

Key Points
FLT3 inhibitor AC220 caused DNA repair defects and sensitized FLT3(ITD)-positive AML stem and progenitor cells to PARP1 inhibitors.
Quiescent and proliferating FLT3(ITD)-positive AML cells were eliminated by the combination of FLT3 and PARP1 inhibitors.
Abstract
Mutations in FMS-like tyrosine kinase 3 (FLT3), such as internal tandem duplications (ITDs), can be found in up to 23% of patients with acute myeloid leukemia (AML) and confer a poor prognosis. Current treatment options for FLT3(ITD)-positive AMLs include genotoxic therapy and FLT3 inhibitors (FLT3i’s), which are rarely curative. PARP1 inhibitors (PARP1i’s) have been successfully applied to induce synthetic lethality in tumors harboring BRCA1/2 mutations and displaying homologous recombination (HR) deficiency. We show here that inhibition of FLT3(ITD) activity by the FLT3i AC220 caused downregulation of DNA repair proteins BRCA1, BRCA2, PALB2, RAD51, and LIG4, resulting in inhibition of 2 major DNA double-strand break (DSB) repair pathways, HR, and nonhomologous end-joining. PARP1i, olaparib, and BMN673 caused accumulation of lethal DSBs and cell death in AC220-treated FLT3(ITD)-positive leukemia cells, thus mimicking synthetic lethality. Moreover, the combination of FLT3i and PARP1i eliminated FLT3(ITD)-positive quiescent and proliferating leukemia stem cells, as well as leukemic progenitors, from human and mouse leukemia samples. Notably, the combination of AC220 and BMN673 significantly delayed disease onset and effectively reduced leukemia-initiating cells in an FLT3(ITD)-positive primary AML xenograft mouse model. In conclusion, we postulate that FLT3i-induced deficiencies in DSB repair pathways sensitize FLT3(ITD)-positive AML cells to synthetic lethality triggered by PARP1i’s. Therefore, FLT3(ITD) could be used as a precision medicine marker for identifying AML patients that may benefit from a therapeutic regimen combining FLT3 and PARP1i’s.
Visual Abstract
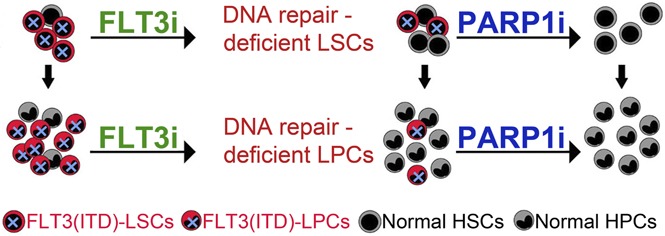
Introduction
Acute myeloid leukemia (AML) represents the deadliest form of acute leukemia among adults. Treatment involves chemotherapy and/or stem cell transplantation (for those who are eligible); however, the strategies are only curative in a fraction (30% to 40%) of younger patients and in <10% of patients older than 65 years. More specific therapies have been developed against AMLs carrying internal tandem duplications (ITDs) in FMS-like tyrosine kinase 3 (FLT3). Combination of chemotherapy with midostaurin, a tyrosine kinase inhibitor, which, among other targets, inhibits FLT3 activity, has shown efficacy in FLT3-mutant AML and has recently been approved by the US Food and Drug Administration.1 However, other FLT3 activity inhibitors (FLT3i’s), such as quizartinib (AC220) or sorafenib, rarely produced remissions when administered alone or in combination with cytotoxic drugs, and these remission are often short-lived and followed by early relapse in almost all cases.2
Leukemia stem cells (LSCs) have a dual role as tumor-initiating and therapy-refractory cells.3,4 Therefore, even if antitumor treatment clears a disease burden consisting mostly of leukemia progenitor cells (LPCs), it usually fails to eradicate LSCs and therapy-refractory residual LPCs. Several experimental approaches have been developed to eradicate LSCs, such as targeting of BCL2,5 glutathione metabolism,6 BCL6,7 mTOR,8 SDF-1,9 HDAC,10 and Wnt11 or involving granulocyte-colony stimulating factor (G-CSF)12 have recently been tested against LSCs. However, their clinical application may produce adverse events, because these proteins/mechanisms are also important in normal cells.13,14 Therefore, it is imperative to identify new therapies that, alone or in combination with traditional treatments, will cure or prolong the remission time and/or be used in refractory AML patients.
Numerous reports indicated that AML cells accumulate high levels of spontaneous and drug-induced DNA lesions, including highly lethal DNA double-strand breaks (DSBs), but they survive because of enhanced/altered DNA repair activities.15-22 DSBs are repaired by 2 major mechanisms: BRCA1/2-mediated homologous recombination (HR) and DNA-PK–mediated nonhomologous end-joining (D-NHEJ).23 In addition, PARP1 plays a central role in preventing/repairing lethal DSBs by activation of base excision repair/single-stranded DNA break repair, by stimulation of fork repair/restart, and by mediating the back-up nonhomolopgous end-joining (NHEJ) repair.24-27
Accumulation of potentially lethal DSBs in AML cells creates an opportunity to eradicate these cells by targeting DNA repair mechanisms. The success of the PARP1 inhibitor (PARP1i) olaparib in BRCA1/2-mutated breast and ovarian cancers established a proof-of-concept for personalized cancer therapy utilizing synthetic lethality to target DNA repair mechanisms.28 Because BRCA1/2 mutations are rare in AMLs,29 markers predicting their sensitivity to DNA repair inhibitors need to be identified. Unfortunately, The Cancer Genome Atlas (TCGA) database analysis did not reveal whether AML-related mutations were associated with specific DSB repair deficiencies (supplemental Figure 1, available on the Blood Web site).
Given the high frequency and poor prognosis of FLT3(ITD) mutations, as well as the cellular stress induced by these mutations,30 therapies targeting FLT3(ITD) mutations may leave AML cells vulnerable to DSB-inducing therapies. In particular, we hypothesized that FLT3i causes inhibition of HR and D-NHEJ (“BRCAness/DNA-PKness” phenotype), which, in combination with PARP1i, causes synthetic lethality in FLT3(ITD)-positive AML cells due to accumulation of lethal DSBs beyond the reparable threshold (Figure 1).
Figure 1.

Proposed model of FLT3i-guided synthetic lethality triggered by PARP1i in FLT3(ITD)-positive AML cells. Synthetic lethality arises when a combination of deficiencies in the expression of 2 or more genes leads to cell death, whereas a deficiency in only 1 of these genes does not. FLT3i downregulates the expression of multiple genes involved in DSB repair causing HR and D-NHEJ deficiency in FLT3(ITD)-positive leukemia cells but not in normal counterparts. This effect causes PARP1i-triggered accumulation of toxic DSBs and synthetic lethality in leukemia cells, whereas normal cells are spared.
