

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
Whole-exome sequencing may identify specific therapeutic opportunities for patients with HLH.
HLH should be conceptualized as a critical illness phenotype driven by toxic activation of immune cells from different underlying mechanisms.
Abstract
The HLH-2004 criteria are used to diagnose hemophagocytic lymphohistiocytosis (HLH), yet concern exists for their misapplication, resulting in suboptimal treatment of some patients. We sought to define the genomic spectrum and associated outcomes of a diverse cohort of children who met the HLH-2004 criteria. Genetic testing was performed clinically or through research-based whole-exome sequencing. Clinical metrics were analyzed with respect to genomic results. Of 122 subjects enrolled over the course of 17 years, 101 subjects received genetic testing. Biallelic familial HLH (fHLH) gene defects were identified in only 19 (19%) and correlated with presentation at younger than 1 year of age (P < .0001). Digenic fHLH variants were observed but lacked statistical support for disease association. In 28 (58%) of 48 subjects, research whole-exome sequencing analyses successfully identified likely molecular explanations, including underlying primary immunodeficiency diseases, dysregulated immune activation and proliferation disorders, and potentially novel genetic conditions. Two-thirds of patients identified by the HLH-2004 criteria had underlying etiologies for HLH, including genetic defects, autoimmunity, and malignancy. Overall survival was 45%, and increased mortality correlated with HLH triggered by infection or malignancy (P < .05). Differences in survival did not correlate with genetic profile or extent of therapy. HLH should be conceptualized as a phenotype of critical illness characterized by toxic activation of immune cells from different underlying mechanisms. In most patients with HLH, targeted sequencing of fHLH genes remains insufficient for identifying pathogenic mechanisms. Whole-exome sequencing, however, may identify specific therapeutic opportunities and affect hematopoietic stem cell transplantation options for these patients.
Visual Abstract
Introduction
Isolated cases of children with fever, fatal systemic inflammation, and bone marrow or tissue specimens demonstrating hemophagocytosis were first described in 1939,1 followed by familial reports that suggested a potential genetic predisposition toward hemophagocytic lymphohistiocytosis (HLH).2 Consensus criteria were developed to standardize clinical trial enrollment (supplemental Table 1, available on the Blood Web site)3 and now serve as de facto diagnostic criteria for HLH. HLH, however, might be better conceptualized as a convergent entity or common final pathway/phenotype characterized by toxic immune activation secondary to a range of diverse underlying molecular diagnoses rather than as a distinct disease.
Historically, patients have long been categorized as having either “primary” HLH from an inherited defect in cytolytic immune function or “secondary” HLH resulting from immune activation incited by various antigenic challenges, including autoimmune disease, persistent infection, or malignancy. For primary or familial HLH (fHLH), mutations in genes needed for cytolytic immune function have been identified and serve as part of the HLH-2004 diagnostic criteria.3,4 The clinical distinctions between primary and secondary HLH may not be this straightforward; in HLH-94, no difference in survival was observed for patients with presumed primary vs secondary HLH.5 Thus, it becomes apparent that clinical fulfillment of the HLH-2004 criteria does not necessarily imply defective cytolytic activity for all patients. Similarly, the spectrum of inherited genetic defects that inform a need for immune suppression or hematopoietic stem cell transplantation (HSCT) are inadequately captured by primary or secondary HLH categories.
The uncertain performance of the HLH-2004 criteria and dire consequences of missed or incorrect HLH diagnosis have led to widely variable approaches to diagnosis and treatment of patients.6,7 If untreated, fHLH is fatal in 90% of children, and prompt initiation of immunochemotherapy in patients with active HLH remains critical for survival.8-10 HSCT is necessary for long-term survival in patients at risk for recurrent episodes of HLH.7 Alternately, it has been proposed that clinical diagnosis of HLH using the HLH-2004 criteria with no confirmed gene mutations may prove dangerous and that these criteria fail to distinguish HLH from sepsis or systemic inflammatory response syndrome, for which immunosuppression or HSCT may be contraindicated.6 Targeted sequencing of fHLH genes in ethnically enriched cohorts has identified disease-causing gene mutations in 34% to 100% of affected individuals with greater enrichment when genetic testing is restricted to patients with abnormal expression of HLH-related proteins or defective cytolytic function.11-14 In this study, we sought to evaluate the genomic spectrum and associated outcomes of a prospectively enrolled, multiethnic cohort of children clinically diagnosed with HLH, using the HLH-2004 criteria at a large tertiary care center.
Methods
Subject enrollment
Patients at Texas Children’s Hospital or collaborating referral centers who met HLH-2004 criteria for the diagnosis of HLH were offered participation in this research study, approved by the Baylor College of Medicine Institutional Review Board. All subjects and family members provided written informed consent to have their clinical and genetic information published in medical or scientific journals. All procedures involving human participants were performed in accordance with institutional and international ethical standards.
Ascertainment of clinical data
Fulfillment of the HLH-2004 diagnostic criteria was confirmed for all subjects. Comorbidities, clinical outcomes, and clinical genetic test results, including targeted individual gene sequencing, HLH gene panel testing, and clinical whole-exome sequencing (WES), were derived from the medical record. Clinical WES was conducted by Baylor Genetics Laboratories (Houston, TX). None of the subjects had known features of or mutations in genes associated with primary immunodeficiency diseases or dysregulated immune activation or proliferation before HLH onset and diagnosis.
Research-based WES and analysis
WES of probands who lacked clinical molecular diagnoses and their family members was conducted within the Baylor-Hopkins Center for Mendelian Genomics, as previously described (supplemental Methods).15,16 For all subjects who underwent HSCT, pretransplantation samples were sequenced.
