, Kim YG, Moon HB, Yoo B. Effects of advanced glycation end products on the expression of COX-2, PGE2 and NO in human osteoarthritic chondrocytes. Rheumatology. 2008;47:425–431. doi: 10.1093/rheumatology/kem376. [DOI] [PubMed] [Google Scholar]
- 17.Eltawil NM, De Bari C, Achan P, Pitzalis C, Dell’Accio F. A novel in vivo murine model of cartilage regeneration. Age and strain-dependent outcome after joint surface injury. Osteoarthritis Cartilage. 2009;17:695–704. doi: 10.1016/j.joca.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alvarez-Garcia O, Rogers NH, Smith RG, Lotz MK. Palmitate has proapoptotic and proinflammatory effects on articular cartilage and synergizes with interleukin-1. Arthritis Rheum. 2014;66:1779–1788. doi: 10.1002/art.38399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sherwood J, Bertrand J, Nalesso G, Poulet B, Pitsillides A, Brandolini L, Karystinou A, DeBari C, Luyten FP, Pitzalis C, Pap T, Dell’Accio F. A homeostatic function of CXCR2 signalling in articular cartilage. Ann Rheum Dis. 2014;74:2207–2215. doi: 10.1136/annrheumdis-2014-205546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patel HB, Kornerup KN, Sampaio ALF, D’Acquisto F, Seed MP, Girol AP, Gray M, Pitzalis C, Oliani SM, Perretti M. The impact of endogenous annexin A1 on glucocorticoid control of inflammatory arthritis. Ann Rheum Dis. 2012;71:1872–1880. doi: 10.1136/annrheumdis-2011-201180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryan SM, McMorrow J, Umerska A, Patel HB, Kornerup KN, Tajber L, Murphy EP, Perretti M, Corrigan OI, Brayden DJ. An intra-articular salmon calcitonin-based nano-complex reduces experimental inflammatory arthritis. J Control Release. 2013;167:120–129. doi: 10.1016/j.jconrel.2013.01.027. [DOI] [PubMed] [Google Scholar]
- 22.Bockermann R, Schubert D, Kamradt T, Holmdahl R. Induction of a B-cell-dependent chronic arthritis with glucose-6-phosphate isomerase. Arthritis Res Ther. 2005;7:R1316–R1324. doi: 10.1186/ar1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boilard E, Nigrovic PA, Larabee K, Watts GFM, Coblyn JS, Weinblatt ME, Massarotti EM, Remold-O’Donnell E, Farndale RW, Ware J, Lee DM. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. 2010;327:580–583. doi: 10.1126/science.1181928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Distler JHW, Jüngel A, Huber LC, Seemayer CA, Reich CF, III, Gay RE, Michel BA, Fontana A, Gay S, Pisetsky DS, Distler O. The induction of matrix metalloproteinase and cytokine expression in synovial fibroblasts stimulated with immune cell microparticles. Proc Natl Acad Sci USA. 2005;102:2892–2897. doi: 10.1073/pnas.0409781102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schelbergen RFP, Blom AB, van den Bosch MHJ, Slöetjes A, Abdollahi-Roodsaz S, Schreurs BW, Mort JS, Vogl T, Roth J, van den Berg WB, van Lent PLEM. Alarmins S100A8 and S100A9 elicit a catabolic effect in human osteoarthritic chondrocytes that is dependent on Toll-like receptor 4. Arthritis Rheum. 2012;64:1477–1487. doi: 10.1002/art.33495. [DOI] [PubMed] [Google Scholar]
- 26.Leoni G, Neumann PA, Kamaly N, Quiros M, Nishio H, Jones HR, Sumagin R, Hilgarth RS, Alam A, Fredman G, Argyris I, Rijcken E, Kusters D, Reutelingsperger C, Perretti M, Parkos CA, Farokhzad OC, Neish AS, Nusrat A. Annexin A1–containing extracellular vesicles and polymeric nanoparticles promote epithelial wound repair. J Clin Invest. 2015;125:1215–1227. doi: 10.1172/JCI76693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Girol AP, Mimura KKO, Drewes CC, Bolonheis SM, Solito E, Farsky SHP, Gil CD, Oliani SM. Anti-inflammatory mechanisms of the annexin A1 protein and its mimetic peptide Ac2-26 in models of ocular inflammation in vivo and in vitro. J Immunol. 2013;190:5689–5701. doi: 10.4049/jimmunol.1202030. [DOI] [PubMed] [Google Scholar]
- 28.Gerke V, Creutz CE, Moss SE. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005;6:449–461. doi: 10.1038/nrm1661. [DOI] [PubMed] [Google Scholar]