Materials and methods
Primary human cells
Peripheral blood and bone marrow samples from patients with newly diagnosed AMLs were obtained from the Department of Internal Medicine III, University of Ulm; the Department of Internal Medicine (Hematology, Oncology, Hemostaseology, and Stem Cell Transplantation), RWTH Aachen University; and the Department of Internal Medicine I, Medical University of Vienna. Clinically relevant mutations for these samples are listed in supplemental Table 1. Samples of normal hematopoietic cells were purchased from Cambrex Bio Science (Walkersville, MD). Lin−CD34+ cells were obtained from mononuclear fractions by magnetic sorting using EasySep negative selection human progenitor cell enrichment cocktail, followed by human CD34 positive selection cocktail (STEMCELL Technologies, Vancouver, BC, Canada), as described previously.31
Primary murine cells
Evi1-GFP–transgenic mice (GFP knockin to Evi1 locus does not affect the function of Evi1) were generously provided by Mineo Kurokawa (University of Tokyo),32 and FLT3(ITD)-knockin mice33 were obtained from The Jackson Laboratory. FLT3(ITD)-knockin mice were crossed to Evi1-GFP mice to generate the FLT3(ITD);Evi1-GFP compound mouse. Mice were genotyped in-house by polymerase chain reaction. As expected, FLT3(ITD);Evi1-GFP mice contained enlarged spleens compared with their Evi1-GFP counterparts. In addition, although spleen and bone marrow organ architecture and cellular composition remained preserved in the Evi1-GFP mice, they were essentially completely replaced by infiltrate of cells with large nuclei and scant occasionally granulated cytoplasm, consistent with AML, in FLT3(ITD);Evi1-GFP mice (supplemental Figure 2A-D). Moreover, we observed an approximately twofold increase in the number of Lin−Sca-1+c-Kit+ (LSK) cells and a modest increase in Lin−Sca-1−c-Kit+ cells in bone marrow of FLT3(ITD);Evi1-GFP mice over Evi1-GFP mice,33 whereas >70% of LSK cells were GFP+ (supplemental Figure 2E-F). The detection of LSK GFP+ cells was performed by incubating with PE anti-mouse Lineage Cocktail antibody, Brilliant Violet 421–conjugated antibody to Sca1 (Ly6A/E; D7), and allophycocyanin-conjugated antibody to c-Kit (ACK2). LSK cells were also obtained from FLT3(ITD)+/+ mice, FLT3(ITD)+/− mice, and their wild-type (wt) littermates.33 All antibodies were purchased from BioLegend and eBioscience. Cells were sorted with a FACSAria (Becton Dickinson) automated cell sorter at the Temple University Lewis Katz School of Medicine flow cytometry core.
Cell lines
Human acute leukemia cell lines carrying homozygous FLT3(ITD) allele (MV-4-11)34 and FLT3(wt) (HL60, REH)35 were maintained in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and antibiotic cocktail. Murine BaF3 parental cell and clones overexpressing wt FLT3 and those expressing various FLT3 mutants36,37 were cultivated in Iscove modified Dulbecco medium supplemented with 10% FBS, interleukin-3 (IL-3), and antibiotic cocktail. Expression of constitutively active FLT3 was confirmed by western blot analysis.
Inhibitors/drugs
The following compounds were used: the FLT3 activity inhibitors AC220, gilteritinib, and crenolanib, the PARP1i’s BMN673 and olaparib (all from Selleckchem), mutT homolog 1 inhibitor (MTH1i) SCH51344 (Tocris), and reactive oxygen species (ROS) scavenger vitamin E (Sigma).
Western blot analyses
Nuclear cell lysates and total cell lysates were obtained and resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis, as previously described.31 Protein expression was analyzed using primary antibodies detecting BRCA1 (MAB22101; R&D Systems), BRCA2 (MAB2476; R&D Systems), RAD51 (sc-6862; Santa Cruz Biotechnology), DNA-PKcs (A300-518A; Bethyl Laboratories), Ku70 (A302-623A; Bethyl Laboratories), Ku80 (MA5-15873; Thermo Fisher Scientific), PARP1 (sc-7150; Santa Cruz Biotechnology), PALB2 (A302-627A; Bethyl Laboratories), Lig3 (GTX70147; GeneTex), Lig4 (ab26039; Abcam), phospho-Y694/T699 STAT5 (sc-11761; Santa Cruz Biotechnology), STAT5 (sc-28685; Santa Cruz Biotechnology), cleaved caspase-3 (9661; Cell Signaling Technology), and β-actin (A5316; Sigma).
Flow cytometry
DSBs were detected by γ-H2AX immunofluorescence (fluorescein isothiocyanate–conjugated anti–γ-H2AX antibody; 580445; BD Pharmingen), and cell viability was determined using fixable dead cell stain (65-0865-14; eBioscience), as described previously.38
Comet assay
The neutral comet assay was performed as described previously.39
Examination of HR and D-NHEJ
HR and D-NHEJ activities in BRCA/DNA-PK–deficient (blue) and -proficient (red) primary AML samples were detected as described previously.40 Briefly, 2-5 × 106 cells were nucleofected with 5 µg of I-SceI–linearized HR or NHEJ reporter plasmid and 2.5 µg of pDsRed plasmid (transfection efficiency control) using Nucleofector (program U-008, Human CD34 Cell Nucleofector Kit; Lonza). HR or D-NHEJ event restores functional GFP expression. After 72 hours, the percentage of GFP+/DsRed+ cells in DsRed+ cells was analyzed by flow cytometry to assess HR and D-NHEJ activity.