Variant selection methods have been previously reported.15,16 Briefly, variants were prioritized using 7 criteria: existence in known HLH-causing genes; appropriate cosegregation with phenotype among family members; allelic frequencies in internal and public (eg, Exome Aggregation Consortium) exome databases less than 0.005 for autosomal and X-linked recessive inheritance models and less than 0.001 for autosomal dominant inheritance models; in silico conservation and damage prediction (eg, Combined Annotation Dependent Depletion score17 greater than the Mutational Significance Cutoff18); expression in immune cells, especially T cells, natural killer (NK) cells, monocytes, and macrophages (eg, http://biogps.org); relevance of the affected gene to known diseases that predispose toward infection, autoimmunity, or malignancy (eg, in the Online Mendelian Inheritance for Man catalog); and known function of the affected protein in immune function and inflammation (eg, in PubMed). Stratified disease-associated variants were confirmed by Sanger sequencing. Variants in common fHLH genes (PRF1, STX11, STXBP2, and UNC13D plus LYST and RAB27A) were scrutinized for possible contributions to disease.
Categorization of variants in primary immunodeficiency disease genes identified by WES
For variants in primary immunodeficiency disease (PIDD)-associated genes, molecular diagnoses were designated as “likely” (supplemental Table 4) if HLH had been previously demonstrated in other individuals with the associated PIDD, the associated PIDD was known to impair cytolytic T or NK cell function, or the molecular diagnosis was consistent with relevant features of the clinical phenotype of the subject. If the molecular diagnosis was otherwise solely supported by the exome data analyses, it was considered a “possible” genetic explanation for HLH in the subject.
Statistical analysis
Demographic and clinical information were abstracted from medical records. The χ-squared test was used if counts exceeded 5; otherwise Fisher’s Exact test was implemented. Logistic regression was used to obtain odds ratios and 95% confidence intervals for comparisons with counts exceeding 5. Kaplan-Meier survival curves were generated to estimate survival from time of diagnosis to end of follow-up, and a log-rank test estimated differences across strata of interest. Subjects whose genetic profiles aligned with multiple categories (n = 3) were excluded from analyses stratified by genetic profile (n = 119). Otherwise, all 122 subjects were assessed. All statistical analyses were conducted in STATA 13.v1.
Results
Demographics
Overall, 122 subjects clinically diagnosed with HLH at Texas Children’s Hospital and referring centers from 1999 to 2016 were enrolled (Table 1). All subjects met HLH-2004 diagnostic criteria for HLH. Median age at diagnosis was 6.1 years (range, 2.1 weeks-18.5 years) with equal proportions of boys (n = 62) and girls (n = 60). Eight subjects had family histories of consanguinity. Six others denied consanguinity, but distant parental kinship could not be excluded after an increased number of rare homozygous variants were observed in the WES data.
Table 1.
HLH patient characteristics
| Patient characteristics | HLH (n = 122) |
|---|---|
| Age at diagnosis, median (range), y | 6.07 (0.04-18.50) |
| Sex, n (%) | |
| Male | 62 (50.82) |
| Female | 60 (49.18) |
| Race/ethnicity, n (%) | |
| Non-Hispanic white | 24 (19.67) |
| Hispanic | 52 (42.62) |
| Non-Hispanic black | 19 (15.57) |
| Non-Hispanic Asian | 6 (4.92) |
| Non-Hispanic other | 2 (1.64) |
| Other | 13 (9.84) |
| Unknown | 7 (5.74) |
| HLH genetic profile, n (%) | |
| fHLH | 19 (15.57) |
| PIDD | 11 (9.02) |
| DIAP | 8 (6.56) |
| PIDD/DIAP | 3 (2.46) |
| Other candidate defects | 5 (4.10) |
| No genetic explanation | 76 (62.30) |
| No biallelic fHLH gene variants | 63 (51.64) |
| One fHLH gene variant | 6 (4.92) |
| Possible digenic fHLH variants | 7 (5.74) |
| HLH-related trigger, n (%) | |
| Infection | 44 (36.07) |
| Autoimmune | 32 (26.23) |
| Malignancy | 14 (11.48) |
| No associated trigger | 32 (26.23) |
| Therapeutic strategy, n (%) | |
| Observation only | 9 (7.38) |
| Biologics/steroids | 18 (14.75) |
| Immunochemotherapy | 54 (44.26) |
| HSCT | 38 (31.15) |
| Unknown | 3 (2.46) |
| Alive at end of follow-up, n (%) | |
| Yes | 76 (62.30) |
| No | 41 (33.61) |
| Unknown | 5 (4.10) |
Genetic testing workflow
Of the 122 subjects, a total of 101 received genetic testing. Clinical genetic testing was performed for 87 subjects, including 60 who were tested only by targeted Sanger sequencing, 15 who were assessed only by HLH gene panel testing, 11 who underwent clinical WES, and 1 who received only chromosomal microarray testing (Figures 1A; supplemental Figure 1). Research-based WES analyses were performed for 48 subjects. Genetic testing was not performed in 21 subjects for various reasons, including early death and clinical testing availability at the time of evaluation.
Figure 1.

Genetic testing reveals diverse pathogenic mechanisms in patients with HLH. (A) Summary of workflow for 48 subjects who underwent research-based WES analyses. (B) Genetic profiles for 101 subjects who met the HLH-2004 criteria and received genetic testing. (C) Dominant NLRC4 and NLRP12 variants and recessive NLRP4, NLRC3, and NLRP13 variants are significantly associated with development of HLH. The number of alleles containing potential protein-altering variants (frameshift insertions/deletions, stop gain/stop loss, splicing defects, nonframeshift insertions/deletions, and missense variants) in NLRC4, NLRP12, NLRP4, NLRP13, and NLRC3 in HLH-affected cases (n = 48) was compared with the number in other samples in the Baylor-Hopkins Center for Mendelian Genomics database (total n = 6677; analyzed n = 5981), which contains exomes from well-phenotyped diseased and healthy individuals (recorded within the PhenoDB database) collected among more than 380 phenotypic cohorts. Unaffected relatives in the HLH cohort (n = 51) and individuals in the Immunodeficiency cohort (n = 645) were excluded from the analysis. Differences in allelic counts between HLH cases and controls (n = 5981) were tested by random sampling without replacement (100 000 iterations).