- 29.Goulding NJ, Dixey J, Morand EF, Dodds RA, Wilkinson LS, Pitsillides AA, Edwards JC. Differential distribution of annexins-I, -II, -IV, and -VI in synovium. Ann Rheum Dis. 1995;54:841–845. doi: 10.1136/ard.54.10.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Damazo AS, Yona S, Flower RJ, Perretti M, Oliani SM. Spatial and temporal profiles for anti-inflammatory gene expression in leukocytes during a resolving model of peritonitis. J Immunol. 2006;176:4410–4418. doi: 10.4049/jimmunol.176.7.4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bena S, Brancaleone V, Wang JM, Perretti M, Flower RJ. Annexin A1 interaction with the FPR2/ALX receptor: Identification of distinct domains and downstream associated signaling. J Biol Chem. 2012;287:24690–24697. doi: 10.1074/jbc.M112.377101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bevers EM, Wiedmer T, Comfurius P, Shattil SJ, Weiss HJ, Zwaal RF, Sims PJ. Defective Ca2+-induced microvesiculation and deficient expression of procoagulant activity in erythrocytes from a patient with a bleeding disorder: A study of the red blood cells of Scott syndrome. Blood. 1992;79:380–388. [PubMed] [Google Scholar]
- 33.Redini F, Galera P, Mauviel A, Loyau G, Pujol JP. Transforming growth factor β stimulates collagen and glycosaminoglycan biosynthesis in cultured rabbit articular chondrocytes. FEBS Lett. 1988;234:172–175. doi: 10.1016/0014-5793(88)81327-9. [DOI] [PubMed] [Google Scholar]
- 34.Scannell M, Flanagan MB, deStefani A, Wynne KJ, Cagney G, Godson C, Maderna P. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J Immunol. 2007;178:4595–4605. doi: 10.4049/jimmunol.178.7.4595. [DOI] [PubMed] [Google Scholar]
- 35.Dalli J, Norling LV, Montero-Melendez T, Federici Canova D, Lashin H, Pavlov AM, Sukhorukov GB, Hinds CJ, Perretti M. Microparticle alpha-2-macroglobulin enhances pro-resolving responses and promotes survival in sepsis. EMBO Mol Med. 2014;6:27–42. doi: 10.1002/emmm.201303503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Norling LV, Spite M, Yang R, Flower RJ, Perretti M, Serhan CN. Cutting edge: Humanized nano-proresolving medicines mimic inflammation-resolution and enhance wound healing. J Immunol. 2011;186:5543–5547. doi: 10.4049/jimmunol.1003865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fredman G, Kamaly N, Spolitu S, Milton J, Ghorpade D, Chiasson R, Kuriakose G, Perretti M, Farokhzad O, Tabas I. Targeted nanoparticles containing the proresolving peptide Ac2-26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci Transl Med. 2015;7:275ra20. doi: 10.1126/scitranslmed.aaa1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee C, Mitsialis SA, Aslam M, Vitali SH, Vergadi E, Konstantinou G, Sdrimas K, Fernandez-Gonzalez A, Kourembanas S. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation. 2012;126:2601–2611. doi: 10.1161/CIRCULATIONAHA.112.114173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andaloussi SEL, Mäger I, Breakefield XO, Wood MJA. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013;12:347–357. doi: 10.1038/nrd3978. [DOI] [PubMed] [Google Scholar]
- 40.Bolukbasi MF, Mizrak A, Ozdener GB, Madlener S, Ströbel T, Erkan EP, Fan JB, Breakefield XO, Saydam O. miR-1289 and “Zipcode”-like sequence enrich mRNAs in micro-vesicles. Mol Ther Nucleic Acids. 2012;1:e10. doi: 10.1038/mtna.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang KV, Hung CY, Aliwarga F, Wang TG, Han DS, Chen WS. Comparative effectiveness of platelet-rich plasma injections for treating knee joint cartilage degenerative pathology: A systematic review and meta-analysis. Arch Phys Med Rehabil. 2014;95:562–575. doi: 10.1016/j.apmr.2013.11.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Tmem16f knockout mice have reduced MV shedding and exacerbated sGAG loss in experimental arthritis.
Fig. S2. Characterization of parent neutrophils used to generate offspring MVs.
Fig. S3. Presence of tetraspanin TSG101 in neutrophil MVs.
Fig. S4. Concentration-dependent chondroprotection of MV against cytokine stimulation of chondrocytes.
Fig. S5. Selective effect of MV treatment on chondrocyte matrix degrading enzyme mRNA expression.
Fig. S6. MV treatment protects from chondrocyte apoptosis.
Fig. S7. MV chemokine receptor and adhesion molecule expression.
Fig. S8. Neutrophils induce ECM accumulation in chondrocytes only when prohibited from direct cell-to-cell contact.
Fig. S9. Generation of neutrophil MVs from AnxA1−/− mice.
Table S1. RA patient demographics.
Table S2. RA patient clinical profiles.
Table S3. RA patient treatment history.
Movie S1. MVs penetrate cartilage ex vivo to deliver AnxA1.