In vitro treatment
Lin−CD34+ human AML primary cells and human bone marrow cells from healthy donors were cultivated in StemSpan SFEM (STEMCELL Technologies) in the presence of recombinant human growth factors (100 ng/mL stem cell factor [SCF]; 10 ng/mL Flt3 ligand; 20 ng/mL IL-3, IL-6, G-CSF, and granulocyte-macrophage colony-stimulating factor [GM-CSF]; 2.5 ng/mL TPO). Murine FLT3(ITD)-positive cells were cultured in Iscove modified Dulbecco medium supplemented with 20% FBS and recombinant growth factors (15 ng/mL mouse [m]IL-3, 20 ng/mL mSCF, 12.5 ng/mL human [h]IL-6). Cells were incubated with AC220, gilteritinib, crenolanib, olaparib, and/or BMN673 for 3 days, followed by counting in trypan blue or fixable dead cell stain. To examine clonogenic activity, 104 cells per 0.1 mL were incubated with the inhibitor(s) or were left untreated for 3 days, followed by plating in MethoCult (STEMCELL Technologies) in the presence of growth factors. Colonies were counted after 7-10 days. For quiescent/proliferating cells, Lin− cells were stained with CellTrace Violet (CTV; eBioscience) and incubated for 5 days in StemSpan SFEM (STEMCELL Technologies) supplemented with the cocktail of growth factors (15 ng/mL [m]IL-3, 20 ng/mL mSCF, 12.5 ng/mL hIL-6) and inhibitors when indicated. Quiescent (CTVmax) and proliferating (CTVlow) leukemia cells were detected by flow cytometry using fluorochrome-conjugated anti-Lin (340546), anti-CD34 (347203), and anti-CD38 (555460) antibodies (all from BD Biosciences), as described previously.41 Lin−CD34+ AML primary cells were also cocultured on HS-5 human bone marrow stromal cells (American Type Culture Collection), seeded at a 1:1 ratio under hypoxia (1% O2), and treated with AC220 and/or olaparib for 3 days, followed by plating in MethoCult in the presence of growth factors.
In vivo treatment
NOD.Rag1−/−;γcnull mice expressing human IL-3, GM-CSF, and SCF (NRGS mice,42 The Jackson Laboratory) were sublethally irradiated (600 Gy) and injected with 1 × 106 Lin−CD34+ primary AML cells expressing FLT3(ITD) (10KM1950 sample; supplemental Table 1). Three weeks later, mice were treated with vehicle, AC220 (10 mg/kg),43 BMN673 (0.33 mg/kg),40 and AC220 + BMN673 for 7 days. hCD45+ AML cells were detected in bone marrow cells, splenocytes, and/or peripheral blood leukocytes at the end of treatment, as described previously.40,41 Moreover, 1 × 106 bone marrow cells were transplanted into the sublethally irradiated secondary recipients to assess the effect of treatment on LSCs. Median survival time of the primary and secondary recipients was determined. To assess the toxicity of the treatment, C57BL/6 mice were treated with vehicle or AC220 + BMN673, as described above. Peripheral blood, bone marrow, and other organs were tested to assess potential toxicity.
Statistical analyses
Data are presented as mean ± standard deviation (SD) from 3 independent experiments and were compared using the unpaired 2-tailed Student t test; P values <.05 were considered significant. The response additivity approach was used to study synergistic effects.44 Median survival time of the mice ± standard error was calculated using Kaplan-Meier log-rank survival analysis. P values <.05 were considered significant.
Study approval
Human studies were approved by the appropriate Institutional Review Boards and met all requirements of the Declaration of Helsinki. Animal studies were approved by the Temple University Institutional Animal Care and Use Committee.
Results
FLT3i inhibited DSB repair activity in FLT3(ITD)-positive leukemia cells
To determine whether AC220-mediated inhibition of FLT3(ITD) activity is associated with inhibition of DSB repair activity, we performed a western blot array to assess expression of key proteins in DSB repair pathways (Figure 2A). FLT3(ITD)-positive BaF3 cells and their parental counterparts were treated with AC220 for 24 hours in the presence of IL-3 to inhibit FLT3(ITD) activity, as documented by downregulated phosphotyrosine-STAT5 content in total cell lysates (Figure 2B). At the same time, caspase-3 activation was minimal, PARP1 was not cleaved, and AC220-treated cells were alive in the trypan blue exclusion test. Selected key proteins in HR (BRCA1, BRCA2, PALB2, and RAD51) and D-NHEJ (LIG4) were downregulated in nuclear lysates from AC220-treated FLT3(ITD)-positive cells (Figure 2B).
Figure 2.

FLT3i AC220 downregulated HR and D-NHEJ proteins and inhibited HR and D-NHEJ activity. (A) DSB repair pathways and some key proteins. (B) Western blot analysis of the indicated proteins in parental BaF3 cells transfected with empty plasmid (Empty) and in those expressing FLT3(ITD) after a 24-hour incubation with 10 nM AC220 or vehicle in the presence of IL-3. Cells were nucleofected with linearized plasmids containing HR (C) or D-NHEJ (D) reporter cassette and pDsRed plasmid (transfection efficiency control). HR or D-NHEJ event restores functional GFP expression. After 72 hours, GFP+/DsRed+ cells in DsRed+ cells were analyzed by flow cytometry to assess HR and D-NHEJ activity in untreated (Control) and AC220-treated (10 nM) REH cells and MV-4-11 cells. Results represent mean (percentage) ± SD of GFP+/DsRed+ cells in DsRed+ cells from triplicates per sample. *P = .02, **P < .001, 2-tailed Student t test.
Next, we examined whether AC220-induced downregulation of HR and D-NHEJ proteins caused reduction of DSB repair activities. Using specific reporter plasmids whereby restoration of GFP expression depends on HR or D-NHEJ, we determined that both DSB repair pathways were inhibited in AC220-treated FLT3(ITD)-positive MV-4-11 cells but not in REH cells expressing FLT3(wt) (Figure 2C-D).
These collective results strongly suggest that inhibition of FLT3(ITD) activity resulted in induction of early defects in DSB repair pathways.
FLT3i enhanced PARP1i-dependent accumulation of DSBs and elimination of leukemia cells bearing FLT3 mutants
BaF3 cells expressing FLT3-activating mutations in juxtamembrane domain (ITD, delEY, delIns) and tyrosine kinase domain 1 (E611V), but not BaF3 cells transfected with empty plasmid and these overexpressing FLT3(wt), were sensitive to the FLT3i’s AC220 and crenolanib and the PARP1i’s olaparib and BMN673 used individually (Figure 3A; supplemental Figure 3). However, the strongest antileukemia effect was exerted by a combination of FLT3i + PARP1i selectively in BaF3 cells expressing FLT3 activating mutations.
Figure 3.

FLT3i’s enhanced the antileukemia effect of PARP1i’s in FLT3(ITD)-positive cell lines. (A) BaF3 parental cells transfected with empty plasmid (Empty) and those expressing FLT3(wt) or FLT3(ITD) mutant (ITD) were left untreated (Control) or were incubated with PARP1i [1.25 μM olaparib (Ola), 12.5 nM BMN673 (BMN)], FLT3i (10 nM AC220, 10 nM crenolanib), and FLT3i+PARP1i for 96 hours in the presence of IL-3. Results from 3 experiments are represented by the mean (percentage) ± SD of living cells detected by trypan blue exclusion compared with untreated counterparts. *P < .001 vs individual treatment, 2-tailed Student t test. (B) FLT3(ITD)-positive BaF3 cells were incubated with the indicated concentrations of BMN673, alone or in combination with 200 μM vitamin E (VE) or 3 μM MTH1i SCH51344, for 96 hours in the presence of IL-3. Results represent mean (percentage) ± SD of living cells detected by trypan blue exclusion in comparison with untreated control from 3 independent experiments. *P < .05 vs BMN673 treatment, 2-tailed Student t test. (C-E) MV-4-11 [FLT3(ITD)-positive] and HL60 and REH [both FLT3(ITD)-negative] human leukemia cell lines were left untreated (Control) or were treated with 5 nM BMN673, 10 nM AC220, or the combination. DSBs were detected after 24 hours by neutral comet assay (C) and γ-H2AX immunofluorescence (D). (E) Dead cells were determined by fixable dead cell stain after 96 hours. Results represent mean ± SD from 3 experiments. *P < .02 vs individual treatment, 2-tailed Student t test (C-E).