Genetic testing confirmed the presence of disease-causing fHLH gene mutations in a limited number of subjects
Nineteen of 101 subjects tested (19%) had biallelic mutations in 6 designated fHLH genes (Figure 1B). Clinical sequencing of individual fHLH genes identified 13 cases. Research-based WES of 20 of the remaining 48 subjects within this group revealed a rare, computationally predicted damaging homozygous RAB27A variant (supplemental Table 2) in a boy who presented with presumed malignancy-associated HLH and lacked the hair hypopigmentation associated with Griscelli syndrome.19,20 Clinical HLH gene panel testing identified 2 cases of Munc13-4 deficiency, and clinical WES successfully diagnosed 2 cases of perforin deficiency and 1 case of Munc13-4 deficiency.
Variants of unknown significance in fHLH genes were observed in 26 subjects who underwent research-based WES (supplemental Table 2). Most of these variants were either observed frequently in public and internal exome databases or not located in protein-encoding regions. The known pathogenic intronic UNC13D c.118-308C>T mutation11,21 was excluded by Sanger sequencing in the 3 subjects who had single heterozygous UNC13D variants (supplemental Table 2). Four subjects had clinical microarray testing performed to exclude copy number variations. Of the 26 subjects, 9 (HLH_014, HLH_016, HLH_017, HLH_063, HLH_087, HLH_097, HLH_109, HLH_117, and HLH_122) had rare and potentially protein sequence-altering single allelic variants of unknown significance in fHLH genes. Similarly, 5 subjects who received only clinical genetic testing carried solitary single heterozygous fHLH gene variants. The possibility that some of these monogenic variants may cause dominant-negative loss of function of the corresponding protein22 cannot be fully excluded.
Seven subjects who underwent research-based WES (HLH_014, HLH_016, HLH_017, HLH_063, HLH_087, HLH_097, and HLH_117) had single allelic variants in 2 fHLH genes that were potentially protein sequence-altering (supplemental Table 2). Computational analyses (supplemental Methods) found that heterozygous variants in the various “digenic” dual-gene combinations, except for one (LYST/RAB27A in subject HLH_117, who carries a disease-associated homozygous RAG1 mutation), were not uncommon (supplemental Table 3). Another subject received only clinical individual gene sequencing and was found to have single allelic variants in STX11 and UNC13D. This variant combination was associated with the HLH phenotype (P = .03). These analyses suggest that select combined variants23 (ie, mutational burden) in fHLH genes may be clinically relevant, but the majority of such variants are present in the general population. Thus, the potential digenic model for variant contribution to HLH lacked overall statistical support.
WES reveals the presence of other molecular diagnoses in subjects who meet HLH-2004 criteria
Excluding the subject discovered to have RAB27A deficiency, 47 subjects underwent research WES analyses and lacked biallelic mutations in fHLH genes, and potential underlying PIDDs were identified in 14 (30%; supplemental Table 4). Four of the 14 subjects had variants in genes relevant for cytolytic function in T and NK cells: WAS,24 TTC7A25/LRBA,26 DOCK8,27 and CARMIL2.28-30 For the subject with compound heterozygous variants in both TTC7A and LRBA, it remains unclear whether defects in both of these genes are necessary for development of HLH. For DOCK8, deletion of the 5′ portion of the gene was discovered through bioinformatic detection of loss of WES reads and confirmed by array comparative genomic hybridization testing (see Stray-Pedersen et al.,15 Figure E4, Proband 30.1). We also identified biallelic mutations in RAG1 and RAG2 in 2 subjects who in hindsight had Omenn syndrome, which shares similar features with HLH.31 Two other subjects had underlying chronic granulomatous disease.31 Both subjects carried variants in second genes relevant toward inflammation or cytolytic immune function that may have further increased the risk for HLH. Four subjects had defects in key T or NK cell-signaling molecules: STAT1,32 STAT2,33 STAT3,34 and PIK3CD.35 WES identified another subject with biallelic variants in MCM3AP and MCM9. MCM3-associated protein deficiency has been reported to cause PIDD associated with a DNA repair defect.36 The precise mechanism for HLH in this subject remains unclear. Finally, 1 subject was discovered to have autoimmune lymphoproliferative syndrome type 2 because of a mutation in CASP10. Patients with autoimmune lymphoproliferative syndrome may present with features that overlap with HLH.37
Here, we introduce a new term, DIAP, or dysregulated immune activation or proliferation, to indicate a group of genes with putative or proven roles in dysregulated immune activation or proliferation. Of the 47 subjects who did not have biallelic mutations in fHLH genes and underwent research WES analyses, 11 (23%) had alterations in DIAP genes (supplemental Table 4). Three of the subjects (HLH_120, HLH_121, and HLH_122) also carried pathogenic or contributing variants in PIDD-associated genes. We bioinformatically tested the significance of differences in allelic counts between cases and controls for variants in these DIAP genes (supplemental Methods). This analysis demonstrated that the single allelic variants in NLRC438 and NLRP1239 and biallelic variants in NLRP4,40 NLRC3,41,42 and NLRP13 were significantly associated with HLH (supplemental Table 4; Figure 1C). The correlation between the NLRC4 variants and HLH corroborates the known link between NLRC4 mutations and HLH.38,43 These novel findings underscore a role for mutations in inflammation-associated genes in producing a phenotype that meets criteria for HLH. Finally, 1 subject was found to have a mutation in NRAS that is known to produce RAS-associated autoimmune leukoproliferative disorder.44 Because the variant exists de novo in this subject and all reported mutations in NRAS that cause RAS-associated autoimmune leukoproliferative disorder are somatic, this case is included in the DIAP and not the PIDD category.
Other potentially novel genetic explanations were identified in 5 subjects (10%; supplemental Table 4). Because none of these candidates have yet been fully biologically validated, assignment of causality remains uncertain. Nonetheless, they remain leading hypotheses because of their relevance in pathways known to be defective in HLH, including cytotoxicity, regulation of immune activation, and control of proliferation of hematopoietic cells.