It has been reported that FLT3(ITD)-positive leukemia cells accumulate ROS-induced DSBs.17 To determine whether oxidative stress–induced DSBs contribute to PARP1i-mediated cytotoxicity, leukemia cells were incubated with BMN673 and ROS scavenger vitamin E or MTH1i SCH51344 (MTH1 sanitizes oxidized 2′-deoxynucleoside 5′-triphosphate pools to prevent incorporation of damaged bases during DNA replication).45 Vitamin E diminished, whereas SCH51344 enhanced, the toxic effect of BMN673 in FLT3(ITD)-positive cells (Figure 3B).
DSBs were examined by neutral comet assay and by detection of Ser139 H2AX phosphorylation (γ-H2AX), which have been shown to give a sensitive and accurate estimation of the number of DSBs within DNA.46,47 The combination of AC220 and BMN673 induced accumulation of DSBs in FLT3(ITD)-positive MV-4-11 cells, but not in FLT3(wt) HL60 and REH cells, 24 hours after the treatment (Figure 3C-D), which was associated with enhanced death of MV-4-11 cells detected 96 hours after treatment (Figure 3D).
Altogether, we postulate that AC220 sensitized FLT3(ITD)-positive leukemia cells to PARP1i’s by promoting accumulation of lethal DSBs, while sparing cells carrying FLT3(wt).
FLT3i’s enhanced the sensitivity of FLT3(ITD)-positive proliferative and quiescent LSC-enriched primary cells to PARP inhibitors
To test the effect of the combination of FLT3i and PARP1i on FLT3(ITD)-positive primary cells, LSK bone marrow cells from FLT3(ITD)+/+, FLT3(ITD)+/−, and FLT3(wt) mice were incubated with AC220 and olaparib, followed by γ-H2AX immunostaining and plating in methylcellulose. The combination of AC220 and olaparib resulted in enhanced accumulation of γ-H2AX–positive FLT3(ITD)+/+ and FLT3(ITD)+/− cells but not FLT3(wt) cells (Figure 4A). This effect was associated with AC220 dose-dependent reduction of clonogenic potential of olaparib-treated FLT3(ITD)+/+ and FLT3(ITD)+/− cells compared with their FLT3(wt) counterparts (Figure 4B).
Figure 4.

FLT3i enhanced the antileukemia effect of PARP1i’s against proliferating and quiescent primary FLT3(ITD)-positive cells. (A-B) LSK bone marrow cells from FLT3(ITD)+/+, FLT3(ITD)+/−, and FLT3(wt) mice (n = 3 per group) were incubated for 24 hours with 100 nM AC220 and/or 2.5 μM olaparib (Ola), followed by γ-H2AX immunostaining, and for 4 days with 2.5 μM Ola and the indicated concentrations of AC220, followed by plating in methylcellulose. Results represent mean (percentage) ± SD of γ-H2AX positive cells (A) and surviving clonogenic cells (B) compared with their Ola-treated counterparts. *P < .05, 2-tailed Student t test. (C) GFP+ FLT3(ITD)-positive LSK murine bone marrow cells [FLT3(ITD)+/+] and GFP+ LSK normal counterparts [FLT3(wt)] obtained from 2 or 3 mice per group were left untreated (0) or were treated with AC220 (low: 20 nM; high: 100 nM), Ola (low: 1 μM; high: 2.5 μM), and the combination of AC220 + Ola in the presence of growth factors for 4 days, followed by plating in methylcellulose. Results represent mean (percentage) ± SD of colonies in comparison with untreated group from triplicate experiments. *P < .001 vs individual treatments using response additivity approach. Lin−CD34+ FLT3(ITD)-positive cells from 5 AML patients (188, 194, 499, 9KM3949, 10KM1950) were stained with CTV and left untreated or were treated with the FLT3i’s AC220 (10 nM), crenolanib (10 nM), and gliteritinib (10 nM), the PARP1i’s Ola (2.5 μM) and BMN673 (25 nM), and the combination of FLT3i + PARP1i in the presence of growth factors for 3 days, followed by assessment of Lin−CD34+ clonogenic cells (D) and immunofluorescent detection of Lin−CD34+CD38−CTVlow cells (E) and Lin−CD34+CD38−CTVmax cells (F). Results represent mean (percentage) ± SD of clonogenic/surviving cells in comparison with untreated cells from triplicate experiments. *P < .05 vs individual treatments, 2-tailed Student t test. (G) Lin−CD34+ FLT3(ITD)-positive cells from 4 AML patients (188, 194, 499, 10KM1950) were left untreated or were treated with AC220 (100 nM), Ola (2.5 μM), and the combination of AC220 + Ola on HS-5 cell monolayers in 1% O2 for 3 days, followed by plating in methylcellulose. Results represent mean (percentage) ± SD of clonogenic cells in comparison with untreated cells from triplicate experiments. *P < .05 vs individual treatment, 2-tailed Student t test.
To test the effect of FLT3i combined with PARP1i on FLT3(ITD)-positive stem cells, we employed hematopoietic precursors harvested from FLT3(ITD)+/+;Evi1-GFP+/− mice. Using Evi1-GFP reporter mice, Kataoka et al had demonstrated that EVI1 is expressed exclusively in the hematopoietic stem cell (HSC) population in the bone marrow and that its expression marks hematopoietic cells with long-term multilineage repopulating activity,32 which was recently reproduced by us.48 We treated HSCs-enriched mouse FLT3(ITD)-positive GFP+ cells with AC220 and olaparib, and found that FLT3(ITD)-positive GFP+ LSK cells were synergistically sensitive to the combination of AC220 and olaparib (Figure 4C). At the same time, FLT3(wt) counterparts were approximately 7-times less sensitive to AC220 + olaparib.
We reported previously that D-NHEJ–deficient quiescent and HR/D-NHEJ–deficient proliferating tumor cells were sensitive to dual cellular synthetic lethality exerted by PARP1i.40 Therefore, we tested whether FLT3i-induced downregulation of D-NHEJ and HR sensitizes FLT3(ITD)-positive quiescent and proliferating AML primary cells, respectively, to PARP1i-mediated synthetic lethality. AC220 caused downregulation of BRCA1, BRCA2, RAD51, and LIG4 in Lin−CD34+ primary AML cells (supplemental Figure 4). AC220, as well as the more recently developed FLT3i’s gilteritinib and crenolanib, enhanced the sensitivity of LSC/LPC-enriched Lin−CD34+ cells to olaparib and BMN673 (Figure 4D). Moreover, AC220 increased the effect of olaparib against LSC-enriched Lin−CD34+CD38−CTVlow proliferating and Lin−CD34+CD38−CTVmax quiescent cells (Figure 4E-F). Under the same conditions, the combination of AC220 and olaparib exerted only a modest effect against normal hematopoietic cells (supplemental Figure 5A).
It has been reported that the bone marrow microenvironment induced resistance of FLT3(ITD)-positive AML cells to the kinase inhibitors and cytotoxic drugs.49 To test the impact of the bone marrow microenvironment on the effectiveness of AC220+/− olaparib, FLT3(ITD)-positive Lin−CD34+ AML primary cells were cocultured under hypoxic conditions (1% O2) with the HS-5 stromal cell line, which secrete significant levels of G-CSF, GM-CSF, macrophage colony-stimulating factor, SCF, macrophage inflammatory protein-1α, IL-6, IL-8, and IL-11 to support the growth of HSCs.50 Under these conditions, olaparib reduced the number of clonogenic leukemia cells not eliminated by the high concentration (100 nM) of AC220 (Figure 4G).
Altogether, these results support the conclusion that inhibition of FLT3(ITD) activity enhanced the sensitivity of proliferating and quiescent LSCs to PARP1i’s in vitro, while sparing significant numbers of normal counterparts.
AC220 enhanced the effect of BMN673 against FLT3(ITD)-positive primary AML xenografts in humanized immunodeficient mice
NRSG mice bearing FLT3(ITD)-positive AML primary cells were treated with vehicle (control), AC220, BMN673, or AC220 + BMN673 (Figure 5A). The therapeutic effect was examined by detection of hCD45+ cells in peripheral blood and bone marrow, and by determination of median survival time (MST) of the treated mice. In addition, 1 × 106 bone marrow cells were retransplanted into secondary recipients to examine LSCs.
Figure 5.

PARP1i BMN673 combined with FLT3i AC220 inhibited the growth in FLT3(ITD)-positive AML primary cells in mice and reduced the number of LSCs. (A) Experimental design. (B) Mean percentage of hCD45+ cells ± SD in peripheral blood cells (PBL, n = 5-6) and bone marrow cells (BMC, n = 3-5). *P < .01, **P = .002 vs individual treatment, Student t test and response additivity approach, respectively. (C) Survival curves and MST of mice bearing a primary AML xenograft and treated as indicated (n = 6-10 mice per group). (D) Survival curves and MST of secondary transplant mice. *P ≤ .002 vs individual treatments, Kaplan-Meier log-rank test.
Leukemic mice treated with AC220 + BMN673 contained significantly fewer hCD45+ leukemia cells in peripheral blood and bone marrow than did those treated with individual drugs (Figure 5B). Moreover, the combination of AC220 + BMN673 exerted a synergistic effect against hCD45+ cells in bone marrow. In addition, MST of the leukemia-bearing mice was significantly prolonged by the combination of AC220 + BMN673 in comparison with mice treated with AC220 or BMN673 (Figure 5C). Importantly, secondary recipients injected with bone marrow cells harvested at the end of AC220 + BMN673 treatment survived longer than did mice injected with cells from individually treated animals (Figure 5D), suggesting that the combination targeted LSCs in vivo.
AC220 + BMN673 treatment did not appear to be significantly toxic for normal cells and tissues, except for the transient reduction (by ∼43%) in the number of white cells in bone marrow (supplemental Table 2; supplemental Figure 6). Because the pharmacokinetic parameters of FLT3i and PARP1i may differ in humans and mice, we examined the prolonged in vitro effect of clinically relevant concentrations of AC220 combined with olaparib on human Lin−CD34+ bone marrow cells obtained from healthy donors. Seven-day treatment with AC220 (535 nM)51 + olaparib (6 μM)52 in 100% FBS inhibited their clonogenic activity by ∼40%, and olaparib did not significantly increase the toxicity of AC220 (supplemental Figure 5B).
Discussion
Although tyrosine kinase inhibitors, such as AC220 (quizartinib), target FLT3(ITD) activity, which functions as a driver oncogene in a large subgroup of AMLs, the treatment, even if combined with cytotoxic drugs, is not curative in the vast majority of cases because leukemia cells are able to deal with DNA damage.2,53-55 Therefore, we postulated that specific inhibitors of DNA repair may dramatically improve the therapeutic outcome of patients with FLT3(ITD)-positive AML. The success of PARP1i’s in patients with BRCA1/2 mutated breast and ovarian carcinomas supported our hypothesis.28
Although inactivating mutations in genes whose products are responsible for DNA repair are rather infrequent in leukemias,29 we and other investigators reported that the presence of certain leukemia-driving oncogenes (eg, AML1-ETO, BCR-ABL1, PML-RARA) is associated with BRCA and/or DNA-PK–deficient status and sensitivity to synthetic lethality induced by PARP1i’s.31,40,56-58 On the other hand, because FLT3(ITD) activity was associated with activation of BRCA1-RAD51–dependent HR, it was unlikely that FLT3(ITD)-positive AML cells will be highly sensitive to PARP1i’s.55,59 In concordance, we detected that FLT3(ITD)-positive cells displayed only modest sensitivity to PARP1i’s, which is probably due to the accumulation of ROS-mediated DNA damage and/or inhibition of Ku70 protein involved in D-NHEJ.17,35
Because AC220 promotes quiescence rather than cytotoxicity,60 it seemed important to combine AC220 with a drug that would be able to eliminate proliferating and nonproliferating cells. Remarkably, inhibition of FLT3(ITD) activity by AC220 caused early and dramatic downregulation of selected proteins in HR (BRCA1, BRCA2, PALB2, and RAD51) and D-NHEJ (LIG4), which resulted in simultaneous inhibition of HR and D-NHEJ activities (“BRCAness/DNA-PKness” phenotype). Numerous major intracellular signaling pathways, including PI3K-AKT, RAF1-MEK, and JAK2-STAT5, are activated in FLT3(ITD)-positive AML cells.61,62 Results obtained with the use of specific inhibitors suggest that JAK2 kinase stimulates expression of BRCA1, RAD51, and LIG4; PI3K promotes expression of RAD51 and LIG4; and RAF1 kinase does not regulate expression of these proteins in FLT3(ITD)-positive cells (supplemental Figure 7). The mechanisms responsible for FLT3i-mediated downregulation of BRCA2 and PALB2 are not known.