Genetic findings are not enriched in subjects with abnormal CD107a mobilization or decreased expression of HLH-related proteins
HLH immunologic characterization was performed in 76 subjects: 33 were tested both for mobilization of CD107a and expression of HLH-related proteins (perforin, signaling lymphocytic activation molecule-associated protein, and/or baculoviral inhibitor of apoptosis repeat-containing protein 4), 37 were assessed for protein expression alone, and 6 received isolated CD107a functional testing. Genetic testing of 39 of the 43 individuals with abnormal results identified likely explanations in only 20 (51%). Although 10 fHLH probands were captured using this approach, subjects with PIDD (n = 3), DIAP (n = 4), and combined PIDD/DIAP (n = 3) were also discovered among the cases.
Correlations identified between genetic findings and clinical presentations
Genetic findings correlated with aspects of clinical presentation (Table 2 and Figure 2). Among the 19 subjects with biallelic fHLH genetic mutations, 58% had no identified trigger for HLH and 37% had associated infections; no fHLH subjects had rheumatologic triggers (Figure 2A). For the 11 subjects who had PIDD-associated gene variants and no variants in DIAP-associated genes, 36% had no identified trigger for HLH and 46% had associated infections (Figure 2A). Among the 8 subjects with DIAP-associated variants and no underlying PIDD, the majority (63%) had rheumatologic triggers (Figure 2A). The incidence of HLH-associated triggers did not vary significantly with age aside from perhaps rheumatologic triggers, which were suggested as increased in subjects older than 12 years compared with subjects younger than 1 year (36% vs 11%; P = .06; Figure 2B). Interestingly, however, subjects with biallelic fHLH mutations were significantly younger (P = .02) than subjects who lacked genetic findings, unlike subjects in the other genetically defined categories (Figure 2C; Table 2). In fact, subjects with fHLH were more likely than other subjects (P < .0001) to present younger than 1 year (Figure 2D). In contrast, all subjects who had underlying variants in DIAP-associated genes presented at older than 1 year, whereas subjects with underlying PIDD-associated gene defects presented with similar frequency at all ages.
Table 2.
Genetic findings and aspects of clinical presentation
| Genetic category | N | Age at diagnosis, median (range), y | P value, median age | Overall survival estimate,* % | Associated trigger, % | Maximum level of therapy, % |
|---|---|---|---|---|---|---|
| fHLH | 19 | 0.51 (0.04-16.07) | .02 | 54 | None: 57.9† | Observation/none: 0.0 |
| Autoimmune: 0.0† | Biologics/steroids: 0.0 | |||||
| Infection: 36.8 | Immunochemotherapy: 26.3‡ | |||||
| Malignancy: 5.3 | HSCT: 73.7† | |||||
| PIDD | 11 | 8.73 (0.14-17.96) | .71 | 88 | None: 36.4 | Observation/none: 0.0 |
| Autoimmune: 9.1 | Biologics/steroids: 0.0 | |||||
| Infection: 45.5 | Immunochemotherapy: 81.8† | |||||
| Malignancy: 9.1 | HSCT: 0.0 | |||||
| Unknown: 18.2 | ||||||
| DIAP | 8 | 10.96 (1.36-18.51) | .06 | 67 | None: 25.0 | Observation/none: 0.0 |
| Autoimmune: 62.5 | Biologics/steroids: 37.5 | |||||
| Infection: 0.0† | Immunochemotherapy: 50.0 | |||||
| Malignancy: 12.5 | HSCT: 12.5 | |||||
| Other candidate defects | 5 | 9.34 (2.79-17.03) | .36 | 67 | None: 20.0 | Observation/none: 20.0 |
| Autoimmune: 0.0 | Biologics/steroids: 0.0 | |||||
| Infection: 40.0 | Immunochemotherapy: 40.0 | |||||
| Malignancy: 40.0 | HSCT: 20.0 | |||||
| Unknown: 20.0 | ||||||
| No genetic explanation | 76 | 6.18 (0.05-17.53) | Reference | 47 | None: 17.1 | Observation/none: 10.5 |
| Autoimmune: 32.9 | Biologics/steroids: 18.4 | |||||
| Infection: 38.2 | Immunochemotherapy: 43.4 | |||||
| Malignancy: 11.8 | HSCT: 27.6 |
By Kaplan-Meier analysis.
Significantly different compared with no genetic explanation group (P < .05).
N = 5, including 3 who died before HSCT and 2 with HSCT anticipated.
Figure 2.

Features of clinical presentation correlate with genetic findings. (A) HLH-associated trigger by genetic profile. Primary triggers for the presenting HLH episode of 122 subjects were defined as infection (blue), malignancy (green), autoimmune disease (red), or no associated trigger (orange). A trigger was identified in 74% of subjects. (B) HLH-associated trigger by age at diagnosis. Subjects (n = 122) were separated into 4 groups by age in years (x-axis) and analyzed by HLH-triggering event. A two-sample test of proportions with a 95% confidence level for each comparison was used to analyze proportional differences in trigger by age. (C) HLH genetic profile by age at diagnosis. Subjects were placed into the same 4 groups by age in years (x-axis), excluding subjects with potential disease-causing variants in PIDD and DIAP genes (n = 3). A two-sample test of proportions with a 95% confidence level for each comparison was used to analyze proportional differences in genetic profile by age (total n = 119). (D) Biallelic fHLH variants are enriched in subjects younger than 1 year. A two-sample test of proportions with a 95% confidence level was used to analyze the proportional difference between fHLH cases diagnosed at younger than 1 year and older than 1 year of age (total n = 122).