We have reported previously that HR- and D-NHEJ–deficient proliferating and quiescent leukemia cells were sensitive to synthetic lethality triggered by PARP1i’s.40 Therefore, PARP1i’s should eliminate proliferating, as well as quiescent, leukemia cells. In concordance, we observed that FLT3i-treated FLT3(ITD)-positive AML cells, including LSC-enriched Lin−CD34+CD38− quiescent and proliferating cells, were extremely sensitive to PARP1i’s in vitro and in vivo. This effect was associated with accumulation of DSBs and induction of cell death. The antileukemic effect of FLT3i AC220 + PARP1i olaparib was enhanced by addition of a standard cytotoxic drug, such as doxorubicin (supplemental Figure 8).
It has been reported that G0-arrested FLT3(ITD)-positive leukemia cells display inappropriate RAD51 expression and HR activity.55 Therefore FLT3i-mediated inhibition of RAD51-mediated HR in these cells may cause the “BRCAness” phenotype and also contribute to PARP1i-mediated synthetic lethality in FLT3(ITD)-positive quiescent AML cells.
We also showed that PARP1i used as a single agent was able to reduce the number of AML-initiating FLT3(ITD)-positive cells, as well as clonogenic cells, in bone marrow, including the hypoxic conditions mimicking the bone marrow microenvironment. This observation is in concordance with another report demonstrating hypoxia-induced HR deficiency and enhanced synthetic lethality triggered by PARP1 inhibition.63 Moreover, combination of FLT3i + PARP1i was more effective than individual treatment. This is particularly important because FLT3(ITD)-positive leukemia cells expand in the bone marrow niche, which also promotes their survival under FLT3i treatment.64,65 Altogether, we postulate that the presence of FLT(ITD) creates a window of opportunity to induce the FLT3i-mediated “BRCAness/DNA-PKness” phenotype promoting a profound PARP1i-mediated synthetic lethal effect in AML quiescent and proliferating stem and progenitor cells. This effect most likely depends on inhibition of FLT3(ITD) activity, because we showed that the FLT3i-mediated “BRCAness/DNA-PKness” phenotype was selectively induced in FLT3(ITD)-positive cells and that 3 FLT3i’s displaying various target repertoires (AC220-FLT3, c-KIT, and PDGFRα; gilteritinib-FLT3 and AXL; and crenolanib-FLT3 and PDGFR)66 enhanced the PARP1i-mediated anti-AML effect.
The degree of sensitivity of FLT3(ITD)-positive AML cells to PARP1i and to a combination of FLT3i + PARP1i could be affected by additional mutations (eg, AML1-ETO and MLL-AF90) and/or overexpression of transcription factors HOXA9 + MEIS1 (supplemental Figure 9). AML1-ETO–driven exceptional sensitivity and HOXA9 + MEIS1–mediated resistance to olaparib could be explained by BRCA deficiency and BRCA proficiency, respectively, as reported by us and other investigators.40,58 On the other hand, strong sensitivity of BRCA-proficient MLL-AF9–expressing cells to AC220 + olaparib may be due to their enhanced overall dependence on PARP1-mediated DNA repair to promote survival under genotoxic stress and on FLT3i-mediated BRCA deficiency.67
In conclusion, we postulate that FLT3i-induced deficiencies in DSB repair pathways sensitize FLT3(ITD)-positive AML cells to synthetic lethality triggered by PARP1i’s. Therefore, FLT3(ITD) could be used as a precision medicine marker to identify patients with AMLs who may benefit from a therapeutic regimen combining FLT3 and PARP1i’s. Moreover, TKI-mediated inhibition of DSB repair, which sensitizes malignant cells to PARP1i-triggered synthetic lethality, may have a more broad application in tumors expressing oncogenic tyrosine kinases. This speculation is supported by our recent study indicating that the JAK2 inhibitor ruxolitinib caused the “BRCAness/DNA-PKness” phenotype, resulting in PARP1i-mediated synthetic lethality in myeloproliferative neoplasms.68
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
This work was supported by National Institutes of Health (NIH), National Cancer Institute (NCI) grant 1R01 CA186238 (T. Skorski) and NIH National Heart, Lung, and Blood Institute grant R00 HL107747 (J.H.). K.S.-R. was supported by NIH/Department of Health and Human Services grant 1F31 CA203161. P.P.-B. was supported by the Kosciuszko Foundation Scholarship for 2016/2017 and by Polish National Science Centre Research grant UMO-2014/15/D/NZ3/05187. M.T. was supported by the Kosciuszko Foundation Scholarship for 2017/2018. B.V.L. has been supported by the European Union's Horizon 2020 Research and Innovation Programme under Marie Sklodowska-Curie grant agreement number 665735 and by the Polish Ministry of Science and Higher Education funds for the implementation of international projects, 2016-2020. L.B. was supported in part by the Deutsche Forschungsgemeinschaft (Heisenberg-Professur BU 1339/8-1). P.V. was supported by Austrian Science Fund SFB grant F4704-B20. N.C. was supported by the START program of the Faculty of Medicine at RWTH Aachen University. T. Stoklosa and M.M.M. were supported by Polish National Science Centre grant HARMONIA 2014/14/M/NZ5/00441.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.M., M.N.-S., Y.D., K.S.-R., B.V.L., Z.L., P.P.-B., E.A.B., M.S., A.N., and M.T. performed experiments; M.M.M. and M.R. prepared next-generation sequencing (NGS) libraries and performed NGS of several patient samples; H.Z. performed statistical analyses; J.J. analyzed The Cancer Genome Atlas (TCGA) database; K.P. supervised B.V.L., M.A.W. supervised E.A.B., and T. Sliwinski supervised M.T.; T. Stoklosa analyzed NGS data and supervised M.M.M.; R.P. analyzed NGS data and supervised M.R.; T.F., S.M.S., N.C., S.K., L.B., P.V., and J.H. provided essential cell lines and primary cells and contributed to the final version of the manuscript; and T. Skorski conceived the idea, supervised the project, and wrote the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Tomasz Skorski, Temple University, School of Medicine, Department of Microbiology and Immunology and Fels Institute for Cancer Research and Molecular Biology, 3400 N. Broad St, MRB 548, Philadelphia, PA 19140; e-mail: tskorski@temple.edu.