Clinical outcomes are associated with genetic findings
Overall survival for the entire cohort was 45% with a median 18.6 months of follow-up (range, 0.04-201.4; Figure 3A). Overall survival estimates were not suggested to differ by HLH genetic profile (Figure 3B; supplemental Table 5). Individuals with fHLH were more likely to receive HSCT than other therapies compared with subjects with no genetic explanation (odds ratio, 7.33; 95% confidence interval, 2.35-22.89). Disease trigger was significantly associated with survival: subjects with rheumatologic disease or no obvious trigger exhibited better survival than subjects with HLH triggered by infection or malignancy (P < .05; Figure 3C; supplemental Table 6). All 9 subjects whose milder clinical course required only observation survived. No significant difference in survival was observed between subjects treated with biologics/steroids, immunochemotherapy, and HSCT, and no deaths occurred after 1 year in subjects treated with biologics/steroids (Table 3).
Figure 3.

Outcome data for HLH subject cohort (Kaplan-Meier survival curves). (A) Overall survival estimate from HLH diagnosis to date of death or last contact in years (n = 122). (B) Survival estimates from HLH diagnosis to date of death or last contact in years by genetic profile (total n = 119). (C) Survival estimates from HLH diagnosis to date of death or last contact in years by associated trigger (n = 122).
Table 3.
Clinical and genetic features by maximum level of therapy received
| Maximum level of therapy | Total | Overall survivors, N | 1-year survival estimate,* % | 3-year survival estimate,* % | Overall survival estimate,* % | Associated trigger, % | Genetic profile, % |
|---|---|---|---|---|---|---|---|
| Observation/none | 9 | 9 | 100 | 100 | 100 | None: 22.2 | fHLH: 0.0 |
| Autoimmune: 11.1 | PIDD: 0.0 | ||||||
| Infection: 55.6 | DIAP: 0.0 | ||||||
| Malignancy: 11.1 | PIDD/DIAP: 0.0 | ||||||
| Other candidate defects: 11.1 | |||||||
| No genetic explanation: 88.9 | |||||||
| Biologics/steroids | 18 | 14 | 70 | 70.0 | 70 | None: 11.1 | fHLH: 0.0 |
| Autoimmune: 55.6 | PIDD: 0.0 | ||||||
| Infection: 33.3 | DIAP: 16.7 | ||||||
| Malignancy: 0.0† | PIDD/DIAP: 5.6 | ||||||
| Other candidate defects: 0.0 | |||||||
| No genetic explanation: 77.8 | |||||||
| Immunochemotherapy | 54 | 32 | 58 | 58 | 39 | None: 18.5 | fHLH: 9.3 |
| Autoimmune: 31.5 | PIDD: 16.7 | ||||||
| Infection: 29.6 | DIAP: 7.4 | ||||||
| Malignancy: 20.4 | PIDD/DIAP: 1.9 | ||||||
| Other candidate defects: 3.7 | |||||||
| No genetic explanation: 61.1 | |||||||
| HSCT | 38 | 21 | 75 | 57 | 40 | None: 47.4† | fHLH: 36.8† |
| Autoimmune: 10.5† | PIDD: 0.0† | ||||||
| Infection: 39.5 | DIAP: 2.6 | ||||||
| Malignancy: 2.6† | PIDD/DIAP: 2.6 | ||||||
| Other candidate defects: 2.6 | |||||||
| No genetic explanation: 55.3 |
By Kaplan-Meier analysis.
Significantly different compared with HLH standard of care Immunochemotherapy group (P < .05).
Discussion
Diagnostic considerations
These results demonstrate that patients clinically diagnosed with HLH using the HLH-2004 criteria represent a distinct category of patients with persistent, toxic immune activation at high risk for death. We found that 80 (66%) of 122 subjects with HLH had likely or potential disease-causing genetic, autoimmune, or oncologic etiologies underlying the phenotype. Importantly, 55 subjects received genetic testing and lacked biallelic fHLH gene mutations or other genetic explanations, yet 35 (64%) were not evaluated by WES. For 48 subjects who underwent research WES analyses, potential disease-causing genetic defects were identified in 28 (58%).
In this ethnically diverse cohort, although the HLH-2004 criteria did not demonstrate the high prevalence of fHLH exhibited in the European studies,11-14 they identified patients with HLH resulting from other genetic conditions. Overall, genetic testing of 101 subjects with HLH secured likely molecular diagnoses in 46 (46%; or 70% excluding the 35 subjects not tested by WES). Biallelic fHLH gene mutations were identified in only 19% of 101 subjects. This result may reflect the fact that genetic testing was performed on the basis of overall fulfillment of HLH-2004 criteria and not simply focused toward subjects with abnormal expression of HLH-related proteins or defective cytolytic function.11-14 Furthermore, modeling of mutational burden in fHLH genes found that most potential digenic combinations were unlikely to arise with greater frequency in HLH than in the general population. Clinical laboratory results reporting digenic variants should therefore be interpreted with caution. Meanwhile, variants associated with PIDDs and DIAP were found in 22% of 101 cases (33% excluding the 35 subjects not tested by WES). Thus, in this cohort, HLH-2004 criteria identified more cases of potential PIDD or DIAP than fHLH. This observation emphasizes the need to conceptualize HLH as a phenotype rather than a single disease. For example, in patients with PIDD, impaired ability to eliminate pathogens can drive formation of the HLH phenotype. This phenomenon has been clearly reported by others.31 In contrast, the HLH phenotype may arise in patients with DIAP because of exaggerated or poorly controlled immunologic responses. Neither of these conditions necessitates defective cytotoxicity. Thus, targeted sequencing or testing of cytolytic function may not be sufficient for identifying inherited causes of HLH.
An emerging concept creates a spectrum of genotype and phenotype rather than dichotomous primary and secondary categories.4 Our results argue that even patients with presumed secondary HLH may benefit from investigations for underlying genetic conditions. Among our subjects with presumed secondary HLH, disease-associated variants in fHLH, PIDD, and DIAP genes were identified in 22% (7 of 32) of subjects with autoimmune disease and 21% (3 of 14) of subjects with malignancy.
Our results also suggest that addition of an age parameter to the HLH-2004 criteria may increase the specificity for fHLH. We observed that fHLH was statistically enriched in subjects who were diagnosed at younger than 1 year of age. This phenomenon is consistent with findings reported by others.12,13,45 Meanwhile, the majority of subjects who had underlying variants in DIAP-associated genes were older. Nevertheless, age ranges for these groups overlap.