REFERENCES
- 1.Stone RM, Mandrekar SJ, Sanford BL, et al. . Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leick MB, Levis MJ. The future of targeting FLT3 activation in AML. Curr Hematol Malig Rep. 2017;12(3):153-167. [DOI] [PubMed] [Google Scholar]
- 3.Frame FM, Maitland NJ. Cancer stem cells, models of study and implications of therapy resistance mechanisms. Adv Exp Med Biol. 2011;720:105-118. [DOI] [PubMed] [Google Scholar]
- 4.Ho TC, LaMere M, Stevens BM, et al. . Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood. 2016;128(13):1671-1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lagadinou ED, Sach A, Callahan K, et al. . BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12(3):329-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pei S, Minhajuddin M, Callahan KP, et al. . Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J Biol Chem. 2013;288(47):33542-33558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duy C, Hurtz C, Shojaee S, et al. . BCL6 enables Ph+ acute lymphoblastic leukaemia cells to survive BCR-ABL1 kinase inhibition. Nature. 2011;473(7347):384-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuwatsuka Y, Minami M, Minami Y, et al. . The mTOR inhibitor, everolimus (RAD001), overcomes resistance to imatinib in quiescent Ph-positive acute lymphoblastic leukemia cells. Blood Cancer J. 2011;1(5):e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nervi B, Ramirez P, Rettig MP, et al. . Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood. 2009;113(24):6206-6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang B, Strauss AC, Chu S, et al. . Effective targeting of quiescent chronic myelogenous leukemia stem cells by histone deacetylase inhibitors in combination with imatinib mesylate. Cancer Cell. 2010;17(5):427-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Emami KH, Nguyen C, Ma H, et al. . A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected] [published correction appears in Proc Natl Acad Sci USA 2004;101(47):16707]. Proc Natl Acad Sci USA. 2004;101(34):12682-12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saito Y, Uchida N, Tanaka S, et al. . Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol. 2010;28(3):275-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Basso K, Dalla-Favera R. Roles of BCL6 in normal and transformed germinal center B cells. Immunol Rev. 2012;247(1):172-183. [DOI] [PubMed] [Google Scholar]
- 14.Proud CG. mTOR signalling in health and disease. Biochem Soc Trans. 2011;39(2):431-436. [DOI] [PubMed] [Google Scholar]
- 15.Petruccelli LA, Pettersson F, Del Rincón SV, Guilbert C, Licht JD, Miller WH Jr. Expression of leukemia-associated fusion proteins increases sensitivity to histone deacetylase inhibitor-induced DNA damage and apoptosis. Mol Cancer Ther. 2013;12(8):1591-1604. [DOI] [PubMed] [Google Scholar]
- 16.Krejci O, Wunderlich M, Geiger H, et al. . p53 signaling in response to increased DNA damage sensitizes AML1-ETO cells to stress-induced death. Blood. 2008;111(4):2190-2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sallmyr A, Fan J, Datta K, et al. . Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: implications for poor prognosis in AML. Blood. 2008;111(6):3173-3182. [DOI] [PubMed] [Google Scholar]
- 18.Hole PS, Zabkiewicz J, Munje C, et al. . Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood. 2013;122(19):3322-3330. [DOI] [PubMed] [Google Scholar]
- 19.Cavelier C, Didier C, Prade N, et al. . Constitutive activation of the DNA damage signaling pathway in acute myeloid leukemia with complex karyotype: potential importance for checkpoint targeting therapy. Cancer Res. 2009;69(22):8652-8661. [DOI] [PubMed] [Google Scholar]
- 20.Abdel-Wahab O, Levine RL. Metabolism and the leukemic stem cell. J Exp Med. 2010;207(4):677-680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boehrer S, Adès L, Tajeddine N, et al. . Suppression of the DNA damage response in acute myeloid leukemia versus myelodysplastic syndrome. Oncogene. 2009;28(22):2205-2218. [DOI] [PubMed] [Google Scholar]
- 22.Jacoby MA, De Jesus Pizarro RE, Shao J, et al. . The DNA double-strand break response is abnormal in myeloblasts from patients with therapy-related acute myeloid leukemia. Leukemia. 2014;28(6):1242-1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47(4):497-510. [DOI] [PubMed] [Google Scholar]
- 24.Karanam K, Kafri R, Loewer A, Lahav G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell. 2012;47(2):320-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng Z, Scott SP, Bussen W, et al. . Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc Natl Acad Sci USA. 2011;108(2):686-691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bryant HE, Petermann E, Schultz N, et al. . PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009;28(17):2601-2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ying S, Chen Z, Medhurst AL, et al. . DNA-PKcs and PARP1 bind to unresected stalled DNA replication forks where they recruit XRCC1 to mediate repair. Cancer Res. 2016;76(5):1078-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lord CJ, Tutt AN, Ashworth A. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu Rev Med. 2015;66(1):455-470. [DOI] [PubMed] [Google Scholar]
- 29.Ley TJ, Miller C, Ding L, et al. ; Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rebechi MT, Pratz KW. Genomic instability is a principle pathologic feature of FLT3 ITD kinase activity in acute myeloid leukemia leading to clonal evolution and disease progression. Leuk Lymphoma. 2017;58(9):1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cramer-Morales K, Nieborowska-Skorska M, Scheibner K, et al. . Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood. 2013;122(7):1293-1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kataoka K, Sato T, Yoshimi A, et al. . Evi1 is essential for hematopoietic stem cell self-renewal, and its expression marks hematopoietic cells with long-term multilineage repopulating activity. J Exp Med. 2011;208(12):2403-2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee BH, Tothova Z, Levine RL, et al. . FLT3 mutations confer enhanced proliferation and survival properties to multipotent progenitors in a murine model of chronic myelomonocytic leukemia. Cancer Cell. 2007;12(4):367-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quentmeier H, Reinhardt J, Zaborski M, Drexler HG. FLT3 mutations in acute myeloid leukemia cell lines. Leukemia. 2003;17(1):120-124. [DOI] [PubMed] [Google Scholar]
- 35.Fan J, Li L, Small D, Rassool F. Cells expressing FLT3/ITD mutations exhibit elevated repair errors generated through alternative NHEJ pathways: implications for genomic instability and therapy. Blood. 2010;116(24):5298-5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arreba-Tutusaus P, Mack TS, Bullinger L, et al. . Impact of FLT3-ITD location on sensitivity to TKI-therapy in vitro and in vivo. Leukemia. 2016;30(5):1220-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chatain N, Perera RC, Rossetti G, et al. . Rare FLT3 deletion mutants may provide additional treatment options to patients with AML: an approach to individualized medicine. Leukemia. 2015;29(12):2434-2438. [DOI] [PubMed] [Google Scholar]
- 38.Nieborowska-Skorska M, Kopinski PK, Ray R, et al. . Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors. Blood. 2012;119(18):4253-4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koptyra M, Falinski R, Nowicki MO, et al. . BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood. 2006;108(1):319-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nieborowska-Skorska M, Sullivan K, Dasgupta Y, et al. . Gene expression and mutation-guided synthetic lethality eradicates proliferating and quiescent leukemia cells. J Clin Invest. 2017;127(6):2392-2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bolton-Gillespie E, Schemionek M, Klein HU, et al. . Genomic instability may originate from imatinib-refractory chronic myeloid leukemia stem cells. Blood. 2013;121(20):4175-4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goyama S, Wunderlich M, Mulloy JC. Xenograft models for normal and malignant stem cells. Blood. 2015;125(17):2630-2640. [DOI] [PubMed] [Google Scholar]
- 43.Zarrinkar PP, Gunawardane RN, Cramer MD, et al. . AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood. 2009;114(14):2984-2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Slinker BK. The statistics of synergism. J Mol Cell Cardiol. 1998;30(4):723-731. [DOI] [PubMed] [Google Scholar]
- 45.Gad H, Koolmeister T, Jemth AS, et al. . MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature. 2014;508(7495):215-221. [DOI] [PubMed] [Google Scholar]
- 46.Fairbairn DW, Olive PL, O’Neill KL. The comet assay: a comprehensive review. Mutat Res. 1995;339(1):37-59. [DOI] [PubMed] [Google Scholar]
- 47.Banáth JP, Olive PL. Expression of phosphorylated histone H2AX as a surrogate of cell killing by drugs that create DNA double-strand breaks. Cancer Res. 2003;63(15):4347-4350. [PubMed] [Google Scholar]
- 48.Wang Y, Tian H, Cai W, et al. . Tracking hematopoietic precursor division ex vivo in real time. Stem Cell Res Ther. 2018;9(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zeng Z, Shi YX, Samudio IJ, et al. . Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113(24):6215-6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roecklein BA, Torok-Storb B. Functionally distinct human marrow stromal cell lines immortalized by transduction with the human papilloma virus E6/E7 genes. Blood. 1995;85(4):997-1005. [PubMed] [Google Scholar]
- 51.Sanga M, James J, Marini J, Gammon G, Hale C, Li J. An open-label, single-dose, phase 1 study of the absorption, metabolism and excretion of quizartinib, a highly selective and potent FLT3 tyrosine kinase inhibitor, in healthy male subjects, for the treatment of acute myeloid leukemia. Xenobiotica. 2017;47(10):856-869. [DOI] [PubMed] [Google Scholar]
- 52.Bundred N, Gardovskis J, Jaskiewicz J, et al. . Evaluation of the pharmacodynamics and pharmacokinetics of the PARP inhibitor olaparib: a phase I multicentre trial in patients scheduled for elective breast cancer surgery. Invest New Drugs. 2013;31(4):949-958. [DOI] [PubMed] [Google Scholar]
- 53.Seedhouse CH, Hunter HM, Lloyd-Lewis B, et al. . DNA repair contributes to the drug-resistant phenotype of primary acute myeloid leukaemia cells with FLT3 internal tandem duplications and is reversed by the FLT3 inhibitor PKC412. Leukemia. 2006;20(12):2130-2136. [DOI] [PubMed] [Google Scholar]
- 54.Santos MA, Faryabi RB, Ergen AV, et al. . DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature. 2014;514(7520):107-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gaymes TJ, Mohamedali A, Eiliazadeh AL, Darling D, Mufti GJ. FLT3 and JAK2 mutations in acute myeloid leukemia promote interchromosomal homologous recombination and the potential for copy neutral loss of heterozygosity. Cancer Res. 2017;77(7):1697-1708. [DOI] [PubMed] [Google Scholar]
- 56.Podszywalow-Bartnicka P, Wolczyk M, Kusio-Kobialka M, et al. . Downregulation of BRCA1 protein in BCR-ABL1 leukemia cells depends on stress-triggered TIAR-mediated suppression of translation. Cell Cycle. 2014;13(23):3727-3741. [DOI] [PubMed] [Google Scholar]
- 57.Deutsch E, Dugray A, AbdulKarim B, et al. . BCR-ABL down-regulates the DNA repair protein DNA-PKcs. Blood. 2001;97(7):2084-2090. [DOI] [PubMed] [Google Scholar]
- 58.Esposito MT, Zhao L, Fung TK, et al. . Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat Med. 2015;21(12):1481-1490. [DOI] [PubMed] [Google Scholar]
- 59.Dai Y, Chen S, Kmieciak M, et al. . The novel Chk1 inhibitor MK-8776 sensitizes human leukemia cells to HDAC inhibitors by targeting the intra-S checkpoint and DNA replication and repair. Mol Cancer Ther. 2013;12(6):878-889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taylor SJ, Dagger SA, Thien CB, Wikstrom ME, Langdon WY. Flt3 inhibitor AC220 is a potent therapy in a mouse model of myeloproliferative disease driven by enhanced wild-type Flt3 signaling. Blood. 2012;120(19):4049-4057. [DOI] [PubMed] [Google Scholar]
- 61.Cook AM, Li L, Ho Y, et al. . Role of altered growth factor receptor-mediated JAK2 signaling in growth and maintenance of human acute myeloid leukemia stem cells. Blood. 2014;123(18):2826-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takahashi S. Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: biology and therapeutic implications. J Hematol Oncol. 2011;4:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chan N, Pires IM, Bencokova Z, et al. . Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Res. 2010;70(20):8045-8054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ghiaur G, Levis M. Mechanisms of resistance to FLT3 inhibitors and the role of the bone marrow microenvironment. Hematol Oncol Clin North Am. 2017;31(4):681-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mead AJ, Neo WH, Barkas N, et al. . Niche-mediated depletion of the normal hematopoietic stem cell reservoir by Flt3-ITD-induced myeloproliferation. J Exp Med. 2017;214(7):2005-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hassanein M, Almahayni MH, Ahmed SO, Gaballa S, El Fakih R. FLT3 inhibitors for treating acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2016;16(10):543-549. [DOI] [PubMed] [Google Scholar]
- 67.Maifrede S, Martinez E, Nieborowska-Skorska M, et al. . MLL-AF9 leukemias are sensitive to PARP1 inhibitors combined with cytotoxic drugs. Blood Adv. 2017;1(19):1467-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nieborowska-Skorska M, Maifrede S, Dasgupta Y, et al. . Ruxolitinib-induced defects in DNA repair cause sensitivity to PARP inhibitors in myeloproliferative neoplasms. Blood. 2017;130(26):2848-2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.