Therapeutic implications
In this cohort, overall survival was 45%, which is comparable to other reports.5 The management of HLH at this time is dictated by the clinical presentation and course of each patient. Patients with minimal or short-lived symptoms typically receive only observation or minimally toxic therapies. The current approach to patients who require immunochemotherapy involves initiation of etoposide and dexamethasone (for patients lacking co-incident autoimmune disease).7 These agents, however, are not feasible for long-term immunosuppression because of cumulative toxicity.46 In general, HSCT is reserved for patients presenting with HLH because of fHLH gene defects, exhibiting persistent or progressive pathologic inflammation after HLH-94 induction, demonstrating persistent cytolytic immune function defects, or having central nervous system inflammation.7 Thus, patients who exhibit greater severity of disease and likelihood for nonsurvival receive more intense therapies that convey increased risk for poor outcomes (Table 3).
The current practice of relying on failure of therapy to dictate escalation of treatment places patients at risk for increased morbidity and mortality. Although our results suggest that WES may be deferred in patients who are managed by observation alone, this study also supports the use of WES to guide long-term clinical decision-making for patients with clinically diagnosed HLH on initiation of therapy. First, these data revealed multiple potential molecular pathways leading to HLH. Early identification of these altered pathways may offer alternative therapeutic opportunities, such as targeted biologic therapy. Second, WES can confirm the presence of fHLH or an underlying PIDD for which HSCT is indicated. Finally, WES can identify unanticipated inherited gene defects that may be shared by potential allograft donors.
Genetic testing in children with HLH
Our genetic findings reflect a very diverse population at a large referral center and differ from results elsewhere that suggest focused use of genetic testing toward subjects who demonstrate decreased mobilization of CD107a or expression of HLH-related proteins.14 Although such an approach led to genetic diagnoses in 85% of subjects in 1 cohort, we found that only 51% of individuals with such laboratory abnormalities in our population had likely genetic explanations. The discrepancy may be partially attributed to differences in study populations and design, as 46 of the subjects in this cohort, including 6 subjects with fHLH, did not receive testing of CD107a mobilization or expression of HLH-related proteins. Nonetheless, the defective genes in our cohort extended well beyond PRF1, UNC13D, STX11, STXBP2, LYST, RAB27A, AP3B1, SH2D1A, and XIAP. Furthermore, such a focused approach would have failed to capture the subject with CASP10 deficiency and another subject with a potential novel genetic diagnosis, as both probands had normal CD107a function and HLH-related protein expression.
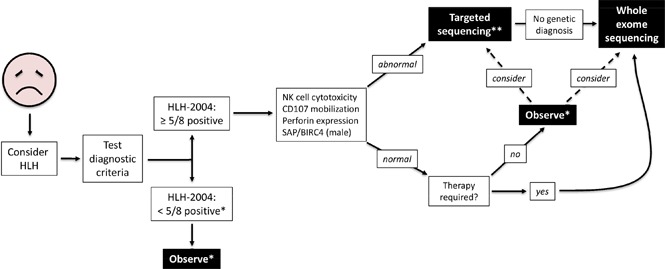
We propose a broader approach for use of WES, if rapidly available, in patients with HLH (Figure 4). This algorithm combines our findings with experiences reported by others.12,14,47 First, HLH must be considered in ill children, and the HLH-2004 diagnostic criteria should be interrogated accordingly. If sufficient criteria are not fulfilled, clinical judgment must be exercised to determine which individuals should be observed closely for potential progression such that the criteria eventually become met. Children who satisfy 5 or more of the 8 criteria should receive immunologic laboratory testing for cytolytic function and expression of HLH-related proteins. If these tests demonstrate abnormalities, targeted gene sequencing should be performed, especially if the patient is younger than 1 year. We argue that WES should follow if no genetic diagnosis has been secured. Children with HLH who have normal or unhelpful immunologic laboratory testing results and require minimal therapy may be observed without genetic testing, although targeted gene sequencing or WES should be considered. We did not find any cases of familial HLH, PIDD, or DIAP in this small subgroup of individuals, but further studies will be needed to determine whether WES should be used to search for other underlying genetic conditions. In contrast, patients with persistent or recurrent HLH or who require initiation of steroids, biologics, or more should receive WES.
Figure 4.

Proposed algorithm for testing of critically ill children. *Decision regarding further evaluation requires clinical judgment. **Strongly recommended in children younger than 1 year. Abbreviations: BIRC4, baculoviral inhibitor of apoptosis repeat-containing protein 4; SAP, signaling lymphocytic activation molecule-associated protein.
It remains important to note that although WES currently serves as the most expedient broad genetic test in terms of cost, analytic difficulty, turnaround time, and availability, other techniques may eventually supplant this method because of several of its limitations. WES is known to miss structural variations, and despite enhanced bioinformatic tools,15,48 identification of gross copy number variations remains difficult. In addition, WES is not designed to interrogate intronic variants that may affect either promoter or enhancer elements, resulting in altered protein expression, or introduce cryptic start codons or splicing sites. These abilities are better captured by whole-genome sequencing and RNA sequencing, which may ultimately become preferred diagnostic tests in the future.
In conclusion, in this study the HLH-2004 criteria identified a cohort of critically ill children with a broad spectrum of genetic conditions, autoimmune disease, and malignancy that result in the HLH phenotype. Clinical suspicion for HLH and application of the HLH-2004 criteria remain the most critical steps in identifying patients with pathologic immune activation. After clinical identification, subsequent evaluations should include studies to identify inherited molecular defects that underlie inability to regulate immunologic homeostasis. Additional prospective trials enrolling at time of suspicion of HLH will prove helpful for determining therapeutic strategies based on pathogenic molecular mechanisms.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
This work was funded in part by grants from the HistioCure Foundation (K.L.M. and C.E.A.), St. Baldrick’s Foundation (to NACHO Consortium, K.L.M., and C.E.A.), National Institutes of Health, National Cancer Institute (R25CA160078: Training Program in Pediatric Cancer Epidemiology and Control Grant to E.C.P.-G.), American Society of Hematology (Scholar Award in Clinical Research to E.C.P.-G.), the Thrasher Research Fund (Early Career Award to E.C.P.-G.), and the Feldman Family. This work was also supported by the following: National Institutes of Health, National Institute of Allergy and Infectious Diseases grants R01AI067946 and R01AI120989 (J.S.O.), National Institutes of Health, National Human Genome Research Institute/National Heart, Lung, and Blood Institute grant UM1HG006542 (to the Baylor-Hopkins Center for Mendelian Genomics), National Institutes of Health, National Institute of Neurological Disorders and Stroke grant R01NS058529 (J.R.L.), and the Jeffrey Modell Foundation Translational Research Program Grant (I.K.C.).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: I.K.C. analyzed data and wrote the manuscript; O.S.E. and B.R.G. analyzed clinical data; E.C.P.-G. and Z.H.C.-A. performed statistical and bioinformatic analyses, respectively, and wrote portions of the manuscript; L.R.F., S.K.N., H.A.A., M.I.D., H.E.H., R.A.K., C.A.M., T.C.N., D.A.B., J.R.G., L.A.P., M.C.P., J.C.A.-B., S.A.M., W.A.-H., A.C., T.N.C., and D.N.H. collected samples and clinical or research data; E.M.M., T.P.V., and A.S.-P. analyzed genetic data; S.N.J., D.M.M., and R.A.G. performed WES; J.R.L., J.S.O., and K.L.M. provided expertise and supervision of the research; and C.E.A. supervised the research study and wrote portions of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Carl E. Allen, Feigin Center, Suite 730.06, 1102 Bates St, Houston, TX 77030; e-mail: ceallen@txch.org; and Ivan K. Chinn, Feigin Center, Suite 330, 1102 Bates St, Houston, TX 77030; e-mail: chinn@bcm.edu.
References
- 1.Bodley Scott R, Robb-Smith AHT. Histiocytic medullary reticulosis. Lancet. 1939;234(6047):194-198. [Google Scholar]
- 2.Farquhar JW, Claireaux AE. Familial haemophagocytic reticulosis. Arch Dis Child. 1952;27(136):519-525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henter J-I, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. [DOI] [PubMed] [Google Scholar]
- 4.Allen CE, McClain KL. Pathophysiology and epidemiology of hemophagocytic lymphohistiocytosis. Hematology Am Soc Hematol Educ Program. 2015;2015:177-182. [DOI] [PubMed] [Google Scholar]
- 5.Henter J-I, Samuelsson-Horne A, Aricò M, et al. ; Histocyte Society. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367-2373. [DOI] [PubMed] [Google Scholar]
- 6.Castillo L, Carcillo J. Secondary hemophagocytic lymphohistiocytosis and severe sepsis/ systemic inflammatory response syndrome/multiorgan dysfunction syndrome/macrophage activation syndrome share common intermediate phenotypes on a spectrum of inflammation. Pediatr Crit Care Med. 2009;10(3):387-392. [DOI] [PubMed] [Google Scholar]
- 7.Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118(15):4041-4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Henter JI, Elinder G. Familial hemophagocytic lymphohistiocytosis. Clinical review based on the findings in seven children. Acta Paediatr Scand. 1991;80(3):269-277. [DOI] [PubMed] [Google Scholar]
- 9.Janka GE. Familial hemophagocytic lymphohistiocytosis. Eur J Pediatr. 1983;140(3):221-230. [DOI] [PubMed] [Google Scholar]
- 10.Imashuku S, Kuriyama K, Teramura T, et al. Requirement for etoposide in the treatment of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. J Clin Oncol. 2001;19(10):2665-2673. [DOI] [PubMed] [Google Scholar]
- 11.Meeths M, Chiang SCC, Wood SM, et al. Familial hemophagocytic lymphohistiocytosis type 3 (FHL3) caused by deep intronic mutation and inversion in UNC13D. Blood. 2011;118(22):5783-5793. [DOI] [PubMed] [Google Scholar]
- 12.Tesi B, Lagerstedt-Robinson K, Chiang SCC, et al. Targeted high-throughput sequencing for genetic diagnostics of hemophagocytic lymphohistiocytosis. Genome Med. 2015;7(1):130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cetica V, Sieni E, Pende D, et al. Genetic predisposition to hemophagocytic lymphohistiocytosis: Report on 500 patients from the Italian registry. J Allergy Clin Immunol. 2016;137(1):188-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ammann S, Lehmberg K, Zur Stadt U, et al. ; HLH study of the GPOH. Effective immunological guidance of genetic analyses including exome sequencing in patients evaluated for hemophagocytic lymphohistiocytosis. J Clin Immunol. 2017;37(8):770-780. [DOI] [PubMed] [Google Scholar]
- 15.Stray-Pedersen A, Sorte HS, Samarakoon P, et al. Primary immunodeficiency diseases: Genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol. 2017;139(1):232-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lupski JR, Gonzaga-Jauregui C, Yang Y, et al. Exome sequencing resolves apparent incidental findings and reveals further complexity of SH3TC2 variant alleles causing Charcot-Marie-Tooth neuropathy. Genome Med. 2013;5(6):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Itan Y, Shang L, Boisson B, et al. The mutation significance cutoff: gene-level thresholds for variant predictions. Nat Methods. 2016;13(2):109-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Netter P, Chan SK, Banerjee PP, et al. A novel Rab27a mutation binds melanophilin, but not Munc13-4, causing immunodeficiency without albinism. J Allergy Clin Immunol. 2016;138(2):599-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cetica V, Hackmann Y, Grieve S, et al. Patients with Griscelli syndrome and normal pigmentation identify RAB27A mutations that selectively disrupt MUNC13-4 binding. J Allergy Clin Immunol. 2015;135(5):1310-1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seo JY, Song J-S, Lee K-O, et al. ; Korea Histiocytosis Working Party. Founder effects in two predominant intronic mutations of UNC13D, c.118-308C>T and c.754-1G>C underlie the unusual predominance of type 3 familial hemophagocytic lymphohistiocytosis (FHL3) in Korea. Ann Hematol. 2013;92(3):357-364. [DOI] [PubMed] [Google Scholar]
- 22.Spessott WA, Sanmillan ML, McCormick ME, et al. Hemophagocytic lymphohistiocytosis caused by dominant-negative mutations in STXBP2 that inhibit SNARE-mediated membrane fusion. Blood. 2015;125(10):1566-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sepulveda FE, Garrigue A, Maschalidi S, et al. Polygenic mutations in the cytotoxicity pathway increase susceptibility to develop HLH immunopathology in mice. Blood. 2016;127(17):2113-2121. [DOI] [PubMed] [Google Scholar]
- 24.Pasic S, Micic D, Kuzmanovic M. Epstein-Barr virus-associated haemophagocytic lymphohistiocytosis in Wiskott-Aldrich syndrome. Acta Paediatr. 2003;92(7):859-861. [DOI] [PubMed] [Google Scholar]
- 25.Lemoine R, Pachlopnik-Schmid J, Farin HF, et al. Immune deficiency-related enteropathy-lymphocytopenia-alopecia syndrome results from tetratricopeptide repeat domain 7A deficiency. J Allergy Clin Immunol. 2014;134(6):1354-1364. [DOI] [PubMed] [Google Scholar]
- 26.Gámez-Díaz L, August D, Stepensky P, et al. The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. J Allergy Clin Immunol. 2016;137(1):223-230. [DOI] [PubMed] [Google Scholar]
- 27.Randall KL, Chan SSY, Ma CS, et al. DOCK8 deficiency impairs CD8 T cell survival and function in humans and mice. J Exp Med. 2011;208(11):2305-2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Ma CS, Ling Y, et al. Dual T cell- and B cell-intrinsic deficiency in humans with biallelic RLTPR mutations. J Exp Med. 2016;213(11):2413-2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sorte HS, Osnes LT, Fevang B, et al. A potential founder variant inCARMIL2/RLTPRin three Norwegian families with warts, molluscum contagiosum, and T-cell dysfunction. Mol Genet Genomic Med. 2016;4(6):604-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roncagalli R, Cucchetti M, Jarmuzynski N, et al. The scaffolding function of the RLTPR protein explains its essential role for CD28 co-stimulation in mouse and human T cells. J Exp Med. 2016;213(11):2437-2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bode SFN, Ammann S, Al-Herz W, et al. ; Inborn Errors Working Party of the EBMT. The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis. Haematologica. 2015;100(7):978-988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Faitelson Y, Bates A, Shroff M, Grunebaum E, Roifman CM, Naqvi A. A mutation in the STAT1 DNA-binding domain associated with hemophagocytic lymphohistocytosis. LymphoSign Journal. 2014;1(2):87-95. [Google Scholar]
- 33.Hambleton S, Goodbourn S, Young DF, et al. STAT2 deficiency and susceptibility to viral illness in humans. Proc Natl Acad Sci USA. 2013;110(8):3053-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Milner JD, Vogel TP, Forbes L, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood. 2015;125(4):591-599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lucas CL, Kuehn HS, Zhao F, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol. 2014;15(1):88-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gatz SA, Salles D, Jacobsen E-M, et al. MCM3AP and POMP mutations cause a DNA-repair and DNA-damage-signaling defect in an immunodeficient child. Hum Mutat. 2016;37(3):257-268. [DOI] [PubMed] [Google Scholar]
- 37.Rudman Spergel A, Walkovich K, Price S, et al. Autoimmune lymphoproliferative syndrome misdiagnosed as hemophagocytic lymphohistiocytosis. Pediatrics. 2013;132(5):e1440-e1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Canna SW, de Jesus AA, Gouni S, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet. 2014;46(10):1140-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jéru I, Duquesnoy P, Fernandes-Alnemri T, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci USA. 2008;105(5):1614-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cui J, Li Y, Zhu L, et al. NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat Immunol. 2012;13(4):387-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schneider M, Zimmermann AG, Roberts RA, et al. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-κB. Nat Immunol. 2012;13(9):823-831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang L, Mo J, Swanson KV, et al. NLRC3, a member of the NLR family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor STING. Immunity. 2014;40(3):329-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang J, Alfano DN, Squires JE, et al. Novel NLRC4 mutation causes a syndrome of perinatal autoinflammation with hemophagocytic lymphohistiocytosis, hepatosplenomegaly, fetal thrombotic vasculopathy, and congenital anemia and ascites. Pediatr Dev Pathol. 2017;20(6):498-505. [DOI] [PubMed] [Google Scholar]
- 44.Calvo KR, Price S, Braylan RC, et al. JMML and RALD (Ras-associated autoimmune leukoproliferative disorder): common genetic etiology yet clinically distinct entities. Blood. 2015;125(18):2753-2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sepulveda FE, Debeurme F, Ménasché G, et al. Distinct severity of HLH in both human and murine mutants with complete loss of cytotoxic effector PRF1, RAB27A, and STX11. Blood. 2013;121(4):595-603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson TS, Terrell CE, Millen SH, Katz JD, Hildeman DA, Jordan MB. Etoposide selectively ablates activated T cells to control the immunoregulatory disorder hemophagocytic lymphohistiocytosis. J Immunol. 2014;192(1):84-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tesi B, Sieni E, Neves C, et al. Hemophagocytic lymphohistiocytosis in 2 patients with underlying IFN-γ receptor deficiency. J Allergy Clin Immunol. 2015;135(6):1638-1641. [DOI] [PubMed] [Google Scholar]
- 48.Gambin T, Akdemir ZC, Yuan B, et al. Homozygous and hemizygous CNV detection from exome sequencing data in a Mendelian disease cohort. Nucleic Acids Res. 2017;45(4):1633-1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.